

Abstract
By use of ESR and UV-vis spectral studies, this work identifies the protonation states of one-electron oxidized G:C (viz. G•+:C, G(N1-H)•:C(+H+), G(N1-H)•:C, and G(N2-H)•:C) in a DNA oligomer d[TGCGCGCA]2. Benchmark ESR and UV-vis spectra from one electron oxidized 1-Me-dGuo are employed to analyze the spectral data obtained in one-electron oxidized d[TGCGCGCA]2 at various pHs. At pH ≥7, the initial site of deprotonation of one-electron oxidized d[TGCGCGCA]2 to the surrounding solvent is found to be at N1 forming G(N1-H)•:C at 155 K. However, upon annealing to 175 K, the site of deprotonation to the solvent shifts to an equilibrium mixture of G(N1-H)•:C and G(N2-H)•:C. For the first time, the presence of G(N2-H)•:C in a ds DNA-oligomer is shown to be easily distinguished from the other prototropic forms, owing to its readily observable nitrogen hyperfine coupling (Azz(N2)= 16 G). In addition, for the oligomer in H2O, an additional 8 G N2-H proton HFCC is found. This ESR identification is supported by a UV-vis absorption at 630 nm which is characteristic for G(N2-H)• in model compounds and oligomers. We find that the extent of photo-conversion to the C1′ sugar radical (C1′•) in the one-electron oxidized d[TGCGCGCA]2 allows for a clear distinction among the various G:C protonation states which can not be easily distinguished by ESR or UV-vis spectroscopies with this order for the extent of photo-conversion: G•+:C > G(N1-H)•:C(+H+) >> G(N1-H)•:C. We propose that it is the G•+:C form that undergoes deprotonation at the sugar and this requires reprotonation of G within the lifetime of exited state.
Introduction
All high energy radiations result in near random ionizations of the bases and the sugar-phosphate backbone of DNA.1 The initial holes or cation radical sites (i.e., G•+, A•+, C•+, T•+, and [sugar-phosphate]•+) are formed roughly in proportion to the electron density at that site with favoring of ionization of the outer valence electrons.8 It has been widely accepted that the holes are the major damaging entity in DNA radiation damage and much work has focused on the transfer and localization of the hole in DNA.1–9 ESR studies at low temperatures have clearly established that, for the holes on the DNA bases, a fast hole transfer process from A•+, C•+, T•+, and [sugar-phosphate]•+ localizes the hole preferentially to a nearby guanine base forming G•+ 1, 8 in agreement with the theoretically predicted ionization potential of guanine in the G:C base pair system.10, 11 Both experimental and theoretical works have established that stacks of Gs, i.e., GG, GGG, are ultimately the most stable hole localization sites after long range hole transfer and these G stacks form loci for DNA base damage. 2 – 6, 12 – 16
Prototropic equilibria critically influence (i) rate and distance of hole transfer and (ii) the final stabilization of the hole either at a G or Gn site or another introduced hole acceptor. Two important proton transfer processes that control the prototropic equilibrium of one-electron oxidized guanine in double stranded (ds) DNA are shown in Scheme 1. They are: the intra-base proton transfer process (represented as 1) within the one-electron oxidized G:C base pair, and the proton transfer processes (2, 3 and 4) from the one-electron oxidized G:C base pair to the surrounding water.1 – 3, 8 – 11, 16 – 20
Scheme 1.

Schematic representation of prototropic equilibria in the one-electron oxidized G:C. The intra-base pair proton transfer within the one-electron oxidized G:C (process 1) and proton transfer to water either from the N1 atom (process 2) or from the nitrogen atom in the exocyclic amine (N2) of the guanine moiety in the one-electron oxidized G:C (process 4) are shown. The interconversion between G(N1-H)•:C and G(N2-H)•:C is represented by process 3. The conformation of the G(N1-H)•:C shown in this scheme has been adopted from the gas phase optimized geometry obtained by Bera et al. by using DFT (see ref. 25 for details) and this conformation of G(N1-H)•:C has been verified in this work for hydrated one-electron oxidized G:C.
The pKa values of the ion-radicals involved control the extent of these proton transfer processes. On the basis of the pKa values of G•+ in dGuo (ca. 3.9) and C(H+) in 2'-deoxycytidine (ca. 4.5), the equilibrium constant for this intra-base pair proton transfer process, Keq, had been estimated by Steenken as ca. 2.5.9,17 From this value of Keq, at ambient temperature, both cation radical (G•+:C) (30%) and deprotonated neutral radical (G(N1-H)•:C(+H+)) (70%) forms should exist.9 This equilibrium constant suggests that ΔGo (intra-base pair proton transfer) = −0.5 kcal/mol at 298 K. 9, 17 It is clear in Scheme 1 that intra-base pair proton transfer (G•+:C to G(N1-H)•:C(+H+)) leads to separation of charge from spin. Measurements of rate constants for hole transfer with the aid of laser flash photolysis in 5-bromocytosine incorporated ds DNA oligomers vis-à-vis in ds DNA oligomers containing cytosine yields results in accordance with this equilibrium between G•+:C and G(N1-H)•:C(+H+).20
Recent electron spin resonance (ESR) investigations have demonstrated that selective deuteration at C-8 on the guanine moiety (G*) in dGuo resulted in an ESR signal from the guanine cation radical (G*•+) which is easily distinguishable from that of the N1-deprotonated radical, G*(N1–H)•.9, 21 Based upon these results, ESR studies at low temperatures employing G* incorporated in the ds DNA-oligomer d[G*CG*CG*CG*C]2 clearly established that one-electron oxidized guanine exists only as G(N1-H)•, or more accurately, G(N1-H)•:C(+H+), see scheme 1.9 These results are in good agreement with the room temperature results since the thermodynamic equilibrium would shift to the more favored species, i.e., G(N1-H)•:C(+H+), at low temperatures.17, 20
Early theoretical studies without the inclusion of bulk solvent (“gas phase calculation”) reported that, in G•+:C, for the intra-base pair proton transfer along N1-H bond shown in scheme 1, ΔGo (proton transfer) = 1.4 kcal/mol at 298 K.10, 11 Thus, theory predicted that G•+:C is thermodynamically more stable than G(N1-H)•:C(+H+); whereas, ESR9 and pulse radiolysis17 results have established that one-electron oxidized G:C exists predominantly as G(N1-H)•:C(+H+). Recently, DFT theoretical calculations which considered the G•+:C pair with 11 waters of hydration have shown that the free energy for intra-base pair proton transfer in G•+:C becomes favorable on inclusion of water (ΔG= −0.65 kcal/mol).22 Thus the theoretical results22 are in accord with the results obtained by ESR studies on one-electron oxidized ds-DNA oligos9 and with the pulse radiolysis studies by Steenken.17
While the site of intra-base pair proton transfer in G•+:C is now well established to be from N1 in guanine producing G(N1-H)•:C(+H+), the site of subsequent deprotonation of G(N1-H)•:C(+H+) to water is still uncertain. A few recent pulse radiolysis studies in aqueous solution at ambient temperature have proposed that the likely site for deprotonation of the one-electron oxidized guanine in DNA to water is at the exocyclic amine N2 (see scheme 1).23, 24 DFT calculations (B3LYP/DZP++) in the gas phase also show the difference between the energies of the optimized geometries of G(N2-H)•:C and G(N1-H)•:C is ca. 1 kcal/mol.25 This very small difference between the energies of the optimized geometries of G(N2-H)•:C and G(N1-H)•:C clearly suggests that the site of deprotonation of one-electron oxidized guanine in ds DNA to the surrounding water could be either at N2 or at N1 (see scheme 1) or more likely a mixture of the two. In this work, employing the B3LYP/6-31+G** method, the geometries of G(N1-H)•:C and G(N2-H)•:C shown in scheme 1 have been calculated in the presence of 11 water molecules and these calculations show that these two forms remain nearly identical in total energy. Thus, calculations employing DFT/B3LYP/6-31+G** method suggest a very little preference for deprotonation from either N1 or N2 site in the guanine moiety of one-electron oxidized G:C to the surrounding solvent.
In this work, we employ UV-visible and ESR spectroscopy to elucidate the prototropic equilibria of one-electron oxidized guanine in a ds DNA oligomer. In scheme 1, we present the four protonation states of the one-electron oxidized G:C base pair, i.e., G•+:C, G(N1-H)•:C(+H+), G(N1-H)•:C, and G(N2-H)•:C. The first two forms, G•+:C and G(N1-H)•:C(+H+), have already been distinguished in our earlier work by G* substitution in ds DNA9 and our results clearly establish that, at low temperatures, only the G(N1-H)•:C(+H+) form exists in one-electron oxidized ds DNA9. In this work, we show that G(N1-H)•:C, and G(N2-H)•:C are easily distinguished both by ESR and UV-vis spectroscopy, and therefore, three of the four protonation states (see scheme 1) are distinguishable.
It is therefore desirable to distinguish the remaining pair of protonation states, i.e., G(N1-H)•:C(+H+) and G(N1-H)•:C (see scheme 1). However, these species cannot be distinguished by ESR or UV-vis spectroscopy owing to the near identical spectra found. Sugar radical formation in dGuo and in Guo via photo-excitation of one-electron oxidized G has been observed to depend on the pH of the solution.26, 27 Sugar radical formation on photo-excitation of the cation radical (G•+) is most efficient with 80 to 100% conversion in dGuo26 and in Guo27. However, at pH ≥9, where only G(N1-H)• is present, only small amounts (ca. ≤10%) sugar radicals are been found on photoexcitation.26, 27 Production of sugar radical in the ss DNA and RNA oligomers have also been reported via photo-excitation of G•+.8, 26, 27, 29 –31 Thus, arguing from the pH dependence in the formation of sugar radicals via photo-excitation of nucleosides discussed above, it is expected that the extent of photo-conversion to sugar radicals via photo-excitation of G(N1-H)•:C(+H+) might be significantly larger that that via photo-excitation of G(N1-H)•:C with the greatest extent of sugar radical formation on photo-excitation of the cation radical G•+:C. This hypothesis is tested in this work.
Important in these analyses are the UV-vis and ESR spectra for the legitimate N1 and N2 deprotonation radicals from G•+. To obtain benchmark information regarding legitimate deprotonation from N2, we have investigated 1-methyl-2'-deoxyguanosine (1-Me-dGuo), and the corresponding RNA analog 1-Me-Guo. In these compounds, the N1-H atom is replaced by a methyl group and deprotonation occurs only from the N2 site as reported in earlier pulse radiolysis studies.17 Our UV-visible spectral data reported in this work, have confirmed these earlier results. In addition, ESR spectral data and calculations performed by employing density functional theory (DFT) in one-electron oxidized 1-Me-dGuo presented in this work further substantiate the identity of G(N2-H)• and its hyperfine couplings are reported. Using these results as benchmarks, the UV-visible and ESR spectral data in one-electron oxidized dsDNA oligomer d[TGCGCGCA]2 at various pHs have been analyzed; it have been shown here that the site of deprotonation of the one-electron oxidized d[TGCGCGCA]2 to the solvent can be monitored by UV-vis and ESR spectroscopy. This allowed us, for the first time, to identify and characterize G(N2-H)• in a ds DNA oligomer d[TGCGCGCA]2 both by ESR spectroscopy and by UV-vis spectral studies. Especially, significant in this work is the correlation of UV-vis spectra of one-electron oxidized ds-DNA oligos found in this work at low temperatures with that found in the pulse radiolysis experiments reported in earlier studies of one-electron oxidized DNA oligomers at ambient conditions.18
Materials and methods
Compounds
1-Methyl-2'-deoxyguanosine (1-Me-dGuo) was procured from Berry & Associates, Inc. (Dexter, MI). 2'-Deoxyguanosine (dGuo), 1-methyl-guanosine (1-Me-Guo) and lithium chloride (99% anhydrous, SigmaUltra) were purchased from Sigma Chemical Company (St Louis, MO). Potassium persulfate (crystal) was obtained from Mallinckrodt, Inc. (Paris, KY). The desalted, dried, and column purified ds DNA oligomer, d[TGCGCGCA]2 and the desalted, dried, and HPLC purified ds DNA oligomer, d[TGGGCCCA]2 - were obtained from SYNGEN, Inc. (Hayward, CA). All the compounds were used without any further purification.
