

Abstract
mPTPB is a virulent phosphatase from Mycobacterium tuberculosis and a promising therapeutic target for tuberculosis. To facilitate mPTPB-based drug discovery, we identified α-sulfophenylacetic amide (SPAA) from cefsulodin, a third generation β-lactam cephalosporin antibiotic, as a novel pTyr pharmacophore for mPTPB. Structure-guided and fragment-based optimization of SPAA led to the most potent and selective mPTPB inhibitor 9, with a Ki of 7.9 nM and more than 10,000-fold preference for mPTPB over a large panel of 25 phosphatases. Compound 9 also exhibited excellent cellular activity and specificity in blocking mPTPB function in macrophage. Given its novel structure, modest molecular mass, and extremely high ligand efficiency (0.46), compound 9 represents an outstanding lead compound for anti-TB drug discovery targeting mPTPB.
Keywords: Protein tyrosine phosphatase, pTyr mimetics, mPTPB inhibitor, antituberculosis
Mycobacterium tuberculosis (Mtb) is the causative agent of tuberculosis (TB), a devastating infectious disease affecting one-third of world population and inflicting 2 million deaths every year.1 Although multidrug regimens are available, the emergence of HIV/AIDS has prompted a resurgence of TB.2−4 The major obstacle to the cure and eradication of TB is antibiotic resistance, due primarily to the lack of compliance from patients during the lengthy (>6 months) and complex course of treatment. Multidrug-resistant (MDR) TB now affects over 50 million people, with an increasing number of extensively drug-resistant (XDR) TB, which is extremely difficult to deal with.1,5,6 Traditional antibiotics act by blocking essential processes in Mtb to stop bacterial growth and replication. This approach inevitably results in the selection of drug resistant mutant strains. Given the prevalence of MDR- and XDR-TB and the AIDS epidemic, there is an urgent need for new targets and innovative strategies to speed up the course of treatment and combat drug resistance TB.2−4,7 An emerging and alternative approach is to identify and target pathogenic virulence factors to compromise TB infection process and persistence.2−4
As an intracellular pathogen, Mtb survives and replicates primarily in host macrophages. To evade the antimicrobial functions of the macrophage, Mtb encodes a number of virulence factors including Mtb protein tyrosine phosphatase B (mPTPB), which is secreted by Mtb into the cytoplasm of the macrophage upon infection.8−10 Deletion of mPTPB impairs intracellular survival of Mtb in macrophages and severely reduces the bacterial load in guinea pig models of TB infection.8−10 We recently showed that mPTPB promotes Mtb survival by subverting both host defense and survival pathways.11 The requirement of mPTPB for Mtb survival in macrophages suggests that specific inhibition of mPTPB activity may augment intrinsic host signal pathways to eliminate TB infection. Moreover, targeting the virulent phosphatase is expected to specifically undermine pathogen–host interactions without adverse effects on bacterial growth, therefore exerting less selective pressure for drug resistance. Several small molecule mPTPB inhibitors have been reported and some of them are capable of reversing the altered host immune responses and reducing intracellular mycobacteria in infected macrophages.11−19 Despite this promising progress, the therapeutic benefits of modulating this anti-TB target have not been realized due to the unavailability of more potent, specific, and drug-like mPTPB inhibitors for preclinical evaluation.
