

Abstract
This Forum Article focuses on recent advances in structural and spectroscopic studies of biosynthetic models of nitric oxide reductases (NORs). NORs are complex metalloenzymes found in the denitrification pathway of Earth’s nitrogen cycle where they catalyze the proton-dependent two-electron reduction of nitric oxide (NO) to nitrous oxide (N2O). While much progress has been made in biochemical and biophysical studies of native NORs and their variants, a clear mechanistic understanding of this important metalloenzyme related to its function is still elusive. We report herein UV–vis and nuclear resonance vibrational spectroscopy (NRVS) studies of mononitrosylated intermediates of the NOR reaction of a biosynthetic model. The ability to selectively substitute metals at either heme or nonheme metal sites allows the introduction of independent 57Fe probe atoms at either site, as well as allowing the preparation of analogues of stable reaction intermediates by replacing either metal with a redox inactive metal. Together with previous structural and spectroscopic results, we summarize insights gained from studying these biosynthetic models toward understanding structural features responsible for the NOR activity and its mechanism. The outlook on NOR modeling is also discussed, with an emphasis on the design of models capable of catalytic turnovers designed based on close mimics of the secondary coordination sphere of native NORs.
Short abstract
New insights into nitric oxide reductases (NORs) are obtained. Using nuclear resonance vibrational spectroscopy, we probe both iron atoms in mononitrosylated intermediates of the NOR reaction in a biosynthetic protein model that reveal new insights into the structural and electronic features responsible for the NOR activity and its likely mechanism.
Introduction
Denitrification is an important process in biology that involves the sequential reduction of nitrate (NO3–) to nitrite (NO2–), nitric oxide (NO), nitrous oxide (N2O), and finally to dinitrogen (N2), carried out by several different metalloenzymes.1,2 Reduction of NO to N2O (2NO + 2e– + 2H+ → N2O + H2O) is a key step of this process and is catalyzed by nitric oxide reductases (NORs).3 NO is an important molecule in biology because it impacts events ranging from blood pressure regulation,4 neurotransmission,5 and immune response6 in mammalian cells to transcriptional regulation7 and biofilm formation in bacteria.8,9 The presence of NORs in pathogenic bacteria such as Pseudomonas aeruginosa helps to detoxify NO and allow the bacteria to survive.10 Furthermore, an increase in N2O production caused by the use of artificial fertilizers generated from artificial nitrogen fixation has disrupted the global nitrogen cycle, as well as highlighted N2O’s potent ability to deplete ozone.2,11 Despite the biochemical, biomedical, and environmental significance of NORs, structural features responsible for its activity and a clear mechanistic understanding of its reaction, particularly the membrane-bound NORs from bacteria, are not well understood.12
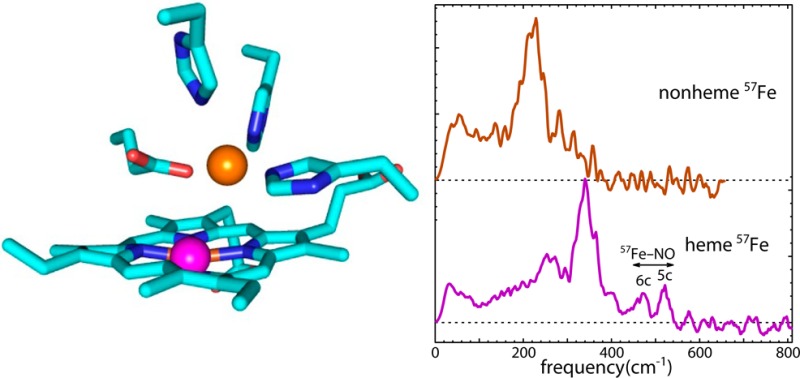
Bacterial NOR is a complex enzyme consisting of a c-type heme, a heme b, and a heme b3/nonheme iron (FeB) center. Electrons are delivered from heme c to heme b and then to the heme b3/FeB active site, where NO is reduced to N2O (Figure 1).13 The active site of NOR consists of a high-spin (HS) heme b3 and FeB coordinated by three histidine and one glutamate residues (Figure 1).14 Three mechanisms of NO reduction by NORs have been proposed (Scheme 1).15 Briefly, the trans mechanism suggests that both heme b3 and FeB sites bind NO, one each, before N–N bond formation, while in the cis heme b3 mechanism, a second NO electrophilically attacks a heme-bound NO. In the cis FeB mechanism, both molecules of NO bind to the FeB site.
Figure 1.

Representation of the electron-transfer pathway from cNOR (left) and the active site structure including FeB (orange sphere) PDB 3O0R (right).
Scheme 1. Proposed Mechanisms for the Reduction of NO by NORs.

Enzymatic and mechanistic studies of native bacterial NORs16−20 are complicated by the presence of several metal sites (three hemes on a nonheme iron; see Figure 1), which makes spectroscopic studies difficult, as well as difficulties in purifying the protein in high yield and homogeneity because NORs are membrane proteins.21 Synthetic models of NOR have been used to complement the study of the native enzyme to great success.1,12,22−30 The recent progress made in synthetic modeling has been summarized by other Articles in this special Forum and will not be duplicated here.
Biosynthetic Modeling Approach
To complement both native enzyme and synthetic modeling approaches, we have used small, stable, easy-to-purify, and well-characterized proteins such as myoglobin (Mb) as “scaffolds” to make biosynthetic models of more complex metalloenzymes. While a great deal of effort has been put forth to understand both the structure and function of native enzymes and their variants using biochemical and biophysical methods,31−33 an ultimate test of our knowledge of this class of enzymes is creating functional models that mimic both the structure and function of native enzymes.34 In contrast to studying native proteins in a “top-down” approach, which can identify necessary structural features responsible for function,35,36 biosynthetic modeling is a “bottom-up” approach that elucidates structural features sufficient for activity. Furthermore, the biosynthetic models may be amenable to investigation in ways that have not yet been developed for the native enzymes (e.g., replacement of the heme in the active site with a non-native cofactor, such as zinc(II) protoporphyrin IX (PPIX)).37 On the other hand, thanks to recent progress in molecular biology and protein biochemistry, protein models can now be more readily prepared than by the chemical synthesis of models of complex metalloenzymes such as NOR because the latter method requires rigorous synthetic skills. For example, it normally takes about 1 week to construct, express, and purify protein models with a yield of ∼100 mg/L of Escherichia coli culture; it would take much longer to prepare heme FeB models chemically with lower yield because the synthesis of porphyrin-containing models is quite challenging and requires multistep synthesis. Despite challenges associated with preparing synthetic analogues of complex enzymes, remarkable progress has been made to understand the structure/function of complex enzymes using synthetic models.1,22,24,26−28,30 Furthermore, it is becoming clear that noncovalent, secondary coordination sphere interactions around the primary coordination sphere, such as hydrophobicity and hydrogen-bonding interactions, often involving structurally well-defined water, can play a key role in enzymatic function.38 Addressing these issues in modeling requires a rigid framework that allows the introduction of these elements at specific locations. Biosynthetic modeling is an ideal choice in addressing this issue because such secondary coordination sphere interactions can be conveniently introduced at a specific location of the rigid protein scaffold, without elaborate synthesis of model compounds that may be more flexible. Therefore, although biosynthetically designed protein models are intermediate in complexity compared to the complex native proteins and synthetic analogues, they contain many features of both the proteins and small-molecule models, providing us with unique constructs to understand the structure, function, and mechanism of complex metalloenzymes.
Nomenclature
In this article we will use the following nomenclature convention to designate various biosynthetic models and their corresponding metallated and nitrosylated derivatives. FeBMb1 and FeBMb2 represents the first and second generations of biosynthetic model proteins, respectively. The corresponding metallated derivatives are represented as MII-FeBMb1(FeII) where the MII represents a metal ion with the designated oxidation state (II) occupies the nonheme FeB center and the FeII represents FeII-protoporphyrin IX (heme) in the heme-binding site. When the FeB center is empty, it will be represented as E-FeBMb1(FeII). When the FeII-protoporphyrin IX (heme) is replaced with ZnII-protoporphyrin IX, it will be designated as MII-FeBMb1(ZnII). Based on this convention, 57FeII-FeBMb1(FeII) and FeII-FeBMb1(57FeII) indicate 57FeII in the nonheme FeB center and heme center, respectively. The corresponding nitrosyl derivatives, 57FeIINO-FeBMb1(ZnII) and FeII-FeBMb1(57FeIINO), indicate NO binding to the nonheme 57FeB center and heme 57Fe center, respectively.
Biosynthetic Models of NORs
In order to complement studies of native NORs and its synthetic models, our group utilizes small, easy-to-purify, and well-characterized proteins like Mb as the scaffold to prepare biosynthetic models of NORs.39 This endeavor was built upon our initial success in using Mb to prepare structural and functional models of heme copper oxidases (HCOs) by introducing a CuB center in the distal pocket of sperm whale myoglobin (swMb) through L29H/F43H mutations (called CuBMb).40−43 Because NORs and HCOs belong to the same superfamily with similar overall structural folds, and some HCOs have been shown to exhibit NOR activity,44−46 we first investigated the cross reactivity of this HCO mimic CuBMb to reduce NO and found that the presence of CuI at the CuB site in CuBMb indeed displayed NOR activity with consumption of 2 mol of NO/mol of CuBMb/min, similar to that of HCO from Thermus thermophilus (3 mol of NO/mol of HCO/min).20,45−47
Encouraged by the above success, we turned our attention to mimic both the structure and function of native NORs. At the time of our pursuit, there was no crystal structure of NOR available to guide the rational design of a NOR model using Mb. However, biochemical studies and sequence homology analysis have indicated that, in addition to the presence of FeII in the FeB center (instead of CuI in the CuB center), NORs contain at least two conserved Glu residues in the active site that are absent in the HCOs.14,48 Because CuBMb did not bind FeII and thus did not show any NOR activity in the absence of a metal ion in the nonheme metal center, we decided to introduce a Glu to the CuBMb. After evaluation of several positions to introduce the Glu through computer modeling and energy minimization, we found the best candidate to be V68E, called E-FeBMb1(FeII) (L29H/F43H/V68E swMb). This protein binds FeII readily (Figure 2a) and the metal bound FeII-FeBMb1(FeII) displays NOR activity, making it the first structural and functional model of NOR.39
Figure 2.

Overlays of cNOR (yellow) with (a) FeII-FeBMb1(FeII) (cyan), (b) FeII-FeBMb2(FeII) (green), and (c) FeII-FeBMb1(ZnII) (magenta). FeB sites are shown as brown spheres and amino acid residues as sticks, and the water molecule involved in hydrogen bonding is shown as a cyan sphere in part b.
