

Abstract
N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) is a ubiquitous tetrapeptide hydrolyzed almost exclusively by angiotensin-converting enzyme (ACE). Chronic treatment with Ac-SDKP decreases cardiac and renal fibrosis and inflammatory cell infiltration in hypertensive rats. However, very little is known about endogenous synthesis of Ac-SDKP, except that thymosin-β4 may be the most likely precursor. Two enzymes are potentially able to release Ac-SDKP from thymosin-β4: prolyl oligopeptidase (POP) and endoproteinase asp-N. POP is widely present and active in several tissues and biological fluids, whereas endoproteinase asp-N appears to be lacking in mammals. Therefore, we hypothesized that POP is the main enzyme involved in synthesizing the antifibrotic peptide Ac-SDKP. We investigated in vitro and in vivo production of Ac-SDKP. Using kidney cortex homogenates, we observed that Ac-SDKP was generated in a time-dependent manner in the presence of exogenous thymosin-β4, and this generation was significantly inhibited by several POP inhibitors (POPi), Z-prolyl-prolinal, Fmoc-prolyl-pyrrolidine-2-nitrile, and S17092. Long-term administration of S17092 in rats significantly decreased endogenous levels of Ac-SDKP in the plasma (from 1.76±0.2 to 1.01±0.1 nM), heart (from 2.31±0.21 to 0.83±0.09 pmol/mg protein), and kidneys (from 5.62±0.34 to 2.86±0.76 pmol/mg protein). As expected, ACE inhibitors significantly increased endogenous levels of Ac-SDKP in the plasma, heart, and kidney, whereas coadministration of POPi prevented this increase. We concluded that POP is the main enzyme responsible for synthesis of the antifibrotic peptide Ac-SDKP.
Keywords: angiotensin-converting enzyme, heart, kidney
N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) is a tetrapeptide normally present in organs and biological fluids of humans and experimental animals.1–3 It was originally described as a natural inhibitor of pluripotent hematopoietic stem cells, preventing entry into the S phase.4 Recently, Ac-SDKP was found to be hydrolyzed almost exclusively by angiotensin-converting enzyme (ACE). After treatment with ACE inhibitors (ACEi), its plasma concentration increased 5-fold.5–7 Thus, some of the protective effects of ACEi may be mediated by an increase in Ac-SDKP. Our laboratory has recently reported that Ac-SDKP prevents fibroblast proliferation and collagen synthesis in vitro8 and that long-term treatment decreases cardiac and renal fibrosis and inflammatory cell infiltration in hypertensive rats without altering blood pressure.9,10 However, very little is known about endogenous synthesis of Ac-SDKP and the enzymes involved in the release of this tetrapeptide from its precursor thymosin-β4. Thymosin-β4, the most abundant β-thymosin in mammals,11 was reported to be the most likely precursor of the tetrapeptide because it possesses the sequence Ac-SDKP in its N-terminus, and thus a single cleavage (Pro4-Asp5) would be sufficient to release the tetrapeptide.12,13 However, we could find no reports addressing the enzymes involved in the production of Ac-SDKP in vivo.
Two enzymes are potentially able to cleave the Pro4–Asp5 bond of thymosin-β4 to release Ac-SDKP: prolyl oligopeptidase (POP), which cleaves the carboxyl end of proline, and endoproteinase asp-N, which cleaves the amino end of aspartate. Lenfant et al previously reported that endoproteinase asp-N is the main enzyme responsible for Ac-SDKP synthesis, based on in vitro studies with a bacterial enzyme.12 However, we know of no evidence that endoproteinase asp-N is present in mammals, which would suggest that it plays little or no role in production of Ac-SDKP in vivo. The other candidate enzyme, POP, belongs to a new class of serine peptidases (clan SC, family S9) unrelated to the well-known trypsin and subtilisin families14 and is able to hydrolyze a wide variety of proline-containing peptides.15 Unlike endoproteinase asp-N, POP is widely present and active in human and animal tissues and biological fluids.16–19 Therefore, we hypothesized that POP is the main enzyme responsible for release of the antifibrotic peptide Ac-SDKP. We tested whether POP is involved in release of Ac-SDKP from thymosin-β4 in vitro, and whether treatment with a specific POP inhibitor (POPi) would lower endogenous levels of Ac-SDKP in vivo.
Methods
Animals
We used 10-week-old male Sprague-Dawley rats (Charles River Laboratories). All surgical procedures were conducted with the animals under pentobarbital anesthesia (50 mg/kg IP). The study was approved by the Institutional Animal Care and Use Committee (IACUC) of Henry Ford Health System.
