

Abstract
Autoimmune pancreatitis (AIP) can be chronic or recurrent, but frequently completely reversible after steroid treatment. A cystic lesion in AIP is a rare finding, and it can mimic a pancreatic cystic neoplasm. Difficulties in an exact diagnosis interfere with treatment, and surgery cannot be avoided in some cases. We report the history of a 63-year-old male presenting with jaundice and pruritus. AIP was confirmed by imaging and elevated IgG4 blood levels, and the patient completely recovered after corticosteroid therapy. One year later, he presented with a recurrent episode of AIP with elevated IgG4 levels, accompanied by the appearance of multiple intrapancreatic cystic lesions. All but 1 of these cysts disappeared after steroid treatment, but the remaining cyst in the pancreatic head was even somewhat larger 1 year later. Pancreatoduodenectomy was finally performed. Histology showed the wall of the cystic lesion to be fibrotic; the surrounding pancreatic tissue presented fibrosis, atrophy and lymphoplasmacytic infiltration by IgG4-positive cells, without malignant elements. Our case illustrates the rare possibility that cystic lesions can be part of AIP. These pseudocysts appear in the pancreatic segments involved in the autoimmune disease and can be a consequence of the local inflammation or related to ductal strictures. Steroid treatment should be initiated, after which these cysts can completely disappear with recovery from AIP. Surgical intervention may be necessary in some exceptional cases.
Key Words: Autoimmune pancreatitis, Pseudocyst, Neoplastic cystic lesion, Intraductal papillary mucinous neoplasm, Pancreatic cancer
Background
Autoimmune pancreatitis (AIP) is a benign disease that was first reported in 1961. It can occur as a primary disease of the pancreas (type 2) or as part of a systemic disease produced by IgG4-positive lymphoplasmacytic infiltration of the involved organs associated with elevated IgG4 blood levels (type 1). AIP-induced morphological changes are quickly reversible by corticosteroids; thus, this response has been used as one of the diagnostic criteria for the disease [1, 2].
The diagnosis can be made clinically based on characteristic images of AIP and elevated serum IgG4 levels; however, the gold standard is histology [1, 2]. The pancreatic disease can be diffuse or focal. In the latter case, it can be difficult to distinguish from pancreatic cancer.
The presence of cystic lesions in AIP is rare; there are only a few reports of AIP presenting with pancreatic cysts [3, 4, 5, 6, 7, 8, 9, 10]. In this paper, we describe a patient with AIP observed at our institute. One year after his documented complete recovery, he presented with a recurrent attack of AIP with multiples cysts in the whole pancreas, with an incomplete recovery after steroid treatment.
Case Presentation
A 63-year-old male with a history of psoriasis and hypothyroidism presented for a medical consultation with slight jaundice and pruritus that had started 5 days previously. The initial laboratory values showed bilirubin at 1.9 mg/dl (normal 0.2–1.3), alkaline phosphatase at 976 U/l (normal 38–126), γ-glutamyltranspeptidase at 565 U/l (normal 15–73), aspartate aminotransferase at 531 U/l (normal 17–59), alanine aminotransferase at 778 U/l (normal 21–72), amylase at 127 U/l (normal 30–110) and lipase at 608 U/l (normal ≤60).
Contrast-enhanced CT showed mild dilatation of the intra- and extrahepatic bile duct and an increased pancreatic volume with ill-defined contours, without focal lesion. AIP was suspected. Abdominal magnetic resonance imaging (MRI) was performed, showing a diffusely enlarged, ‘sausage-shaped’ pancreas without pancreatic duct dilatation (fig. 1). The IgG4 level was 804 mg% (normal <135); other autoantibodies were not present (antinuclear, anti-DNA or rheumatoid factor).
Fig. 1.

Initial MR image. a, b Diffuse enlargement of the pancreas with a ‘sausage-shaped’ aspect (arrows), but without pancreatic duct dilatation. c, d Diffuse parenchymal hypointensity on a T1-weighted image, showing heterogeneous enhancement with associated involvement of the intrapancreatic common bile duct (c), which in cholangiographic sequences demonstrates a marked irregularity and stenosis by compression (d, arrow). No cysts are to be seen.