Because there are at least two conserved Glu in the active site of NORs, we decided to investigate the role of the second Glu in the biosynthetic models. While there is no room to introduce the second Glu in the primary coordination sphere of the FeB center, we evaluated introducing the second Glu in the secondary coordination sphere of the FeB center. We found I107E to be at an ideal location to provide an extended hydrogen-bonding network around the FeB center; thus, a “second-generation” model (I107E-FeBMb1(FeII), called E-FeBMb2(FeII) was prepared, which also bound Fe(II) in the FeB site and the metallated derivative FeII-FeBMb2(FeII) improved the NOR activity over FeII-FeBMb1(FeII) by nearly 100%.
Both FeII-FeBMb1(FeII) and FeII-FeBMb2(FeII) were prepared before the publication of the first crystal structure of cytochrome c dependent NOR (cNOR).14 After the crystal structure of cNOR became available, we overlaid the crystal structures of FeII-FeBMb1(FeII) and FeII-FeBMb2(FeII) with that of native NOR (Figures 2a,b) and were pleased to see that, in addition to displaying NOR activity, both biosynthetic models mimic native cNOR structurally.39 This accomplishment, achieved through computational modeling guided by homology modeling with structurally related proteins and by activity that mimics those of native enzymes, demonstrated the immense potential of biosynthetic approach in making close structural and functional models of native enzymes.
Early Studies of the Biosynthetic Models
Spectroscopic studies of FeII-FeBMb1(FeII) and FeII-FeBMb2(FeII) using Fourier transform infrared (FTIR) and resonance Raman (rR) have shown that heme-bound NO adopts a strong nitroxyl character through interactions with the nonheme iron,49 and time-resolved rapid-mixing experiments provided evidence for both heme and nonheme nitrosyl complexes, supporting the trans mechanism.50 Additionally, electron paramagnetic resonance (EPR) studies of FeII-FeBMb2(FeII) reacted with excess NO showed the formation of a five-coordinate low-spin (5cLS) ferrous heme species due to cleavage of the proximal histidine bond.51 EPR measurements taken below 30 K of FeII-FeBMb1(FeII) and FeII-FeBMb2(FeII) upon the addition of 1 equiv of NO show signals at g = 6.1, which likely arise from exchange coupling of an S = 1/2 6cLS {FeNO}7 heme and S = 2 nonheme FeII.49
Heme nitrosyl species have been spectroscopically probed,52−55 but a more complete understanding of the nonheme nitrosyl was limited to only a few studies12 because the spectroscopic signals of the heme often dominate the spectra of NORs over the nonheme iron center, hampering our understanding of the role of nonheme iron in NOR reactivity. Recently, we have replaced the native heme (iron protoporphyrin IX, FePPIX) in FeII-FeBMb1(FeII) with ZnPPIX, which also bound Fe(II), and the FeII-FeBMb1(ZnII) derivative allowed for a thorough spectroscopic and computational investigation into the FeB nitrosyl complex selectively, without interference from the heme nitrosyl. By using UV–vis absorbance, EPR, and Mössbauer spectroscopies, as well as X-ray crystallography (Figure 2c) and density functional theory (DFT) calculations, the nonheme nitrosyl was characterized as an S = 3/2 {FeNO}7 complex, using the Enemark–Feltham notation,56 best described as a HS ferrous iron antiferromagnetically coupled to an NO radical.37
New Results To Provide Deeper Insights
To further probe the interaction of our NOR model with NO, we report here UV–vis and nuclear resonance vibrational spectroscopy (NRVS) measurements. Using this information, we can systematically characterize the intermediates illustrated in Scheme 1 because they are likely to have distinctly different spectroscopic signatures. NRVS gives a complete and quantitative vibrational frequency spectrum for 57Fe-enriched nuclei. It offers a selectivity similar to that of rR spectroscopy but is not bound by the optical selection rules of rR or IR spectroscopy.57 This is especially important in the study of NORs, given that the Fe–NO stretching mode vibrations are IR-silent and decompose upon laser irradiation.12 Unfortunately, performing NRVS requires extremely concentrated protein samples (>5 mM) that are impractical when working with native enzymes such as NOR. However, studies have already been performed on Mb and its mutants,58,59 thus offering our NOR mimics a unique opportunity to utilize this advanced spectroscopic technique that would otherwise be inaccessible to native proteins.
UV–Vis Spectroscopy
UV–vis spectroscopy was used to monitor NO binding to FeII-FeBMb1(57FeII) and ZnII-FeBMb1(57FeII) during NRVS sample preparation. Representative spectra are shown in Figure 3. Upon the addition of 1 equiv of NO to FeII-FeBMb1(57FeII), where the nonheme site was reconstituted with FeII, the Soret peak at 433 nm (Figure 3, black curve) underwent a blue shift to 419 nm (Figure 3, red curve), corresponding to the formation of a 6cLS {FeNO}7 species and another broad peak at 398 nm corresponding to a 5cLS {FeNO}7 species (Figure 3, red curve and inset), and this assignment is confirmed by NRVS results (vide infra). The addition of 1 equiv of NO to the ZnII-FeBMb1(57FeII) sample also caused a blue shift of the Soret peak from 434 nm (Figure 3, cyan curve) to 403 nm (Figure 3, purple curve), corresponding to the formation of a 5cLS {FeNO}7 species.
Figure 3.
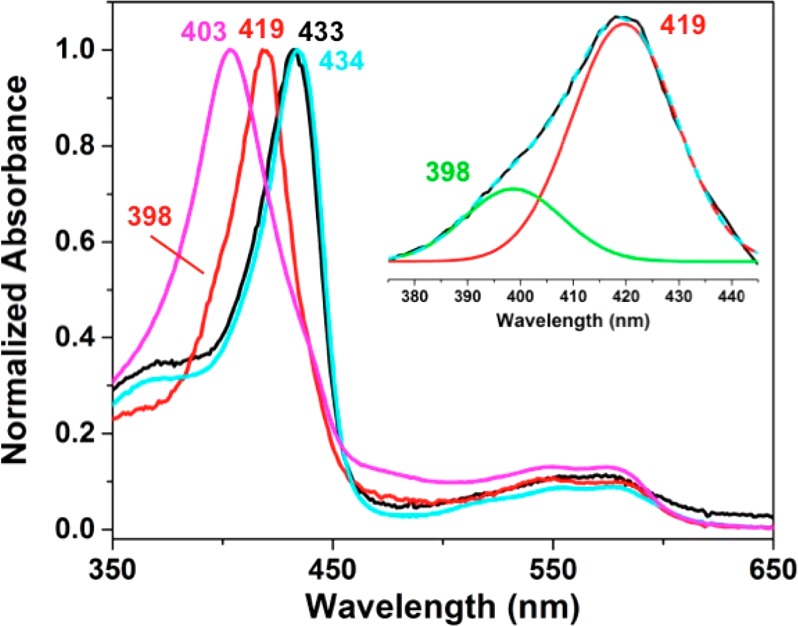
UV–vis spectra of FeII-FeBMb1(57FeII) (black curve), ZnII-FeBMb1(57FeII) (cyan curve), and the corresponding mononitrosyl derivatives FeII-FeBMb1(57FeIINO) (red curve) and ZnII-FeBMb1(57FeIINO) (purple curve). The inset shows deconvolution of the Soret region of FeII-FeBMb1(57FeIINO) demonstrating two components: one peak at 419 nm corresponding to the 6cLS {FeNO}7 species and a second broader peak at 398 nm corresponding to the 5cLS {FeNO}7 species. The presence of both 6cLS and 5cLS {FeNO}7 species is consistent with the NRVS results (vide infra). In the case of the ZnII-FeBMb1(57FeIINO) sample, the presence of the Soret peak at 403 nm is also consistent with the presence of 5cLS {FeNO}7 also observed by NRVS (vide infra).
NRVS
Site-Selective Probe of Individual Iron Atoms in Complex Systems
NRVS exploits technology developed at third-generation synchrotron light sources to monitor the vibrational properties of Mössbauer nuclei, including 57Fe.60 Tuning of a monochromatic X-ray beam in the vicinity of the nuclear resonance reveals vibrational sidebands displaced from the recoilless resonance observed in conventional Mössbauer spectroscopy. A growing number of NRVS applications exploit its exclusive and quantitative sensitivity to vibrational motions of the probe nucleus.61,62 Specifically, each vibrational mode contributes to the measured signal in direct proportion to the mean-squared displacement of the probe nucleus along the beam direction, and well-established data analysis methods63 directly extract a partial vibrational density of states (VDOS) for that measurement.
For a randomly oriented ensemble of molecules containing 57Fe, each vibrational normal mode contributes to the 57Fe VDOS an area equal to the fraction eFe2 of the mode’s kinetic energy associated with motion of the 57Fe nucleus. The information content of the VDOS is quantitative, allowing direct comparison with vibrational predictions on an absolute scale.64,65 The VDOS is comprehensive because all vibrations involving 57Fe contribute, without the artificial restrictions imposed by selection rules in more familiar vibrational spectroscopies (IR and Raman). Finally, the VDOS is uniquely site-selective because only motion of the 57Fe will contribute, even in a macromolecule containing thousands of other atoms.
On the basis of these characteristics, NRVS is a uniquely valuable probe of protein active sites containing iron. Several investigations have reported the vibrational dynamics of iron in heme proteins58,66,67 and iron porphyrins65,68−70 using NRVS. NRVS measurements on oriented single crystals of iron porphyrins exploit the sensitivity to motion along the direction of the X-ray beam to provide additional insights into the interpretation of the results on heme proteins.71 Proteins with nonheme iron sites are equally amenable to NRVS investigations,72,73 which have informed the structural characterization of reaction intermediates in nonheme iron enzymes.74
Vibrational spectra resulting from previous NRVS measurements on proteins containing multiple iron atoms contain superposed contributions from all sites.75
NRVS Independently Quantifies Forces Acting on Heme and Nonheme Iron in FeII-FeBMb1(FeII)
Here, we demonstrate for the first time that we can specifically label either the heme or nonheme iron sites of FeII-FeBMb1(FeII) with 57Fe, allowing us to independently monitor vibrations of iron at each site. Partial unfolding of the protein at low pH allows removal of the heme and reconstitution of the protein with 57Fe-enriched PPIX, following the same procedure as that used for previous NRVS investigations on native Mb.76 Heme vibrations will dominate the NRVS signal of the reconstituted protein, even after the incorporation of natural abundance iron (or another metal) into the nonheme site because the natural abundance of 57Fe is only 2%. Similarly, the incorporation of 57Fe into the nonheme site of E-FeBMb1(FeII) reconstituted with natural abundance heme should allow us to specifically monitor the vibrations of the nonheme iron.