Ac-SDKP Determinations
Ac-SDKP in plasma, urine, and tissue was measured as described by Azizi et al,6 using a commercially available enzyme immunoassay kit (Cayman Chemicals). Blood was collected in a prechilled syringe containing 10 μL heparin and 10 μL 10−3 mol/L lisinopril per mL of blood, and the plasma was separated. The purpose of adding an ACEi was to prevent Ac-SDKP degradation during sample processing. Urine was collected directly from the bladder in a prechilled syringe containing 10 μL 10−3 mol/L lisinopril. Tissue samples were homogenized with cold 50 mmol/L phosphate-saline buffer (pH=7.4) containing 10−5 mol/L lisinopril. Ac-SDKP was extracted from an aliquot of plasma or tissue homogenate (150 and 100 μL, respectively) and processed according to the manufacturer’s instructions. Urine was assayed without purification in 1:100 dilutions. We observed no cross-reactivity with thymosin-β4. Total proteins were measured by the Bradford method and creatinine by the Jaffé reaction.
Effect of POPi on Ac-SDKP Synthesis In Vitro
Homogenates of kidney cortex were chosen because we have evidence that Ac-SDKP has renal antifibrotic effects.10 Tissue was desegregated and diluted up to 150 μg/mL total protein with cold phosphate–saline buffer; 100-μL aliquots of the homogenate were incubated at 37°C with: (1) thymosin-β4 vehicle and POPi vehicle (time control); (2) thymosin-β4 (2 μmol/L) plus POPi vehicle; (3) thymosin-β4 plus POPi (0.01 μmol/L); or (4) thymosin-β4 plus POPi (0.1 μmol/L). Thymosin-β4, purified from bovine spleen (gift of D. Safer, University of Pennsylvania, Philadelphia, Pa),20 was dissolved in water. Three different POPi were used: Z-prolyl-prolinal (Z-Pro-Pro, Biomol), Fmoc-prolyl-pyrrolidine-2-nitrile (Fmoc-Pro-PyrrCN, a gift of S. Wilk, Mount Sinai School of Medicine, NY),21 and S17092 (a gift of P. Vanhoutte, Institut de Recherches Internationales Servier, France).22 Stock solutions of POPi were made with 20% DMSO, then diluted to their final concentration with water. POPi vehicle had the same percentage of DMSO. Ac-SDKP content was measured at 0, 20, 40, 80, and 160 minutes of incubation.
Effect of an Oral POPi on Endogenous Ac-SDKP
We tested whether long-term S17092 treatment would decrease endogenous Ac-SDKP by systemically blocking POP and whether it would also prevent the tetrapeptide from increasing during ACE inhibition. Rats were randomly divided into: (1) vehicle (n=10); (2) POPi (S17092, 20 mg/kg per day; n=7);23 (3) ACEi (captopril, 100 mg/kg per day; n=7); and (4) ACEi plus POPi (n=7). S17092 was mixed with 0.5 mL peanut oil to obtain an emulsion with the proper dose according to the body weight. POPi or vehicle was administered once daily by gavage. ACEi was given in drinking water. After 7 days of treatment, rats were anesthetized and blood, urine, heart, and kidneys were collected and kept at −70°C until Ac-SDKP assay.
Effect of S17092 on ACE Activity In Vivo
Rats were randomly divided into: (1) vehicle (n=4); (2) POPi (S17092, 40 mg/kg per day; n=4); and (3) ACEi (captopril, 100 mg/kg per day; n=5). We used a higher dose of S17092 than in the previous protocol to ensure maximum effect. After 5 days of treatment, rats were anesthestized and the femoral artery and vein catheterized for mean arterial pressure (MAP) measurement and drug administration, respectively. After a 20-minute stabilization period, MAP was recorded for 5 minutes (basal), followed by different doses of bradykinin (Bk) (25, 50, and 100 ng) or angiotensin I (Ang I) (12, 25, and 50 ng). After each dose, MAP was allowed to stabilize for 15 minutes.
Data Analysis
In vitro experiments and changes in MAP were analyzed using ANOVA, and the Fisher protected least significant difference (Fisher LSD) was used to adjust for multiple comparisons. Ac-SDKP levels in vivo were analyzed using the Wilcoxon nonparametric rank-sum test. P<0.05 was considered significant. Data are expressed as mean±SE.
Results
Effect of POPi on Ac-SDKP Synthesis In Vitro
Figure 1 shows time-dependent production of Ac-SDKP in vitro. Kidney cortex homogenates were incubated with exogenous thymosin-β4 in the presence or absence of different POPi (n=7). Panels A, B, and C show production of Ac-SDKP in the presence of Z-Pro-Pro, Fmoc-Pro-PyrrCN, and S17092, respectively. When homogenates were incubated in the presence of exogenous thymosin-β4, Ac-SDKP increased significantly in a time-dependent manner compared with time control (all panels). The highest concentration of Z-Pro-Pro (0.1 μmol/L) significantly blocked Ac-SDKP synthesis, whereas a lower dose (0.01 μmol/L) was ineffective (panel A); 0.1 μmol/L Fmoc-Pro-PyrrCN significantly inhibited Ac-SDKP production, whereas 0.01 μmol/L gave only partial inhibition (panel B). S17092 at high and low concentrations completely inhibited Ac-SDKP synthesis (panel C).
Figure 1.