Corticosteroid therapy was initiated after a diagnosis of AIP type 1 at a dose of 40 mg/day, corresponding to approximately 0.6 mg/kg/day. At the 2-month follow-up visit, the control laboratory tests and abdominal CT images revealed normal liver enzymes and normal intra- and extrahepatic bile ducts as well as a morphologically normal pancreas. The steroid was tapered and stopped after 20 weeks.
One year later, the patient presented again with epigastric pain, associated with the history of an 8-kg loss of body weight (from an initial 74 to 66 kg) in 2 months. The laboratory tests showed normal liver values, a mild rise in pancreatic enzymes (amylase at 282 U/I and lipase at 163 U/I). MRI was performed; moderate atrophy of the pancreatic parenchyma was seen, and multiple intrapancreatic cysts had appeared in the whole pancreas – without communication with the main pancreatic duct, which was not dilated. The IgG4 level was again high (820 mg%).
Based on his previous history and the elevated IgG4 level, we hypothesized that cystic lesions formed part of the recurrence of AIP. Corticosteroid therapy was thus started at a similar dose of 40 mg/day. A marked reduction, as well as an almost complete disappearance of the small cystic lesions, was found on a control MR image of the abdomen 6 weeks later (fig. 2), albeit with persistence of the major lesion in the pancreatic head. Continuation of steroid treatment was indicated – which was finally suspended by the patient 4 months later.
Fig. 2.
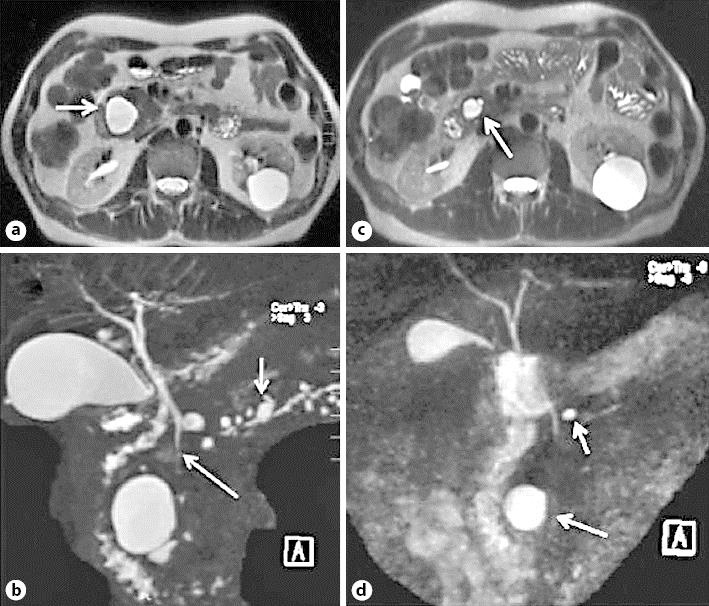
MR image 1 year after the initial diagnosis. a, b Signs of atrophy of the pancreatic parenchyma and appearance of multiple cysts, the largest in the head (arrow) and several smaller cystic lesions also in the body and tail (arrows), not communicating with the pancreatic duct. c, d Control images after 6 weeks of corticosteroid treatment showing almost complete regression of the cysts displayed in a and b; only the cyst in the head persists, with a significant decrease in size (arrows).
One year later, he was free of any abdominal symptoms at the follow-up. MRI showed signs of pancreatic atrophy and a 4-cm cyst in the pancreatic head that was slightly larger when compared to the previous images (fig. 3). This time, the patient became anxious; he wanted to exclude any other origin of the growing cyst than AIP, and he did not want to receive steroid (and immunosuppressive) therapy again without histological examination. Endoscopic ultrasound was performed, which showed a large cystic lesion of about 3.9 × 3.6 cm with a thick wall. Unfortunately, the attempt to puncture it with a fine needle to access the cyst through the duodenal wall failed. Serum tumor markers including carbohydrate antigen 19-9 and carcinoembryonic antigen were normal.
Fig. 3.

MR image 2 years after the diagnosis. In half-Fourier acquisition single-shot turbo spin-echo sequences, the pancreas is markedly atrophic without pancreatic duct dilatation. A dominant cystic lesion persists in the head (b, arrow), and it is slightly larger than on the previous images.