The VDOS determined from NRVS measurements on reduced FeII-FeBMb1(FeII) (Figure 4) demonstrate that specific labeling of the heme and nonheme iron sites with 57Fe allows us to distinguish vibrations at distinct sites within the same protein. The vibrational signal from the heme iron strongly resembles that reported for native Mb.58 The dominant feature of the VDOS includes contributions from vibrations of the axial Fe–NHis bond to His 93 and of the equatorial Fe–Npyr bonds to the four heme pyrrole nitrogen atoms at approximately 230 and 250 cm–1, respectively. The Fe–NHis frequency is well-known from rR measurements on heme proteins,77−79 where this vibration is strongly enhanced upon excitation into the Soret band. The NRVS signal is determined by the relative amplitude of iron motion and also includes the Fe–Npyr vibrations, which are not easily observable using other spectroscopies.
Figure 4.

Site-selective enrichment of FeII-FeBMb1(FeII) with 57Fe allows independent monitoring of iron vibrations at either the heme or nonheme site (57FeII-FeBMb1(FeII)). The upper and lower traces present the partial VDOS of the heme and nonheme iron of the reduced proteins, respectively, derived from such measurements and reflect the distinct coordination of iron at each site. The heme VDOS is nearly identical with that reported58 for reduced native Mb from horse heart, where contributions from the Fe–NHis vibration perpendicular to the heme and vibrations of the in-plane Fe–Npyr bonds to the heme pyrrole nitrogen atoms were identified.58 Although individual vibrations are not resolved for the less symmetric nonheme site, the stiffness derived from the VDOS (Table 1), nevertheless, reflects the lower coordination of iron in this environment. Here and in subsequent figures, error bars reflect experimental uncertainties determined from counting statistics, while solid traces represent a five-point running average of the experimental VDOS.
The nonheme iron of reduced 57FeII-FeBMb1(FeII) displays a clearly distinct vibrational signal dominated by a broad feature with a peak near 230 cm–1. The crystallographic model includes His 29, His 43, His 64, Glu 68, and a water molecule as ligands to the nonheme iron.39 The relatively featureless signal observed for the nonheme iron apparently masks the vibrational structure that one might expect in light of this diverse ligand field, and the reduced symmetry in comparison with the heme site may further increase the vibrational complexity. We identified multiple unresolved vibrational modes contributing to a similar broad vibrational feature in reduced cytochrome c, based on a quantitative comparison with 54Fe/57Fe isotope shifts observed in rR measurements, and attributed this complexity to the reduced symmetry of iron coordination in the distorted heme.81 Conformational heterogeneity is well-documented for native Mb and may also broaden vibrational features. Regardless of the reasons, the unresolved vibrational complexity may hinder the identification of well-defined iron-ligand vibrations.
Fortunately, the vibrational information revealed by the NRVS measurement yields a quantitative measure of the coordination strength even in the absence of detailed vibrational frequency assignments. The VDOS D(ν̅) determines the stiffness,
| 1 |
an effective force constant that directly measures the force required to displace the iron with its coordination environment held fixed.82 The stiffness for both sites is much lower than that for the LS heme iron in reduced cytochrome c,81 where the stiffness was more than 300 pN/pm. The stiffness is consistent with the presence of a HS iron at both sites in reduced FeII-FeBMb1(57FeII) and 57FeII-FeBMb1(FeII). This contrasts with evidence for a LS heme reported for cNOR from Ps. Nautica.83
Within experimental uncertainty, the stiffness of the heme iron in reduced FeII-FeBMb1(57FeII) is the same as that in native Mb (Table 1), confirming the expectation that the introduction of the nonheme metal site does not significantly affect the coordination strength of the heme iron. However, the stiffness of the nonheme iron environment is significantly lower than that determined for the heme iron. The slightly lower force restraining the iron in the nonheme site presumably reflects its reduced coordination.
Table 1. Averaged Experimental Force Constants of Ferrous Iron Sites in Proteins.
Similar conclusions follow from data recorded on oxidized FeIII-FeBMb1(57FeIII) (Figure 5). A feature near 270 cm–1 dominates the heme iron VDOS, which strongly resembles that previously reported for native swMb.80 Because iron ligand vibrations are undoubtedly the primary contribution to this feature, this indicates that the ligation of the heme iron, to His 93 and to a neutral water molecule, is the same in FeII-FeBMb1(57FeII) as it is in native Mb. In particular, these data provide no indication for an oxo group bridging the two iron sites, as observed14 for the oxidized state of NOR (Scheme 1), and the HS NRVS signal from the heme iron contrasts with the LS heme iron reported for cNOR from Ps. Nautica.83
Figure 5.

The iron VDOS of oxidized FeIII-FeBMb1(FeIII), shown in the upper trace, strongly resembles that reported80 for native Mb from sperm whale, indicating that coordination of the heme iron is unaffected by the presence of the additional nonheme iron engineered in the distal pocket. In spite of the limited signal, the nonheme iron VDOS in 57FeIII-FeBMb1(FeIII) (lower trace) clearly reports vibrations from a distinct iron site characterized by a reduced coordination strength.
As seen above for reduced 57FeII-FeBMb1(FeII), the VDOS for the nonheme iron is clearly distinct from that for the heme iron, in spite of a relatively low 57Fe concentration and a consequently reduced signal in the 57FeIII-FeBMb1(FeIII) sample. This dual confirmation of successful site-specific labeling of each site illustrates the opportunity to probe the reactivity of each iron independently.
Resilience: An “Outer-Sphere” Force Constant
The iron VDOS determines an additional averaged force constant, the resilience84
| 2 |
which provides information distinct from the stiffness. As defined more generally by Zaccai,85 the resilience
| 3 |
measures the rate at which the mean-squared displacement ⟨xFe2⟩ of the probe atom (here, iron) increases with temperature. NRVS lacks the energy resolution to capture highly anharmonic motions that contribute to temperature-dependent measurements of ⟨xFe2⟩ using techniques such as inelastic neutron scattering or Mössbauer spectroscopy above a “dynamical transition” near 200 K. On the other hand, at temperatures below 200 K, we have shown quantitative agreement between determinations of ⟨xFe2⟩ from Mössbauer measurements on oxidized cytochrome c at a series of temperatures86 and the values expected on the basis of the iron VDOS determined using NRVS at a single temperature.84 The vibrational contribution to the resilience (eq 2) captures this temperature variation in a single parameter, with lower values of the resilience characterizing environments with large fluctuations of the 57Fe probe atom.
Low-frequency vibrations play the primary role in determining the resilience, as we illustrate by directly plotting the integrand
| 4 |
in eq 2 as a “resilience spectrum” in Figure 6. The resilience is equal to the inverse of the area of this spectrum and is primarily determined by vibrations below 100 cm–1. Molecular dynamics simulations on Mb and cytochrome c show similar mean-squared displacements for all heme atoms including iron,84 supporting our suggestion58 that translation of the heme in response to fluctuations of the embedding protein matrix make the primary contribution to the NRVS signal below 100 cm–1.
Figure 6.
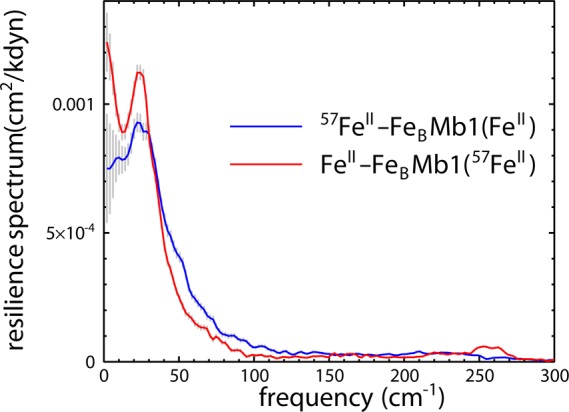
The resilience spectrum (eq 4) suppresses contributions from localized iron ligand vibrations and highlights low-frequency oscillations of the protein that drive translational motion of both iron sites in reduced 57FeII-FeBMb1(FeII) and FeII-FeBMb1(57FeII). Quantitative agreement between the areas determined for both sites yields values for the resilience that are identical, within experimental uncertainty (Table 1). Nevertheless, comparison as a function of frequency reveals subtle differences in the coupling of long-range protein fluctuations to these two sites.
As a result, we interpret the resilience as a measure of the elastic properties of the protein environment. Previously, we found a significant increase of the resilience in cytochrome c in comparison with Mb.84 Here, we find that the resiliences of reduced FeII-FeBMb1(57FeII) and native Mb are the same, within experimental uncertainty, suggesting that the introduction of the additional nonheme metal site does not seriously perturb the elastic properties of the protein. Moreover, small differences in the coupling of the protein fluctuations to the heme and nonheme iron sites, apparent from examination of Figure 6, average out to yield values for the resilience that agree quantitatively for the two distinct sites, within the experimental uncertainty. This further supports the notion that the resilience quantifies global properties of the embedding protein and contrasts with the sensitivity of the stiffness to differences in the coordination of the two iron sites. In short, we propose that the resilience can be interpreted as an “outer-sphere” force constant that probes the elasticity of the embedding protein, in contrast with the probe of the immediate coordination environment, as quantified by the stiffness.
Effect of Nonheme Metal on the Coordination of Heme Iron
The vibrational dynamics of the heme iron undergo noticeable changes upon exposure to NO. The vibrational signal from FeII-FeBMb1(57FeIINO) containing 57Fe-enriched hemes covers a wider frequency range than that in the absence of NO, with significant features resolved beyond 500 cm–1 (Figure 7). The experimentally determined stiffness for ZnII-FeBMb1(57FeIINO) exceeds 300 pN/pm (Table 1), indicating a substantial increase in the coordination forces exerted on the iron. The enhanced coordination strength is consistent with a LS iron. We observed stiffnesses exceeding 300 pN/pm for the LS heme iron in reduced cytochrome c.81
Figure 7.