Time-dependent generation of Ac-SDKP in vitro in the presence of exogenous thymosin-β4 and various POP inhibitors; 100-μL aliquots of the kidney homogenate were incubated at 37°C with: (1) thymosin-β4 vehicle and POPi vehicle (time control); (2) thymosin-β4 (2 μmol/L)+POPi vehicle; (3) thymosin-β4+POPi (0.01 μmol/L); or (4) thymosin-β4+POPi (0.1 μmol/L). A, Ac-SDKP generation in kidney homogenates containing exogenous thymosin-β4 in the presence or absence of Z-prolyl-prolinal (Z-pro-pro) at 0.01 and 0.1 μmol/L; **P<0.001 vs time control; †P<0.01 and ‡P<0.001 vs thymosin-β4. B, Ac-SDKP generation in kidney homogenates containing exogenous thymosin-β4 in the presence or absence of Fmoc-prolyl-pyrrolidine-nitrile (Fmoc-Pro-PyrrCN) at 0.01 and 0.1 μmol/L; **P<0.001 vs time control; †P<0.05 and ‡P<0.001 vs thymosin-β4. C, Ac-SDKP generation in kidney homogenates containing exogenous thymosin-β4 in the presence or absence of S17092 at 0.01 and 0.1 μmol/L; **P<0.001 vs time control; †P<0.01 and ‡P<0.001 vs thymosin-β4. Data are expressed as mean±SE.
Effect of an Oral POPi on Endogenous Ac-SDKP
Figure 2 shows concentration of Ac-SDKP in plasma and urine, and Figure 3 shows Ac-SDKP content in the heart and kidney after 7 days of S17092 with or without captopril. Plasma levels of Ac-SDKP were slightly lower in rats treated with S17092 alone compared with vehicle but did not reach statistical significance, whereas urinary levels were similar in both groups. Plasma and urinary Ac-SDKP increased significantly after captopril treatment, and this increase was significantly prevented by coadministration of S17092. Chronic treatment with S17092 alone significantly decreased endogenous levels of Ac-SDKP in the heart and renal cortex compared with vehicle. Captopril significantly increased endogenous Ac-SDKP, whereas coadministration of S17092 significantly prevented this increase.
Figure 2.
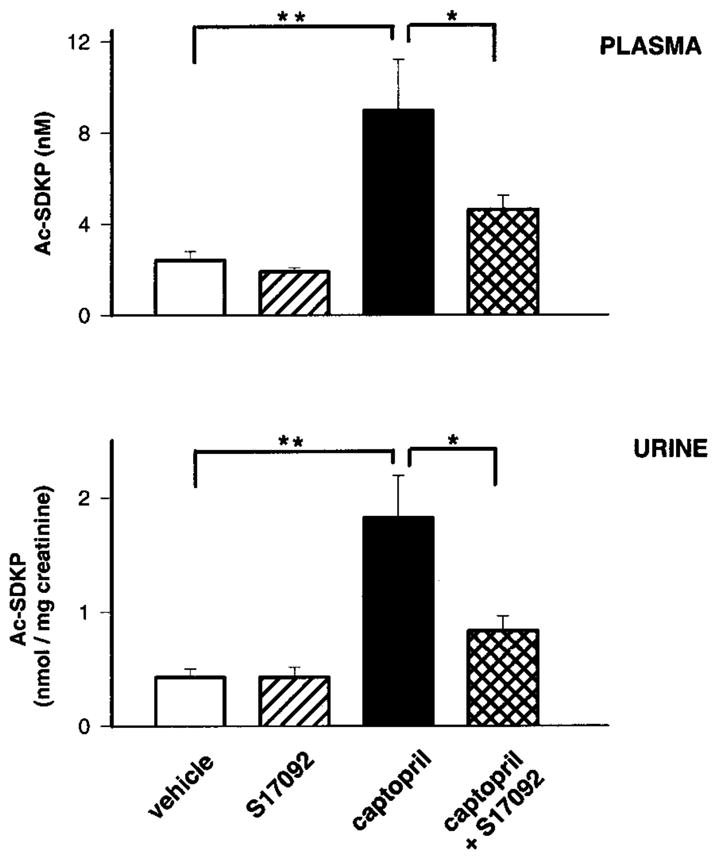
Plasma and urinary levels of Ac-SDKP in rats chronically treated with POPi. Ac-SDKP concentrations in the plasma and urine of rats chronically treated with vehicle (peanut oil), S17092 (20 mg/kg per day), captopril (100 mg/kg per day), or captopril+S17092. **P<0.01 and *P<0.05. Data are expressed as mean±SE.
Figure 3.
