

Abstract
Important changes occur in the cardiovascular system with advancing age, even in apparently healthy individuals. Thickening and stiffening of the large arteries develop due to collagen and calcium deposition and loss of elastic fibers in the medial layer. These arterial changes cause systolic blood pressure to rise with age, while diastolic blood pressure generally declines after the sixth decade. In the left ventricle, modest concentric wall thickening occurs due to cellular hypertrophy, but cavity size does not change. Although left ventricular systolic function is preserved across the age span, early diastolic filling rate declines 30–50% between the third and ninth decades. Conversely, an age-associated increase in late diastolic filling due to atrial contraction preserves end-diastolic volume. Aerobic exercise capacity declines approximately 10% per decade in cross-sectional studies; in longitudinal studies, however, this decline is accelerated in the elderly. Reductions in peak heart rate and peripheral oxygen utilization but not stroke volume appear to mediate the age-associated decline in aerobic capacity. Deficits in both cardiac b-adrenergic receptor density and in the efficiency of postsynaptic b-adrenergic signaling contribute significantly to the reduced cardiovascular performance during exercise in older adults. Although these cardiovascular aging changes are considered “normative”, they lower the threshold for the development of cardiovascular disease, which affects the majority of older adults.
Keywords: Cardiovascular, Aging, Arterial, Ventricular function, Exercise, b adrenergic
Introduction
Diseases and disabilities associated with aging are of increasing global importance as longevity increases. In the United States alone, it is estimated that there will be over 70 million people over the age of 65 by the year 2030, representing almost 25% of the population [1]. Cardiovascular (CV) disease is the leading cause of death in those aged 65 and above (40%), while 80% of all deaths from CV disease occur in this age group. It is important, therefore, that clinicians and researchers understand the physiologic changes that occur with aging if new approaches to disease identification and treatment and health maintenance are to be devised that not only increase longevity but also improve the quality of life at advanced ages.
The field of aging research has undergone a number of significant changes in the past few decades. While knowledge gleaned from autopsy-based studies has formed an indispensible foundation for our understanding of the aging process, there has been an inherent difficulty in separating the effect of aging per se from those of the comorbid illnesses that caused a subject’s death. The development of modern CV imaging techniques, however, has allowed studies like the Baltimore Longitudinal Study of Aging (BLSA), Framingham, and others to recruit healthy populations that can be studied non-invasively over long spans of time and thereby better separate the effects of aging from those of CV disease or lifestyle variables. Supplementation of these studies with animal and cell culture models has permitted significant insights into the complex changes that occur in the aging cardiovascular system.
Vascular aging
In studying the effects of aging on the CV system, it is important not to consider the heart as an isolated organ since it is in series with the vascular system. In fact, many researchers now feel that the greatest risk factor for the development of CV disease is “unsuccessful” age-associated arterial aging. Rather than acting as simple conduits for blood flow, blood vessels are dynamic structures that adapt, repair, remodel, and govern their structural and function properties using complex signaling pathways in response to load, stress, and age.
Arterial structural and functional changes
Macroscopic
A number of age-associated structural changes occur in the arterial system, including thickening and dilation of large arteries [2] (Table 1). Echocardiographic studies show that the aortic root dilates modestly with age, approximating 6% between the fourth and eighth decades [3] (Fig. 1a). In serial echocardiography over 16 years in Framingham participants, predicted aortic root diameter increased by 0.89 mm in men and 0.68 mm in women after adjustment for blood pressure and antihypertensive therapy for each 10 years increase in age [4]. Similar increases in aortic knob diameter have been observed in serial chest X-rays. Such aortic root dilation provides an additional stimulus for LV hypertrophy because the larger volume of blood in the proximal aorta leads to a greater inertial load against which the senescent heart must pump. Autopsy reports published as early as 1910 described age-associated aortic thickening. Cross-sectional studies using ultrasound imaging have demonstrated that the intimal-medial layer of the carotid artery thickens nearly threefold between the ages of 20 and 90 years in apparently healthy individuals [5] (Fig. 1b). Both the average and range of intimal-medial thickness measurements are greater at higher ages, suggesting a variable response to chronological age that merits further study to identify the components of “successful aging”.
Table 1.
Relationship of cardiovascular aging in healthy humans to cardiovascular disease
| Age-associated changes | Plausible mechanisms | Possible relationship to disease |
|---|---|---|
| CV structural remodeling | ||
| : Vascular intimal thickness | : VSMC migration and matrix production | Early stages of atherosclerosis |
| : Vascular stiffness | Elastin fragmentation : Elastase activity : Collagen production and cross-linking |
Systolic hypertension |
| Altered growth factor regulation and tissue repair | Atherosclerosis | |
| : LV wall thickness | : LV myocyte size ; Myocyte number |
; Early LV diastolic filling : LV filling pressure/dyspnea |
| Focal collagen deposition | ||
| : L. atrial size | : L. atrial volume/pressure | : Risk of atrial fibrillation |
| CV functional changes | ||
| Altered vascular tone | ; NO production/effects ; bAR responses |
Vascular stiffening/hypertension |
| ; CV reserve | : Vascular load ; Intrinsic myocardial contractility ; b-adrenergic modulation of heart rate, LV contractility and vascular tone |
Lower threshold for heart failure |
| ; Physical activity | Comorbidities ; Skeletal muscle mass |
Accelerated aging changes in CV structure and function; : Risk of CV disease |
bAR beta-adrenergic receptor, CV cardiovascular, LV left ventricular, NO nitric oxide, VSMC vascular smooth muscle cell. From reference 32
Fig. 1.