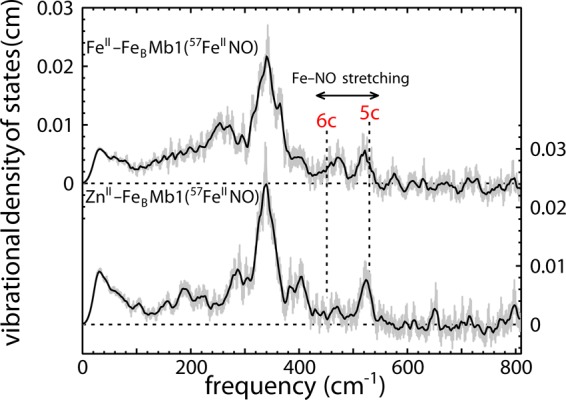
The heme iron VDOS reveals that the presence of a second metal in the nonheme site influences NO binding to FeII-FeBMb1(57FeII) and ZnII-FeBMb1(57FeII). Fe–NO stretching vibrations, clearly resolved above 400 cm–1, probe the axial ligation. For reference, dashed lines indicate Fe–NO stretching frequencies reported for native horse heart MbNO (452 cm–1),93 characteristic of a six-coordinate complex with NO coordinated trans to a histidine ligand, and for Fe(DPIX)(NO) (528 cm–1),88 a typical five-coordinate heme NO complex. A substantial fraction of hemes exhibit an Fe–NO stretching frequency characteristic of five-coordinate heme nitrosyls when either ZnII or FeII is present in the nonheme site. This contrasts with previous measurements on native MbNO,92 which revealed a NRVS signal consistent with six-coordinate heme NO.
The presence of well-resolved features yields more specific information on individual Fe–ligand bonds. In particular, previous NRVS measurements have identified an Fe–NO stretching mode in the 520–530 cm–1 range in five-coordinate heme NO complexes,65,71,87,88 which is also observed in rR measurements.52,89,90 Binding of an imidazole ligand trans to NO weakens the Fe–NO bond,54,91 and we observe this mode at lower frequencies in the 450–460 cm–1 range for these six-coordinate heme NO complexes.69,92−95 One well-characterized six-coordinate heme NO complex is native MbNO, where this Fe–NO vibration appears at 452 cm–1 and contributes to both the rR and NRVS signals.92,93
Unlike native MbNO, Fe–NO stretching frequencies near 530 cm–1 characteristic of five-coordinate heme NO contribute to the experimental VDOS of FeII-FeBMb1(57FeIINO) and ZnII-FeBMb1(57FeIINO) upon the introduction of a divalent metal in the nonheme site (Figure 7). This result supports previous evidence for formation of a five-coordinate heme NO complex in FeII-FeBMb1(FeIINO) exposed to excess NO, which was based on observation of the same Fe–NO vibration at 522 cm–1 using rR spectroscopy as well as the presence of a 1660 cm–1 N–O stretching frequency in IR measurements.50 The altered heme ligation in response to the neighboring nonheme metal contrasts with the insensitivity of the unligated heme to the nonheme metal noted above (Figures 4 and 5) and demonstrates that the nonheme metal specifically influences the structure of the heme ligand complex.
In the presence of 1 equiv of NO, the FeII-FeBMb1(57FeIINO) VDOS (Figure 7) also has a feature with a 480 cm–1 frequency that we attribute to six-coordinate heme NO. Because the NRVS signal depends only on the mean-squared vibrational amplitude of the iron and on the relative population of contributing species, the FeII-FeBMb1(57FeIINO) VDOS suggest comparable amounts of five- and six-coordinate heme NO (Figure 7). Interestingly, the Fe–NO frequency is significantly increased with respect to that observed for native MbNO,92,93 providing additional information on how the nonheme metal influences the electronic structure of the neighboring heme NO complex. The contribution of a vibrational signal attributable to six-coordinate heme NO is significantly reduced for ZnII-FeBMb1(57FeIINO) in the presence of 1 equiv of NO.
Another Fe–NO vibration undergoes a large shift from ca. 380 cm–1 in five-coordinate to ca. 560 cm–1 in six-coordinate heme NO complexes.71,92 Experimental characterization of its kinetic energy distribution based on isotope shifts indicates that this vibrational mode primarily involved motion of the central nitrogen atom of the FeNO unit.92,96,97 On this basis, this N-centered vibration can be qualitatively described as an FeNO bending mode to distinguish it from the Fe–NO stretching mode that contributes more strongly to the NRVS signal. However, it must be emphasized that neither mode can exhibit pure FeNO bending or Fe–NO stretching character for the nonlinear FeNO unit. Both modes exhibit rather modest Soret enhancement in Raman scattering from six-coordinate heme NO complexes, but the FeNO bending frequency is more reliably detected in heme proteins because of its large sensitivity to 14N/15N substitution and is thus more often reported. Although the iron amplitude and thus the NRVS signal is necessarily smaller for the FeNO bending vibration, the FeII-FeBMb1(57FeIINO) VDOS includes minor features near 380 and 580 cm–1 consistent with contributions from the FeNO bending vibration of five- and six-coordinate heme NO complexes, respectively, supporting conclusions based on the stronger Fe–NO stretching frequency discussed above.
Raman and IR measurements on FeII-FeBMb1(FeIINO) resulting from reaction with stoichiometric NO also identify FeNO bending and N–O stretching frequencies that are 15–20 cm–1 higher and 70–80 cm–1 lower, respectively, than those typically observed for six-coordinate heme NO.49 Together, the unusual values for all three vibrations of the FeNO fragment suggest that the nonheme FeII strongly perturbs the electronic structure of heme NO. In particular, it is conceivable that the FeII cation electrostatically predisposes the heme-bound NO to the electron transfer that will ultimately be required for reactivity.
Effect of Heme Metal on Coordination of Nonheme Iron
As found above, the nonheme iron influences the coordination of the heme, strengthening the Fe–NO bond and weakening the Fe–His linkage to the protein. In contrast, the vibrational dynamics of the nonheme iron in 57FeII-FeBMb1(FeII) exposed to 1 equiv of NO do not differ significantly from those observed in the absence of NO (Figure 8). This indicates that the ligation and electronic structure of the nonheme iron is insensitive to the structural and electronic changes that take place upon NO binding to the heme iron. Moreover, it indicates that the nonheme iron has a much lower affinity for NO than the heme iron does.
Figure 8.

The VDOS of the nonheme iron atom reveals perturbations in 57FeII-FeBMb1(ZnII) when the nonheme site is saturated with excess NO (see Materials and Methods for details of sample preparation). When FeII is present in the heme site, on the other hand, the nonheme iron VDOS exhibits no significant change upon reaction with stoichiometric NO, in contrast with the clear signatures for NO binding to the heme iron seen in Figure 7.
Replacement of the heme iron with ZnPPIX eliminates the possibility of NO binding to the heme and allows the investigation of NO binding to the nonheme iron selectively. The VDOS of the nonheme iron in the resulting 57FeII-FeBMb1(ZnII) (Figure 8) in the absence of NO strongly resembles that observed for 57FeII-FeBMb1(FeII) under the same conditions (Figure 4), indicating that the structure of the nonheme iron site is insensitive to the substitution of the heme metal. However, noticeable changes in the nonheme VDOS of 57FeII-FeBMb1(ZnII) take place in the presence of excess NO (Figure 9), which we attribute to the binding of NO to the nonheme iron forming the 57FeIINO-FeBMb1(ZnII) under these conditions.
Figure 9.
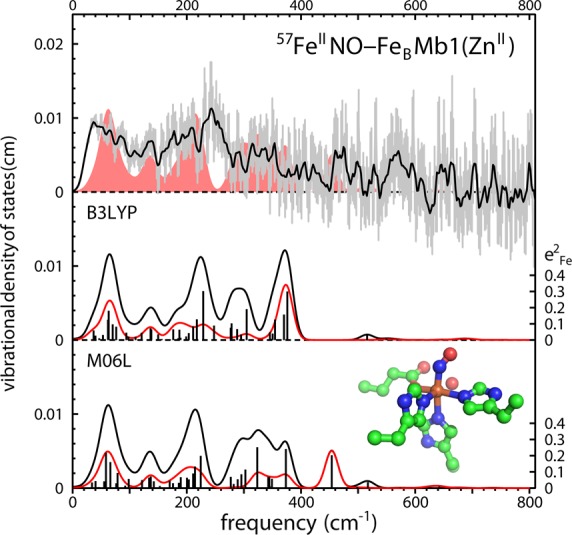
Computational models for the NO-ligated nonheme FeB site in 57FeIINO-FeBMb1(ZnII) using differing functionals yield quantitative predictions for the iron VDOS. Comparison with the experimental VDOS for 57FeIINO-FeBMb1(ZnII) in the presence of excess NO is consistent with a substantial contribution from NO-ligated iron. The red trace indicates the contribution from iron motion along the Fe–NO bond direction and highlights the variability of the predicted Fe–NO stretching frequency, which shifts from 376 cm–1 using B3LYP to 454 cm–1 using M06L. The image excludes hydrogen atoms.
One significant advantage of the NRVS method is the relative ease of quantitative comparison with DFT predictions.65,68,98 Overall, such comparisons provide useful guidance for interpreting experimental results, but we have found that predicted vibrational frequencies for the FeNO fragment exhibit significant dependence on the functional used for DFT calculations.65,69,98 For five-coordinate heme NO complexes, predicted Fe–NO stretching frequencies vary by nearly 200 cm–1.65,69,98 DFT investigations of nonheme FeNO complexes also reveal significant variability of the electronic structure predicted using different functionals.99,100 It remains to be established whether any currently available functional adequately accounts for electron correlation in iron nitrosyl complexes.
Examination of a wide variety of functionals found that M06L101 gave the best overall account of the iron VDOS for the five-coordinate nitrosyl heme complex Fe(OEP)(NO).98 The VDOS predicted using this functional is presented both as the lower trace in Figure 9 and, for comparison, as a filled area behind the experimental VDOS in the upper trace of Figure 9. The M06L prediction does exhibit significant correspondence with the experimental signal, supporting the conclusion that direct NO ligation accounts for the observed vibrational changes. Unfortunately, because of the limited 57Fe concentration, the relatively low signal level from this sample precludes an experimental identification of the Fe–NO stretching frequency.
Insights into NOR Gained from Biosynthetic Studies: Structural Features Responsible for High NOR Activity
The biosynthetic models have allowed us to provide insight into NORs that is otherwise difficult to obtain in studying native enzymes. For instance, to the best of our knowledge, the nonheme FeB in native NORs has not been replaced or removed, making it difficult to assess the role of FeB in the activity of NORs. In contrast, because the biosynthetic models are purified without a nonheme metal ion, investigations into the role of iron or other nonheme metals is greatly simplified by changing the nonheme metal source that is used (e.g., FeCl2 vs ZnCl2). Therefore, our biosynthetic model allowed us to answer the previously unaddressed question of what would happen if FeB was replaced with CuB. Given the structural homology between HCOs and NORs and their known cross reactivity, a fascinating issue arises as to the role of each class of enzymes’ different nonheme metal.44,45,102 Activity assays using iron or copper as the nonheme metal ion both demonstrated NOR activity, while controls using redox-inactive zinc did not possess NOR activity.51 This study demonstrated a critical insight that a redox-active metal ion was needed to confer NOR activity, an insight that would not be possible when studying the native enzymes.