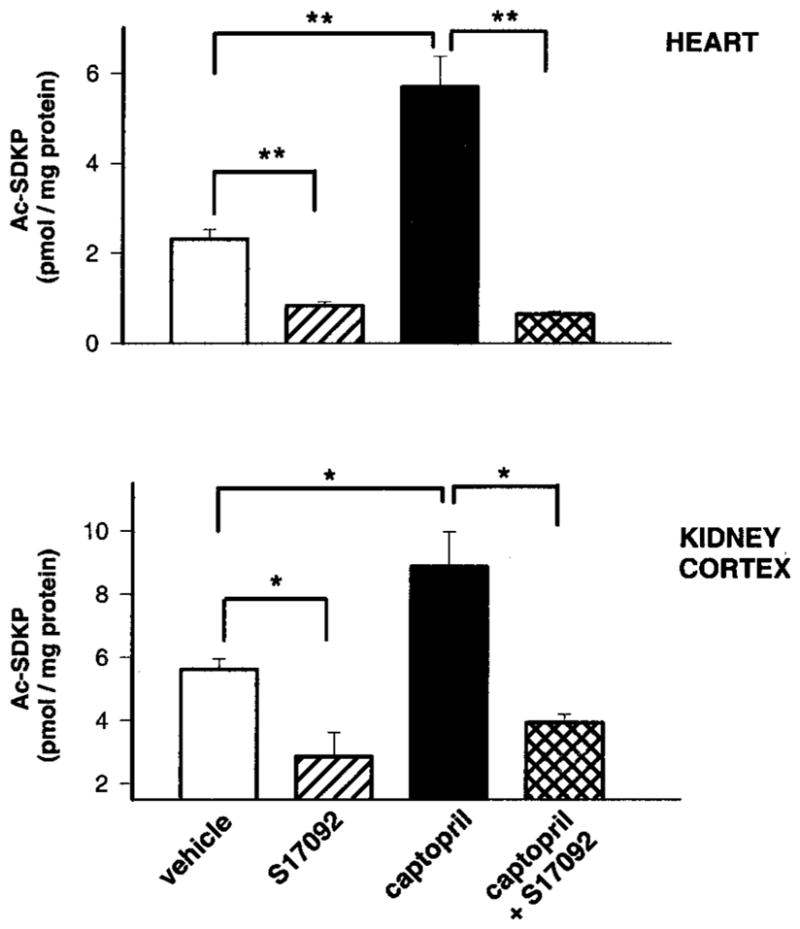
Endogenous levels of Ac-SDKP in rats treated with POPi long-term. Amount of Ac-SDKP per mg protein in heart and kidney cortex of rats chronically treated with vehicle (peanut oil), S17092 (20 mg/kg per day), captopril (100 mg/kg per day), or captopril+S17092. **P<0.01 and *P<0.05. Data are expressed as mean±SD.
Effect of S17092 on ACE Activity In Vivo
We tested whether a high dose of S17092 can inhibit ACE. In this protocol, we doubled the dose of S17092 used to measure endogenous Ac-SDKP to maximize possible effects of POPi on ACE activity and avoid problems related to insufficient dosage. Figure 4 shows changes in MAP caused by bolus injection of increasing amounts of Bk (panel A) and Ang I (panel B) in rats treated with vehicle, captopril, or S17092. Because Bk and Ang I are ACE substrates, they were used as indicators of ACE activity. We found that in rats treated with captopril, Bk-induced hypotension was significantly enhanced and the Ang I-induced pressor effect was significantly inhibited compared with vehicle. However, the changes in MAP seen in rats treated with S17092 were no different from vehicle. Plasma Ac-SDKP was also measured after the experiments, and we found that POPi alone at 40 mg/kg per day significantly decreased Ac-SDKP compared with vehicle (1.76±0.2 versus 1.01±0.1 nM; P<0.05).
Figure 4.

Changes in mean arterial blood pressure of rats treated with POPi. Arterial blood pressure was monitored and changes in mean arterial pressure plotted during bolus injections of bradykinin (A) or angiotensin I (B) in rats treated long-term with vehicle (peanut oil), S17092 (40 mg/kg per day), or captopril (100 mg/kg per day). **P<0.01 and *P<0.05. Data are expressed as mean±SE.
Discussion
We found that: (1) Ac-SDKP generation in kidney homogenates increased in a time-dependent manner in the presence of exogenous thymosin-β4, and this process was blocked significantly by several POPi; (2) rats that received POPi long-term showed significantly lower levels of endogenous Ac-SDKP in the heart and kidney; and (3) ACEi significantly increased endogenous Ac-SDKP in the plasma, urine, heart, and kidney, whereas coadministration of POPi significantly prevented this increase. We believe this is the first study showing that POP is the enzyme responsible for production of the antifibrotic peptide Ac-SDKP in vitro and in vivo. Thymosin-β4 was postulated to be the most likely precursor for the tetrapeptide, because it possesses the sequence Ac-SDKP in its N-terminus. Thymosin-β4 was originally found in the thymus but is widely present in mammals and distributed ubiquitously in tissue and in the circulation.11 Its structure is the same in all mammals studied so far, including humans, rats, dogs, mice, cats, calves, pigs, sheep, and horses, except for the rabbit, which has Ala4 instead of Pro4. Although we have no direct evidence that Ac-SDKP is released from thymosin-β4 in vivo, precursor and product peptides have been colocalized to many tissues and their concentration shows a positive correlation.2 Moreover, in the present study we found that addition of exogenous thymosin-β4 increased Ac-SDKP production in vitro, supporting this assumption.