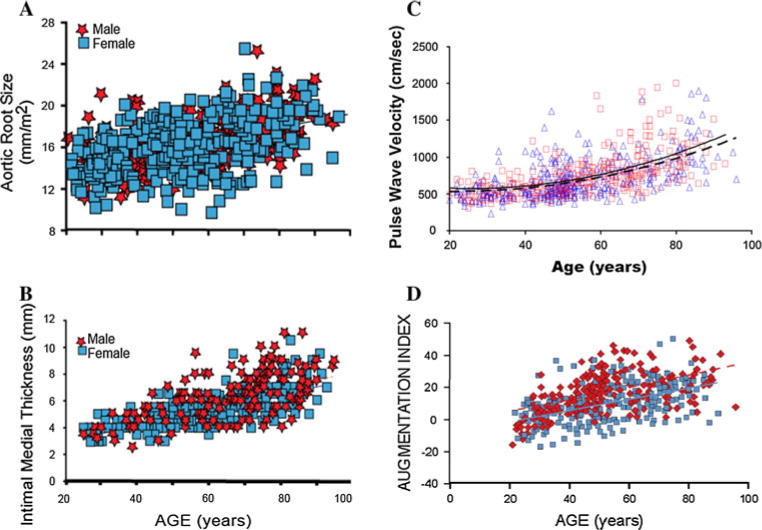
Age-associated changes in arterial structure and stiffness in healthy normotensive volunteers from the Baltimore Longitudinal Study of Aging (BLSA). Panel A Aortic root echocardiographic diameter, r = 0. 65, P \ 0.001 (from reference 3). Panel B Carotid intimal-medial thickness, r = 0.68, P \ 0.001 (from reference 5). Panel C Aortofemoral pulse wave velocity, men, dashed line and squares, r = 0.50, P \ 0.001; women, solid line and triangles, r = 0.63, P \ 0.001 (from reference 14). Panel D Carotid artery augmentation index, men, solid line and squares, r = 0. 63, P \ 0.001; women, dashed line and diamonds, r = 0.61, P \ 0.001 (from reference 14)
Microscopic and biochemical
Aging is associated with a number of structural and functional changes of the arterial wall media (hypertrophy, extracellular matrix accumulation, calcium deposits) and the vascular endothelium (decrease in the release of vasodilators and increased synthesis of vasoconstrictors) that are associated with increased vascular stiffness [6, 7]. Collagen and elastin provide the strength and elasticity, respectively, of the arterial wall and are normally stabilized by enzymatic cross-linking. With aging, an increase in collagen content, collagen cross-linking, and fraying of elastin fibrils occur in the medial layer [8], all of which reduce arterial distensibility and increase stiffness. The occurrence of irreversible non-enzymatic glycation-based cross-linking of collagen to form advanced glycation end products (AGEs) increases with age and is associated with increased arterial stiffness in elderly people [9]. These AGEs can interact with their receptors (RAGE) to stimulate a number of inflammatory and stress responses.
Through its secretion of nitric oxide (NO) and endothelin, the endothelium is a powerful regulator of arterial tone. Endothelial dysfunction has been identified in a number of CV disorders, including hypertension, hypercholesterolemia, and coronary and peripheral atherosclerosis. Human and animal studies have revealed that aging is associated with a reduction in endothelial dependent vasodilatation, thought secondary to reduced NO production [8]. A number of animal studies have found that NO production and NO levels decline with aging, likely as a result of a decline in the level of endothelial nitric oxide synthase (eNOS) [10]. Conversely, there is a 1,000-fold increase in angiotensin II (Ang-II) levels as well as significantly increased Ang-II signaling in aged arterial walls [11], both of which are thought to play a pivotal role in arterial aging, given the potent pressor and mitogenic effect of Ang-II. The marked increase in Ang-II in the aged arterial wall appears to offset the effect of reduced plasma levels of Ang-II in the elderly.
Systemic arterial function: pulse wave velocity and reflected pulse waves
Central arterial stiffening occurs with aging even in the absence of clinical hypertension. Systolic blood pressure (SBP), which is influenced by both arterial stiffness and cardiac function, rises with age even in normotensive cohorts [12]. In contrast, diastolic blood pressure (DBP) typically increases until the sixth decade and declines in later years. Thus, hypertension in the elderly is characterized by isolated or predominant SBP elevation. Pulse pressure, the difference between SBP and DBP, is a useful clinical index of arterial stiffness and the pulsatile load on the arterial tree. Some studies have suggested that pulse pressure is a more powerful predictor of future CV events than either SBP or DBP in middle-aged and older adults [13].
Two additional measures of arterial stiffness include augmentation index (AI) and pulse wave velocity (PWV). Pulse wave velocity (PWV) is a Doppler-based method that measures the speed with which an arterial pressure wave travels along the arterial tree, typically from the carotid region to the femoral artery. Multiple studies have shown that aortofemoral PWV increases with age, generally two-to threefold across the adult lifespan (Fig. 1c) [14, 15]. Importantly, PWV has been shown both in clinically healthy cohorts and in those with CV disease to be a predictor of future CV events, independent of blood pressure [16].
Another common method of arterial stiffness assessment, AI, illustrates the importance of cardiac-vascular interaction. When the forward pulse wave reaches an area of impedance mismatch (vessel bifurcation or movement to a higher resistance vessel), a reflected wave is generated that travels back up the arterial tree toward the central aorta. This reflected wave is identified as a small notch, inflection point, in the carotid and radial pulse waveforms, measured by arterial applanation tonometry. Similar to PWV, AI increases with age (Fig. 1d), even in clinically healthy volunteers [14]. The clinical significance of these AI changes with age is that in young subjects, the reflected wave typically arrives back at the proximal aorta in diastole and may assist in coronary artery diastolic filling. However, in older individuals, the reflected waves travel faster, thus arriving at the proximal aorta during late systole, thereby creating an increased load for the ventricle, a failure to augment DBP, and a potential compromise of coronary blood flow. Several studies in cohorts with CV disease have observed that higher AI is associated with adverse clinical outcomes [17].
Pulmonary arterial changes
Although less studied than the systemic arterial tree, the pulmonary vessels also appear to undergo age-associated remodeling, leading to increased pulmonary arterial stiffness and pulmonary artery systolic pressure (PASP). Among 1,413 Olmsted County, MN, residents aged 45 years and older, PASP estimated from Doppler echocardiography increased modestly from 26 ± 4 mmHg in persons 45–54 years old to 30 ± 6 mmHg in those aged 72–96 years [18]. Of note, higher PASP was an independent predictor of mortality (HR 1.46 per 10 mmHg). Other studies have shown an exaggerated rise in PASP and pulmonary artery mean pressure with age during cycle exercise in healthy adults [19].