Preparation of solutions of dGuo, 1-Me-dGuo and 1-Me-Guo
Following our earlier works with DNA- and RNA-nucleosides and nucleotides, 8, 9, 21, 26 – 29 homogeneous solutions of dGuo, 1-Me-dGuo and 1-Me-Guo were prepared by dissolving ca. 3 mg of each compound in 1 mL of 7.5 M LiCl in H2O or D2O with ca. 5 mg K2S2O8. Some experiments were carried out using 15 M LiCl in H2O to test the effect of increasing ionic strength. Note that addition of K2S2O8 as an electron scavenger eliminates formation of anion radicals and simply results in an increase in the extent of the one-electron oxidant Cl2•−.26 Therefore, presence of K2S2O8 ensures formation of only one-electron oxidized species, i.e., the guanine cation radical thereby enabling us to study the subsequent prototropic reactions of the guanine cation radical (scheme 1) as reported in this work.
Preparation of solutions of the DNA-oligomers
As done in our works with DNA-oligomers,8, 9, 30, 31 about 1.5 mg of each of the ds DNA-oligomers d[TGCGCGCA]2 and d[TGGGCGCA]2 were dissolved in either in 0.35 mL of 7.5 M LiCl in D2O or in 0.35 mL of 7.5 M LiCl in H2O at room temperature. Occasional vortexing was performed to ensure formation of the homogeneous solution of the ds DNA-oligomer in 7.5 M LiCl in D2O or in H2O. Subsequently, an electron scavenger, ca. 2.5 – 3 mg K2S2O8 were added to each solution.
pH/pD Adjustments
As per our earlier work with dGuo 21 and dAdo 28, pH/pD values of the solutions of dGuo, 1-Me-dGuo, 1-Me-Guo, and the dsDNA-oligomers had been adjusted by addition of μL volumes of 0.1 to 1 M NaOH or HCl under ice-cooled condition. To measure pH/pD values of these solutions, pH papers were used. Note that due to the high ionic strength of solutions required to prepare glassy samples and because of the use of pH papers, the pH/pD values reported in this study are approximate.
Measurement of melting point of the DNA-oligomer
To measure the melting point of the ds DNA oligomer d[TGCGCGCA]2 in 7.5 M LiCl in D2O, the solution was first warmed in a suprasil quartz cuvette to 70°C; subsequently, by measuring the absorbance at 274 nm while this solution was gradually cooled up to room temperature (23°C), the melting point of this ds DNA oligomer was determined to be ca. 48°C. The temperature of the solution containing this DNA-oligomer was recorded directly by inserting the probe (Thermocouple Thermometer Type T, Oakton Instruments, Vermon Hills, IL) into the solution in the cuvette outside of the light path.
The renaturation curve of d[TGCGCGCA]2 (see results and discussion) shows that this DNA-oligomer is double stranded in 7.5 M LiCl in D2O up to 48°C. We note that in homogeneous aqueous (H2O) glasses (10 M LiCl), the dsDNA oligomers have been reported to be in the B-conformation. 32
Preparation of glassy sample and their storage
All solutions were bubbled thoroughly with nitrogen to remove dissolved oxygen and then were drawn in 4 mm suprasil quartz tubes (cat. no 734-PQ-8, WILMAD Glass co., Inc. Buena, NJ). Immediate cooling of these tubes containing the solutions to 77 K resulted in the formation of transparent glassy samples. As mentioned in our previous work, 9, 21, 26 – 31 these transparent glassy samples are supercooled homogeneous solutions and not crystalline solids. Because of thermal annealing at higher temperatures, these supercooled solutions soften on warming and allow molecular migration and solution phase chemistry. These glassy samples were stored in the dark at 77 K.
γ-irradiation of these glassy samples and their storage
These glassy samples were γ-irradiated at 77 K in an 109 GR 9 irradiator with a shielded 60Co source. Following our earlier work,9 the glassy samples of the nucleosides (dGuo, 1-Me-dGuo, and 1-Me-Guo) were γ-irradiated at 77 K with an absorbed dose of 2.5 kGy, whereas, the glassy samples of d[TGCGCGCA]2, and of T[GGGCCCA]2 were γ-irradiated at 77 K with an absorbed dose of 5 kGy. All the γ-irradiated samples were stored at 77 K in the dark.9
Formation of one-electron oxidized dGuo, 1-Me-dGuo, 1-Me-Guo, and DNA-oligomers by thermal annealing of glassy samples
One-electron oxidized dGuo, 1-Me-dGuo, 1-Me-Guo, and the dsDNA-oligomers were produced via annealing these γ-irradiated glassy samples in the dark at 155± 2 K for 20 – 30 min in a variable temperature assembly (Air Products) employing cooled nitrogen gas.9 Thermal annealing of these glassy samples leads to the softening of the aqueous (D2O or H2O) glass which allows for migration of Cl2•− in these samples. One-electron oxidation of the solutes in these viscous solutions by Cl2•− results in the formation of one-electron oxidized dGuo, 1-Me-dGuo, 1-Me-Guo, and of the dsDNA-oligomers. Owing to these one-electron oxidation processes, the intensity of the ESR spectrum of Cl2•− decreases and this decrease is accompanied by the simultaneous increase in the intensity of the ESR spectrum of one-electron oxidized guanine in dGuo, 1-Me-dGuo, 1-Me-Guo, and of the dsDNA-oligomers. Subsequent reactions of the one-electron oxidized solute (see scheme 1) were followed by ESR and UV-vis spectroscopies on annealing these samples in the temperature range of 155 – 175 K. Similar to our already published work in 2'-deoxyribonucleosides/tides and in DNA-oligomers, and also in Guo and in RNA-oligomers,8, 9, 21, 26 – 31 we do not observe sugar radical formation by the direct attack of Cl2•− on the sugar moiety in these glassy samples.
UV-visible spectroscopy
A Cary 50 UV-vis spectrophotometer was employed to obtain the UV-vis spectra of the one-electron oxidized samples (dGuo, 1-Me-dGuo, 1-Me-Guo, and ds DNA oligomers) in liquid nitrogen (77 K). The same suprasil quartz dewars used for recording the ESR spectra were employed for this purpose. For recording the UV-vis spectra of one-electron oxidized 1-Me-dGuo or 1-Me-Guo, the glassy (7.5 M LiCl) aqueous (D2O or H2O) solutions of 1-Me-dGuo (or 1-Me-Guo) in 4 mm suprasil quartz tubes (cat. no 734-PQ-8, WILMAD Glass co., Inc. Buena, NJ) were used. To record the UV-vis spectrum of one-electron oxidized d[TGCGCGCA]2, the glassy (7.5 M LiCl) solutions of d[TGCGCGCA]2 in H2O were used in 1mL polystyrene cuvettes.
Photo-excitation
The temperature assembly which has already been employed for thermal annealing of the glassy samples was used here for photo-excitation of glassy samples of one-electron oxidized guanine in d[TGCGCGCA]2.
By employing a photoflood lamp (250 W), the glassy samples of d[TGCGCGCA]2 were photo-excited at 148 K (±2 K). During photo-excitation, the IR and UV components of this light were cut off by a water filter and a 310 nm cut off filter, respectively. The glassy samples of d[TGCGCGCA]2 were also photo-excited at 148 K (±2 K) by employing a thermoelectrically cooled blue laser (TECBL-20G-405, World Star Tech., Lot 6880) (λ = 405 nm, 20 mW). The 405 nm laser has been observed to be several times more effective than the photoflood lamp in causing sugar radical formation as the 405 nm light is in the optimal wavelength region 310 – 480 nm which is in low intensity in the photoflood lamp.26
Electron spin resonance
A Varian Century Series ESR spectrometer operating 9.2 GHz with an E-4531 dual cavity, 9-in. magnet, and with a 200 mW klystron, was employed to record at 77 K and at 45 dB (6.3 μW) the ESR spectra of γ-irradiated and annealed samples. The three Fremy's salt (potassium nitrosodisulphonate) ESR lines (g (center of the spectrum) = 2.0056, AN = 13.09 G) were used for field calibration following our work.33
Analyses of ESR spectra
Each ESR spectrum was stored in a 1000-point array along with field calibration marks from the three ESR line's of the Fremy's salt following our earlier work.9, 33 We have estimated the fractional contribution of a radical species in an experimentally obtained ESR spectrum by fitting the benchmark spectrum of that particular radical to the experimentally obtained ESR spectrum of interest using least squares analyses by employing programs (ESRADSUB, ESRPLAY) developed in our laboratory. 9, 21, 26 – 31, 33 Based on our previous error analyses,9 the error limit of these above-mentioned analyses has been estimated as ±10% with good quality signals. We have subtracted a small singlet “spike” from irradiated quartz at g = 2.0006 from the recorded spectra for our analyses.
Simulations of ESR spectra
Simulations of the experimentally obtained ESR spectra of one-electron oxidized 1-Me-Guo at different protonation states were carried out by employing the WIN-EPR and SIMFONIA (Bruker) programs. Anisotropic hyperfine coupling constants (HFCC) of nitrogens (N1 and N3 in the guanine moiety), of the N1-H-atom, of the N2-H atom, and of the C8-H atom were found by the DFT method (B3LYP/6-31G(d)) (vide infra). These values were then employed to carry out the simulations of the 1-Me-Guo spectra; the theoretically obtained HFCC values and the g-values have been adjusted to fit these experimentally observed ESR spectra. The radicals in the two prototropic forms were calculated by DFT to be only slightly nonplanar. Therefore, we assume that Azz for the guanine moiety nitrogen atoms are colinear with Azz for C8(H). We carried out several simulations of the experimentally observed ESR spectrum for each species shown in scheme 2 and have been able to ascertain error limits of our fits of ±2 G for the largest nitrogen anisotropic couplings (Azz) and ±1 G for each of the three components of the C8(H) anisotropic coupling (see supplemental information Figure S1 for examples of simulations which support this). The perpendicular nitrogen anisotropic couplings (Axx and Ayy) are not observed as they are small and unresolved and predicted to be so by theory (Table 1). We have assumed these HFCC values as zero in our simulations and the line-width was adjusted to account for unresolved couplings. Note that the N2-H is an exchangeable proton, thus we find that the N2-H hyperfine couplings are observed only in H2O and not in D2O. The Axx and Ayy values for the N2-H hyperfine are not well determined by experiment and reasonable estimates from the published literature (ESR/ENDOR studies of the X-ray irradiated single crystals 40 and theoretical studies 21, 41a (see supplemental information Table T1) were employed in the simulations. For the Axx, Ayy tensors, principal axes (x and y) were not collinear for the C8-H and N2-H protons and this has been taken into account in the simulation.
Scheme 2.

Prototropic equilibria of one-electron oxidized guanine in 1-methyl guanine including the cation radical (G•+), the mono- deprotonated species, G(N2-H)•, in syn and anti- conformers with respect to the N3 atom). The numbering scheme shown here is followed in this work involving the theoretical calculations with 1-Me-dGuo.