While the whole clinical history, alterations in laboratory values and good response to repeated steroid treatment argued for AIP and its complications, without any cyst fluid analysis it was impossible to exclude a premalignant true cystic lesion, eventually intraductal papillary mucinous neoplasms (IPMN). Thus, after a conversation with the patient, it was finally decided to conduct pancreatic resection.
At surgery, a solid cystic lesion was found in the pancreatic head. Its content was a brownish fluid, but no mucin was found. A rapid biopsy did not reveal any suspicion of malignancy; only inflammatory cells were detected. Pancreatoduodenectomy was performed, with resection in the normal pancreatic tissue.
A nodular mass of about 6 cm was found in the resected pancreatic head, with a cyst inside of this nodule. Diffuse lymphoplasmacytic infiltration was found in the pancreatic tissue, with an increased number of eosinophils and some histiocytes. This inflammatory infiltration was more marked in the nodular mass, with atrophy of the pancreatic parenchyma and fibrosis. Inside this nodule, the cystic cavity was surrounded by a fibrotic wall, without epithelial cells. A slight, irregular ductal dilation was observed in the whole resected specimen, without identifying any relationship of the ductal changes to the cystic lesion. The inflammatory cells showed a strong positivity for IgG4. These findings clearly confirmed that the lesion was a focal manifestation of the AIP, without malignant elements (fig. 4). No maintenance therapy was started after the surgery.
Fig. 4.

a Macroscopic photo showing serial sections of a solid cystic pancreatic lesion, with a thick fibrotic wall (arrows). b Histological picture showing pancreatic atrophy, lymphoplasmacytic infiltration and storiform fibrosis. Some collapsed remnants of small ducts can be seen (arrows). HE. ×40. c Histological picture. Dense inflammatory lymphoplasmacytic infiltration, with some histiocytes and eosinophils (arrows). HE. ×400. d Plasma cells showing positivity for IgG4 (arrows). ×400.
At present, 3 years later, the patient is doing well, without symptoms and in occasional follow-up, with normal pancreatic exocrine and endocrine function, and without focal lesions in the pancreatic remnant. Serum total IgG and IgG4 levels are normal as well.
Discussion
AIP is a rare and benign disease of the pancreas. Two types of AIP have been identified: type 1 is associated with extrapancreatic manifestations and elevated levels of IgG4-positive cells, while type 2 is mainly a pancreas-specific disorder with a lack of IgG4-positive cells. Its clinical presentation varies depending on the subtype; the most common presenting symptom is obstructive jaundice, like in our patient [1, 2].
In 2010, an international panel of experts developed a consensus on the diagnostic criteria for AIP. A diagnosis of type 1 AIP can be made with a dynamic CT and/or MRI showing typical features of diffuse pancreatic enlargement with delayed enhancement and pancreatic ductal strictures without significant dilatation, associated with elevated serum IgG4 levels. In contrast, a definitive diagnosis of type 2 AIP requires histology. The traditional ‘gold standard’ for the diagnosis of AIP is characteristic histology: in type 1 AIP, the pancreas demonstrates periductal lymphoplasmacytic infiltration rich in IgG4, storiform fibrosis and obliterative venulitis. In type 2 AIP, the affected pancreas demonstrates dominant neutrophilic infiltration in the ductal epithelium with duct destruction [1, 2].
There have only been a few reports of AIP presenting with pancreatic cysts, all of them from after 2000. These cysts are rare, probably due to the absence of severe tissue necrosis and/or lack of stasis of the pancreatic juice in this condition [3, 4, 7]. The pathomechanism of their formation is not clear. In our patient, the cystic lesion was intrapancreatic; it had a fibrotic wall without epithelial cells and contained a brownish fluid and some debris, resembling the usual findings in noninfected pseudocysts and not those in true neoplastic cystic lesions. Pseudocysts were classified by Sarles et al. [11] in 1963 and later on by D'Egidio and Schein [12] in 1991, who distinguished 3 types: postnecrotic pseudocysts (I) in acute pancreatitis, (II) in acute-to-chronic inflammation and (III) always related to ductal stricture, by a mechanism of retention and communication with the strictured duct, characteristic in chronic pancreatitis. The classification of our patient's lesion is not easy; it can be classified as type II, or eventually type III, which means it fulfilled criteria for a pseudocyst in a chronic process. On one hand, AIP is considered to be a subtype of chronic pancreatitis, and the complication of a chronic process is consistent with this classification. On the other hand, AIP itself can be reversible, and patients may recover without residual pancreatic damage. In our patient, cystic lesions appeared parallel to the recurrence of AIP, but removal of these cystic lesions was quickly achieved by steroid treatment, possibly due to the acute strictures on the pancreatic ductal system during the acute flare-up of AIP and their rapid response to steroid treatment, with consecutive resolution of the stasis and retention of pancreatic secretion.