Changes in cardiac structure and resting function with aging
Macroscopic
Left ventricular mass
The understanding of the aging-induced changes in left ventricular (LV) mass has undergone a number of changes over time as researchers have made improvements in technological approach, exclusion criteria, and statistical analysis. An autopsy-based study without specific screening criteria suggested that cardiac mass increased significantly with aging [20]. Initial M-mode echocardiographic studies in normal volunteers appeared to corroborate these findings [3]. However, in autopsies on subjects free from hypertension and coronary heart disease (CHD), Kitzman et al. found an increase in cardiac mass with age only for women, with no change in men [21]. A later autopsy study of hospitalized patients free of CHD found an age-associated decrease in cardiac mass of men and no change for women [22]. These latter findings have received support from a magnetic resonance imaging (MRI)-based study of healthy participants in the BLSA [23] as well as recent 3-dimensional echocardiographic studies. Based on these studies, it now appears that there is no change in LV mass in women but a decrease in LV mass in men with aging among individuals without CHD.
Left ventricular wall thickness, cavity size, and shape
Despite the absence of an increase in cardiac mass with aging, there is a significant increase in myocardial thickness [23] as a result of increased cardiomyocyte size. Although there is concentric LV hypertrophy, the interventricular septum increases in thickness more than the free wall [19], and there is a change in LV shape. The MRI study in BLSA volunteers demonstrated a shortening of the LV along its long axis and a shift from an elongated prolate ellipsoid geometry to a more spherical left ventricle with age [23]. Since a more spherical ventricle is subject to higher wall stress, the age-associated change in cardiac shape has important implications for overall contractile efficiency. Left ventricular diastolic and systolic dimensions, indexed to body surface area, are not age-related in healthy adults [3, 23]. However, a recent cardiac MRI study in 5,004 apparently healthy volunteers observed an age-related decline in both LV diastolic and systolic volumes and increase in LV mass/volume ratio in both sexes [24].
Microscopic
The heart of a young adult is composed of approximately 25% cardiomyocytes and a complex structure of connective tissue. Over time, there is a decrease in the total number of cardiomyocytes, likely due to apoptosis, but an increase in their individual size, i.e., hypertrophy [22]. In both animal and human studies, apoptotic myocytes were more prevalent in the hearts of older males compared to females, paralleling the age-related decline of LV mass in men but not in women [22]. Within the connective tissue, there is an increase in collagen content, fibrosis, and deposition of cardiac amyloid and lipofuscin [25].
While it has been traditionally thought that all cardiomyocytes are terminally differentiated, carbon dating methods have established that cardiomyocytes continue to be synthesized throughout life [26]. The cardiac myocyte-to-collagen ratio in the older heart either remains constant or increases. Studies have generally found that the heart becomes more fibrotic [27, 28] and stiffer, i.e., greater passive and active tension, [29] with age.
In all parts of the conduction system, there is an increase in elastic and collagenous tissue with age. Fat accumulates around the sinoatrial node (SA), sometimes separating the node from the atrial musculature. A marked decrease in the number of pacemaker cells in the SA node generally occurs after age 60; by 75 years, there is more than a 90% reduction in the cell number found in the young adult. These changes may predispose the older heart to sick sinus syndrome. With aging, the left side of the cardiac skeleton undergoes a variable degree of calcification; this may affect the aortic and mitral annuli, the central fibrous body, and the summit of the interventricular septum. If the atrioventricular (AV) node, AV bundle, bifurcation, and proximal left and right bundle branches are involved in this process, AV or intraventricular block may develop.
Resting cardiac function
Echocardiographic LV shortening fraction [3] and radio-nuclide LV ejection fraction (LVEF) [30], the two most common measures of global LV systolic performance, are not affected by age in healthy normotensive persons at rest. Prolonged contractile activation of the thickened LV wall [31] maintains a normal ejection time and compensates for the late systolic augmentation of BP, preserving systolic cardiac pump function despite increased arterial stiffness (Fig. 2) [32]. In contrast to systolic LV function, however, LV diastolic performance is prominently altered by aging. Whereas diastolic filling of the ventricles of younger adults mostly occurs in early diastole, pulsed echocardiographic Doppler [33] and radionuclide [34] techniques show that transmitral early diastolic peak-filling rate declines by 30–50% between ages 20 and 80 years (Fig. 3). Conversely, peak A-wave velocity, which represents late LV filling facilitated by atrial contraction, increases with age [33] via a modest age-associated increase in left atrial size [35]. Tissue Doppler echocardiography, which is less dependent on preload and afterload effects than pulsed Doppler, confirms the age-associated reduction in early diastolic filling rate and increased late filling of the LV [36]. Thus, the pattern of predominant early diastolic LV filling among younger healthy adults is reversed with advanced age.
Fig. 2.
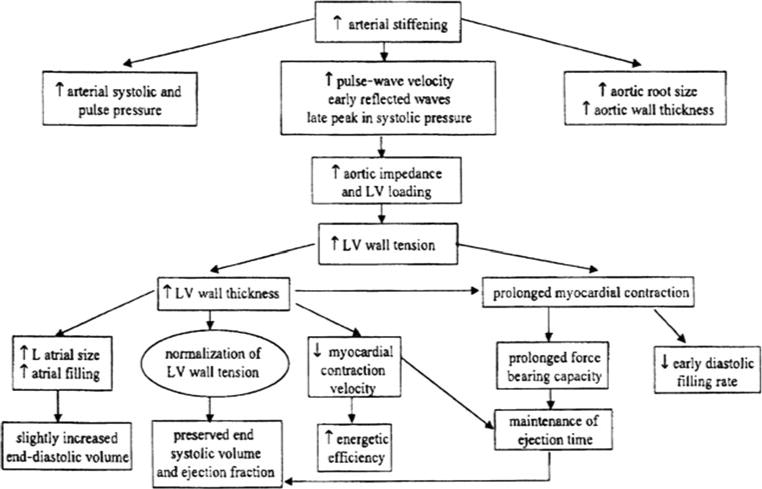
Conceptual framework for age-associated changes in cardiovascular structure and function (from reference 32)
Fig. 3.