In addition, we also demonstrated that the glutamate ligand to FeB is essential for both iron binding at the nonheme site and NOR activity.39 Finally, an extended hydrogen-bonding network was shown as a critical component in improved NOR activity in our biosynthetic models when a glutamate residue (I107E), was introduced at the secondary coordination sphere into our model to facilitate proton transfer to the active site.51
Insights into NOR Gained from Biosynthetic Studies: Mechanistic Insights
The ultimate goal of studying native enzymes and their models is to unravel the details of how they work and apply that understanding to other related enzymes as well as biomedical and biotechnological applications. This goal can best be achieved by thorough mechanistic characterization, which has been achieved to great success in our biosynthetic models of NORs. For example, although progress has been made in elucidating the structural aspects of NORs aided by recent success in solving the X-ray structure of cNOR, understanding the mechanism of the enzyme continues to be problematic due to several technical barriers. To illustrate, even though the isolated enzyme cNOR is reactive to NO with a moderate turnover rate under steady-state conditions, in the reduced form the enzyme shows very slow turnover in pre-steady-state conditions because of the presence of an obscure structural form of the enzyme. Flash-flow experiments with the carbonyl complex of cNOR can result in fast reaction kinetics.103−105 However, these studies are primarily dependent on monitoring UV–vis changes of the protein, which is dominated by the signals from the high-affinity heme site and does not provide any information on the events occurring at the FeB site.
Apart from these experimental challenges, the presence of multiple configurations of the oxidized cNOR further hinders our understanding of the mechanistic aspects. In one such configuration where the enzyme exists as a μ-oxodiferric complex, strong magnetic coupling between the five-coordinate heme FeIII (His is dissociated from heme iron in this form) and nonheme FeBIII is observed,18 while in other cases, only weak magnetic coupling was observed.83,106 In addition, there are no experimentally accessible methods to selectively probe the NO complex of the FeB site because the high-affinity heme site dominates spectroscopic signatures including UV–vis, EPR, Mössbauer, and NRVS.
Owing to these practical problems, a unified mechanism of NORs is lacking. Several reaction routes have been proposed, leading to N–N bond formation from cleavage of the N–O bond18,83,106,107 (Scheme 1). In the first route, the so-called “trans mechanism”, one NO molecule binds each of the heme iron and nonheme FeB sites in a trans configuration, where both iron centers are present as {FeNO}7 complexes. This step is followed by the reductive activation of the dinitrosyl moiety, leading to the formation of a hyponitrite dianion intermediate, where both iron centers are now oxidized to FeIII. In the “cis heme b3 mechanism”,108,109 supported by theoretical studies, the first NO binds to the heme FeII, followed by reductive activation of the NO complex, which is stabilized by electrostatic interactions with FeBII. Next, a second NO electrophilically attacks the first heme-bound NO, leading to the formation of a hyponitrite dianion, which is electrostatically stabilized by FeBIII. Finally, in the “cis FeB mechanism”, both NO units bind to the FeB site and the hyponitrite dianion form is stabilized by electrostatic interactions with the heme FeIII. In all of these proposed mechanisms, it is also unclear how the dianion leads to the product formation, e.g., whether this hyponitrite intermediate becomes protonated, followed by chemical rearrangement of the complex, is also not well understood.
With these hurdles in understanding the mechanism of NOR using native enzymes, simpler protein-based model systems that are stable, easy-to-prepare, and well-characterized are needed. To this end, engineered E-FeBMb1(FeII) and E-FeBMb2(FeII) and their corresponding metallated and nitrosyl derivatives have provided the much needed insight into the mechanistic aspects of NORs, as summarized below.
Resonance Raman studies have shown that in the reduced form both these models exist as 5cHS heme in both the absence and presence of FeII in the FeB site.49 However, in the presence of 1 equiv of NO, both proteins loaded with the FeB site form stable 6cLS {FeNO}7 complexes at the heme site. One important revelation from these studies was the presence of exceptionally low ν(NO) stretching and high ν(FeNO) frequencies compared to all reported 6cLS heme nitrosyl complexes. These results were attributed to ferric heme iron(III) nitroxyl (FeIIINO–) complex, where NO– was stabilized by electrostatic interactions with the FeB site. Strong back-donation from heme iron caused an increase in the ν(FeNO) frequency, while the negative charge on NO resulted in a decrease in the ν(NO) frequency. In the event of excess NO addition, both proteins form a [{FeNO}7]2 trans nitrosyl dimer, leading to N2O formation, supporting the so-called “trans mechanism”.50 Under single-turnover conditions, using FTIR studies, no N2O production was observed in FeBMb1, suggesting that the presence of the FeB site is not enough to reduce NO to N2O. However, in FeII-FeBMb2(FeII), 50% N2O production was observed, suggesting that the presence of the second glutamate is critical for N2O formation, presumably by facilitating proton transfer via a hydrogen-bonding network during turnover. Unproductive complexes in both the proteins are characterized by a trans dinitrosyl complex, where the heme iron is present as a 5cLS {FeNO}7 species with a dissociated heme–His bond and a second NO bound to the FeB site. Surprisingly, from stopped-flow and rapid freeze quench experiments, NO binding to the FeB site was observed to be kinetically favored with a t1/2 of ∼1 ms, followed by binding of the second NO to the heme iron, leading to the trans dinitrosyl 6cLS {FeNO}7-FeBNO complex. This finding provided experimental evidence that FeB binds NO before it is bound to heme b3, which was suggested previously, but not confirmed, in a study of Ps. nautica NOR.83
In FeIINO-FeBMb1(FeIINO), the dinitrosyl complex leads to the formation of a dead-end 5cLS {FeNO}7 species, where the heme–His bond is dissociated, but in FeIINO-FeBMb2(FeIINO), the presence of the second glutamate residue leads to ∼50% effective turnover, which results in a decreased rate of dissociation of the proximal heme–His bond, leading to the formation of the dead-end complex. During the decay of the trans dinitrosyl complex (6cLS heme {FeNO}7/FeB-NO), the hyponitrite intermediate was not observed in either protein derivative, in contrast to proposed mechanisms. Furthermore, when excess NO was added after formation of the 6cLS {FeNO}7 complex, the same dinitrosyl complexes of FeIINO-FeBMb1(FeIINO) and FeIINO-FeBMb2(FeIINO) were observed (vide supra), ruling out the formation of any electrophilically attached second NO. This observation, therefore, does not support the so-called “cis heme b3”, as proposed by theoretical studies.
As stated above, a major barrier to understanding the NOR mechanism is the difficulty associated with isolating pure FeB-NO complexes due to the presence of a high-affinity heme site. To circumvent this critical methodological barrier in native NORs, in a recent effort we spectroscopically probed NO binding to the FeB site after replacing the high-affinity heme with ZnPPIX.37 Such a strategy cannot be easily applied with native NORs since the heme cannot be selectively replaced because of the complex nature of the enzyme. From EPR, Mössbauer, and quantum mechanics/molecular mechanics calculations on the NO derivative of FeIINO-FeBMb1(ZnII), the electronic state of FeB-NO could be best described as HS S = 3/2 Fe2+-NO• having a high ferrous character and a radical nature on NO. The radical nature on NO would promote N–N bond formation by radical coupling with a heme-bound NO and thus would support the trans mechanism. These results highlight the usefulness of biosynthetic models of complex enzymes within easy-to-produce and well-characterized proteins. Taken together, the engineered NOR models have provided important insights into the reaction mechanism of NOR and support the proposed “trans mechanism” of NO reduction by NORs.
Summary and Outlook
Results presented here exploit the ability to replace metals at either site, illustrating an important opportunity enabled by the biosynthetic approach. Selective substitution with 57Fe provides an independent structural probe for either of the two metal sites in FeII-FeBMb1(FeII). NRVS measurements quantify the forces exerted on 57Fe by its coordination environment and indicate the presence of HS iron at both sites in the absence of substrate. Reduced and oxidized proteins serve to model the initial and final states, respectively, in Scheme 1, although the vibrational signals provide no evidence for a solvent-derived ligand bridging the metals in the oxidized state. Rather, vibrations of the heme iron are comparable to those reported for native Mb, confirming that the heme is unperturbed by the engineered nonheme site. On the other hand, the observation of distinct vibrational dynamics for the nonheme iron confirms successful site-specific labeling with 57Fe.
Substitution of a redox-inactive ZnII ion for FeII allows the preparation of stable mononitrosylated intermediates that would precede the formation of the putative (and unstable) dinitrosylated intermediates depicted in Scheme 1. The presence of ZnII in the nonheme site perturbs the vibrational properties of the adjacent heme NO complex, in a manner consistent with electron transfer to the NO ligand. This suggests that the electrostatic influence of the nonheme FeII in NORs could act to promote the enzymatic reaction in either the trans or cis heme mechanism. With ZnII in the heme site, alteration of the nonheme iron vibrations upon exposure to NO confirms that NO can bind to the nonheme FeII if the heme site is unavailable, as would be required in the trans mechanism.
Considerable advances in the biosynthetic modeling of NORs have been achieved recently, and given that both the resting state14,15 and ligand-bound and reduced forms of cNOR have been crystallized,110 our understanding of NOR modeling should only improve moving forward. Importantly, models that can perform enzymatic turnover will be critical because we are unaware of any model, biosynthetic or otherwise, that is capable of reducing NO with turnover numbers comparable to those of the native enzymes. In conjunction with this, new models that more closely replicate the secondary coordination sphere of native NORs must be developed because these interactions are critical for improving activity.38,51 With improved structural information into the active site of NOR, fine-tuning of factors such as the heme–nonheme distance will also be important. The creation of such models will provide further insights into the reaction mechanism and activity of NORs.
Materials and Methods
All samples were prepared in a 50 mM Bis-Tris pH 7.3 buffer after chelexing overnight, followed by pH adjustment and filtration to remove chelex beads. Buffers were degassed in a Schlenk line for ∼5 h by several cycles of freeze–pump–thaw prior to their transfer into an anaerobic chamber (Coy Laboratories, Inc.) for sample preparations. Dry Sephadex G25 beads (GE Healthcare) were suspended in a buffer solution and degassed in the Schlenk line for several hours before transfer into the glovebag. All solid materials were kept under vacuum overnight in the antechamber prior to transfer into the glovebag. All protein solutions were exchanged from a 100 mM phosphate pH 7 buffer to a 50 mM Bis-Tris pH 7.3 buffer outside the glovebag using small size-exclusion columns (PD 10 columns, GE Healthcare) preequilibrated in an exchangeable buffer. The protein solutions were then degassed by three cycles of freeze–pump–thaw in a Schlenk line and brought into the glovebag. A diethylamine NONOate (DEA-NONOate; ε250 nm = 6.5 mM–1 cm–1; Cayman Chemicals) solution prepared in 10 mM NaOH and used as the NO source was degassed by three cycles of freeze–pump–thaw before transfer into the glovebag. A stock solution of FeCl2 was prepared inside the glovebag by dissolving solid FeCl2 in degassed water.