Lenfant et al previously reported that endoproteinase asp-N was the enzyme responsible for release of Ac-SDKP from thymosin-β4 in vitro.12 They arrived at this conclusion by incubating pure bacterial endoproteinase asp-N and POP with 1 mg thymosin-β4 for 18 hours. However, we believe the in vitro conditions used in their study are questionable for the following reasons: (1) bacterial enzymes could not properly reproduce the in vivo mammalian enzymatic and biochemical environment; and (2) incubation of extremely high amounts of substrate for a long time could lead to loss of enzyme specificity. In the present study, we clearly demonstrated that POP is responsible for Ac-SDKP production both in vitro and in vivo, because generation of the tetrapeptide was significantly blocked and endogenous levels were significantly decreased by specific POP inhibitors.
One particular characteristic of POP is that it only cleaves oligopeptides no larger than 30 amino acids.17 Its catalysis is controlled by a gating filter mechanism that only allows small peptides to gain access to the active site; in this way, larger structural peptides and proteins are protected from proteolysis.24 Because thymosin-β4 has 43 amino acids, we believe it must be hydrolyzed into a smaller peptide by some other peptidases, resulting in truncation of thymosin-β4 before POP hydrolysis and release of Ac-SDKP from the N-terminus. Although we have not tested whether the real substrate for POP would be a truncated rather than an intact thymosin-β4, it is still important to show that POP is responsible for biosynthesis of Ac-SDKP. This could explain why in Lenfant’s studies of pure POP did not generate Ac-SDKP from thymosin-β4, because the incubates only contained the pure bacterial POP and thymosin-β4, and the absence of other peptidases made it impossible to produce a truncated thymosin-β4 as a substrate for POP and finally release Ac-SDKP. In contrast, our in vitro studies using tissue homogenates have the advantage of providing the proper enzymatic environment for hydrolysis and release of Ac-SDKP from thymosin-β4, because peptidases normally found in the kidney are still present in the homogenate.
We blocked Ac-SDKP synthesis using 3 different POPi, which have been developed to treat mental disorders such as depression, anorexia and bulimia nervosa, and Alzheimer disease.23,25–27 In general, peptide aldehyde analogs of the acyl portion of protease substrates are potent competitive inhibitors of serine and sulfhydryl proteases, because the aldehyde interacts with the active site serine to form a tetrahedral transition-state analog. Based on this, S. Wilk and M. Orlowski synthesized Z-prolyl-prolinal (Ki 14 nM).28 Fmoc-Pro-PyrrCN (Ki 5 nM) was also synthesized by S. Wilk;21 contrary to Z-pro-prolinal, this is a reversible inhibitor that could easily be synthesized from commonly available intermediates using a simple 2-step procedure. S17092 (Ki 1.5 nM) was developed by the Institut de Recherches Internationale Servier22 and was described as a highly potent, specific, and cell-permeant inhibitor of proline endopeptidase. All of these inhibitors were tested for their capacity to block several other proteolytic enzymes at a concentration >1000-fold greater than their Ki for POP, and all failed to do so. Although these inhibitors are reportedly highly specific and do not block other peptidases, by using several compounds we increased the degree of certainty that proteolytic release of Ac-SDKP from thymosin-β4 is caused by POP.
Endogenous Ac-SDKP in the heart and kidney was significantly lower in rats treated with POPi alone (lower dose) compared with vehicle, but not in the plasma. However, plasma levels of Ac-SDKP were significantly lower when a higher dose was used. It could be argued that Ac-SDKP in plasma did not decrease significantly because plasma ACE activity is inhibited by POPi; if so, Ac-SDKP degradation is blocked and its concentration would not decrease (Figure 5 shows clarification of enzymatic reactions). Therefore, we tested whether ACE activity is affected during chronic POPi treatment. We found that in rats treated with ACEi, Bk-induced vasodilatation was enhanced and Ang I-induced vasoconstriction was inhibited; however, in rats treated with a high dose of S17092, changes in MAP caused by acute administration of Bk and Ang I were no different from vehicle, suggesting that ACE activity is not affected by POPi. This confirms the findings of Barelli et al, who tested a saturating concentration of S17092 in vitro and observed that peptidase activity, including ACE, was not affected.22
Figure 5.