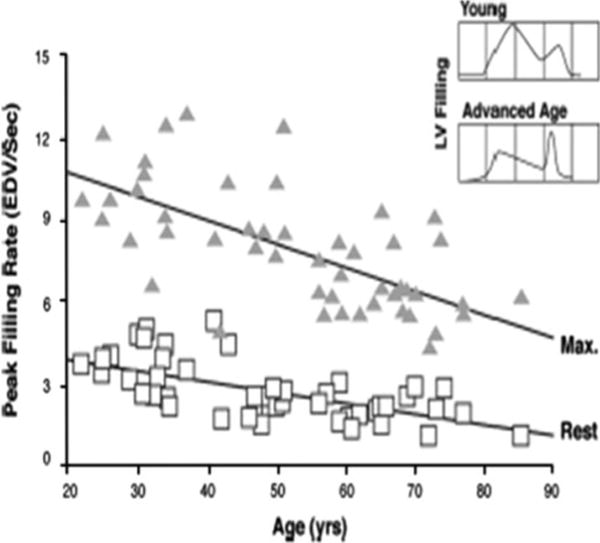
Reduction in radionuclide-derived early left ventricular diastolic filling with age at rest and during maximal upright cycle exercise in healthy BLSA volunteers. The decline in peak filling rate with age was significant, both at rest and during exercise, r = − 0.64, P \ 0.05, for each condition. The inset displays a typical transmitral Doppler echocardiographic flow profile of a young and older adult (from reference 34)
Delays in LV filling with age may derive in part from mechanical loads induced by reduced LV diastolic compliance and increased LV wall thickness. However, animal studies show prolonged isovolumic relaxation (the interval between end systole and mitral valve opening) and increased myocardial diastolic stiffness in both left and right ventricles of older animals, even though pulmonary artery pressure rises only slightly with age. Therefore, aging changes in cardiac isovolumic relaxation may also result from intrinsic age-related impairment of calcium accumulation by the sarcoplasmic reticulum [37]. Figure 2 provides a conceptual framework for the age-associated changes in CV structure and resting function.
Although age-related delays in early diastolic filling rate will usually not compromise LV end-diastolic volume and stroke volume at rest, stress-induced tachycardia (e.g., with exercise, fever, or other physiologic stress) is likely to exacerbate diastolic filling abnormalities. Tachycardia not only disproportionately foreshortens the time available for diastolic filling but also exacerbates impaired energy-dependent uptake of calcium into the sarcoplasmic reticulum. Therefore, fast heart rates are commonly associated with diastolic filling abnormalities in the elderly, and the higher LV diastolic pressure is transmitted into the lungs. Thus, blood is more likely to back into the lungs despite normal resting LV systolic function. Echocardiographic assessment has evolved to better identify older adults with such diastolic-based congestive heart failure [38].
The atrial enlargement that occurs as a function of age and diastolic dysfunction occurs primarily after 70 years of age [35] and increases susceptibility of older adults to atrial fibrillation (AF). Although AF is relatively innocuous in many younger adults, it is more likely to provoke symptoms and clinical events among the elderly [39]. Not only is AF commonly associated with poorly tolerated fast ventricular rates, but the loss of the atrial boost to diastolic filling that occurs with AF aggravates age-related diastolic filling limitations. Thus, older patients with AF are more likely to suffer reduced cardiac output and resultant dyspnea and fatigue than younger individuals.
Age-associated myocardial changes also predispose some older adults to ischemia and heart failure. LV mural thickening predisposes to subendocardial ischemia by increasing the distance between the epicardial coronary arteries and the subendocardial myocardial cells. Furthermore, in contrast to cardiac hypertrophy in young athletes, capillary growth and flow regulation in older hearts may not match the oxygen demands of the hypertrophied myocytes [40]. These intramyocardial changes in capillarity and flow-dynamics are compounded by the peripheral arterial stiffening and accelerated PWV, i.e., faster reflected pressure waves now arriving in systole, such that subendocardial perfusion is no longer bolstered by augmented pressures in diastole.
Due to the age-associated changes in the vasculature and heart described above, often compounded by a long exposure to other CV risk factors, hypertension, coronary events, and heart failure all become more common with aging (Table 1) [8]. Intrinsic vulnerability to atherosclerosis in the vasculature predisposes to myocardial ischemia and infarction and to stroke and peripheral arterial disease. Systolic heart failure may develop as the result of ischemic coronary events or prolonged hypertension, either of which can lead to deterioration of LV systolic function.
Heart failure with preserved LV systolic function is another common cardiac disorder in the elderly, resulting, at least in part, from the age-associated impairment of early diastolic filling [38]. Although LV filling is delayed in most older adults, it becomes more conducive to heart failure when accentuated by hypertension, diabetes, CHD, and AF, all common comorbidities seen with aging.
Cardiovascular response to exercise
The CV response to physical exercise in older individuals has considerable relevance in clinical medicine. First, the ability of older adults to maintain functional independence depends on their ability to perform tasks requiring both aerobic capacity and muscle strength. In addition, the CV response to exercise stress is important in assessing the ability of older individuals to respond to disease states. Finally, the CV response to exercise has considerable value in the diagnosis and treatment of patients with CV disease. Exercise testing is frequently utilized to detect and quantify the severity of CV disease. Clearly, the utility of such diagnostic exercise testing depends on precise information regarding normal limits relative to age.
Aerobic exercise capacity
The performance of oxygen-utilizing (i.e., aerobic) activities is a fundamental requirement of independent living and is probably the best-studied CV stressor. The accepted standard for aerobic fitness, maximal oxygen consumption (VO2max), is the product of cardiac output (the central component) and arteriovenous oxygen difference (the peripheral component). In healthy younger adults, VO2max is up to 15 times greater than resting VO2. This increase is accomplished by a four- to fivefold increase in cardiac output and a threefold widening of the arteriovenous oxygen (A–V) O2 difference. The increase in cardiac output is achieved by a tripling of heart rate and 30–75% increase in stroke volume. The increase in (A–V) O2 difference is due to both a marked increase in the relative proportion of cardiac output delivered to working muscles and an augmented oxygen extraction by these muscles. Given that total body VO2max is strongly influenced by body size and muscle mass, VO2max is typically compared across individuals by normalizing for body weight or fat-free mass.