Protein Expression and Purification
E-FeBMb1(FeIII) was purified using a known protocol, as reported previously.39,51 A similar protocol was employed for E-FeBMb1(57FeIII) purification except that in this case 57Fe-labeled heme (Frontier Scientific) was used during the protein refolding step. The identity of each of the purified proteins was confirmed by electrospray ionization mass spectrometry. The r/z of pure protein was >4 in a 100 mM potassium phosphate pH 7 buffer. E-FeBMb1(ZnII) was prepared and purified as previously described.37 Pure proteins were stored at −80 °C until further use. Molar extinction coefficients ε406 = 175 mM–1 cm–1 for Met E-FeBMb1(FeIII), ε433 = 143 mM–1 cm–1 for deoxy E-FeBMb1(FeII), and ε427 of 136.2 mM–1 cm–1 for E-FeBMb1(ZnII) were used to determine the concentrations of the corresponding proteins.37
Synthesis of 57FeCl2
57FeCl2 synthesis was performed inside the glovebag. A total of 300 mL of deionized water and 1 mL of 9.14% methanolic HCl (285.6 μL of 32% HCl + 714.4 μL of methanol) were degassed and transferred into the glovebag. A total of 25 mg of 57Fe metal (0.44 mmol; Cambridge Isotope Lab) was taken into a small dry NMR tube and transferred into the glovebag. The degassed water was transferred into a small water bath and heated to ∼60 °C using a hot plate equipped with a stir bar. The NMR tube containing 57Fe was immersed into the water bath, and 350 μL of 9.14% methanolic HCl (0.88 mmol) was added to the tube. H2 evolution started immediately. The reaction was allowed to proceed for 3–4 h until the gas evolution ceased. The solution was carefully transferred into a tared Schlenk flask with an adaptor. The Schlenk flask was removed from the bag, immersed in a dry ice/ethanol slush bath, and slowly opened to vacuum in a Schlenk line. The flask was then slowly warmed to 100 °C using a water bath while in vacuum. After the solvent evaporated and the solid turned from green to white, the water bath was replaced with an oil bath and heated to 160 °C, allowing the residual methanol to evaporate. The product was cooled to room temperature slowly, purged with argon, sealed, and weighed. The yield of 57FeCl2 was ∼60–65%.
NRVS
NRVS data were collected at the Advanced Photon Source on beamline 3ID-D, as described in detail elsewhere.79 Briefly, the X-ray energy was scanned in the vicinity of the 57Fe nuclear resonance at 14.4125 keV in steps of 0.25 meV. Data were recorded on frozen solutions at 5–10 mM protein concentration at temperatures of 60–80 K, and each measurement was the average of 15–35 scans. A comparison of early and late scans confirmed the absence of spectroscopic changes during X-ray exposure. The VDOS was extracted from the measured data using the program PHOENIX.52
Met E-FeBMb1(FeIII) was reduced inside the glovebag with excess dithionite and passed through a small hand-packed size-exclusion column (Sephadex G25) preequilibrated with a 50 mM Bis-Tris pH 7.3 buffer to remove excess dithionite and eluted with the same buffer. The eluted protein was then concentrated to 1 mM (ε433 nm = 143 mM–1 cm–1), and the nonheme site was reconstituted with 1.0 equiv of either 57FeCl2 or ZnCl2 (prepared freshly in the inert-atmosphere bag) added in aliquots of 0.25 equiv with 15 min between each addition. E-FeBMb1(ZnII) was transferred into the glovebag after degassing and concentrated to 1 mM (ε427 nm = 136.2 mM–1 cm–1), and the FeB site was reconstituted with 57FeII without reducing the protein because ZnPPIX is redox-inactive. Reconstitution of the FeB site with 57FeII or ZnII, as desired, was ensured by checking the UV–vis spectrum after metal addition, as evidenced by shifting of the Soret peak from 427 nm in E-FeBMb1(ZnII) and 433 nm in E-FeBMb1(FeII), in the absence of the nonheme metal to 429 nm in 57FeII-FeBMb1(ZnII) and 434 nm in 57FeII-FeBMb1(FeII) or FeII-FeBMb1(57FeII) in the reconstituted protein, respectively. When applicable, oxidation of the heme iron and nonheme iron was achieved after reconstitution by the addition of excess ferricyanide and then passage through a small size-exclusion column. The FeB reconstituted proteins were then concentrated to ∼10–15 mM before loading 15 μL of the samples into the well of a high-density polyethylene block sample holder inside the glovebag, transferred outside, and frozen immediately where other components (sapphire window, copper block, brass screws) were assembled.
Nitrosyl derivatives were prepared using the following protocol. For E-FeBMb1(57FeII), 1 equiv of NO was added to the FeII or ZnII reconstituted proteins present at 1 mM concentration. At each step, 0.25 equiv of NO was added to the reconstituted proteins in the form of DEA-NONOate, allowing enough time to for NO release between each addition (t1/2 = 16 min at pH 7.3). NO binding to the proteins was confirmed by measuring UV–vis spectra of NO-bound samples. Similarly, for 57FeII reconstituted 57FeII-FeBMb1(ZnII), the protein was kept at 1 mM concentration before NO addition. Excess NO was added, in the form of DEA-NONOate as described above, to 57FeII-FeBMb1(ZnII) to saturate the FeB site with NO. After NO binding, the samples were further purified by another PD10 column equilibrated in a 50 mM Bis-Tris pH 7.3 buffer to remove any trace impurities including the decay product of DEA-NONOate. All of the nitrosyl complexes thus prepared were then concentrated to ∼10–15 mM and loaded into NRVS cells described above. The protocol of adding NO at 1 mM of the reconstituted proteins followed by concentrating to higher concentrations has proven to be a successful strategy in our studies, as we have recently reported.37 An aliquot of each of the above concentrated samples was diluted, and their UV–vis spectra were checked inside the glovebag to ensure that no changes in NO coordination occurred during the final step of sample preparation.
Calculations
DFT calculations were performed using Gaussian 09 with the B3LYP111,112 and M06L101 functionals. The VTZ basis set113 was used for the iron orbitals and 6-31G* for all other atoms. The computational model for the nitrosyl complex of the nonheme site of 57FeIINO-FeBMb1(ZnII) used the atomic coordinates deposited in the Protein Data Bank under access code 3K9Z(39) for His 29, His 43, His 64, Glu 68 (with α-carbon atoms replaced by terminal methyl groups), the iron and a ligated water, and added NO as a sixth ligand. Energy optimization of the experimentally observed S = 3/2 state yielded nearly octahedral coordination for the iron, with an Fe–N–O angle varying from 142.8° (B3LYP) to 143.2° (M06L). The atomic displacements of the vibrational normal modes were used to calculate the iron VDOS as described previously.61
Acknowledgments
This work was supported by the U.S. National Institutes of Health (Grant GM06221 to Y.L. and Grant 5T32-GM070421 to J.R), and use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract DE-AC02-06CH11357.
Author Present Address
⊥ Center for Integrated Nanotechnologies, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, United States
The authors declare no competing financial interest.
After this paper was posted ASAP on August 14, 2015, additional corrections were made to Table 1, and the paper was reposted on August 24, 2015.