Table 1.
Theoreticala ESR Parameters for 1-Me-dG•+ and 1-Me-dG(N2-H)• and adjustments of these parameters to obtain the simulated parameters.
| 1-Me-Guo |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Radical | 14N3 couplings(G) | 14N2 couplings(G) | C8(H) couplings (G) | N2-H couplings (G) | g-values (experimental) | ||||||||
| Azz | Axx | Ayy | Azz | Axx | Ayy | Azz | Axx | Ayy | Azz | Axx | Ayy | ||
| 1-Me-dG • + b,c | In H2O | ||||||||||||
| Theory+ 7 H2O | 11.1 | 0.6 | 0.7 | 8.7 | 0.6 | 0.7 | −7.5 | −9.9 | −2.3 | Not determined | gzz=2.0021 | ||
| In D2O | gxx=2.0045 | ||||||||||||
| Exp b | 13.0 | ~0 | ~0 | 6.5 | ~0 | ~0 | −7.5 | −10.5 | −3.5 | --------------------- | gyy=2.0045 | ||
|
| |||||||||||||
| 1-Me-dG(N2-H) • cd | |||||||||||||
| Theory | |||||||||||||
| G(N2-H)• + 7 H2O (syn) | 15.8 | 1.0 | 1.1 | 17.7 | 0.4 | 0.5 | −6.1 | −8.2 | −2.1 | −11.5 | −15.6 | −0.6 | |
| G(N2-H)• + 7 H2O (anti) | 16.3 | 1.0 | 1.1 | 17.5 | 0.2 | 0.3 | −5.5 | −7.3 | −1.9 | −11.5 | −17.0 | −0.2 | gzz=2.0019 |
| In D2O | gxx=2.0041 | ||||||||||||
| Exp b | 12.5 | ~0 | ~0 | 15.7 | ~0 | ~0 | −6.0 | −8.2 | −2.1 | ----------------------- | gyy=2.0041 | ||
| In H2Od | |||||||||||||
| 12.5 | ~0 | ~0 | 15.7 | ~0 | ~0 | −6.0 | −8.2 | −2.1 | −8.0 | −12.0 | −2.0 | ||
Structures optimized and hyperfine couplings calculated using DFT (B3LYP) with a 6-31G(d) basis set.
These values were taken from ref. 21 and these values were used for simulation of G•+ spectrum in dGuo. The ESR spectra of cation radical of 1-Me-dGuo and dGuo are found to be identical (see supplemental information Figures S2 and S3).
Experimental value of Axx and Ayy nitrogen hyperfine couplings are ca. zero; these values are within the line-width and are too small to be characterized. Theoretically calculated values confirm the small values of these couplings.
For 1-Me-dG•+ in D2O, the line-widths used (7.1, 5.7, 5.7) G and Lorentzian/Gaussian =5; For 1-Me-dG(N2-H)• in D2O, the line-widths used (4.8, 5.5, 5.3)G and Lorentzian/Gaussian =0.8; For 1-Me-dG(N2-H)• in H2O, the line-widths used (4.0, 4.0, 6.0) G, Lorentzian/Gaussian =0.8. The Axx, Ayy tensor principal axes differ for the C-H and N-H protons and this has been taken into account in the simulation.
Studies using density functional theory (DFT)
In the present study, the geometries of 1-Me-Guo cation radical and its N2-deprotonated radicals (in anti- and syn-conformations with respect to N3 atom of the guanine (see scheme 2)) were fully optimized in the presence of seven water molecules using B3LYP functional and 6-31G(D) basis set.
We also calculated the geometries of G•+:C and its N1H and N2H′ deprotonated froms - G(N1-H)•:C(+H+) and G(N2-H)•:C (see in scheme 1) in the presence of 11 water molecules using B3LYP/6-31+G** level of theory. The numbering scheme for these calculations in the base pair is shown in scheme 3 below.
Scheme 3.
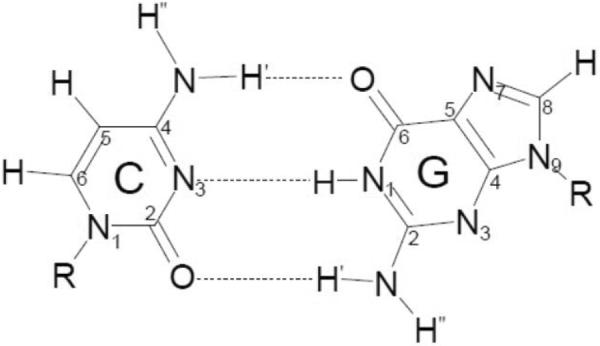
The numbering scheme for the calculations of one-electron oxidized G:C structures shown in scheme 1.
In calculating the various properties of the radical species (e.g., HFCC values), inclusion of discrete water molecules has already been found to allow us to obtain a good fit to the corresponding experimental observations.21, 22,28, 34 To calculate the HFCC values of cation radical of 1-Me-dGuo and its deprotonated species, 1-Me-dG(N2-H)• (see scheme 2), the B3LYP/6-31G(d) method was employed and these calculated HFCC values were used for the simulation of experimentally obtained ESR spectra. The B3LYP functional is a Becke's three parameter exchange functional (B3)35 with Lee, Yang and Parr's correlational functional (LYP)36. All calculations were performed by employing the Gaussian 03 suites of programs.37 The applicability of the B3LYP functional for radical species is well established in the literature.21, 22, 38 The structures were drawn using JMOL molecular modeling program.39
Results and Discussion
A. Organization of the results
Our approach is as follows:
In order to elucidate the prototropic equilibrium between DNA and solvent, the deprotonation site from the G:C base pair cation radical to the solvent must be identified. There are two possibilities - deprotonation either from N1 or from N2 in the guanine moiety to the surrounding solvent as shown in Scheme 1. The ESR spectrum of the N1 deprotonated species, G(N1-H)• and its UV-vis spectrum are now well established,9, 21 but the ESR spectrum of the N2 deprotonated species in aqueous systems (scheme 1) is not. We note here that the N2 deprotonated species is well-documented in previous ESR/ENDOR studies of X-ray irradiated single crystals.40, 41a Our ESR work with dGuo in glassy system at low temperature 21 as well as pulse radiolysis studies with dGuo in aqueous solution at ambient temperature 17, 18 shows that G•+ naturally deprotonates from N1 first to produce G(N1-H)• and subsequently from N2 to form G(−2H)•−. Therefore, to produce an authentic ESR benchmark spectrum of the neutral N2 deprotonated species, we employ 1-Me-dGuo or 1-Me-Guo, which can only deprotonate from N2 as the N1-H atom here has been replaced by the CH3 group.
While the ESR and UV-vis spectrum of G(N1-H)•:C(+H+) is well established, we are not able to distinguish the two forms of G(N1-H)•, i.e., G(N1-H)•:C(+H+) vs. G(N1-H)•:C, shown in Scheme 1 either by ESR or UV-vis in DNA oligos. However, in this work we show that we find that visible exposure of light converts substantially the G(N1-H)•:C(+H+) form to sugar radicals and allows for the assignment of protonation state in the DNA-oligo.
Finally, we present the results that the protonation state G(N2-H)•:C of the one-electron oxidized G:C shown in scheme 1, can be easily distinguished in a ds DNA-oligo from the other protonation states viz. G•+:C, G(N1-H)•:C(+H+), and G(N1-H)•:C as shown in scheme 1.
B. ESR and UV-vis studies of the cation radical (1-Me-G•+) and its deprotonated form (1-Me-G(N2-H)•) in 1-Me-dGuo and 1-Me-Guo
(a) Studies in D2O – annealing at 155 K
In Figures 1A and B, the ESR spectra of the cation radical 1-Me-G•+ (red) and the corresponding deprotonated cation radical assigned as 1-Me-G(N2-H)• (blue) (vide infra) formed in 1-Me-Guo samples at pDs ca. 5 and ca. 8.5 respectively in 7.5 M LiCl in D2O after one-electron oxidation by Cl2•− via annealing at 155 K in the dark are presented. We find that 1-Me-G•+ does not deprotonate in the pD range ca. 3 to 5. The characteristics of the ESR spectrum (overall hyperfine splitting, g-value at the center of the spectrum, and the lineshape) shown in Figure 1A match very well (see supplemental information Figures S2 and S3) with the already published ESR spectrum of G•+ in dGuo 9, 21 or Guo27. This result points out that for 1-Me-G•+, (i) the methyl group at N1 does not have a significant effect on the spin distribution and hyperfine couplings at other sites, and (ii) the N1 site has a low spin density and no significant methyl coupling is observed; this is further supported by the theoretically (DFT/B3LYP/6-31G(d)) predicted HFCC values (see Table 1) (vide infra).
Figure 1.

ESR spectra obtained at 77 K for the one-electron oxidized 1-Me-Guo by Cl2•− (A, B) at various pDs in 7.5 M LiCl glasses in D2O in the presence of the electron scavenger K2S2O8. Figure (C) represents the UV-visible absorption spectra of the same samples of one-electron oxidized 1-Me-Guo – 1-Me-G•+ (red) and 1-Me-G(N2-H)• respectively at 77 K in 7.5 M LiCl glass/D2O. In Table 1, the ESR parameters, hyperfine couplings and g-values, used for the simulated spectra (black) are given. Our results clearly show that 1-Me-G•+ is found at pDs ≤5 and 1-Me-G(N2-H)• is formed at pDs 8–12. The ESR spectra as well as the UV-vis spectra were recorded at 77 K. The three reference markers showing the ESR spectra in this Figure and in subsequent Figures containing ESR spectra are Fremy's salt resonances with central marker is at g= 2.0056, and each of three markers is separated from one another by 13.09 G.
Computer simulations (black) of the experimentally obtained ESR spectra for both 1-Me-G•+ (Figure 1A (red)), and in 1-Me-G(N2-H)• (Figure 1B, blue) were performed using the hyperfine couplings and g-values given in Table 1. The hyperfine couplings first used to simulate the spectra where the theoretically calculated (DFT/B3LYP/6-31G(d)) ones for the 1-Me-dG•+ and 1-Me-dG(N2-H)• shown in scheme 2 in the presence of 7 water molecules (vide infra) shown in Table 1. These theoretically predicted values were then adjusted to improve the fit to experiment (see supplemental information Figure S1) and these values are the “experimental values” in Table 1. The HFCC values used for the simulation of the experimental spectrum of 1-Me-dG•+ (red) closely match those used previously for G•+ in dGuo21. This establishes that the methyl group at N1 does not have a significant effect on the hyperfine couplings and hence on the spin distribution at N1 and at other sites (see Table 1).