Over the past half century, numerous studies have demonstrated that weight-adjusted treadmill VO2max declines with age. In cross-sectional studies, the decline is approximately 50% from the third to ninth decade (Table 2) [41]. Nevertheless, the extent of the VO2max decline with aging varies among studies, depending on age ranges, body weight and composition, and habitual physical activity patterns among the populations tested. A more pronounced age-associated decline in VO2max is reported in longitudinal than in cross-sectional studies. A typical conclusion of cross-sectional studies is that VO2max declines linearly with age. However, a recent analysis in the BLSA demonstrated that the longitudinal decline in aerobic capacity is not constant across adulthood as assumed by cross-sectional studies but accelerates markedly with time, especially in men, regardless of physical activity levels (Fig. 4) [42].
Table 2.
Normal changes in maximal aerobic capacity and its determinants between ages 20 and 80 years
| Exercise variable | Aging change |
|---|---|
| Oxygen consumption | ; 50% |
| AV oxygen difference | ; 20% |
| Cardiac output | ; 30% |
| Heart rate | ; 30% |
| LV stroke volume | No change |
| LV end-diastolic volume | : 30% |
| LV end-systolic volume | : 100% |
| LV ejection fraction | ; 15% |
| LV contractility | ; 60% |
| Systemic vascular resistance | : 30% |
| Plasma catecholamines | : |
| CV b-adrenergic responses | ; |
AV arteriovenous, CV cardiovascular, LV left ventricular. From reference 32
Fig. 4.
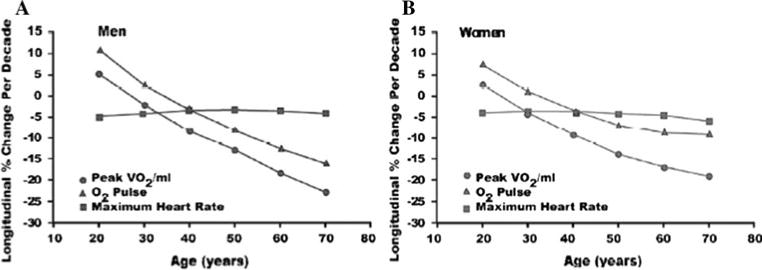
Longitudinal changes in peak oxygen consumption (VO2) and its components, maximal heart rate and oxygen pulse, in healthy BLSA volunteers. Whereas the decline in maximal heart rate across successive decades remains relatively constant at * 5%/decade, there is an accelerated age-associated decline in oxygen pulse that parallels that for peak VO2 (from reference 42)
Examining the components of VO2max revealed that the longitudinal decline in oxygen pulse (VO2 per heart beat) mirrored that of VO2max, whereas maximal heart rate decreased only 4–6% per decade regardless of initial age (Fig. 4). The pattern of accelerated VO2max decline with age persisted even after normalizing it for fat-free mass rather than body weight [42]. These data in healthy older BLSA volunteers represent a “best case scenario.” The superimposition of CV or pulmonary disease plus the deconditioning induced by the sedentary lifestyle common to many older adults accentuates this decline in VO2max. Although the age-associated decline in VO2max was similar regardless of physical activity level, it should be emphasized that the more active quartiles maintain a higher VO2max than their sedentary peers at all ages [39].
An accelerated decline of aerobic capacity with age has important implications regarding functional independence and quality of life. Given that activities of daily living typically require a fixed aerobic expenditure, they require a significantly larger percent of VO2max in an older than in a younger person. When the energy required for an activity approaches or exceeds the aerobic capacity of an elderly individual, he or she will likely be unable to perform it. Thus, a low aerobic capacity characterizes one of the five components of the “frailty phenotype” [43].
Because the heart is difficult to image during treadmill exercise, radionuclide cardiac scanning during cycle ergometry has been used to examine the mechanisms for the age-associated decline in aerobic capacity (Table 2). The peak VO2 of healthy BLSA participants during upright cycle ergometry averages about 80% of that during treadmill exercise, regardless of age. Leg fatigue is the primary factor limiting the duration and intensity of cycle exercise. In healthy, non-athletic BLSA men and women, peak cycle work rate and VO2 decline by * 50% between ages 20 and 80 years secondary to declines of * 30% in cardiac output and 20% in (A–V) O2 difference (Table 2) [32]. The decrease in cardiac index with age at maximal effort during upright cycle exercise is due entirely to a reduction in heart rate, as the LV stroke volume (SV) does not decline with age in either sex [30]. However, aging dramatically affects the process by which SV is achieved during maximal exercise. Whereas older individuals have a blunted capacity to reduce LV end-systolic volume (ESV) and to increase LVEF, this deficit is offset by a larger end-diastolic volume (EDV) [30]. Thus, normal aging is not characterized by a “stiff heart” that prohibits sufficient filling between beats during exercise. The larger EDV in healthy older versus younger individuals during vigorous aerobic exercise is achieved by a longer diastolic interval (i.e., slower heart rate), allowing greater time for LV filling, and by a greater amount of blood remaining in the heart at end systole [30].
Despite the accelerated decline in VO2max with age, it has been amply documented that physical conditioning of older persons can substantially increase their maximum aerobic work capacity. In a meta-analysis of 41 trials in 2,102 individuals aged 60 and older, aerobic training elicited a 16.3% mean increase in VO2max [44]. The degree to which this conditioning effect derives from enhanced cardiac performance versus augmented peripheral mechanisms likely varies with the characteristics of the population studied, the type and degree of conditioning achieved, gender, body position during study, and genetic factors.
Physical conditioning of older individuals does not appear to offset the age-associated deficit in sympathetic modulation. Rather, increased LV ejection fraction from aerobic training in this age group [45] appears to result from the reduction in arterial afterload, as reflected in a reduced PWV and carotid AI [46], with possible contribution from augmented maximum intrinsic myocardial contractility. Additionally, aerobic training in sedentary older adults reduced their oxygen debt immediately postexercise by nearly 30%, translating into an 18% increase in exercise efficiency; in younger persons, however, efficiency did not change after training [47].