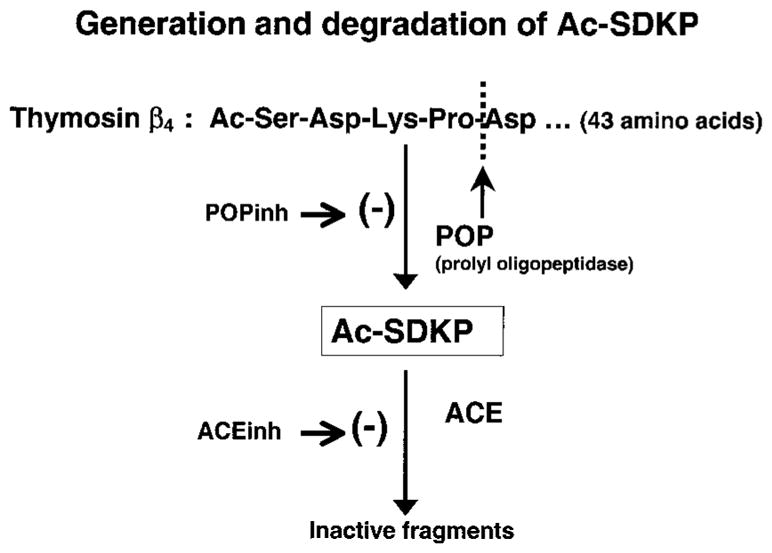
Schematic diagram illustrating enzymatic generation and degradation of Ac-SDKP.
Rats that received long-term ACEi treatment showed significantly increased endogenous levels of Ac-SDKP in the plasma, urine, heart, and kidney. This was as expected, because Ac-SDKP degradation is blocked by ACEi, and this was in accord with previous studies.5,6,29 By increasing Ac-SDKP levels with an ACEi, we wanted to show that simultaneous inhibition of tetrapeptide synthesis by coadministration of POPi could prevent Ac-SDKP accumulation.
Peptidases that are able to hydrolyze adjacent proline bonds are considered to have great metabolic importance, because the presence of a proline residue confers certain conformational restrictions to the peptide chain along with increased resistance to hydrolysis.30,31 POP is a member of the prolyl oligopeptidase family, a relatively new class of serine peptidases. It is involved in the maturation and degradation of many peptide hormones and neuropeptides such as angiotensins, kinins, vasopressin, and substance P,15,17 and degrades bradykinin (Bk) into inactive peptides while breaking down Ang I and II to form angiotensin 1–7. In this study, we have shown that POP is also involved in hydrolysis of thymosin-β4 to release the antifibrotic peptide Ac-SDKP.
Because POP has been described as a ubiquitous enzyme that is widely distributed in various tissues,17,19 Ac-SDKP can be produced locally in any organ and released into the circulation. Ac-SDKP was also found to be synthesized by bone marrow cells in long-term murine32 and human33 cultures, although we know of no direct evidence showing POP expression in bone marrow. We performed some preliminary experiments in vitro, incubating rabbit bone marrow homogenates with thymosin β4, and found that production of Ac-SDKP was blocked in the presence of POP inhibitors, similar to what we report in the present article involving kidney homogenates. Although we decided to use the kidney in future experiments because of its relevance to our research, based on our preliminary data we believe POP is present in bone marrow.
Perspectives
Increased extracellular matrix deposition is an important feature of target organ damage in hypertension, and we have reported that Ac-SDKP reduces collagen deposition in the heart and kidney of hypertensive rats by inhibiting fibroblast proliferation and decreasing macrophage infiltration.9,10 However, biosynthesis of Ac-SDKP has been poorly studied. We found that POP is the major enzyme involved in endogenous production of Ac-SDKP. Therefore, future studies in vivo involving long-term inhibition of Ac-SDKP synthesis will help us determine: (1) whether Ac-SDKP plays a physiological role in antagonizing pro-fibrotic stimuli such as Ang II and endothelin-1; and (2) whether endogenous Ac-SDKP is an important mediator in the protective effects of ACEi in hypertension, acting as an anti-inflammatory cytokine to prevent fibrosis and inflammation. Our present findings will help us understand some of the mechanisms by which fibrosis develops in diseases characterized by collagen deposition, such as hypertension, diabetes, and heart failure. Now that we know how Ac-SDKP is synthesized, we will be able to study whether POP activity and/or Ac-SDKP levels are affected during the development of these diseases, allowing fibrosis to progress.
Acknowledgments
We thank Dr Daniel Safer of the University of Pennsylvania, Philadelphia, who generously provided thymosin-β4; Dr Sherwin Wilk of the Mount Sinai School of Medicine, NY, who kindly provided Fmoc-prolyl-pyrrolidine-2-nitrile; and Dr Paul Vanhoutte, Institut de Recherches Internationales Servier, France, for his gift of S17092. This work was supported by NIH grants HL28982-21 (O.A.C.) and HL71806-01 (N.E.R.) and AHA grant 0335378Z (M.A.C.).