The experimentally obtained ESR spectrum of the deprotonated neutral radical (blue) assigned to 1-Me-G(N2-H)• (Figure 1 B) differs significantly from that (red) of 1-Me-G•+ (Figure 1A). For the experimental spectrum of 1-Me-G(N2-H)• shown in Figure 1B, the hyperfine couplings of the nitrogens N2 and N3 are readily apparent in the wings. This allows for an easy distinction between these two prototropic forms - 1-Me-G•+ and 1-Me-G(N2-H)• (scheme 2). The simulated spectrum (black) match very well with those of the experimental spectrum (blue) of 1-Me-G(N2-H)• (see Figure 1B). We note here that the HFCC values for the C8-H atom used to simulate the experimental ESR spectrum of 1-Me-G(N2-H)• in the aqueous (D2O) glassy system (see Table 1) match very well with those of the already reported from experimental (ESR/ENDOR studies of X-ray irradiated single crystals40) and theoretical (DFT/B3LYP/6-31G(d)) 21, 41a for the N2-deprotonated neutral radical (see Table T1 in supplemental information). These results establish that for the N2-deprotonated neutral radical (i) the N1 and N7 nitrogen atoms have very small HFCC couplings which could be neglected, and (ii) the methyl group at N1 does not have a significant effect on the hyperfine couplings and hence on the spin distribution at N1 and at other sites. We note the large N2 hyperfine coupling of 15.7G (Table 1) readily distinguishes G(N2-H)• from G(N1-H)• for which the N2 hyperfine coupling is not observed experimentally.21
Clearly, it is evident from Table 1 that, we cannot strictly distinguish between the syn and the anti- conformations (see scheme 2) of the 1-Me-G(N2-H)•, although the theoretically predicted HFCC values found for the syn conformation appears to be slightly closer to the corresponding experimental values (see Table 1).
As the experimentally obtained ESR spectrum of 1-Me-G(N2-H)• (blue) (Figure 1 B) is found to be significantly different from that (red) of 1-Me-G•+ (Figure 1A), this has also been observed to be so for the corresponding UV-visible spectra (Figure 1C). In Figure 1C, the UV-visible spectra of one-electron oxidized guanine in 1-Me-Guo in aqueous (D2O) glasses and recorded at 77 K are shown at two representative pD values (ca. 5 and 8.5). The UV-visible spectrum at pD ca. 5 corresponds to the protonated (1Me-G•+) form, whereas the UV-visible spectrum at pD ca. 8.5 corresponds to the deprotonated (1-Me-G(N2-H)•) form. In particular, there is a broad absorption band at ca. 630 nm found in the UV-visible spectrum of 1-Me-G(N2-H)•. Note that, a very similar broad absorption band at ca. 640 nm has also been observed for the di-deprotonated G(−2H)•− form in dGuo (see Figure 1 in ref. 21 and Figure 1 in ref. 17 (a)). Since the only site in 1-Me-Guo available for proton loss is at N2, it is clear that the broad absorption band observed in these UV-visible spectra at and above ca. 630 nm is a characteristic for the deprotonation from N2.
Previous pulse radiolysis results for the UV-visible spectra for 1-Me-G•+ (pH 3) and 1-Me-G(N2-H)• (pH > 5) in aqueous solution at room temperature (see Figure 3 in ref. 17 (a)) match with these spectra shown in Figure 1C. Moreover, the UV-vis spectrum of 1Me-G•+ in 1-Me-Guo (red color) in Figure 1C and the ESR spectrum in Figure 1A matches the already published spectra of G•+ in dGuo in the glassy system at 77 K (see Figure 1 in ref. 21). This as expected owing to their near identical electronic structure and spin density distribution (See Table 1, this work and Table 3 in ref. 21).
Figure 3.

(A) ESR spectra of authentic 1-Me-dG(N2-H)• obtained from glassy (7.5 M LiCl) in D2O (blue) and in H2O (green) samples of 1-Me-dGuo by one-electron oxidation via annealing at 155 K in the dark. Expansion of the wings by factor of 5 shows the Azz component of N2-H coupling in H2O – which is lost in D2O. (B) Comparison of the spectrum of 1-Me-dG(N2-H)• obtained from glassy (7.5 M LiCl) in H2O (green) samples of 1-Me-dGuo with the simulated spectrum (black). In Table 1, the ESR parameters, hyperfine couplings and g-values, used for the simulated spectrum (black) are provided. Our results clearly show that in 1-Me-dG(N2-H)• in H2O, the additional line components due to the HFCC value of N2-H atom are clearly observable at the wings of the spectrum. All the ESR spectra were recorded at 77 K.
We note that ESR spectra of 1-Me-dG•+ (i.e., cation radical in 1-Me-dGuo) is identical with the spectra 1-Me-G•+ (see supplemental information Figure S3). This is also found for the ESR spectra of 1-Me-dG(N2-H)• and 1-Me-G(N2-H)• (see supplemental information Figure S3).
(b) Studies in D2O – annealing at 175 K and estimation of the pKa of 1-Me-G•+
At pDs ca. 6.5 to 7, one-electron oxidation of 1-Me-Guo samples via annealing at 155 K show the ESR spectrum of the cation radical (see Figure 2A). Upon further annealing of the same sample up to ca. 175 K, the deprotonation from the N2-atom is observed; this is evidenced by the development of line components of (see Figure 2B) 1-Me-G(N2-H)•. However for samples at and below pD ca. 5, upon further annealing to 175 K, the ESR spectra were found to be unchanged for 1-Me-G•+. As expected in the pD range ca. 7.6 to 12 only 1-Me-G(N2-H)• is found after annealing to 155K and above. Identical results were found for the samples of 1-Me-dGuo (see supplemental information Figure S4). These results indicated that the pKa of the 1-Me-dG•+ or 1-Me-G•+ in our system (7.5 M LiCl/D2O at low temperature) is about 6.5 (see Figure 2C).44, 45
Figure 2.

ESR spectra of: authentic (A) 1-Me-G•+ (red) and (B) 1-Me-G(N2-H)• (blue) obtained from glassy (7.5 M LiCl in D2O) samples of 1-Me-Guo via annealing at 155 K in the dark. The match of these experimentally observed spectra of 1-Me-G•+ and 1-Me-G(N2-H)• as benchmarks with those obtained using one-electron oxidized 1-Me-Guo (black) at pD ca. 6.8 first annealed at 155 K and further annealed at 175 K indicate that for one-electron oxidized 1-Me-Guo at pD 6.8, annealing at 155 K produces first the cation radical 1-Me-G•+ and further annealing at 175 K leads to its deprotonation to form G(N2–H)•. From these results, we estimate the pKa of 1-Me-G•+ as ca. 6.6 in our system (7.5 M LiCl in D2O at low temperature) as shown in Figure (C).
(c) Studies in H2O including annealing at 175 K
The ESR spectral studies of G(N2–H)• in homogenous H2O glasses (7.5 M LiCl/H2O) are presented in Figure 3. Figure 3A shows the ESR spectrum of 1-Me-dG(N2–H)• (green color) formed via one-electron oxidation by Cl2•− of 1-Me-dGuo in 7.5 M LiCl (H2O). For comparison, the ESR spectrum of 1-Me-G(N2–H)• in D2O (7.5 M LiCl) (blue color) from Figure 1B, has been superimposed on it. In the spectrum (green) presented in Figure 3A, the expanded wings show the 8G Azz hyperfine coupling of the N2-H proton in H2O (green color) which is lost in D2O glasses (blue color).
The hyperfine couplings of 1-Me-dG(N2-H)• have been theoretically calculated (DFT/B3LYP/6-31G(d)) in the presence of 7 water molecules (Table 1). We have adjusted these theoretically predicted HFCC values to improve the fit of the simulated ESR spectrum (black) of 1-Me-dG(N2-H)• in H2O towards the corresponding experimentally observed ESR spectrum (green) of 1-Me-dG(N2-H)• in H2O glass already shown in Figure 3A. Comparison of the simulated spectrum (black) with the experimentally recorded spectrum (green) shows that the hyperfine coupling components in the wings (A∥) of the simulated spectrum match very well with those of the experimental spectrum. The HFCC values for the N2-H and C8-H atoms used to simulate the experimental ESR spectrum of 1-Me-dG(N2-H)• in the aqueous (H2O) glassy system (see Table 1) match very well with those of the already reported (ESR/ENDOR studies of the X-ray irradiated single crystals 40 and theoretical studies 21, 41a (see supplemental information Table T1)).
It is evident from Table 1 that the distinction between the syn and the anti- conformations (see scheme 2 and also Figure 8) of the 1-Me-dG(N2-H)• is not possible even in H2O glass.
Figure 8.

The fully optimized geometries of (a) 1-Me-dG•+, (b) 1-Me-dG(N2-H)•syn, and (c) 1-Me dG(N2-H)•anti in the presence of seven water molecules. The optimization was carried out with the aid of DFT/B3LYP/6-31G(d) method. The relative stabilities of 1-Me-dG(N2-H)•syn and 1-Me-dG(N2-H)•anti in kcal/mol are provided in parentheses.
As expected, the UV-vis spectrum of the 1-Me-G(N2-H)• in an H2O glass containing 1-Me-Guo at pH ca. 9 has been found to be identical with the corresponding UV-vis spectrum of the 1-Me-G(N2-H)• in D2O glass (see Figure 1C). 46
Thus, the ESR, UV-vis studies shown in Figures 1 to 3 and the agreement between the experimentally observed and theoretically calculated HFCC values in 1-Me-dG(N2-H)• (see Table 1) clearly establish that the methyl group in 1-Me-dGuo (or in 1-Me-Guo) is not involved in the deprotonation reaction of the 1-Me-dG•+. Deprotonation from the methyl group would yield the radical, 1-CH2•-dG. This species would have a very distinct spectrum with a 1:2:1 pattern resulting from two α-proton couplings from the CH2 group– and this pattern is not at all observed in Figures 1 to 3.
C. ESR and UV-vis studies of the one-electron oxidized guanine (G(N1-H)•:C(H+)) and its deprotonated forms (G(N1-H)•:C) and (G(N2-H)•:C) in the ds DNA-oligomers
We have extended theses studies to the DNA-oligomers, viz. d[TGCGCGCA]2 and d[TGGGCCCA]2.
(a) Renaturation studies
Before carrying out the ESR and UV-vis studies, renaturation experiments were performed to ensure that these DNA-oligomers are in their double stranded form in aqueous solution of 7.5 M LiCl at ambient temperature.
The absorbance of the homogeneous solution of d[TGCGCGCA]2 in 7.5 M LiCl in H2O, preheated to 70°C, was measured at 274 nm47 while this solution was allowed to cool gradually down to room temperature (23°C). The plot of the renaturation curve of this DNA-oligomer at 7.5 M LiCl in H2O is shown in supplemental information Figure S6 as the absorbance at 274 nm against the temperature of the solution. The renaturation curve of d[TGCGCGCA]2 clearly shows that the 7.5 M LiCl solution increases the thermal stabilitiy of the DNA-oligomer in the double stranded form and shows clearly that at room temperature and at lower temperatures, the DNA-oligomer is double stranded in 7.5 M LiCl.
(b) ESR and UV-vis spectral studies before photo-excitation
ESR spectral studies in D2O
In Figures 4A to C, we show the ESR spectra of the one-electron oxidized guanine in d[TGCGCGCA]2 in 7.5 M LiCl/ D2O at different pDs ca. 3, 5, 7, and 9. The one-electron oxidant employed here is Cl2•− and the one-electron oxidation of d[TGCGCGCA]2 was carried out via annealing in the dark for 15-20 min at ca. 155 K, as per our earlier work 9 with the dsDNA oligomers.
Figure 4.