Mechanisms of age-associated reduced left ventricular performance during maximal aerobic exercise
The LVEF at maximal exercise and its increase from rest are useful diagnostic tools to detect the presence and quantify the severity of cardiac disease, particularly CHD. Thus, exercise LVEF has considerable clinical importance. A blunted reduction in ESV in older adults during exercise accounts for their smaller increase in LVEF from rest and their lower maximal value compared to younger individuals [30]. An attenuated LVEF response during exercise is even more pronounced in older adults with exercise-induced silent myocardial ischemia, due to a greater impairment in reducing ESV [48]. The underlying mechanisms for the age-associated reduction in maximal LVEF are multifactorial and probably include (1) reduced intrinsic myocardial contractility, (2) increased arterial afterload, (3) arterial–ventricular load mismatching, and (4) decreased effectiveness of autonomic modulation of both LV contractility and arterial afterload. Whereas these age-associated changes in CV reserve are not sufficient by themselves to produce symptoms and signs of heart failure, they appear to lower the threshold for developing clinical heart failure and adversely influence its severity and prognosis (Table 1).
Sympathetic modulation
Acute exercise and other stressors stimulate sympathetic modulation of the CV system, which increases heart rate, augments myocardial contractility and relaxation, reduces LV afterload, and redistributes blood to working muscles and skin to dissipate heat. Each of the factors known to contribute to the deficient CV regulation with aging, i.e., heart rate, afterload (both cardiac and vascular), myocardial contractility, and redistribution of blood flow, exhibits a deficient sympathetic modulatory component.
It is noteworthy that the apparent deficits in sympathetic modulation of cardiac and arterial functions with aging occur in the presence of elevated neurotransmitter levels. During any perturbation from the supine basal state, plasma levels of norepinephrine and epinephrine increase to a greater extent in older than in younger healthy individuals [49]. This increase in plasma catecholamines appears to be a compensatory response to the reduced cardiac b-receptor density with advancing age [50]. The age-associated increase in plasma norepinephrine derives from an increased cardiac spillover into the circulation and, to a lesser extent, from reduced plasma clearance. Deficient norepinephrine reuptake at nerve endings has been suggested as the primary mechanism for increased spillover. However, during prolonged submaximal exercise in older adults, decreased neurotransmitter reuptake might also be associated with reduced release and spillover, thereby contributing to the blunted cardio-acceleration and LV systolic performance seen with age during such exercise [51].
Deficits in cardiac beta-adrenergic receptor signaling
The increase in neurotransmitter spillover into the circulation that occurs during acute stress in older individuals implies a greater heart and vascular receptor occupancy by these substances. Laboratory studies indicate that this condition leads to desensitization of the postsynaptic signaling components of sympathetic modulation. The deficits in b-adrenergic signaling with aging are attributable in part to reduction in b-receptor numbers, deficient G-protein coupling of receptors to adenyl cyclase, and, possibly, age-associated reductions in the amount or activation of adenyl cyclase, leading to a reduced ability to augment cellular cAMP in response to b-receptor stimulation in the older heart.
The concept that efficiency of postsynaptic b-adrenergic signaling declines with aging is supported by multiple lines of evidence. One line derives from the observation that acute b-receptor blockade converts the exercise hemodynamic profile of younger persons to resemble that of older ones. Thus, during exhaustive aerobic exercise in the presence of acute b-adrenergic blockade, the reduction in heart rate is greater in younger than in older individuals, and significant b-adrenergic blockade–induced LV dilatation occurs only in the younger group [52]. Furthermore, the age-associated deficits in early LV diastolic filling rate, both at rest and during exercise, are abolished by acute b-adrenergic blockade [34]. However, SV increases to a greater extent in younger than in older individuals during acute b-adrenergic blockade, due largely to the greater increase in LV filling time in the young, secondary to greater reduction in their maximal heart rate [52].
When perspectives from subcellular biochemistry in animal models to intact humans are integrated, a diminished responsiveness to b-adrenergic modulation is among the most consistently observed CV changes that occur with advancing age. Perturbations in CV function that exceed the identified limits for healthy elderly individuals most likely represent interactions of aging per se with physical deconditioning and/or CV disease, both of which are highly prevalent among older adults.
Conclusion
The CV system undergoes multiple changes with age, even in individuals without evident CV disease. Stiffening of the larger arteries causes increases in systolic and pulse pressures that, in turn, lead to LV wall thickening and reduced early diastolic filling rate. Aerobic exercise capacity declines * 10% per decade in cross-sectional studies, mediated by reductions in maximal heart rate and peripheral oxygen utilization. In longitudinal studies, the decline in aerobic capacity accelerates at older ages.
Stroke volume during maximal exercise is preserved with age through greater use of the Frank–Starling mechanism to offset reduced systolic emptying, similar to the effects of b-adrenergic blockade of younger adults. Although these age-associated CV changes are not considered pathologic per se, they lower the threshold for symptoms and functional limitations when superimposed upon the CV diseases so highly prevalent in the elderly.
Footnotes
Disclaimer: The views expressed in this review are those of the authors and do not necessarily represent those of the National Institutes of Health or the Department of Health and Human Services.
Contributor Information
Jerome L. Fleg, Email: flegj@nhlbi.nih.gov, Division of Cardiovascular Sciences, National Heart, Lung, and Blood Institute, 6701 Rockledge Drive, Room 8150, Bethesda, MD 20892, USA.
James Strait, Email: strait.j@nia.nih.gov, Laboratory of Cardiovascular Science, National Institute on Aging, Baltimore, MD, USA.
References
- 1.U. S. Census Bureau. National population projections. link: http://www.census.gov/population/www/projections/summarytables.html.