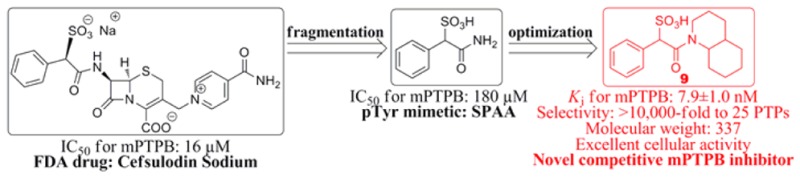
Drug discovery efforts targeting PTPs have historically been hindered by difficulties in overcoming inhibitor selectivity and bioavailability issues. The highly conserved active site or pTyr-binding pocket presents considerable challenge to obtaining compounds that can selectively inhibit the PTP of interest without adversely hitting other family members. Moreover, the positively charged PTP active site, necessary to accommodate pTyr-containing substrates, gives rise to a preponderance of negatively charged molecules in high throughput screening campaigns that suffer from poor cell membrane permeability. Since known drugs already possess acceptable pharmacokinetic properties, we sought to harness the privileged structural space of existing drugs for novel PTP inhibitory scaffolds that can be reconfigured into potent and selective mPTPB inhibitors with improved drug-like properties. To this end, we screened the Johns Hopkins Drug Library20 and identified cefsulodin (Figure 1) as an inhibitor of mPTPB with an IC50 of 16 μM. Interestingly, cefsulodin has also been found to exhibit inhibitory activity against SHP2 and PTP1B.21,22
Figure 1.

Structure and activity of cefsulodin and its fragments.
Cefsulodin is a third generation cephalosporin β-lactam antibiotic with a narrow spectrum restricted to Pseudomonas aeruginosa.23−27 No anti-TB activity has been reported for cefsulodin. Given the chemical reactivity and liability associated with cefsulodin23−27 and its relatively bulky structure, further chemical elaboration of the parent cefsulodin structure may not prove the most efficient strategy to improve its potency and selectivity for mPTPB. Instead, we carried out a fragmentation analysis of cefsulodin in order to identify the minimal mPTPB inhibiting pharmacophore, which can then be elaborated into more potent and selective mPTPB inhibitors with improved drug-like properties. We divided cefsulodin into three major fragments (Figure 1), and chemical synthesis and kinetic characterization of each fragment revealed that α-sulfophenylacetic amide (SPAA) has an IC50 of 180 μM for mPTPB, whereas neither 7-aminocephalosporanic acid (7-ACA) nor isonicotinamide exhibited any measurable inhibition at 200 μM concentration, indicating that SPAA is largely responsible for cefsuodin’s mPTPB inhibitory activity. Detailed kinetic analyses indicated that SPAA is a reversible and competitive inhibitor for mPTPB with a Ki value of 73.4 ± 2.5 μM. The result suggests that SPAA binds mPTPB active site and may function, like for SHP2,21 as a sulfonic acid based nonhydrolyzable pTyr mimetic.
An effective strategy for the acquisition of potent and selective PTP inhibitors is to link appropriately functionalized nonhydrolyzable pTyr mimetics with additional binding elements to engage both the active site and its adjacent unique structural features.28,29 For traditional pTyr mimetics such as phosphonodifluoromethyl phenylalanine and sulfamic acid, structural elaboration is usually introduced opposite to the acid groups on the aromatic ring. In contrast, the location of the carboxamide moiety in SPAA enables sampling of molecular diversity from the same side of the benzene ring where the sulfonate resides. This offers a unique opportunity to capture previously unexplored interactions in the vicinity of the active site.
To guide our effort in developing potent and selective SPAA-based mPTPB inhibitors, we generated a binding model of SPAA with mPTPB using AutoDock4.2.6.30 As shown in Figure 2A, SPAA binds into the active site pocket of mPTPB, with the sulfonate making extensive van der Waals and polar interactions with the P-loop residues (i.e., C160, F161, A162, G163, K164, D165, and R166), and the benzene ring mainly engaged in hydrophobic interactions with Y125, M126, and F133. Compared to its mammalian PTP counterparts, mPTPB has a relatively deeper and broader active site. Thus, there are several unoccupied spaces within the pocket in the presence of SPAA, namely, cavity 1 around the amide nitrogen, cavity 2 near the carbonyl oxygen, and cavity 3 boarding the phenyl ring. Given the well-established tractability of the carboxylic acid-amide chemistry, we focused on cavity 1, which is primarily constituted by hydrophobic residues including F98, L101, L199, I203, L227, V231, and L232 (Figure 2B). We envisioned that an appropriate hydrophobic group attached to the amide nitrogen might capture additional interactions with mPTPB through structural complementarity to cavity 1. Based on this rationale, a panel of amines with diverse hydrophobic groups of varying size and shape including propyl, cyclopropyl, benzene, piperidine, dichloro-ethylbenzene, biphenyl, naphthalene, benzylpiperidine, and decahydroquinoline was selected and attached to the SPAA core. Compounds 1–9 were synthesized by reacting phenyl sulfoacetyl chloride with the corresponding amines in the presence of DIEA and purified by HPLC, and their IC50 values against mPTPB catalyzed hydrolysis of para-nitrophenyl phosphate were determined and shown in Figure 3.