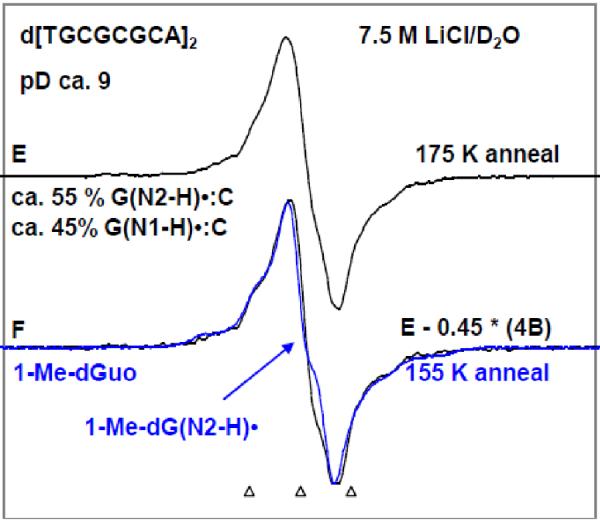
ESR spectra of one-electron oxidized guanine in d[TGCGCGCA]2 in 7.5 M LiCl/D2O (black) in the presence of K2S2O8 as an electron scavenger at various pDs (A) at pD ca. 3, (B) at pD ca. 5, (C, E) at pD ca. 9 obtained by annealing in the dark at 155 K for 15 – 20 min. Authentic G•+ spectrum (red) from 1-Me-Guo (Figure 1A) has been superimposed on spectrum (A). Authentic G(N1-H)•:C(+H+) spectrum (green) obtained from one-electron oxidized d[GCGCGC]2 from our previous work9 has been superimposed on spectrum (B). Spectrum (D) is obtained by subtraction (ca. 20%) of 1-Me-G(N2-H)• (Spectrum 1B). As shown this resultant spectrum matches that of spectrum G(N1-H)• from dGuo in pink from our previous work21 where the deprotonation occurs from N1 site in one-electron oxidized dGuo to the solvent. Spectrum (E) is obtained by further annealing the sample used to obtain spectrum (C) at 175 K in the dark for 15 min. Subtraction (ca. 45%) of spectrum 4B from spectrum 4E resulted in spectrum 4F. Authentic spectrum of G(N2-H)• obtained from 1-Me-Guo (Figure 1B) is superimposed on spectrum 4F. Spectrum (A) is assigned to G•+, spectrum (B) to G(N1-H)•:C(+H+), spectrum (D) is assigned to G(N1-H)•:C. and spectrum (F) to G(N2-H)•:C. All the spectra were recorded at 77 K.
Figure 5A represents the ESR spectrum of the one-electron oxidized guanine in d[TGCGCGCA]2 in 7.5 M LiCl/ D2O at pD ca. 3 (black); the overall hyperfine splitting, g value at the center (g⊥) and the lineshape of this spectrum matches very well with that of the 1-Me-G•+ (red), this work shown in Figure 1A or that of G•+ in dGuo which has already been published21. We assign this ESR spectrum of the one-electron oxidized guanine in d[TGCGCGCA]2 at pD ca. 3 shown in Figure 4A to the guanine cation radical (or G•+) of d[TGCGCGCA]2. Based upon the reported pKa value for protonation at N3 in cytosine in aqueous solution (ca. 4.5),17 the cytosine moiety may also be protonated to some extent although this is not clear as that would disrupt the base pairing. G•+:C cannot be distinguished from G•+:C(+H+) on the basis of ESR or UV-vis spectra. Thus, the protonation state of cytosine in spectrum 4A is uncertain at pH 3.
Figure 5.
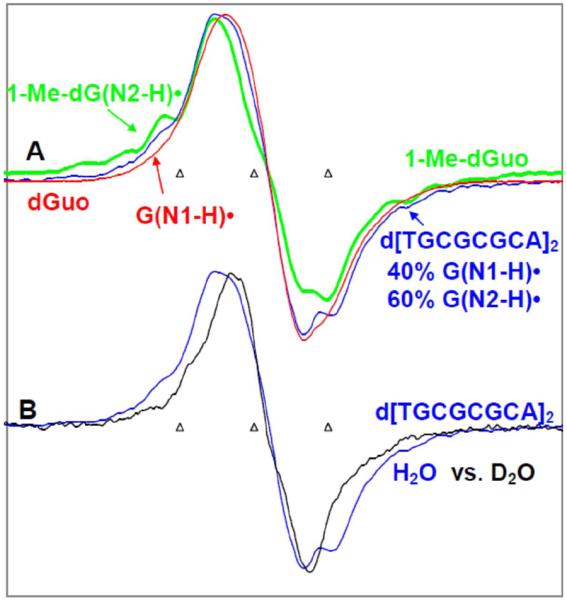
ESR spectra of one-electron oxidized d[TGCGCGCA]2 and model compounds in glassy 7.5 M LiCl solutions at pH ca. 9. (A) ESR spectrum of G(N2-H)• obtained from d[TGCGCGCA]2 in H2O (blue) formed after annealing to ca. 173 K. The authentic G(N2-H)• spectrum (green) from 1-Me-dGuo and the authentic G(N1-H)• spectrum (red) obtained from dGuo are superimposed on it. The wings show the Azz component of the exchangeable N2-H coupling in ds oligomer in H2O. (B) Comparison of the spectrum of one-electron oxidized d[TGCGCGCA] in H2O from A (blue) with that found in D2O (black) for an otherwise identical sample. The Azz component of the remaining N2-H proton hyperfine coupling of G(N2-H)• is clearly visible in the wings spectrum (blue) in Figure (B). All ESR spectra were recorded at 77 K.
The ESR spectrum (black) of the one-electron oxidized guanine moiety of d[TGCGCGCA]2 at pD ca. 5 in 7.5 M LiCl/ D2O is shown in Figure 4B. The ESR characteristics of this spectrum (g value at the center (g⊥), the overall hyperfine splitting, and the lineshape) matches very well with already published ESR spectrum (green) 9 of (G(N1-H)•:C(+H+)) in d[GCGCGC]2 recorded at 77 K at the native pD (ca. 5) in 7.5 M LiCl/D2O. Hence, the spectrum of one-electron oxidized guanine in d[TGCGCGCA]2 found at pD ca. 5 is assigned to (G(N1-H)•:C(+H+)) (see scheme 1).
The one-electron oxidized guanine moiety of d[TGCGCGCA]2 at pDs ca. 7 and ca. 9 in 7.5 M LiCl/ D2O formed on annealing to 155 K in the dark, yielded nearly identical spectra, and hence the spectrum (black) of the one-electron oxidized guanine moiety of d[TGCGCGCA]2 at pD ca. 9 is shown in Figure 4C. Subtraction (ca. 20%) of the spectrum of 1-Me-G(N2-H)• (shown in spectrum 1B) (or of 1-Me-dG(N2-H)• shown in Figure 3A) from this spectrum yields spectrum D (black). The ESR characteristics of this spectrum (g value at the center (g⊥), the overall hyperfine splitting, and the lineshape) matches very well with that of the already published 21 G(N1-H)• spectrum (pink) in dGuo where deprotonation occurs from N1 site to the solvent. Moreover, we find a smaller extent of conversion to sugar radicals via photo-excitation of the one-electron oxidized guanine in d[TGCGCGCA]2 at pD ca. 7 and at pD ca. 9 (see Figure 6, vide infra) in comparison to one-electron oxidized guanine in d[TGCGCGCA]2 at pD ca. 5. Therefore, we assign the spectrum 4B at pD ca. 5 to (G(N1-H)•:C(+H+)). The spectrum of one-electron oxidized guanine in d[TGCGCGCA]2 at pD ca. 9 (or at pD ca. 7, spectrum 4C) has been assigned to the corresponding deprotonated radical (G(N1-H)•:C). The deprotonation of the one-electron oxidized guanine occurs from N1 to the surrounding solvent (see scheme 1).
Figure 6.

The UV-visible absorption spectra of one-electron oxidized guanine in oligos and model compounds. The pink line is that of one-electron oxidized d[TGCGCGCA]2 in glassy (7.5 M LiCl) in H2O at pH 9 at 180K and is assigned to G(N2-H)•:C. The ESR spectrum is shown in Figure 5A. In blue the UV-vis spectrum of an authentic G(N2-H)• from 1-Me-Guo (blue) at 77 K in 7.5 M LiCl glass/H2O at pH 9 whose ESR spectrum is also shown in Figure 5A. The green data points are taken from existing pulse radiolysis spectrum of one-electron oxidized guanine containing oligo (sequence G1AA, see Figure 4 in Ref. 18b) in aqueous solution at ambient temperature. The spectra in the pink, green, and the blue spectrum have been multiplied by 2.5, 4, and 0.5 respectively. The spikes in the blue UV-vis spectra are due to bubbles from liquid nitrogen.
As found for the monomers 1-Me-Guo or 1-Me-dGuo (see Figure 2B), we find that upon further annealing of one-electron oxidized guanine in d[TGCGCGCA]2 (either at pD ca. 7 or at pD ca. 9, spectrum 4C) in 7.5 M LiCl/D2O up to ca. 175 K, spectrum 4E is resulted. In spectrum 4E, we find the development of line components of the anisotropic HFCC of the N2 atom and as found for the monomers, the development of line components of the anisotropic HFCC of the N2 atom is related to the deprotonation to solvent. Subtraction (ca. 45%) of spectrum 4B from spectrum 4E resulted in spectrum 4F and superimposition of an authentic spectrum of 1-Me-G(N2-H)• (blue) (Figure 2B) upon spectrum 4F confirms that spectrum 4F is due to (G(N2-H)•:C). Thus, in spectrum 4E, both the radical species (G(N1-H)•:C) and (G(N2-H)•:C) are found. It is likely that they exist in equilibrium as shown in scheme 1. The annealing at higher temperature (ca. 175 K) suggests that this equilibrium favors the formation of (G(N2-H)•:C) with increase in temperature.
We note here that annealing an identically prepared sample of one-electron oxidized guanine in d[TGGGCCCA]2 at pD ca. 7 in 7.5 M LiCl/D2O up to ca. 177 K yielded an ESR spectrum which is identical to the spectrum 4E (see supplemental information Figure S7).
ESR spectral studies in H2O
ESR studies of one-electron oxidized guanine formed in d[TGCGCGCA]2 in homogenous H2O glasses (7.5 M LiCl/H2O) were carried out at pH ca. 9 following the same procedures for D2O glasses. Also, the UV-vis absorption spectrum was recorded at ca. 175 K and this absorption spectrum has been compared with the existing pulse radiolysis spectrum of one-electron oxidized guanine (sequence G1AA (see Figure 5 in Ref. 18b) in aqueous solution at ambient temperature. These results are presented in Figure 5.
In Figure 5A, the ESR spectrum of one-electron oxidized d[TGCGCGCA]2 (blue) in homogenous glassy (7.5 M LiCl/H2O) solution is shown. One-electron oxidized d[TGCGCGCA]2 has been produced by the one-electron oxidation reaction between Cl2•− and d[TGCGCGCA]2 via annealing in the dark for 45 min at ca. 173 K. The ESR spectrum of authentic G(N2–H)• (green color) in 1-Me-dGuo in H2O (7.5 M LiCl/H2O, Figure 3A) has been superimposed on it for comparison. Also, we have superimposed the authentic G(N1-H)• spectrum (red) obtained from glassy (7.5 M LiCl) in H2O samples of dGuo obtained by one-electron oxidation via annealing at 155 K in the dark. Using the G(N2–H)• (green) and the G(N1-H)• (red) spectra as benchmarks, we find that the ESR spectrum of one-electron oxidized d[TGCGCGCA]2 (blue) is composed of ca. 60% G(N2–H)• and ca. 40% G(N1-H)•. Thus, comparison of this analysis with that shown in Figures 4E and 4F, we conclude that contributions of G(N1-H)• and G(N2–H)• to the spectrum of one-electron oxidized d[TGCGCGCA]2 are very similar in H2O (Figure 5A) and in D2O (Figure 5E) glasses.