- 2.Lakatta E, Wang M, Najjar SS. Arterial aging and subclinical arterial disease are fundamentally intertwined at macroscopic and molecular levels. Med Clin North Am. 2009;93:583–604. doi: 10.1016/j.mcna.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerstenblith G, Frederiksen J, Yin FC, et al. Echocardiographic assessment of a normal adult aging population. Circulation. 1977;56:273–278. doi: 10.1161/01.cir.56.2.273. [DOI] [PubMed] [Google Scholar]
- 4.Lam CSP, Xanthakis V, Sullivan LM, et al. Aortic root remodeling over the adult life course. Longitudinal data from the Framingham heart study. Circulation. 2010;122:884–890. doi: 10.1161/CIRCULATIONAHA.110.937839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagai Y, Metter EJ, Earley CJ, et al. Increased carotid artery intimal-medial thickness in asymptomatic older subjects with exercise-induced myocardial ischemia. Circulation. 1998;98:1504–1509. doi: 10.1161/01.cir.98.15.1504. [DOI] [PubMed] [Google Scholar]
- 6.Ungvari Z, Kaley G, de Cabo R, Sonntag WE, Csiszar A. Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci. 2010;65:1028–1041. doi: 10.1093/gerona/glq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. 2005;25:932–943. doi: 10.1161/01.ATV.0000160548.78317.29. [DOI] [PubMed] [Google Scholar]
- 8.Lakatta E, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107:139–146. doi: 10.1161/01.cir.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- 9.Semba RD, Najjar SS, Sun K, Lakatta E, Ferrucci L. Serum carboxymethyl-lysine, an advanced glycation end product, is associated with increased aortic pulse wave velocity in adults. Am J Hypertens. 2009;22:74–79. doi: 10.1038/ajh.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cernadas MR, et al. Expression of constitutive and inducible nitric oxide synthases in the vascular wall of young and aging rats. Circ Res. 1998;83:279–286. doi: 10.1161/01.res.83.3.279. [DOI] [PubMed] [Google Scholar]
- 11.Wang M, Monticone R, Lakatta E. Arterial aging: a journey into subclinical arterial disease. Curr Opin Nephrol Hypertens. 2010;19:201–207. doi: 10.1097/MNH.0b013e3283361c0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pearson JD, Morrell CH, Brant LJ, Landis PK, Fleg JL. Age-associated changes in blood pressure in a longitudinal study of healthy men and women. J Gerontol A Biol Sci Med Sci. 1997;52:M177–M183. doi: 10.1093/gerona/52a.3.m177. [DOI] [PubMed] [Google Scholar]
- 13.Roman MJ, et al. High central pulse pressure is independently associated with adverse cardiovascular outcome the strong heart study. J Am Coll Cardiol. 2009;54:1730–1734. doi: 10.1016/j.jacc.2009.05.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaitkevicius PV, Fleg JL, Engel JH, et al. Effects of age and aerobic capacity on arterial stiffness in healthy adults. Circulation. 1993;88:1456–1462. doi: 10.1161/01.cir.88.4.1456. [DOI] [PubMed] [Google Scholar]
- 15.Mitchell GF, et al. Changes in arterial stiffness and wave reflection with advancing age in healthy men and women: the Framingham heart study. Hypertension. 2004;43:1239–1245. doi: 10.1161/01.HYP.0000128420.01881.aa. [DOI] [PubMed] [Google Scholar]
- 16.Willum-Hansen T, Staessen JA, Torp-Pedersen C, et al. Prognostic value of aortic pulse wave velocity as index of arterial stiffness in the general population. Circulation. 2006;113:664–670. doi: 10.1161/CIRCULATIONAHA.105.579342. [DOI] [PubMed] [Google Scholar]
- 17.Weber T, et al. Increased arterial wave reflections predict severe cardiovascular events in patients undergoing percutaneous coronary interventions. Eur Heart J. 2005;26:2657–2663. doi: 10.1093/eurheartj/ehi504. [DOI] [PubMed] [Google Scholar]
- 18.Lam CSP, Borlaug BA, Kane GC, et al. Age-associated increases in pulmonary artery systolic pressure in the general population. Circulation. 2009;119:2663–2670. doi: 10.1161/CIRCULATIONAHA.108.838698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J. 2009;34:888–894. doi: 10.1183/09031936.00145608. [DOI] [PubMed] [Google Scholar]
- 20.Linzbach AJ, Akuamoa-Boateng E. Changes in the aging human heart. I. Heart weight in the aged. Klin Wochenschr. 1973;51:156–163. doi: 10.1007/BF01468338. [DOI] [PubMed] [Google Scholar]
- 21.Kitzman DW, Scholz DG, Hagen PT, Ilstrup DM, Edwards WD. Age-related changes in normal human hearts during the first 10 decades of life. Part II (Maturity): a quantitative anatomic study of 765 specimens from subjects 20–99 years old. Mayo Clin Proc. 1988;63:137–146. doi: 10.1016/s0025-6196(12)64946-5. [DOI] [PubMed] [Google Scholar]
- 22.Olivetti G, Giordano G, Corridi D, et al. Gender differences and aging: effects in the human heart. J Am Coll Cardiol. 1995;26:1068–1079. doi: 10.1016/0735-1097(95)00282-8. [DOI] [PubMed] [Google Scholar]
- 23.Hees PS, Fleg JL, Lakatta EG, Shapiro EP. Left ventricular remodeling with age in normal men versus women: novel insights using three-dimensional magnetic resonance imaging. Am J Cardiol. 2002;90:1231–1236. doi: 10.1016/s0002-9149(02)02840-0. [DOI] [PubMed] [Google Scholar]
- 24.Cheng S, Fernandes VRS, Bluemke DA, et al. Age-related left ventricular remodeling and associated risk for cardiovascular outcomes. The multi-ethnic study of atherosclerosis. Circulation Cardiovasc Imaging. 2009;2:191–198. doi: 10.1161/CIRCIMAGING.108.819938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burgess ML, McCrea JC, Hedrick HL. Age-associated changes in cardiac matrix and integrins. Mech Ageing Dev. 2001;122:1739–1756. doi: 10.1016/s0047-6374(01)00296-2. [DOI] [PubMed] [Google Scholar]