Figure 2.
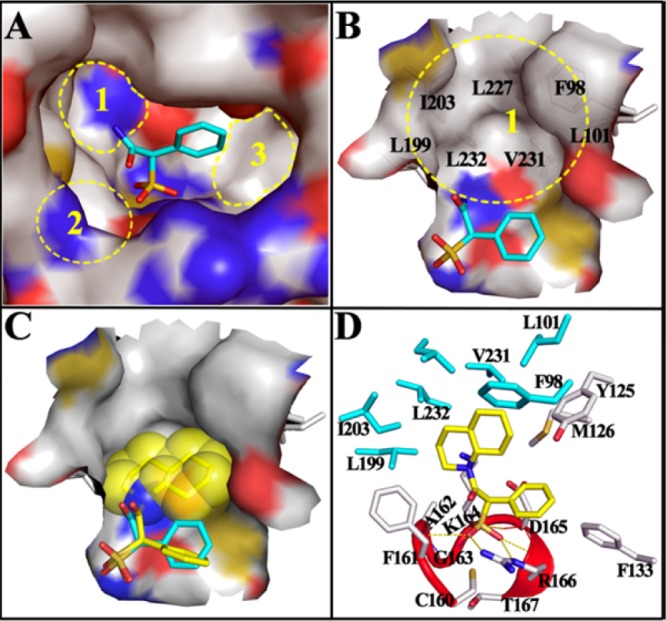
Docking models of SPAA and 9 with mPTPB. (A) SPAA binds into the active site of mPTPB, but there are still multiple free spaces around it. (B) Close-up view of the unoccupied cavity 1, which is mainly constituted by the labeled hydrophobic residues. (C) The binding model of 9 (stick with yellow carbon) with mPTPB. The bound SPAA (stick with cyan carbon) was shown for comparison, and the van der Waals surface of decahydroquinoline was shown in spheres. (D) The interaction details of 9 with mPTPB. The residues within 5 Å distance of 9 are shown in stick, and residues interacting only with 9 but not SPAA are highlighted in cyan. The catalytic P-loop is highlighted in red. H-bonds are indicated by yellow dash lines.
Figure 3.

Structure and activity of mPTPB inhibitors.
In accordance with the modeling prediction that added interaction with cavity 1 should increase mPTPB inhibitor binding affinity, the IC50 values of compounds 1 and 2 were 11- and 32-fold lower than that of SPAA (Figure 3). Replacing simple aliphatic groups with more bulky ring structures further increased the potencies of compounds 3 and 4, which are 180–190-fold better than that of SPAA. Interestingly, adding a terminal aromatic ring to the propyl amine 1 gave compound 5 with an IC50 of 0.29 μM, which represents a 55-fold increase in activity. Likewise, adding a benzene ring to the para-position of aniline in 3 afforded compound 6 with a 5-fold increase in binding affinity, although replacement of the phenyl group with a 1-naphthyl ring only slightly improved potency (compound 7 vs 3). The most striking results were obtained with optimization of compound 4. Compound 8 with an additional benzyl group had an IC50 of 40 nM, a 24-fold improvement over compound 4; and compound 9 with a fused cyclohexane ring had an IC50 of 18 nM, a 53-fold increased affinity over compound 4. Indeed, compounds 8 and 9 are the most potent mPTPB inhibitors reported to date.12−19 Collectively, the data indicate that SPAA-based mPTPB inhibitor binding affinity is strongly influenced by the amine structure and suggests that further increase in potency can be achieved by tailoring the size and shape complementarity of the attached amine to cavity 1.