In Figure 5B, we have compared the spectrum of the one-electron oxidized d[TGCGCGCA]2 (blue color) obtained in H2O glass (see Figure 5A) with the spectrum of the one-electron oxidized d[TGCGCGCA]2 found in D2O glass (black) (seeFigure 4E). As found for 1-Me-dGuo (see Figure 3A), the wings of the G(N2–H)• spectrum of the one-electron oxidized d[TGCGCGCA]2 (blue color) obtained in H2O glass show clearly the Azz component of hyperfine coupling of the exchangeable N-H atom in d[TGCGCGCA]2. In addition the spectrum in D2O shows the collapse of these features expected from the loss of the N2-H coupling.
In Figure 6, we present the UV-visible spectrum of a sample of one-electron oxidized d[TGCGCGCA]2 (pink) in a H2O glass at pH 9 recorded at ca. 175 K. These conditions corresponds those for the ESR spectra found in Figures 4E and 5A where ca. 60% G(N2-H)• was found. The UV-visible spectrum in Figure 6 has an absorption at 630 nm similar to the UV-visible spectrum of the G(N2–H)• in 1-Me-dGuo in H2O glass at pH 9 (blue) recorded at 77 K. This similarity agrees with our more definitive ESR spectral assignment that the sample of one-electron oxidized d[TGCGCGCA]2 in homogenous H2O glass has substantial extent of G(N2–H)• in it.
We find that the UV-visible spectrum of the one-electron oxidized d[TGCGCGCA]2 (pink) and that of the G(N2–H)• found in 1-Me-Guo (black) in Figure 6 are also similar to the reported pulse radiolysis spectrum of one-electron oxidized guanine in homogenous aqueous solution of a ds DNA oligomer containing isolated guanine moiety (sequence G1AA = (5'AAAAAAGAAAAAA3': 3'TTTTTTCTTTTTT5'), seeFigure 4 in ref. 18b) at ambient temperature. Owing to the very close similarities in the UV-visible spectra shown in Figure 6, and on the basis of our ESR spectral results shown in Figure 5, the pulse radiolysis spectrum of the one-electron oxidized G1AA is likely is largely from G(N2–H)•.
(c) ESR studies of photo-excitation of G•+:C, G(N1-H)•:C(+H+) and G(N1-H)•:C in d[TGCGCGCA]2
Results in Figures 4 and 5 very clearly establish that the ESR spectrum of G(N2–H)•:C (see scheme 1) can be very easily distinguished from that of the other prototropic species, i.e., G•+:C, G(N1-H)•:C(+H+) or G(N1-H)•:C either in D2O or in H2O glasses. However, the ESR spectra of G(N1-H)•:C(+H+) or G(N1-H)•:C are not distinguishable by ESR in D2O or in H2O glasses. As mentioned earlier G•+:C, G(N1-H)•:C(+H+) are distinguishable by ESR but only in 8-deuteroG substituted oligos.
As discussed in the introduction, it has been observed that sugar radical formation in dGuo26 and in Guo27 via photo-excitation of one-electron oxidized G depends on the protonation state of the one-electron oxidized guanine, and hence, on the pH of the solution. Substantial sugar radical formation (>80%) via photo-excitation of the cation radical (G•+) but, at pH ≥9, only G(N1-H)• is present and only small amounts (ca. ≤10%) sugar radicals are been found on photoexcitation.26, 27 We also note here that in the homogenous frozen aqueous solution of highly polymerized ds salmon testes DNA, substantial formation of C1′• sugar radical (ca. 50%) via photo-excitation of the guanine cation radical in the wavelength range 310 – 480 nm has been observed.26
These results lead us to test whether the initial rate of photo-conversion to C1′• sugar radical via photo-excitation of G•+ in the ds DNA-oligomer and of G(N1-H)•:C(+H+) might be different from that via photo-excitation of G(N1-H)•:C. These results are presented in Figure 7 below.
Figure 7.

(A) ESR spectra of one-electron oxidized d[TGCGCGCA]2 in 7.5 M LiCl/H2O in the presence of K2S2O8 as an electron scavenger at three pHs - at pH ca. 3 (red), ca. 5 (black), and at ca. 9 (blue). After 1h of photo-excitation by a 250 W photoflood lamp at 148 K, sugar radical formation indicated by the arrows is shown in Figure (B). The outer components are due to the C1′-radical. The extent of sugar radical formation via photo-excitation of one-electron oxidized d[TGCGCGCA]2 at pH ca. 3 is found to be higher by a factor of 7.5, and at pH ca. 5 is found to be higher by a factor of 3 than the corresponding extent of sugar radical formation at pH ca. 9. On this basis, spectrum at pH ca. 3 (red) is assigned to the cation radical G•+ in the ds DNA-oligomer, spectrum at pH ca. 5 (black) in (A) to G(N1-H)•:C(+H+) whereas the spectrum at pH ca. 9 (red) in (A) is assigned to G(N1-H)•:C. All spectra were recorded at 77 K.
In Figure 7A, the ESR spectrum of the one-electron oxidized guanine in d[TGCGCGCA]2 in 7.5 M LiCl/H2O at pHs ca. 3 (red), ca. 5 (black), and ca. 9 (blue) are presented. As per the results from Figure 4, we expect G•+ in the ds DNA-oligomer to be present at pH ca. 3, G(N1-H)•:C(+H+) at pH 5, and G(N1-H)•:C at pH 9. All these samples were identically prepared and handled. The one-electron oxidant employed here is Cl2•− and the one-electron oxidation of d[TGCGCGCA]2 was carried out via annealing in the dark for 15–20 min at ca. 155 K. The ESR characteristics (g value at the center (g⊥), the overall hyperfine splitting, and the lineshape) of the ESR spectrum (red) of the one-electron oxidized d[TGCGCGCA]2 at pH ca. 3, 5 and 9 are near identical even though the GC one electron oxidized species are in differing protonation states which are thus not readily distinguishable in H2O. This result is in agreement with earlier reported results obtained in dGuo samples (see Figure 1 in ref. 9).
After 1h of photo-excitation of one-electron oxidized guanine in d[TGCGCGCA]2 with a 250 W photoflood lamp, the formation of sugar radicals, predominantly C1′•, via photo-excitation at pH ca. 3 (Figure 7B red) is seen to be substantially more that at pH ca. 5 (see Figure 7B, black), and even more than that at ca. 9 (see Figure 7B blue). Analysis of the spectra show that sugar radical production at pH ca. 3 is found to be higher by factor of 2.5 over that at pH ca. 5 and of 7.5 over that at pH ca. 9. Thus, we find that photoconversion occurs most readily for G•+ in the ds DNA-oligomer at pH 3, followed by G(N1-H)•:C(+H+) at pH 5 with the least extent for G(N1-H)•:C at pH 9. Hence the results shown in Figure 7B clearly suggest that G•+:C, G(N1-H)•:C(+H+), and G(N1-H)•:C can be distinguished from one another, based on the extent of photo-conversion to predominantly C1′• sugar radical.
To test the responsiveness of G(N2-H)•:C to sugar radical formation on photoexcitation, we have also photo-excited a glassy sample of one-electron oxidized d[TGCGCGCA]2 in a D2O glass at pH 9 which was annealed to ca. 175 K (see supplemental information Figure S8). The conditions before photo-excitation correspond to those for the ESR spectra found in Figures 4E and 5A where ca. 60% G(N2-H)• was observed. Photo-excitation of this glassy sample using a 20 mW 405 nm laser for 45 min at 148 K caused only a minor (ca. 20 %) conversion to C1'-sugar radical (see supplemental information Figure S8). For an identically prepared (D2O) glassy sample of one-electron oxidized d[TGCGCGCA]2 in which one-electron oxidized guanine is in G(N1-H)•:C(+H+) state, only 30 min of photo-excitation using the same 405 nm laser at 148 K resulted in near complete conversion (ca. 90%) to sugar radicals (see supplemental information Figure S8). These results and those presented above clearly show that G(N1-H)•:C(+H+) is far more sensitive to photoconversion that either G(N2-H)•:C or G(N1-H)•:C.
These results lead us to suggest a mechanism for sugar radical formation on photo-excitation in dsDNA-oligomers containing one-electron oxidized G. Arguing from the results for the free deoxyribonucleotide, dGuo, where G•+ was photosensitive and G(N1-H)• was not photosensitive, one-electron oxidized guanine base should be protonated for sugar radical formation to take place.26 Thus, the substantial photo-conversion of one-electron oxidized d[TGCGCGCA]2 at pH ca. 3 where G•+ is present (see Figure 7B, red) is the expected result. To account for the photo-conversion to sugar radical at pH ca. 5, we propose that reprotonation of the guanine base in G(N1-H)•:C(+H+) in the excited state precedes deprotonation from the sugar (see scheme 4).
Scheme 4.

Plausible mechanism for the sugar radical formation via excited G(N1-H)•:C(+H+).
For photolysis of G(N1-H)•:C at pH ca. 9 (Figure 7B, red), reprotonation of the guanine base in the excited state is hindered as the proton must return from the surrounding solvent and thus the very low extent of photo-conversion to sugar radical is expected. For G(N2-H)•:C is missing the N2 proton and this is also found to deactivate the species to photoconversion to sugar radicals.
Theoretical (DFT) studies
(a) Hydrated 1-Me-dGuo
The optimized geometries of 1-Me-dG•+, 1-Me-dG(N2-H)•syn and 1-Me-dG(N2-H)•anti in the presence of seven waters are shown in Figure 8. The calculations based on DFT/B3LYP/6-31G(d) method show that the 1-Me-dG(N2-H)•syn conformation is more stable than the 1-Me-dG(N2-H)•anti conformation by ca. 7 kcal/mol. This is in agreement with our recent study in one-electron oxidized dGuo21, in which G(N2-H)•syn was found to be more stable than its corresponding anti conformer by 3.6 kcal/mol. In that study, we employed experiment and theory to show that the N1-H of G•+ in dGuo is the preferred site of deprotonation from G•+. However, in 1-Me-dGuo (or 1-Me-Guo), the H atom at the N1-site has been substituted by the methyl group, and the site of deprotonation in 1-Me-dG•+ in 1-Me-dGuo (or, in 1-Me-G•+ in 1-Me-Guo) must be at N2 as Steenken and co-workers17a pointed out earlier. The B3LYP/6-31G(D) calculated HFCCs of 1-Me-dG•+, and 1-Me-dG(N2-H)• in syn- and anti-conformations along with the experimental HFCCs are presented in Table 1. From Table 1, it is evident that the theoretically obtained values of HFCCs of 1-Me-dG•+ are in good agreement with the corresponding experimental values. From the present study, we also found that substitution of the H-atom at N1 site of G•+ by the methyl group in 1-Me-dG•+ does not have any influence on the HFCCs values (see Table 1 (present study) and Table 1 in Ref. 21).
The theoretically obtained HFCCs of 1-Me-dG(N2-H)•syn and 1-Me-dG(N2-H)•anti are found to be very similar and matched reasonably well with the experimental values (Table 1). However, on the basis of stability, we suggest that experimentally observed spectrum of 1-Me-dG(N2-H)• in either 1-Me-dGuo (or of 1-Me-G(N2-H)• in 1-Me-dGuo) has the predominant contribution from the syn-conformation of 1-Me-dG(N2-H)• (Figure 8b)).
(b) Hydrated GC base pair
The intra-base pair proton transfer reactions in one-electron oxidized G:C (see Scheme 1 and Scheme 3 for atom numbering) is quite important and proposed to control the hole transfer in DNA. Recently, the intra-base pair proton transfer from G(N1-H) to C in G•+:C has been studied using experiment9 and B3LYP/6-31+G** level of theory22 and G(N1-H)•:C(+H+) was found to be more stable in the presence of the aqueous environment.