References
- Wasser I. M.; de Vries S.; Moënne-Loccoz P.; Schröder I.; Karlin K. D. Chem. Rev. 2002, 102, 1201–1234 10.1021/cr0006627. [DOI] [PubMed] [Google Scholar]
- Canfield D. E.; Glazer A. N.; Falkowski P. G. Science 2010, 330, 192–196 10.1126/science.1186120. [DOI] [PubMed] [Google Scholar]
- Zumft W. G. Microbiol. Mol. Biol. Rev. 1997, 61, 533–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman J. S. In Nitric Oxide; Lancaster J., Ed.; Academic Press: San Diego, 1996; pp 1–82. [Google Scholar]
- Prast H.; Philippu A. Prog. Neurobiol. 2001, 64, 51–68 10.1016/S0301-0082(00)00044-7. [DOI] [PubMed] [Google Scholar]
- Bogdan C. Nat. Immunol. 2001, 2, 907–916 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- Klink A.; Elsner B.; Strube K.; Cramm R. J. Bacteriol. 2007, 189, 2743–2749 10.1128/JB.01865-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougald D.; Rice S. A.; Barraud N.; Steinberg P. D.; Kjelleberg S. Nat. Rev. Microbiol. 2011, 10, 39–50 10.1038/nrmicro2695. [DOI] [PubMed] [Google Scholar]
- Barraud N.; Kelso M. J.; Rice S. A.; Kjelleberg S. Curr. Pharm. Des 2015, 21, 31–42. [DOI] [PubMed] [Google Scholar]
- Kakishima K.; Shiratsuchi A.; Taoka A.; Nakanishi Y.; Fukumori Y. Biochem. Biophys. Res. Commun. 2007, 355, 587–591 10.1016/j.bbrc.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Ravishankara A. R.; Daniel J. S.; Portmann R. W. Science 2009, 326, 123–125 10.1126/science.1176985. [DOI] [PubMed] [Google Scholar]
- Berto T. C.; Hoffman M. B.; Murata Y.; Landenberger K. B.; Alp E. E.; Zhao J.; Lehnert N. J. Am. Chem. Soc. 2011, 133, 16714–16717 10.1021/ja111693f. [DOI] [PubMed] [Google Scholar]
- Liu J.; Chakraborty S.; Hosseinzadeh P.; Yu Y.; Tian S.; Petrik I.; Bhagi A.; Lu Y. Chem. Rev. 2014, 114, 4366–4469 10.1021/cr400479b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino T.; Matsumoto Y.; Nagano S.; Sugimoto H.; Fukumori Y.; Murata T.; Iwata S.; Shiro Y. Science 2010, 330, 1666–1670 10.1126/science.1195591. [DOI] [PubMed] [Google Scholar]
- Shiro Y.; Sugimoto H.; Tosha T.; Nagano S.; Hino T. Philos. Trans. R. Soc., B 2012, 367, 1195–1203 10.1098/rstb.2011.0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moënne-Loccoz P.; de Vries S. J. Am. Chem. Soc. 1998, 120, 5147–5152 10.1021/ja973671e. [DOI] [Google Scholar]
- Grönberg K. L. C.; Roldán M. D.; Prior L.; Butland G.; Cheesman M. R.; Richardson D. J.; Spiro S.; Thomson A. J.; Watmough N. J. Biochemistry 1999, 38, 13780–13786 10.1021/bi9916426. [DOI] [PubMed] [Google Scholar]
- Moënne-Loccoz P.; Richter O.-M. H.; Huang H.; Wasser I. M.; Ghiladi R. A.; Karlin K. D.; de Vries S. J. Am. Chem. Soc. 2000, 122, 9344–9345 10.1021/ja0016295. [DOI] [Google Scholar]
- Field S. J.; Prior L.; Roldán M. D.; Cheesman M. R.; Thomson A. J.; Spiro S.; Butt J. N.; Watmough N. J.; Richardson D. J. J. Biol. Chem. 2002, 277, 20146–20150 10.1074/jbc.M112202200. [DOI] [PubMed] [Google Scholar]
- Pinakoulaki E.; Ohta T.; Soulimane T.; Kitagawa T.; Varotsis C. J. Am. Chem. Soc. 2005, 127, 15161–15167 10.1021/ja0539490. [DOI] [PubMed] [Google Scholar]
- Lin S.-H.; Guidotti G.. Guide to Protein Purification. In Methods in Enzymology,2nd ed.; Richard B. R., Deutscher M. P., Eds.; Academic Press: New York, 2009; Vol. 463, pp 619–629. [DOI] [PubMed] [Google Scholar]
- Holm R. H.; Kennepohl P.; Solomon E. I. Chem. Rev. 1996, 96, 2239–2314 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- Wasser I. M.; Huang H.; Moënne-Loccoz P.; Karlin K. D. J. Am. Chem. Soc. 2005, 127, 3310–3320 10.1021/ja0458773. [DOI] [PubMed] [Google Scholar]
- Collman J. P.; Yang Y.; Dey A.; Decréau R. A.; Ghosh S.; Ohta T.; Solomon E. I. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 15660–15665 10.1073/pnas.0808606105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Schopfer M. P.; Sarjeant A. A. N.; Karlin K. D. J. Am. Chem. Soc. 2009, 131, 450–451 10.1021/ja8084324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopfer M. P.; Wang J.; Karlin K. D. Inorg. Chem. 2010, 49, 6267–6282 10.1021/ic100033y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich L. E.; Paulat F.; Praneeth V. K. K.; Lehnert N. Inorg. Chem. 2010, 49, 6293–6316 10.1021/ic902304a. [DOI] [PubMed] [Google Scholar]
- Berto T. C.; Speelman A. L.; Zheng S.; Lehnert N. Coord. Chem. Rev. 2013, 257, 244–259 10.1016/j.ccr.2012.05.007. [DOI] [Google Scholar]
- Berto T. C.; Xu N.; Lee S. R.; McNeil A. J.; Alp E. E.; Zhao J.; Richter-Addo G. B.; Lehnert N. Inorg. Chem. 2014, 53, 6398–6414 10.1021/ic5002573. [DOI] [PubMed] [Google Scholar]
- Lee S. C.; Lo W.; Holm R. H. Chem. Rev. 2014, 114, 3579–3600 10.1021/cr4004067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontecilla-Camps J. C.; Amara P.; Cavazza C.; Nicolet Y.; Volbeda A. Nature 2009, 460, 814–822 10.1038/nature08299. [DOI] [PubMed] [Google Scholar]
- Lill R. Nature 2009, 460, 831–838 10.1038/nature08301. [DOI] [PubMed] [Google Scholar]
- Waldron K. J.; Rutherford J. C.; Ford D.; Robinson N. J. Nature 2009, 460, 823–830 10.1038/nature08300. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Yeung N.; Sieracki N.; Marshall N. M. Nature 2009, 460, 855–862 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S.; Hosseinzadeh P.; Lu Y.. Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons, Ltd.: New York, 2011. [Google Scholar]
- Lu Y.; Chakraborty S.; Miner K. D.; Wilson T. D.; Mukherjee A.; Yu Y.; Liu J.; Marshall N. M. In Comprehensive Inorganic Chemistry II, 2nd ed.; Poeppelmeier J. R., Ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp 565–593. [Google Scholar]
- Chakraborty S.; Reed J.; Ross M.; Nilges M. J.; Petrik I. D.; Ghosh S.; Hammes-Schiffer S.; Sage J. T.; Zhang Y.; Schulz C. E.; Lu Y. Angew. Chem., Int. Ed. 2014, 53, 2417–2421 10.1002/anie.201308431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K. M.; van der Donk W. A.; Lu Y.. Non-Natural Amino Acids. In Methods in Enzymology; Muir T. W., Abelson J. N., Eds.; Academic Press: New York, 2009; Vol. 462, pp 97–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung N.; Lin Y.-W.; Gao Y.-G.; Zhao X.; Russell B. S.; Lei L.; Miner K. D.; Robinson H.; Lu Y. Nature 2009, 462, 1079–1082 10.1038/nature08620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigman J. A.; Kwok B. C.; Lu Y. J. Am. Chem. Soc. 2000, 122, 8192–8196 10.1021/ja0015343. [DOI] [Google Scholar]
- Sigman J. A.; Kim H. K.; Zhao X.; Carey J. R.; Lu Y. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 3629–3634 10.1073/pnas.0737308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; Yeung N.; Wang Z.; Guo Z.; Lu Y. Biochemistry 2005, 44, 1210–1214 10.1021/bi0479151. [DOI] [PubMed] [Google Scholar]
- Miner K. D.; Mukherjee A.; Gao Y.-G.; Null E. L.; Petrik I. D.; Zhao X.; Yeung N.; Robinson H.; Lu Y. Angew. Chem., Int. Ed. 2012, 51, 5589–5592 10.1002/anie.201201981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T.; Fukumori Y. J. Bacteriol. 1996, 178, 1866–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffre A.; Stubauer G.; Sarti P.; Brunori M.; Zumft W. G.; Buse G.; Soulimane T. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 14718–14723 10.1073/pnas.96.26.14718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forte E.; Urbani A.; Saraste M.; Sarti P.; Brunori M.; Giuffrè A. Eur. J. Biochem. 2001, 268, 6486–6491 10.1046/j.0014-2956.2001.02597.x. [DOI] [PubMed] [Google Scholar]
- Zhao X.; Yeung N.; Russell B. S.; Garner D. K.; Lu Y. J. Am. Chem. Soc. 2006, 128, 6766–6767 10.1021/ja058822p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butland G.; Spiro S.; Watmough N. J.; Richardson D. J. J. Bacteriol. 2001, 183, 189–199 10.1128/JB.183.1.189-199.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T.; Miner K. D.; Yeung N.; Lin Y.-W.; Lu Y.; Moënne-Loccoz P. Biochemistry 2011, 50, 5939–5947 10.1021/bi200409a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura H.; Hayashi T.; Chakraborty S.; Lu Y.; Moënne-Loccoz P. J. Am. Chem. Soc. 2014, 136, 2420–2431 10.1021/ja410542z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.-W.; Yeung N.; Gao Y.-G.; Miner K. D.; Tian S.; Robinson H.; Lu Y. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 8581–8586 10.1073/pnas.1000526107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle C. M.; Vogel K. M.; Rush T. S.; Kozlowski P. M.; Williams R.; Spiro T. G.; Dou Y.; Ikeda-Saito M.; Olson J. S.; Zgierski M. Z. Biochemistry 2003, 42, 4896–4903 10.1021/bi026395b. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Gossman W.; Oldfield E. J. Am. Chem. Soc. 2003, 125, 16387–16396 10.1021/ja030340v. [DOI] [PubMed] [Google Scholar]
- Wyllie G. R. A.; Schulz C. E.; Scheidt W. R. Inorg. Chem. 2003, 42, 5722–5734 10.1021/ic034473t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford P. C.; Laverman L. E. Coord. Chem. Rev. 2005, 249, 391–403 10.1016/j.ccr.2004.04.006. [DOI] [Google Scholar]
- Enemark J. H.; Feltham R. D. Coord. Chem. Rev. 1974, 13, 339–406 10.1016/S0010-8545(00)80259-3. [DOI] [Google Scholar]
- Scheidt W. R.; Durbin S. M.; Sage J. T. J. Inorg. Biochem. 2005, 99, 60–71 10.1016/j.jinorgbio.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Sage J. T.; Durbin S. M.; Sturhahn W.; Wharton D. C.; Champion P. M.; Hession P.; Sutter J.; Alp E. E. Phys. Rev. Lett. 2001, 86, 4966–4969 10.1103/PhysRevLett.86.4966. [DOI] [PubMed] [Google Scholar]