References
- 1.Pradelles P, Frobert Y, Créminon C, Liozon E, Massé A, Frindel E. Negative regulator of pluripotent hematopoietic stem cell proliferation in human white blood cells and plasma as analysed by enzyme immunoassay. Biochem Biophys Res Commun. 1990;170:986–993. doi: 10.1016/0006-291x(90)90489-a. [DOI] [PubMed] [Google Scholar]
- 2.Pradelles P, Frobert Y, Créminon C, Ivonine H, Frindel E. Distribution of a negative regulator of haematopoietic stem cell proliferation (AcSDKP) and thymosin β4 in mouse tissues. FEBS Lett. 1991;289:171–175. doi: 10.1016/0014-5793(91)81062-d. [DOI] [PubMed] [Google Scholar]
- 3.Stephan J, Melaine N, Ezan E, Hakovirta H, Maddocks S, Toppari J, Garnier D, Wdzieczak-Bakala J, Jegou B. Source, catabolism and role of the tetrapeptide N-acetyl-ser-asp-lys-Pro within the testis. J Cell Sci. 2000;113:113–121. doi: 10.1242/jcs.113.1.113. [DOI] [PubMed] [Google Scholar]
- 4.Lenfant M, Wdzieczak-Bakala J, Guittet E, Prome JC, Sotty D, Frindel E. Inhibitor of hematopoietic pluripotent stem cell proliferation: purification and determination of its structure. Proc Natl Acad Sci U S A. 1989;86:779–782. doi: 10.1073/pnas.86.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azizi M, Rousseau A, Ezan E, Guyene T-T, Michelet S, Grognet J-M, Lenfant M, Corvol P, Ménard J. Acute angiotensin-converting enzyme inhibition increases the plasma level of the natural stem cell regulator N-acetyl-seryl-aspartyl-lysyl-proline. J Clin Invest. 1996;97:839–844. doi: 10.1172/JCI118484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azizi M, Ezan E, Nicolet L, Grognet JM, Menard J. High plasma level of N-acetyl-seryl-aspartyl-lysyl-proline: a new marker of chronic angioten-sin-converting enzyme inhibition. Hypertension. 1997;30:1015–1019. doi: 10.1161/01.hyp.30.5.1015. [DOI] [PubMed] [Google Scholar]
- 7.Junot C, Gonzales M-F, Ezan E, Cotton J, Vazeux G, Michaud A, Azizi M, Vassiliou S, Yiotakis A, Corvol P, Dive V. RPX 407, a selective inhibitor of the N-domain of angiotensin I-converting enzyme, blocks in vivo the degradation of hemoregulatory peptide acetyl-Ser-Asp-Lys-Pro with no effect on angiotensin I hydrolysis. J Pharmacol Exp Ther. 2001;297:606–611. [PubMed] [Google Scholar]
- 8.Rhaleb N-E, Peng H, Harding P, Tayeh M, LaPointe MC, Carretero OA. Effect of N-acetyl-seryl-aspartyl-lysyl-proline on DNA and collagen synthesis in rat cardiac fibroblasts. Hypertension. 2001;37:827–832. doi: 10.1161/01.hyp.37.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rhaleb N-E, Peng H, Yang X-P, Liu Y-H, Mehta D, Ezan E, Carretero OA. Long-term effect of N-acetyl-seryl-aspartyl-lysyl-proline on left ventricular collagen deposition in rats with 2-kidney, 1-clip hypertension. Circulation. 2001;103:3136–3141. doi: 10.1161/01.cir.103.25.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng H, Carretero OA, Raij L, Yang F, Kapke A, Rhaleb N-E. Anti-fibrotic effects of N-acetyl-seryl-aspartyl-lysyl-proline on the heart and kidney in aldosterone-salt hypertensive rats. Hypertension. 2001;37:794–800. doi: 10.1161/01.hyp.37.2.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huff T, Müller CSG, Otto AM, Netzker R, Hannappel E. β-thymosins, small acidic peptides with multiple functions. Int J Biochem Cell Biol. 2001;33:205–220. doi: 10.1016/s1357-2725(00)00087-x. [DOI] [PubMed] [Google Scholar]
- 12.Lenfant M, Grillon C, Rieger KJ, Sotty D, Wdzieczak-Bakala J. Formation of acetyl-Ser-Asp-Lys-Pro, a new regulator of the hematopoietic system, through enzymatic processing of thymosin beta 4. Ann NY Acad Sci. 1991;628:115–125. doi: 10.1111/j.1749-6632.1991.tb17229.x. [DOI] [PubMed] [Google Scholar]
- 13.Huang WQ, Wang QR. Bone marrow endothelial cells secrete thymosin β 4 and AcSDKP. Exp Hematol. 2001;29:12–18. doi: 10.1016/s0301-472x(00)00634-2. [DOI] [PubMed] [Google Scholar]
- 14.Polgár L. Prolyl oligopeptidases. Methods Enzymol. 1994;244:188–200. doi: 10.1016/0076-6879(94)44016-6. [DOI] [PubMed] [Google Scholar]
- 15.Welches WR, Brosnihan KB, Ferrario CM. A comparison of the properties and enzymatic activities of three angiotensin processing enzymes: angiotensin converting enzyme, prolyl endopeptidase and neutral endopeptidase 24.11. Life Sci. 1993;52:1461–1480. doi: 10.1016/0024-3205(93)90108-f. [DOI] [PubMed] [Google Scholar]