While true cystic lesions are premalignant in many cases, pseudocysts have no tendency toward malignant transformation; thus, an exact diagnosis is very important and could eventually avoid unnecessary surgery. However, this is difficult to achieve, because of a reported coexistence of IPMN with AIP [13]. The best method of diagnosis is puncture during endoscopic ultrasound and analysis of the cystic fluid. Unfortunately, we failed to obtain such material for technical reasons. However, in the case reported by Büchler and colleagues [5], carbohydrate antigen 19-9 and carcinoembryonic antigen levels were elevated in the pseudocystic fluid, a finding which does not permit to exclude a mucinous cystic lesion. Differentiating cystic pancreatic lesions continues to be a clinical challenge; therefore, resection of cystic lesions continues to be the treatment of choice if malignant cystic neoplasms cannot be ruled out [5].
There is general agreement that corticosteroid treatment should be started immediately after any appearance of pseudocysts in AIP because of the possibility of their regression with treatment [6, 7]. In effect, all but 1 lesion disappeared in our patient after short steroid treatment, and probably the largest one would have regressed with longer treatment.
One of the particularities of our case is the synchronous appearance of multiple cystic lesions in the whole pancreas. The first episode of AIP treated successfully 1 year previously permitted us to suspect recurrent AIP, which was confirmed by markedly elevated IgG4 levels. On the other hand, we found a practically normal pancreas at the end of the treatment 1 year earlier, without any cystic lesion, demonstrating that the cysts had recently formed, parallel to a recurrent AIP, as part of an acute process. All but 1 cyst disappeared quickly with steroid treatment.
There are some papers concerned with the association of AIP with IPMN [13] as well as with pancreatic malignancy [14, 15]. Unfortunately, we were unable to obtain material for cytological and biochemical analyses of the cyst's content, and the patient declined to agree to a repeated intervention. Therefore, treatment was not prolonged and we decided to conduct the surgical resection. It may be worth noting that the patient's AIP was recurrent a couple of times. However, after surgical resection of the only active focus of the disease, no more recurrences have been observed.
Conclusion
Our case highlights the clinical problem of differentiating cystic pancreatic lesions in a patient with AIP. Cyst formation can be part of the autoimmune disease, and in consequence, these pseudocysts can completely disappear under steroid treatment. If the lesion persists in spite of adequate treatment, definitive exclusion of neoplastic characteristics can be difficult.
Statement of Ethics
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964 and later revisions. Retrospective evaluation of the clinical records was approved by the Ethics Committee of the Hospital Clínico Universidad de Chile. Informed consent was obtained from the present patient for being included in the study and report.
Disclosure Statement
The authors have nothing to declare.