Regarding the subsequent deprotonation of one-electron oxidized G:C to the surrounding solvent, there are two possibilities – (i) the deprotonation at N1-H leading to G(N1-H)•:C, and (ii) the deprotonation at N2-H (see scheme 3) leading to G(N2-H)•:C (see scheme 1). Therefore, we optimized the structures of G(N1-H)•:C and G(N2-H)•:C in the presence of 11 waters using B3LYP/6-31+G** method (Figure 9). It has been observed that the HFCC values are independent of the two methods - B3LYP/6-31G(d) and B3LYP/6-31+G**.38
Figure 9.

The fully optimized geometries of (a) G(N1-H)•:C and (d) G(N2-H)•:C in the presence of 11 water molecules. The optimization was carried out with the aid of DFT/B3LYP/6-31+G** method. The relative stabilities between G•+: C and G(N1-H)•:C(+H+) and G(N1-H)•:C and G(N2-H)•:C are given in parentheses.
Using B3LYP/DZP++ method, Bera and Schaefer25 had also studied the deprotonation from various sites of G:C base pair. They had found that structure shown in Figure 9b due to loss of N2H” is ca. 1 kcal/mol more stable than the corresponding structure obtained due to the loss of N2-H' (see Scheme 3). The calculations reported here show that both G(N1-H)•:C and G(N2-H)•:C (Figure 9) are nearly isoenergetic with a difference of 0.4 kcal/mol, while, in gas phase G(N2-H)•:C was found to be more stable than G(N1-H)•:C by 1 kcal/mol as found by Bera and Schaefer.25 Our calculated optimized structure, shown in Figure 8a, is in accord with the Bera and Schaefer's calculation (see Figure 2 in Ref. 25). From theoretical calculations, it is predicted that at ambient temperature both structures (G(N1-H)•:C and G(N2-H)•:C) should be present in equilibrium in near equal amounts. Our ESR results, see Figures 4E and 5A, at 175 K show near equal amounts of the two forms with N2 slightly favored; pulse radiolysis data at ambient temperature suggest N2 may be the predominant site of deprotonation for a DNA oligomer with one G (See Figure 6).
Conclusions
The experimental and theoretical studies reported here, lead to the following salient findings:
1. Change of site of deprotonation of one-electron oxidized G:C with temperature
The initial site of deprotonation of one-electron oxidized guanine in double stranded DNA to the surrounding solvent is at N1. However, upon increasing the temperature via progressive annealing to 175 K (see Figures 4E, 4F, and Figure 5), the site of deprotonation shifts to an equilibrium mixture of G(N1-H)•:C and G(N2-H)•:C. We note for another ds oligomer, d[TGGGCCCA]2, we have also found an equilibrium mixture of G(N1-H)•:C and G(N2-H)•:C at ca. 175 K (see supplemental information Figure S7). Our theoretical calculations for a single GC base pair, suggest a very slight favoring of G(N2-H)•:C by 0.4 kcal/mol over G(N1-H)•:C. Our UV-vis spectra (Figure 6) when compared to pulse radiolysis studies carried out at room temperature in aqueous solution by Tagawa et al. 18b which show loss of absorption at 630 nm with the increasing extent of continuous G moieties in one electron oxidized oligos. This suggests that G(N1-H)•:C is favored over G(N2-H)•:C with the extent of continuous G moieties. This base sequence dependence needs to be confirmed in other studies.
2. Identification of G(N2–H)•:C in the ds oligomer
By employing the ESR and UV-vis spectral studies of one-electron oxidized 1-Me-dGuo (or 1-Me-Guo) as benchmarks, we have been able to analyze the UV-visible and ESR spectral data from one-electron oxidized d[TGCGCGCA]2 at various pHs. These results clearly show that, for the first time, G(N2-H)•:C is easily identified in the one-electron oxidized ds DNA-oligomer, from the other prototropic forms shown in scheme 1 owing to its distinctly observable exocyclic nitrogen HFCC (Azz(N-2) =16 G), the exchangeable N2-H proton HFCC (Azz = 8G), and by the characteristic UV-vis absorption at 630 nm (see Figures 1C and 6). Pulse radiolysis studies by Tagawa et al. 18 for an one-electron oxidized ds DNA oligomer with one G also show an absorption at 630 nm and thus we suggest provide evidence of formation of G(N2-H)•:C at ambient temperature. Our theoretical calculations performed by employing density functional theory (DFT) add substantial confidence in our identification of G(N2-H)• from the closeness of computed hyperfine couplings to experiment (see Table 1 and Figure 8).
3. Distinction of the protonation states via photo-conversion of one-electron oxidized guanine in the ds DNA oligomer
The four protonation states of the one-electron oxidized G:C in d[TGCGCGCA]2, i.e., G•+:C, G(N1-H)•:C(+H+), G(N1-H)•:C, and G(N2-H)•:C are schematically presented in the scheme 1. It is clearly evident from Figure 7B that, the extent of photo-conversion to sugar radicals (mainly C1′•) for G•+:C, G(N1-H)•:C(+H+) and G(N1-H)•:C in the oligomer allows for a clear distinction between these various protonation states. To explain this difference in sugar radical formation on photoexcitation, we propose that proton transfer to N1 in the G moiety of G(N1-H)•:C(+H+) within the lifetime of excited state is required for the subsequent deprotonation step from the sugar ring (Scheme 4). The presence of the N1-H proton on G in G•+:C accounts for the greater sugar radical formation than for G(N1-H)•:C(+H+). Further, the lack of the N1–H proton on C in G(N1-H)•:C accounts for the lesser amount of sugar radical formation than found for G(N1-H)•:C(+H+). The fact that G(N2-H)•:C also shows little photosensitivity shows that deprotonation at N2 also deactivates photoconversion to sugar radicals. We note that recent work by Kohler and coworkers48 on the excited states in GC oligomer also suggests that proton transfer in ion-radical exciplexes occurs within the time frame of the excitation.
Thus, in this work, we have been able to identify or distinguish each of the four protonation states of one-electron oxidized G:C shown in scheme 1.
4. Relevance of the prototropic to hole transfer studies in DNA
Figures 4 A to D clearly show that up to 155 K, the site of deprotonation of one-electron oxidized guanine in double stranded DNA to the surrounding solvent is at N1. On progressive annealing to 175 K (see Figures 4E, 4F, and Figure 5), the site of deprotonation shifts from G(N1-H)•:C to an equilibrium mixture of G(N1-H)•:C and G(N2-H)•:C. In the homogenous glassy solution of the DNA-oligomers up to 175 K, the characteristic line components for 8-hydroxy-guanyl-7-yl radical (•GOH) which would result from the nucleophilic addition of water to the guanine cation radical33 have not been observed. Furthermore, the sharp singlet at g = 2.0048 for the subsequent 2 electron oxidation product of •GOH which has been assigned to 8-oxo-G cation radical33 was not observed after annealing to 175 K. From our previous work using hydrated DNA pellets,33 it was found that the nucleophilic addition of water to guanine cation radical and the subsequent multiple one-electron oxidation processes to 8-oxo-G cation radical occur above ca. 200 K. However, for the glassy systems used in this work, at temperatures above 180 K, the glass becomes a liquid and radicals are quickly lost. Thus, the glassy system used in this report is most suitable for ESR studies of radical intermediates which are involved in the prototropic equilibria of one electron oxidized guanine radical.
5. Implication of prototropic equlibria of guanine cation radical to oxidative stress
One implication of this work that may lead to further investigations is the effect of the protonation state on subsequent reactions of the one electron oxidized DNA. It is well known that in the presence of oxygen, substantial formation of imidazolone lesions has been reported in both ss and ds DNA-oligomers via attack of O2•− at the C5 carbon atom in the guanine of G(−H)•. 1, 49 – 51 We find that G(N2-H)• has a higher spin density on the heteroatoms (N2 and N3) and less on the carbon sites in the guanine moiety than we find for G(N1-H)•. This is evidenced both from the theoretical calculations and experimental hyperfine couplings reported in this work. Thus, the reaction of O2•− should add to G(N1-H)• more readily than to G(N2-H)•. Thus GGG moieties which are suggested in point 1 to favor G(N1-H)• are predicted to be more prone to oxidative stress than a single G sites which favor G(N2-H)•.
Supplementary Material
Acknowledgments
This work was supported by the NIH NCI under grant no. R01CA045424. Computational studies were supported by a computational facilities grant NSF CHE-0722689.