In order to enable pharmacological validation of mPTPB as a novel anti-TB target, the specificity of mPTPB inhibitor is of utmost importance. With the best potency in class, compound 9 was subjected to specificity profiling for mPTPB versus mPTPA, the only other PTP in the Mtb genome,8 and a large panel of mammalian PTPs, including receptor-like, cytoplasmic, dual specificity phosphatases, low molecular weight PTP, and serine/threonine phosphatase PP5. As shown in Table 1, compound 9 showed no inhibition against the 25 phosphatases tested at 200 μM compound concentration, indicating at least 10,000-fold selectivity for mPTPB. This level of phosphatase inhibitor specificity is unprecedented, to the best of our knowledge. Kinetic analysis revealed that 9 is a reversible and competitive inhibitor of mPTPB with a Ki value of 7.9 ± 1.0 nM (Figure 4), which is in line with the expectation that compound 9 binds the active site of mPTPB.
Table 1. Specificity of 9 for mPTPB over a Panel of PTPs.
| PTP | IC50 |
|---|---|
| mPTPB | 18 ± 2 nM |
| mPTPA, PTP1B, TC-PTP, LYP, HePTP, SHP1, SHP2, PTPH1, Meg2, FAP1, CD45, LAR, PTPα, PTPβ, PTPγ, PTPμ, PTPε, PTPσ, MKP5, VHR, VHX, CDC14A, Laforin, LMWPTP, PP5 | no inhibition at 200 μM |
Figure 4.

Lineweaver–Burk plot for 9 mediated mPTPB inhibition.
To understand the potency and selectivity of compound 9 against mPTPB, we analyzed the most likely binding mode of 9 with mPTPB. As shown in Figure 2C, the SPAA motif in 9 resides in the exact location where the SPAA molecule binds, and the decahydroquinoline tail fills into the hydrophobic cavity 1 and seamlessly complements its topology. In detail (Figure 2D), the SPAA head forms numerous van der Waals interactions with F133 and all P-loop residues (C160 to T167), and the sulfonate makes six H-bonds with the backbone amide of A162, K164, D165, and R166 and the side chain of R166. The decahydroquinoline tail has strong hydrophobic interactions with F98, L101, Y125, M126, L199, I203, V231, and L232, which significantly enhances the binding affinity of compound 9. More importantly, residues comprising cavity 1 are completely unique to mPTPB, which likely contributes to the unprecedented specificity of compound 9 for mPTPB.
Given the excellent potency and specificity, we proceeded to evaluate compound 9’s cellular efficacy. We had previously shown that once inside the macrophage, mPTPB activates Akt signaling and simultaneously blocks ERK1/2 and p38 activation to prevent macrophage apoptosis and cytokine production.11 Thus, inhibition of mPTPB should counteract the effects of mPTPB on ERK1/2, p38, and Akt signaling inside the cell. As expected, compound 9 increased the phosphorylation of ERK1/2 and p38, but reduced Akt phosphorylation in mPTPB transfected Raw264.7 cells in a dose dependent manner (Figure 5). In contrast, it has no appreciable effect on the phosphorylation of ERK1/2, p38, and Akt in vector transfected Raw264.7 cells. This result coupled with the fact that compound 9 phenocopied several structurally unrelated mPTPB inhibitors11,17,18 in cell-based assays strongly indicate that the observed cellular effects of 9 in mPTPB transfected cells are indeed from specific inhibition of mPTPB. Of particular note is the observation that compound 9’s cellular activity tracked very well with its IC50 value measured with isolated enzyme. Collectively, these results indicate that compound 9 is highly efficacious in blocking mPTPB activity inside the cell.