References
- 1.O'Reilly D, Malde D, Duncan T, Rao M, Filobbos R. Review of the diagnosis, classification and management of autoimmune pancreatitis. World J Gastrointest Pathophysiol. 2014;5:71–81. doi: 10.4291/wjgp.v5.i2.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ketwaroo G, Sheth S. Autoimmune pancreatitis. Gastroenterol Rep (Oxf) 2013;1:27–32. doi: 10.1093/gastro/got011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nishimura T, Masaoka T, Suzuki H, Aiura K, Nagata H, Ishii H. Autoimmune pancreatitis with pseudocysts. J Gastroenterol. 2004;39:1005–1010. doi: 10.1007/s00535-004-1436-4. [DOI] [PubMed] [Google Scholar]
- 4.Muraki T, Hamano H, Ochi Y, Arakura N, Takayama M, Komatsu K, Komiyama Y, Kawa S, Uehara T, Kiyosawa K. Corticosteroid-responsive pancreatic cyst found in autoimmune pancreatitis. J Gastroenterol. 2005;40:761–766. doi: 10.1007/s00535-005-1622-z. [DOI] [PubMed] [Google Scholar]
- 5.Welsch T, Kleeff J, Esposito I, Büchler M, Friess H. Autoimmune pancreatitis associated with a large pancreatic pseudocyst. World J Gastroenterol. 2006;12:5904–5906. doi: 10.3748/wjg.v12.i36.5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamisawa T, Anjiki H, Egawa N. Rapid disappearance of a pancreatic cyst after steroid therapy in a patient with autoimmune pancreatitis. Clin Gastroenterol Hepatol. 2008;6:e33–e34. doi: 10.1016/j.cgh.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 7.Sohn JW, Cho CM, Jung MK, Park SY, Jeo SW. A case of autoimmune pancreatitis manifested by a pseudocyst and IgG4-associated cholangitis. Gut Liver. 2012;6:132–135. doi: 10.5009/gnl.2012.6.1.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawakami H, Kuwatani M, Shinada K, Yamato H, Hirano S, Kondo S, Yonemori A, Itoh T, Matsuno Y, Asaka M. Autoimmune pancreatitis associated with hemorrhagic pseudocysts: a case report and literature review. Intern Med. 2008;47:603–608. doi: 10.2169/internalmedicine.47.0731. [DOI] [PubMed] [Google Scholar]
- 9.Kraemer A, Lewin M, Balladur P, Mourra N, Tiret E, Paye F. Autoimmune pancreatitis mimicking an intra-ductal mucinous papillary neoplasm of the pancreas: an original case (in French) Gastroenterol Clin Biol. 2008;32:635–639. doi: 10.1016/j.gcb.2007.12.027. [DOI] [PubMed] [Google Scholar]
- 10.Takita M, Itoh T, Matsumoto S. Autoimmune chronic pancreatitis with IgG4-related pancreatic pseudocyst in a patient undergoing total pancreatectomy followed by autologous islet transplantation: a case report. Pancreas. 2013;42:175–177. doi: 10.1097/MPA.0b013e3182546e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarles H, Martin M, Camatte R, Sarles JC. Le démembrement des pancréatites: les pseudokystes des pancréatites aigues et des pancréatites chroniques. Presse Med. 1963;5:237–240. [PubMed] [Google Scholar]
- 12.D'Egidio A, Schein M. Pancreatic pseudocysts: a proposed classification and its management implications. Br J Surg. 1991;78:981–984. doi: 10.1002/bjs.1800780829. [DOI] [PubMed] [Google Scholar]
- 13.Urata T, Naito Y, Izumi Y, Takekuma Y, Yokomizo H, Nagamine M, Fukuda S, Notohara K, Hifumi M. Localized type 1 autoimmune pancreatitis superimposed upon preexisting intraductal papillary mucinous neoplasms. World J Gastroenterol. 2013;19:9127–9132. doi: 10.3748/wjg.v19.i47.9127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shiokawa M, Kodama Y, Yoshimura K, Kawanami C, Mimura J, Yamashita Y, Asada M, Kikuyama M, Okabe Y, Inokuma T, Ohana M, Kokuryu H, Takeda K, Tsuji Y, Minami R, Sakuma Y, Kuriyama K, Ota Y, Tanabe W, Maruno T, Kurita A, Sawai Y, Uza N, Watanabe T, Haga H, Chiba T. Risk of cancer in patients with autoimmune pancreatitis. Am J Gastroenterol. 2013;108:610–617. doi: 10.1038/ajg.2012.465. [DOI] [PubMed] [Google Scholar]
- 15.Ikeura T, Miyoshi H, Uchida K, Fukui T, Shimatani M, Fukui Y, Sumimoto K, Matsushita M, Takaoka M, Okazaki K. Relationship between autoimmune pancreatitis and pancreatic cancer: a single-center experience. Pancreatology. 2014;14:373–379. doi: 10.1016/j.pan.2014.04.029. [DOI] [PubMed] [Google Scholar]