- 16.Irazusta J, Silveira PF, Gil J, Varona A, Casis L. Effects of hydrosaline treatments on prolyl endopeptidase activity in rat tissues. Regul Pept. 2001;101:141–147. doi: 10.1016/s0167-0115(01)00277-4. [DOI] [PubMed] [Google Scholar]
- 17.Polgár L. The prolyl oligopeptidase family. Cell Mol Life Sci. 2002;59:349–362. doi: 10.1007/s00018-002-8427-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kimura A, Matsui H, Takahashi T. Expression and localization of prolyl oligopeptidase in mouse testis and its possible involvement in sperm motility. Zool Sci. 2002;19:93–102. doi: 10.2108/zsj.19.93. [DOI] [PubMed] [Google Scholar]
- 19.Goossens F, De Meester I, Vanhoof G, Scharpé S. Distribution of prolyl oligopeptidase in human peripheral tissues and body fluids. Eur J Clin Chem Clin Biochem. 1996;34:17–22. doi: 10.1515/cclm.1996.34.1.17. [DOI] [PubMed] [Google Scholar]
- 20.Safer D, Sosnick TR, Elzinga M. Thymosin β4 binds actin in an extended conformation and contacts both the barbed and pointed ends. Biochemistry. 1997;36:5806–5816. doi: 10.1021/bi970185v. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Wilk E, Wilk S. Inhibition of prolyl oligopeptidase by Fmoc-aminoacylpyrrolidine-2-nitriles. J Neurochem. 1996;66:2105–2112. doi: 10.1046/j.1471-4159.1996.66052105.x. [DOI] [PubMed] [Google Scholar]
- 22.Barelli H, Petit A, Hirsch E, Wilk S, De Nanteuil G, Morain P, Checler F. S 17092–1, a highly potent, specific and cell permeant inhibitor of human proline endopeptidase. Biochem Biophys Res Commun. 1999;257:657–661. doi: 10.1006/bbrc.1999.0366. [DOI] [PubMed] [Google Scholar]
- 23.Morain P, Lestage P, De Nanteuil G, Jochemsen R, Robin J-L, Guez D, Boyer P-A. S 17092: a prolyl endopeptidase inhibitor as a potential therapeutic drug for memory impairment. Preclinical and clinical studies. CNS Drug Rev. 2002;8:31–52. doi: 10.1111/j.1527-3458.2002.tb00214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fülöp V, Szeltner Z, Polgár L. Catalysis of serine oligopeptidases is controlled by a gating filter mechanism. EMBO Rep. 2000;11:277–281. doi: 10.1093/embo-reports/kvd048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kahyaoglu A, Haghjoo K, Kraicsovitz F, Jordan F, Polgar L. Benzyloxy-carbonylprolylprolinal, a transition-state analogue for prolyl oligopeptidase, forms a tetrahedral adduct with catalytic serine, not a reactive cysteine. Biochem J. 1997;322:839–843. doi: 10.1042/bj3220839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miura N, Shibata S, Watanabe S. Increase in the septal vasopressin content by prolyl endopeptidase inhibitors in rats. Neurosci Lett. 1995;196:128–130. doi: 10.1016/0304-3940(95)11821-d. [DOI] [PubMed] [Google Scholar]
- 27.Morain P, Robin JL, De Nanteuil G, Jochemsen R, Heidet V, Guez D. Pharmacodynamic and pharmacokinetic profile of S 17092, a new orally active prolyl endopeptidase inhibitor, in elderly healthy volunteers. A phase I study. Br J Clin Pharmacol. 2000;50:350–359. doi: 10.1046/j.1365-2125.2000.00270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilk S, Orlowski M. Inhibition of rabbit brain prolyl endopeptidase by N-benzyloxycarbonyl-prolyl-prolinal, a transition state aldehyde inhibitor. J Neurochem. 1983;41:69–75. doi: 10.1111/j.1471-4159.1983.tb11815.x. [DOI] [PubMed] [Google Scholar]
- 29.Junot C, Nicolet L, Ezan E, Gonzales M-F, Menard J, Azizi M. Effects of angiotensin-converting enzyme inhibition on plasma, urine, and tissue concentrations of hemoregulatory peptide acetyl-Ser-Asp-Lys-Pro in rats. J Cardiovasc Pharmacol. 1999;291:982–987. [PubMed] [Google Scholar]
- 30.Fischer G. Chemical aspects of peptide bond isomerization. Chem Soc Rev. 2000;29:119–127. [Google Scholar]
- 31.Vanhoof G, Goossens F, De Meester I, Hendriks D, Scharpé S. Proline motifs in peptides and their biological processing. FASEB J. 1995;9:736–744. [PubMed] [Google Scholar]
- 32.Wdzieczak-Bakala J, Fache MP, Lenfant M, Frindel E, Sainteny F. AcSDKP, an inhibitor of CFU-S proliferation is synthesized in mice under steady-state conditions and secreted by bone marrow in long term culture. Leukemia (Baltimore) 1990;4:235–237. [PubMed] [Google Scholar]
- 33.Li J, Volkov L, Comte L, Herve P, Praloran V, Charbord P. Production and consumption of the tetrapeptide AcSDKP, a negative regulator of hematopoietic stem cells, by hematopoietic microenvironmental cells. Exp Hematol. 1997;25:140–146. [PubMed] [Google Scholar]