Figure 5.

Cellular activity of 9 in blocking mPTPB-mediated signaling.
In summary, we found from an FDA drug collection cefsulodin as an mPTPB inhibitor with moderate affinity. Fragmentation analysis of cefsulodin identified SPAA as the mPTPB inhibiting pharmacophore and a novel pTyr mimetic. Structure-guided and fragment-based optimization of SPAA led to the discovery of a novel class of highly potent inhibitors for mPTPB. Among them, compound 9 has a Ki of 7.9 nM and with greater than 10,000-fold preference over 25 PTPs. It also exhibits excellent cellular activity and specificity in blocking mPTPB’s function in the macrophage. The results highlight the feasibility of developing highly potent and selective PTP inhibitors by exploiting specific structural features of the active sites of different PTPs for efficient ligand binding interaction. In addition, the disclosed mPTPB inhibitors also possess highly compact structures with molecular weight in the range of 300–400 and very favorable drug-like properties.31,32 Given its novel structure, modest molecular weight, and extremely high ligand efficiency (0.46),32 compound 9 represents an excellent lead compound for anti-TB drug discovery targeting mPTPB. We note that the synthesized SPAA compounds are racemic, whereas cefsulodin exists as a specific enantiomer. Future studies will evaluate the dependence of SPAA chirality on PTP inhibition activity. Finally, because of the generality of the described approach,21 SPAA could serve as a good starting point for the development of small molecular inhibitors targeting other PTPs.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00373.
Detailed experiment procedures and compound characterizations (PDF)
This work was supported in part by National Institutes of Health Grants RO1CA152194 and RO1CA69202.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO Report 2010 on Global TB Control, 2010.
- Fox W.; Mitchison D. A. Short-course chemotherapy for tuberculosis. Lancet 1976, 2, 1349–1350. 10.1016/S0140-6736(76)91989-9. [DOI] [PubMed] [Google Scholar]
- Neff M. ATS, CDC, and IDSA update recommendations on the treatment of tuberculosis. Am. Fam. Phys. 2003, 68, 1857–1858. [PubMed] [Google Scholar]
- Zhang Y. The magic bullets and tuberculosis drug targets. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 529–564. 10.1146/annurev.pharmtox.45.120403.100120. [DOI] [PubMed] [Google Scholar]
- Harries A. D.; Zachariah R.; Corbett E. L.; Lawn S. D.; Santos-Filho E. T.; Chimzizi R.; Harrington M.; Maher D.; Williams B. G.; Cock K. M. De. The HIV-associated tuberculosis epidemic--when will we act?. Lancet 2010, 375, 1906–1919. 10.1016/S0140-6736(10)60409-6. [DOI] [PubMed] [Google Scholar]
- Gandhi N. R.; Nunn P.; Dheda K.; Schaaf H. S.; Zignol M.; van Soolingen D.; Jensen P.; Bayona J. Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. Lancet 2010, 375, 1830–1843. 10.1016/S0140-6736(10)60410-2. [DOI] [PubMed] [Google Scholar]
- Marais B. J.; Raviglione M. C.; Donald P. R.; Harries A. D.; Kritski A. L.; Graham S. M.; El-Sadr W. M.; Harrington M.; Churchyard G.; Mwaba P.; Sanne I.; Kaufmann S. H. E.; Whitty C. J. M.; Atun R.; Zumla A. Lancet 2010, 375, 2179–2191. 10.1016/S0140-6736(10)60554-5. [DOI] [PubMed] [Google Scholar]