- Zeng W.; Barabanschikov A.; Wang N.; Lu Y.; Zhao J.; Sturhahn W.; Alp E. E.; Sage J. T. Chem. Commun. 2012, 48, 6340–6342 10.1039/c2cc31239e. [DOI] [PubMed] [Google Scholar]
- Alp E. E.; Sturhahn W.; Toellner T. S.; Zhao J.; Hu M.; Brown D. E. Hyperfine Interact. 2002, 144/145, 3–20 10.1023/A:1025452401501. [DOI] [Google Scholar]
- Chumakov A. I.; Sergueev I.; van Bürck U.; Schirmacher W.; Asthalter T.; Rüffer R.; Leupold O.; Petry W. Phys. Rev. Lett. 2004, 92, 245508. 10.1103/PhysRevLett.92.245508. [DOI] [PubMed] [Google Scholar]
- Lin J.-F.; Sturhahn W.; Zhao J.; Shen G.; Mao H.; Hemley R. J. Science 2005, 308, 1892–1894 10.1126/science.1111724. [DOI] [PubMed] [Google Scholar]
- Sturhahn W. Hyperfine Interact. 2000, 125, 149–172 10.1023/A:1012681503686. [DOI] [Google Scholar]
- Sage J. T.; Paxson C.; Wyllie G. R. A.; Sturhahn W.; Durbin S. M.; Champion P. M.; Alp E. E.; Scheidt W. R. J. Phys.: Condens. Matter 2001, 13, 7707. 10.1088/0953-8984/13/34/315. [DOI] [Google Scholar]
- Leu B. M.; Zgierski M. Z.; Wyllie G. R. A.; Scheidt W. R.; Sturhahn W.; Alp E. E.; Durbin S. M.; Sage J. T. J. Am. Chem. Soc. 2004, 126, 4211–4227 10.1021/ja038526h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W.; Barabanschikov A.; Zhang Y.; Zhao J.; Sturhahn W.; Alp E. E.; Sage J. T. J. Am. Chem. Soc. 2008, 130, 1816–1817 10.1021/ja077823+. [DOI] [PubMed] [Google Scholar]
- Moeser B.; Janoschka A.; Wolny J. A.; Paulsen H.; Filippov I.; Berry R. E.; Zhang H.; Chumakov A. I.; Walker F. A.; Schünemann V. J. Am. Chem. Soc. 2012, 134, 4216–4228 10.1021/ja210067t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu B. M.; Silvernail N. J.; Zgierski M. Z.; Wyllie G. R. A.; Ellison M. K.; Scheidt W. R.; Zhao J.; Sturhahn W.; Alp E. E.; Sage J. T. Biophys. J. 2007, 92, 3764–3783 10.1529/biophysj.106.093773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidt W. R.; Barabanschikov A.; Pavlik J. W.; Silvernail N. J.; Sage J. T. Inorg. Chem. 2010, 49, 6240–6252 10.1021/ic100261b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder D. P.; Silvernail N. J.; Barabanschikov A.; Zhao J.; Alp E. E.; Sturhahn W.; Sage J. T.; Scheidt W. R.; Rodgers K. R. J. Am. Chem. Soc. 2014, 136, 9818–9821 10.1021/ja503191z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlik J. W.; Barabanschikov A.; Oliver A. G.; Alp E. E.; Sturhahn W.; Zhao J.; Sage J. T.; Scheidt W. R. Angew. Chem., Int. Ed. 2010, 49, 4400–4404 10.1002/anie.201000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y.; Wang H.; George S. J.; Smith M. C.; Adams M. W. W.; Jenney E.; Francis J.; Sturhahn W.; Alp E. E.; Zhao J.; Yoda Y.; Dey A.; Solomon E. I.; Cramer S. P. J. Am. Chem. Soc. 2005, 127, 14596–14606 10.1021/ja042960h. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Wang H.; Xiao Y.; Vogt S.; Thauer R. K.; Shima S.; Volkers P. I.; Rauchfuss T. B.; Pelmenschikov V.; Case D. A.; Alp E. E.; Sturhahn W.; Yoda Y.; Cramer S. P. Inorg. Chem. 2008, 47, 3969–3977 10.1021/ic701251j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell C. B.; Wong S. D.; Xiao Y.; Klinker E. J.; Tenderholt A. L.; Smith M. C.; Rohde J.-U.; Que L.; Cramer S. P.; Solomon E. I. Angew. Chem., Int. Ed. 2008, 47, 9071–9074 10.1002/anie.200803740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra D.; George S. J.; Guo Y.; Kamali S.; Keable S.; Peters J. W.; Pelmenschikov V.; Case D. A.; Cramer S. P. J. Am. Chem. Soc. 2013, 135, 2530–2543 10.1021/ja307027n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teale F. W. J. Biochim. Biophys. Acta 1959, 35, 543. 10.1016/0006-3002(59)90407-X. [DOI] [PubMed] [Google Scholar]
- Kitagawa T.; Nagai K.; Tsubaki M. FEBS Lett. 1979, 104, 376–378 10.1016/0014-5793(79)80856-X. [DOI] [PubMed] [Google Scholar]
- Argade P. V.; Sassardi M.; Rousseau D. L.; Inubushi T.; Ikeda-Saito M.; Lapidot A. J. Am. Chem. Soc. 1984, 106, 6593–6596 10.1021/ja00334a024. [DOI] [Google Scholar]
- Wells A. V.; Sage J. T.; Morikis D.; Champion P. M.; Chiu M. L.; Sligar S. G. J. Am. Chem. Soc. 1991, 113, 9655–9660 10.1021/ja00025a034. [DOI] [Google Scholar]
- Achterhold K.; Keppler C.; Ostermann A.; van Bürck U.; Sturhahn W.; Alp E. E.; Parak F. G. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top. 2002, 65, 051916. 10.1103/PhysRevE.65.051916. [DOI] [PubMed] [Google Scholar]
- Leu B. M.; Ching T. H.; Zhao J.; Sturhahn W.; Alp E. E.; Sage J. T. J. Phys. Chem. B 2009, 113, 2193–2200 10.1021/jp806574t. [DOI] [PubMed] [Google Scholar]
- Lipkin H. J. Phys. Rev. B: Condens. Matter Mater. Phys. 1995, 52, 10073–10079 10.1103/PhysRevB.52.10073. [DOI] [PubMed] [Google Scholar]
- Timóteo C. G.; Pereira A. S.; Martins C. E.; Naik S. G.; Duarte A. G.; Moura J. J. G.; Tavares P.; Huynh B. H.; Moura I. Biochemistry 2011, 50, 4251–4262 10.1021/bi101605p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu B. M.; Zhang Y.; Bu L.; Straub J. E.; Zhao J.; Sturhahn W.; Ercan Alp E.; Timothy Sage J. Biophys. J. 2008, 95, 5874–5889 10.1529/biophysj.108.138198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccai G. Science 2000, 288, 1604–1607 10.1126/science.288.5471.1604. [DOI] [PubMed] [Google Scholar]
- Frolov E. N.; Gvosdev R.; Goldanskii V. I.; Parak F. G. JBIC, J. Biol. Inorg. Chem. 1997, 2, 710–713 10.1007/s007750050187. [DOI] [Google Scholar]
- Rai B. K.; Durbin S. M.; Prohofsky E. W.; Sage J. T.; Wyllie G. R. A.; Scheidt W. R.; Sturhahn W.; Alp E. E. Biophys. J. 2002, 82, 2951–2963 10.1016/S0006-3495(02)75636-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlik J. W.; Peng Q.; Silvernail N. J.; Alp E. E.; Hu M. Y.; Zhao J.; Sage J. T.; Scheidt W. R. Inorg. Chem. 2014, 53, 2582–2590 10.1021/ic4028964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel K. M.; Kozlowski P. M.; Zgierski M. Z.; Spiro T. G. J. Am. Chem. Soc. 1999, 121, 9915–9921 10.1021/ja990042r. [DOI] [Google Scholar]
- Thomas M. R.; Brown D.; Franzen S.; Boxer S. G. Biochemistry 2001, 40, 15047–15056 10.1021/bi011440l. [DOI] [PubMed] [Google Scholar]
- Traylor T. G.; Sharma V. S. Biochemistry 1992, 31, 2847–2849 10.1021/bi00126a001. [DOI] [PubMed] [Google Scholar]
- Zeng W.; Silvernail N. J.; Wharton D. C.; Georgiev G. Y.; Leu B. M.; Scheidt W. R.; Zhao J.; Sturhahn W.; Alp E. E.; Sage J. T. J. Am. Chem. Soc. 2005, 127, 11200–11201 10.1021/ja051052x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W.; Silvernail N. J.; Scheidt W. R.; Sage J. T.. Nuclear Resonance Vibrational Spectroscopy (NRVS). In Applications of Physical Methods to Inorganic and Bioinorganic Chemistry; Scott R. A., Lukehart C. M., Eds.; Wiley: New York, 2007; pp 401–421. [Google Scholar]
- Silvernail N. J.; Barabanschikov A.; Pavlik J. W.; Noll B. C.; Zhao J.; Alp E. E.; Sturhahn W.; Sage J. T.; Scheidt W. R. J. Am. Chem. Soc. 2007, 129, 2200–2201 10.1021/ja066869k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnert N.; Sage J. T.; Silvernail N.; Scheidt W. R.; Alp E. E.; Sturhahn W.; Zhao J. Inorg. Chem. 2010, 49, 7197–7215 10.1021/ic1010677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benko B.; Yu N. T. Proc. Natl. Acad. Sci. U. S. A. 1983, 80, 7042–7046 10.1073/pnas.80.22.7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulat F.; Berto T. C.; DeBeer George S.; Goodrich L.; Praneeth V. K. K.; Sulok C. D.; Lehnert N. Inorg. Chem. 2008, 47, 11449–11451 10.1021/ic801626w. [DOI] [PubMed] [Google Scholar]
- Peng Q.; Pavlik J. W.; Scheidt W. R.; Wiest O. J. Chem. Theory Comput. 2012, 8, 214–223 10.1021/ct2006456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conradie J.; Ghosh A. J. Phys. Chem. B 2007, 111, 12621–12624 10.1021/jp074480t. [DOI] [PubMed] [Google Scholar]
- Hopmann K. H.; Ghosh A.; Noodleman L. Inorg. Chem. 2009, 48, 9155–9165 10.1021/ic9008784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. J. Chem. Phys. 2006, 125, 194101. 10.1063/1.2370993. [DOI] [PubMed] [Google Scholar]
- Zhao X. J.; Sampath V.; Caughey W. S. Biochem. Biophys. Res. Commun. 1995, 212, 1054–1060 10.1006/bbrc.1995.2076. [DOI] [PubMed] [Google Scholar]
- Hendriks J.; Warne A.; Gohlke U.; Haltia T.; Ludovici C.; Lübben M.; Saraste M. Biochemistry 1998, 37, 13102–13109 10.1021/bi980943x. [DOI] [PubMed] [Google Scholar]
- Flock U.; Thorndycroft F. H.; Matorin A. D.; Richardson D. J.; Watmough N. J.; Ädelroth P. J. Biol. Chem. 2008, 283, 3839–3845 10.1074/jbc.M704615200. [DOI] [PubMed] [Google Scholar]
- Lachmann P.; Huang Y.; Reimann J.; Flock U.; Adelroth P. J. Biol. Chem. 2010, 285, 25531–25537 10.1074/jbc.M110.123984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries S.; Strampraad M. J. F.; Lu S.; Moënne-Loccoz P.; Schröder I. J. Biol. Chem. 2003, 278, 35861–35868 10.1074/jbc.M300857200. [DOI] [PubMed] [Google Scholar]
- Moënne-Loccoz P. Nat. Prod. Rep. 2007, 24, 610–620 10.1039/b604194a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg L. M.; Blomberg M. R. A.; Siegbahn P. E. M. Biochim. Biophys. Acta, Bioenerg. 2006, 1757, 240–252 10.1016/j.bbabio.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Blomberg M. R. A.; Siegbahn P. E. M. Biochemistry 2012, 51, 5173–5186 10.1021/bi300496e. [DOI] [PubMed] [Google Scholar]
- Sato N.; Ishii S.; Sugimoto H.; Hino T.; Fukumori Y.; Sako Y.; Shiro Y.; Tosha T. Proteins: Struct., Funct., Genet. 2014, 82, 1258–1271 10.1002/prot.24492. [DOI] [PubMed] [Google Scholar]
- Becke A. D. J. Chem. Phys. 1993, 98, 5648–5652 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Schäfer A.; Horn H.; Ahlrichs R. J. Chem. Phys. 1992, 97, 2571–2577 10.1063/1.463096. [DOI] [Google Scholar]