References
- 1.von Sonntag C. Free-radical-induced DNA Damage and Its Repair. Springer-Verlag; Berlin, Heidelberg: 2006. pp. 335–447. [Google Scholar]
- 2.Wagenknecht H-A, editor. Charge Transfer in DNA: From Mechanism to Application. Wiley-VCH Verlag GmbH & Co.; KGaA, Weiheim: 2005. [Google Scholar]
- 3.Schuster GB, editor. Topics in Current Chemistry. Springer-Verlag; Berlin, Heidelberg: 2004. Long Range Charge Transfer in DNA. I and II. [Google Scholar]
- 4.Wang Q, Fiebig T. In: Charge Migration in DNA Physics, Chemistry and Biology Perspectives. Chakraborty T, editor. Springer-Verlag; Berlin, Heidelberg: 2007. pp. 221–248. [Google Scholar]
- 5.Giese B. Ann. Rev. Biochem. 2002;71:51–70. doi: 10.1146/annurev.biochem.71.083101.134037. [DOI] [PubMed] [Google Scholar]
- 6.Delaney S, Barton JK. J. Org. Chem. 2003;68:6475–6483. doi: 10.1021/jo030095y. [DOI] [PubMed] [Google Scholar]
- 7.Lewis FD. Photochem. Photobiol. 2005;81:65–72. doi: 10.1562/2004-09-01-ir-299.1. [DOI] [PubMed] [Google Scholar]
- 8.(a) Becker D, Adhikary A, Sevilla MD. In: Charge Migration in DNA: Physics, Chemistry and Biology Perspectives. Chakraborty T, editor. Springer-Verlag; Berlin, Heidelberg, New York: 2007. pp. 139–175. [Google Scholar]; (b) Becker D, Adhikary A, Sevilla MD. In: Recent Trends in Radiation Chemistry. Rao BSM, Wishart J, editors. World Scientific Publishing Co.; Singapore, New Jersey, London: In Press. [Google Scholar]; (C) Becker D, Adhikary A, Sevilla MD. In: Charged Particle and Photon Interactions with Matter - Recent Advances, Applications, and Interfaces. Hatano Y, Katsumura Y, Mozumder A, editors. CRC Press; Taylor & Francis, Boca Raton: In Press. [Google Scholar]
- 9.Adhikary A, Khanduri D, Sevilla MD. J. Am. Chem. Soc. 2009;131:8614–8619. doi: 10.1021/ja9014869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Bertran J, Oliva A, Rodriguez-Santiego L, Sodupe M. J. Am. Chem. Soc. 1998;120:8159–8167. [Google Scholar]; (b) Hutter M, Clark T. J. Am. Chem. Soc. 1996;118:7574. [Google Scholar]; (C) Li XF, Sevilla MD. Adv. Quantum Chem. 2007;52:59–87. [Google Scholar]
- 11.Kumar A, Sevilla MD. In: Radiation Induced Molecular Phenomena in Nucleic Acid: A Comprehensive Theoretical and Experimental Analysis. Shukla MK, Leszczynski J, editors. Springer-Verlag; Berlin, Heidelberg, New York: 2008. pp. 577–617. [Google Scholar]
- 12.Arkin MR, Stemp EDA, Pulver SC, Barton JK. Chem. Biol. 1997;4:389–400. doi: 10.1016/s1074-5521(97)90129-0. [DOI] [PubMed] [Google Scholar]
- 13.Gasper SM, Schuster GB. J. Am. Chem. Soc. 1997;119:12762–12771. [Google Scholar]
- 14.Núñez ME, Hall DB, Barton JK. Chem. Biol. 1999;6:85–97. doi: 10.1016/S1074-5521(99)80005-2. [DOI] [PubMed] [Google Scholar]
- 15.Saito I, Nakamura T, Nakatani K, Yoshioka Y, Yamaguchi K, Sugiyama H. J. Am. Chem. Soc. 1998;120:12686–12687. [Google Scholar]
- 16.Lee YA, Durandin A, Dedon PC, Geacintov NE, Shafirovich V. J. Phys. Chem. B. 2008;112:1834–1844. doi: 10.1021/jp076777x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Steenken S, Candeias LP. J. Am. Chem. Soc. 1989;111:1094–1099. [Google Scholar]; (b) Steenken S. Chem. Rev. 1989;89:503–520. [Google Scholar]; (C) Steenken S. Free Radical Res Commun. 1992;16:349–379. doi: 10.3109/10715769209049187. [DOI] [PubMed] [Google Scholar]; (d) Steenken S. Biol. Chem. 1997;378:1293–1297. [PubMed] [Google Scholar]
- 18.(a) Kobayashi K, Tagawa S. J. Am. Chem. Soc. 2003;125:10213–10218. doi: 10.1021/ja036211w. [DOI] [PubMed] [Google Scholar]; (b) Kobayashi K, Yamagami R, Tagawa S. J. Phys. Chem. B. 2008;112:10752–10757. doi: 10.1021/jp804005t. [DOI] [PubMed] [Google Scholar]
- 19.Shafirovich V, Dourandin A, Geacintov NE. J. Phys. Chem. B. 2001;105:8431–8435. [Google Scholar]
- 20.Kawai K, Osakada Y, Majima T. ChemPhysChem. 2009;10:1766–1769. doi: 10.1002/cphc.200900148. [DOI] [PubMed] [Google Scholar]
- 21.Adhikary A, Kumar A, Becker D, Sevilla MD. J. Phys Chem. B. 2006;110:24170–24180. doi: 10.1021/jp064361y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar A, Sevilla MD. J. Phys. Chem. B. 2009;113:11359–11361. doi: 10.1021/jp903403d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chatgilialoglu C, Caminal C, Altieri A, Vougioukalakis GC, Mulazzani QG, Gimisis T, Guerra M. J. Am. Chem. Soc. 2006;128:13796–13805. doi: 10.1021/ja062636h. [DOI] [PubMed] [Google Scholar]
- 24.Anderson RF, Shinde SS, Maroz A. J. Am. Chem. Soc. 2006;128:15966–15967. doi: 10.1021/ja0658416. [DOI] [PubMed] [Google Scholar]
- 25.Bera PP, Schaefer HF., III Proc. Natl. Acad. Sci. USA. 2005;102:6698–6703. doi: 10.1073/pnas.0408644102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adhikary A, Malkhasian AYS, Collins S, Koppen J, Becker D, Sevilla MD. Nucleic Acids Res. 2005;33:5553–5564. doi: 10.1093/nar/gki857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khanduri D, Collins S, Kumar A, Adhikary A, Sevilla MD. J. Phys. Chem. B. 2008;112:2168–2178. doi: 10.1021/jp077429y. [DOI] [PubMed] [Google Scholar]
- 28.Adhikary A, Kumar A, Khanduri D, Sevilla MD. J. Am. Chem. Soc. 2008;130:10282–10292. doi: 10.1021/ja802122s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adhikary A, Becker D, Collins S, Koppen J, Sevilla MD. Nucleic Acids Res. 2006;34:1501–1511. doi: 10.1093/nar/gkl026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adhikary A, Kumar A, Sevilla MD. Radiation Research. 2006;165:479–484. doi: 10.1667/rr3563.1. [DOI] [PubMed] [Google Scholar]
- 31.Adhikary A, Collins S, Khanduri D, Sevilla MD. J. Phys. Chem. B. 2007;111:7415–7421. doi: 10.1021/jp071107c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O'Neill M, Barton JK. J. Am. Chem. Soc. 2004;126:13234–13235. doi: 10.1021/ja0455897. [DOI] [PubMed] [Google Scholar]
- 33.Shukla LI, Adhikary A, Pazdro R, Becker D, Sevilla MD. Nucleic Acids Res. 2004;32:6565–6574. doi: 10.1093/nar/gkh989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Witwicki M, Jezierska J, Ozarowski A. Chem. Phys. Lett. 2009;473:160. [Google Scholar]
- 35.(a) Becke AD. J. Chem. Phys. 1993;98:1372. [Google Scholar]; (b) Stephens PJ, Devlin FJ, Frisch MJ, Chabalowski CF. J. Phys. Chem. 1994;98:11623. [Google Scholar]
- 36.Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 37.Frisch MJ, et al. Gaussian 03, revision B.04. Gaussian, Inc.; Pittsburgh, PA: 2003. For complete Reference. [Google Scholar]
- 38.(a) Hermosilla L, Calle P, García, de la Vega JM, Sieiro C. J. Phys. Chem. A. 2005;109:1114–1124. doi: 10.1021/jp0466901. [DOI] [PubMed] [Google Scholar]; (b) Hermosilla L, Calle P, García, de la Vega JM, Sieiro C. J. Phys. Chem. A. 2006;110:13600–13608. doi: 10.1021/jp064900z. [DOI] [PubMed] [Google Scholar]
- 39.Jmol development team, An Open-Science Project © 2004. http://jmol.sourceforge.net.
- 40.(a) Close DM, Sagstuen E, Nelson WH. J. Chem. Phys. 1985;82:4386–4388. [Google Scholar]; (b) Hole E, Nelson WH, Close DM, Sagstuen E. J. Chem. Phys. 1987;88:5218–5219. [Google Scholar]; (C) Close DM, Nelson WH, Sagstuen E. Radiat. Res. 1987;112:283–301. [PubMed] [Google Scholar]; (d) Hole EO, Sagstuen E. Radiat. Res. 1987;109:190–205. [PubMed] [Google Scholar]; (e) Ravkin B, Herak JN, Voit K, Hüttermann J. Radiat. Environ. Biophys. 1987;26:1–12. doi: 10.1007/BF01211360. [DOI] [PubMed] [Google Scholar]; (f) Nelson WH, Hole EO, Sagstuen E, Close DM. Int. J. Radiat. Biol. 1988;54:963–986. doi: 10.1080/09553008814552361. [DOI] [PubMed] [Google Scholar]; (g) Hole EO, Sagstuen E, Nelson WH, Close DM. Free Radical Res Commun. 1989;6:87–90. doi: 10.3109/10715768909073434. [DOI] [PubMed] [Google Scholar]; (h) Kim H, Budzinski EE, Box HC. J. Chem. Phys. 1989;90:1448–1451. [Google Scholar]; (i) Hole EO, Sagstuen E, Nelson WH, Close DM. Radiat. Res. 1991;125:119–128. [PubMed] [Google Scholar]; (j) Hole EO, Sagstuen E, Nelson WH, Close DM. Radiat. Res. 1992;129:1–10. [PubMed] [Google Scholar]; (k) Bachler V, Hildenbrand K. Radiat. Phys. Chem. 1992;40:59–68. [Google Scholar]; (l) Hole EO, Sagstuen E, Nelson WH, Close DM. Radiat. Res. 1992;129:119–138. [PubMed] [Google Scholar]
- 41.(a) Wetmore SD, Boyd RJ, Eriksson LA. J. Phys. Chem. B. 1998;102:9332–9343. [Google Scholar]; (b) Mundy CJ, Colvin ME, Quong AA. J. Phys. Chem. A. 2002;106:10063–10071. [Google Scholar]; (C) Gervasio FL, Laio A, Iannuzzi M, Parrinello M. Chem. Eur. J. 2004;10:4846–4852. doi: 10.1002/chem.200400171. [DOI] [PubMed] [Google Scholar]; (d) Luo Q, Li QS, Xie Y, Schaefer HF., III Collect. Czech. Chem. Commun. 2005;70:826–836. [Google Scholar]; (e) Naumov S, von Sonntag C. Radiat. Res. 2008;169:364–372. doi: 10.1667/RR1082.1. [DOI] [PubMed] [Google Scholar]
- 42.Adhikary A, Khanduri D, Kumar A, Sevilla MD. J. Phys. Chem. B. 2008;112:15844–15855. doi: 10.1021/jp808139e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hiraoka W, Kuwabara M, Sato F. Int. J. Radiat. Biol. 1989;55:51–58. doi: 10.1080/09553008914550061. and the references therein. [DOI] [PubMed] [Google Scholar]
- 44.Typically, comparison of pKa value of G•+ in the glassy sample at low temperature with that of G•+ in aqueous solution at ambient temperature shows that pKa values in our system (i.e., in the glassy sample at low temperature) are increased by about 1.5 units.42 By employing pulse radiolysis in aqueous solution at ambient temperature, Candeias et al., had determined the pKa value of 1-Me-G•+ as 4.7. 17a Therefore, we predict that the pKa value of the 1-Me-G•+ in our system would be about 6.2 and this value is close to the experimental result (about 6.5)
- 45.The effect of increasing the LiCl concentration on the prototropic equilibrium of 1-Me-dGuo•+ has been investigated. The following observations were found: (i) one-electron oxidized guanine in 1-Me-dGuo at 15 M LiCl/ H2O provided spectrum of 1-Me-dG(N2-H)• which is nearly identical with that shown in Figure 3A, and (ii) similar to what has been observed already in 7.5 M LiCl/ D2O (see Figure 2), 1-Me-dGuo•+ is converted to 1-Me-dG(N2-H)• at pH ca. 6.5 upon annealing at 15 M LiCl/H2O (see supplemental information Figure S5). Therefore, these observations have led us to conclude that the prototropic equilibrium of 1-Me-dG(N2-H)• in 1-Me-dGuo is not greatly affected by the concentration of LiCl
- 46.This result shows that protonation (deuteration) state of 1-Me-G(N2-H)• is not affected by solvent (D2O or H2O) of the supercooled (glassy) solution
- 47.Sprecher CA, Johnson WC., Jr. Biopolymers. 1977;16:2243–2264. doi: 10.1002/bip.1977.360161012. [DOI] [PubMed] [Google Scholar]
- 48.(a) de la Harpe K, Crespo-Hernández CE, Kohler B. J. Am. Chem. Soc. 2009 doi: 10.1021/ja9076364. DOI 10.1021/ja9076364. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) de la Harpe K, Crespo-Hernández CE, Kohler B. ChemPhysChem. 2009;10:1421–1425. doi: 10.1002/cphc.200900004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Misiaszek R, Crean C, Joffe A, Geacintov NE, Shafirovich V. J. Biol. Chem. 2004;279:32106–32115. doi: 10.1074/jbc.M313904200. [DOI] [PubMed] [Google Scholar]
- 50.Yun BH, Lee YA, Kim SK, Kuzmin V, Kolbanovskiy A, Dedon PC, Geacintov NE, Shafirovich V. J. Am. Chem. Soc. 2007;129:9321–9332. doi: 10.1021/ja066954s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cadet J, Douki T, Ravanat JL. Acc. Chem. Res. 2008;41:1075–1083. doi: 10.1021/ar700245e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.