- Koul A.; Choidas A.; Treder M.; Tyagi A. K.; Drlica K.; Singh Y.; Ullrich A. Cloning and characterization of secretory tyrosine phosphatases of Mycobacterium tuberculosis. J. Bacteriol. 2000, 182, 5425–5432. 10.1128/JB.182.19.5425-5432.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.; Rao V.; Shakila H.; Gupta R.; Khera A.; Dhar N.; Singh A.; Koul A.; Singh Y.; Naseema M.; Narayanan P. R.; Paramasivan C. N.; Ramanathan V. D.; Tyagi A. K. Disruption of mptpB impairs the ability of Mycobacterium tuberculosis to survive in guinea pigs. Mol. Microbiol. 2003, 50, 751–762. 10.1046/j.1365-2958.2003.03712.x. [DOI] [PubMed] [Google Scholar]
- Koul A.; Herget T.; Klebl B.; Ullrich A. Interplay between mycobacteria and host signalling pathways. Nat. Rev. Microbiol. 2004, 2, 189–202. 10.1038/nrmicro840. [DOI] [PubMed] [Google Scholar]
- Zhou B.; He Y.; Zhang X.; Xu J.; Luo Y.; Wang Y.; Franzblau S. G.; Yang Z.; Chan R. J.; Liu Y.; Zheng J.; Zhang Z.-Y. Targeting mycobacterium protein tyrosine phosphatase B for antituberculosis agents. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 4573–4578. 10.1073/pnas.0909133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren-Muller A.; Reis-Correa I. Jr.; Prinz H.; Rosenbaum C.; Saxena K.; Schwalbe H.; Vestweber D.; Cagna G.; Schunk S.; Schwarz O.; Schiewe H.; Waldmann H. Discovery of protein phosphatase inhibitor classes by biology-oriented synthesis. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 10606–10611. 10.1073/pnas.0601490103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beresford N. J.; Mulhearn D.; Szczepankiewicz B.; Liu G.; Johnson M. E.; Fordham-Skelton A.; Abad-Zapatero C.; Cavet J. S.; Tabernero L. Inhibition of MptpB phosphatase from Mycobacterium tuberculosis impairs mycobacterial survival in macrophages. J. Antimicrob. Chemother. 2009, 63, 928–936. 10.1093/jac/dkp031. [DOI] [PubMed] [Google Scholar]
- Noren-Muller A.; Wilk W.; Saxena K.; Schwalbe H.; Kaiser M.; Waldmann H. Discovery of a new class of inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatase B by biology-oriented synthesis. Angew. Chem., Int. Ed. 2008, 47, 5973–5977. 10.1002/anie.200801566. [DOI] [PubMed] [Google Scholar]
- Soellner M. B.; Rawls K. A.; Grundner C.; Alber T.; Ellman J. A. Fragment-based substrate activity screening method for the identification of potent inhibitors of the Mycobacterium tuberculosis phosphatase PtpB. J. Am. Chem. Soc. 2007, 129, 9613–9615. 10.1021/ja0727520. [DOI] [PubMed] [Google Scholar]
- Tan L. P.; Wu H.; Yang P.-Y.; Kalesh K. A.; Zhang X.; Hu M.; Srinivasan R.; Yao S. Q. High-throughput discovery of Mycobacterium tuberculosis protein tyrosine phosphatase B (MptpB) inhibitors using click chemistry. Org. Lett. 2009, 11, 5102–5105. 10.1021/ol9023419. [DOI] [PubMed] [Google Scholar]
- Chen l.; Zhou B.; Zhang S.; Wu L.; Wang Y.; Franzblau S. G.; Zhang Z.-Y. Identification and characterization of novel inhibitors of mPTPB, an essential virulent phosphatase from Mycobacterium tuberculosis. ACS Med. Chem. Lett. 2010, 1, 355–359. 10.1021/ml1001135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Xu J.; Yu Z.-H.; Gunawan A. M.; Wu L.; Wang L.; Zhang Z.-Y. Discovery and evaluation of novel inhibitors of mycobacterium protein tyrosine phosphatase B from the 6-Hydroxy-benzofuran-5-carboxylic acid scaffold. J. Med. Chem. 2013, 56, 832–842. 10.1021/jm301781p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L.-F.; Xu J.; He Y.; Wu L.; Gynawan A. M.; Zhang Z.-Y. A facile hydroxyindole carboxylic acid based focused library approach for potent and selective inhibitors of Mycobacterium protein tyrosine phosphatase B. ChemMedChem 2013, 8, 904–908. 10.1002/cmdc.201300115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong C. R.; Chen X.; Shi L.; Liu J. O.; Sullivan D. J. A clinical drug library screen identifies astemizole as an antimalarial agent. Nat. Chem. Biol. 2006, 2, 415–416. 10.1038/nchembio806. [DOI] [PubMed] [Google Scholar]
- He R.; Yu Z.-H.; Zhang R.-Y.; Wu L.; Gunawan A. M.; Lane B. S.; Shim J. S.; Zeng L.-F.; He Y.; Chen L.; Wells C. D.; Liu J. O.; Zhang Z.-Y. Exploring the Existing Drug Space for Novel pTyr Mimetic and SHP2 Inhibitors. ACS Med. Chem. Lett. 2015, 6, 782–786. 10.1021/acsmedchemlett.5b00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller O. J.; El Harrak A.; Maggeat T.; Baret J. C.; Frenz L.; El Debs B.; Mayot E.; Samuels M. L.; Rooney E. K.; Dieu P.; Galvan M.; Link D. R.; Griffiths A. D. High-resolution dose-response screening using droplet-based microfluidics. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 378–383. 10.1073/pnas.1113324109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd D. B.; Lunn W. H. W. Electronic structures of cephalosporins and penicillins. 9. Departure of a leaving group in cephalosporins. J. Med. Chem. 1979, 22, 778–784. 10.1021/jm00193a006. [DOI] [PubMed] [Google Scholar]
- Itakura K.; Aoki I.; Kasahara F.; Nishikawa M.; Mizushima Y. Structures of polymers formed in aqueous solutions of cefsulodin sodium. Chem. Pharm. Bull. 1981, 29, 1655–1661. 10.1248/cpb.29.1655. [DOI] [Google Scholar]
- Coene B.; Schanck A.; Dereppe J.-M.; VanMeerssche M. Substituent effects on reactivity and spectral parameters of cephalosporins. J. Med. Chem. 1984, 27, 694–700. 10.1021/jm00371a025. [DOI] [PubMed] [Google Scholar]
- Faraci W. S.; Pratt R. F. Elimination of a good leaving group from the 3′-position of a cephalosporin need not be concerted with.beta.-lactam ring opening: TEM-2.beta.-lactamase-catalyzed hydrolysis of pyridine-2-azo-4′-(N′,N′-dimethylaniline) cephalosporin (PADAC) and of cephaloridine. J. Am. Chem. Soc. 1984, 106, 1489–1490. 10.1021/ja00317a053. [DOI] [Google Scholar]
- Sugioka T.; Asano T.; Chikaraishi Y.; Suzuki E.; Sano A.; Kuriki T.; Shirotsuka M.; Saito K. Stability and degradation pattern of cefpirome (HR 810) in aqueous solution. Chem. Pharm. Bull. 1990, 38, 1998–2002. 10.1248/cpb.38.1998. [DOI] [PubMed] [Google Scholar]
- He R.; Zeng L.-F.; He Y.; Zhang S.; Zhang Z.-Y. Small molecule tools for functional interrogation of protein tyrosine phosphatases. FEBS J. 2013, 280, 731–750. 10.1111/j.1742-4658.2012.08718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialy L.; Waldmann H. Inhibitors of protein tyrosine phosphatases: next-generation drugs?. Angew. Chem., Int. Ed. 2005, 44, 3814–3839. 10.1002/anie.200461517. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings J. Adv. Drug Delivery Rev. 1997, 23, 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Schultes S.; de Graaf C.; Haaksma E. E. J.; de Esch I. J. P.; Leurs R.; Kramer O. Ligand efficiency as a guide in fragment hit selection and optimization. Drug Discovery Today: Technol. 2010, 7, e157–e162. 10.1016/j.ddtec.2010.11.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.