- 26.Bergmann O, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eghbali M, Eghbali M, Robinson TF, Seifter S, Blumenfeld OO. Collagen accumulation in heart ventricles as a function of growth and aging. Cardiovasc Res. 1989;23:723–729. doi: 10.1093/cvr/23.8.723. [DOI] [PubMed] [Google Scholar]
- 28.Lakatta EG, Yin FC. Myocardial aging: functional alterations and related cellular mechanisms. Am J Physiol. 1982;242:H927–H941. doi: 10.1152/ajpheart.1982.242.6.H927. [DOI] [PubMed] [Google Scholar]
- 29.Lakatta EG. Cardiovascular regulatory mechanisms in advanced age. Physiol Rev. 1993;73:413–467. doi: 10.1152/physrev.1993.73.2.413. [DOI] [PubMed] [Google Scholar]
- 30.Fleg JL, O’Connor FC, Gerstenblith G, et al. Impact of age on the cardiovascular response to dynamic upright exercise in healthy men and women. J Appl Physiol. 1995;78:890–900. doi: 10.1152/jappl.1995.78.3.890. [DOI] [PubMed] [Google Scholar]
- 31.Lakatta EG, Gerstenblith G, Angell CS, et al. Prolonged contraction duration in aged myocardium. J Clin Invest. 1975;55:61–68. doi: 10.1172/JCI107918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fleg JL, Lakatta EG. Normal aging of the cardiovascular system. In: Aronow WS, Fleg JL, editors. Cardiovascular disease in the elderly. 4th. Informa Healthcare USA, Inc.; New York: 2008. pp. 1–43. [Google Scholar]
- 33.Downes TR, Nomeir AM, Smith KM, Stewart KP, Little WC. Mechanism of altered pattern of left ventricular filling with aging in subjects without cardiac disease. Am J Cardiol. 1989;64:523–527. doi: 10.1016/0002-9149(89)90433-5. [DOI] [PubMed] [Google Scholar]
- 34.Schulman SP, Lakatta EG, Fleg JL, et al. Age-related decline in left ventricular filling at rest and exercise. Am J Physiol. 1992;263:H1932–H1938. doi: 10.1152/ajpheart.1992.263.6.H1932. [DOI] [PubMed] [Google Scholar]
- 35.Boyd AC, Schiller NB, Leung D, Ross DL, Thomas L. Atrial dilation and altered function are mediated by age and diastolic function but not before the eighth decade. J Am Coll Cardiol Img. 2011;4:234–242. doi: 10.1016/j.jcmg.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 36.Hees PS, Fleg JL, Dong S-J, et al. MRI and echocardiographic assessment of the diastolic dysfunction of normal aging: altered LV pressure decline or load? Am J Physiol Heart Circ Physiol. 2004;286:H782–H788. doi: 10.1152/ajpheart.01092.2002. [DOI] [PubMed] [Google Scholar]
- 37.Froehlich JP, Lakatta EG, Beard E, et al. Studies of sarcoplasmic reticulum function and contraction duration in young and aged rat myocardium. J Mol Cell Cardiol. 1978;10:427–438. doi: 10.1016/0022-2828(78)90364-4. [DOI] [PubMed] [Google Scholar]
- 38.Oh JK, Hatle L, Tajik AJ, Little WC. Diastolic heart failure can be diagnosed by comprehensive two-dimensional and Doppler echocardiography. J Am Coll Cardiol. 2006;47:500–506. doi: 10.1016/j.jacc.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 39.Tsang TSM, Gersh BJ, Appleton CP, et al. Left ventricular diastolic dysfunction as a predictor of the first nonvalvular atrial fibrillation in 840 elderly men and women. J Am Coll Cardiol. 2002;40:1636–1644. doi: 10.1016/s0735-1097(02)02373-2. [DOI] [PubMed] [Google Scholar]
- 40.Hachamovitch R, Wicker P, Capasso JM, Anversa P. Alterations of coronary blood flow and reserve with aging in Fischer 344 rats. Am J Physiol. 1989;256:H66–H73. doi: 10.1152/ajpheart.1989.256.1.H66. [DOI] [PubMed] [Google Scholar]
- 41.Talbot LA, Metter EJ, Fleg JL. Leisure-time physical activities and their relationship to cardiorespiratory fitness in healthy men and women 18–95 years old. Med Sci Sports Exer. 2000;32:417–425. doi: 10.1097/00005768-200002000-00024. [DOI] [PubMed] [Google Scholar]
- 42.Fleg JL, Morrell CH, Bos AG, et al. Accelerated longitudinal decline of aerobic capacity in healthy older adults. Circulation. 2005;112:674–682. doi: 10.1161/CIRCULATIONAHA.105.545459. [DOI] [PubMed] [Google Scholar]
- 43.Fried LP, Taugen CM, Walston J, For the CHS Collaborative Research Group et al. Frailty in older adults: evidence for a phenotype. J Gerontol. 2001;56A:M158–M166. doi: 10.1093/gerona/56.3.m146. [DOI] [PubMed] [Google Scholar]
- 44.Huang G, Gibson CA, Tran ZV, et al. Controlled endurance exercise training and VO2max changes in older adults: a meta-analysis. Prev Cardiol. 2005;8:217–225. doi: 10.1111/j.0197-3118.2005.04324.x. [DOI] [PubMed] [Google Scholar]
- 45.Schulman SP, Fleg JL, Goldberg AP, et al. Continuum of cardiovascular performance across a broad range of fitness levels in healthy older men. Circulation. 1996;94:359–367. doi: 10.1161/01.cir.94.3.359. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka H, DeSouza CA, Seals DR. Absence of age-related increase in central arterial stiffness in physically active women. Arterioscler Thromb Vasc Biol. 1998;18:127–132. doi: 10.1161/01.atv.18.1.127. [DOI] [PubMed] [Google Scholar]
- 47.Woo JS, Derleth C, Stratton JR, et al. The influence of age, gender, and training on exercise efficiency. J Am Coll Cardiol. 2006;47:1049–1057. doi: 10.1016/j.jacc.2005.09.066. [DOI] [PubMed] [Google Scholar]
- 48.Fleg JL, Schulman SP, Gerstenblith G, et al. Additive effects of age and silent myocardial ischemia on the left ventricular response to upright cycle exercise. J Appl Physiol. 1993;75:499–504. doi: 10.1152/jappl.1993.75.2.499. [DOI] [PubMed] [Google Scholar]
- 49.Fleg JL, Tzankoff SP, Lakatta EG. Age-related augmentation of plasma catecholamines during dynamic exercise in healthy males. J Appl Physiol. 1985;59:1033–1039. doi: 10.1152/jappl.1985.59.4.1033. [DOI] [PubMed] [Google Scholar]
- 50.White M, Roden R, Minobe W, et al. Age-related changes in beta- adrenergic neuroeffector systems in the human heart. Circulation. 1994;90:1225–1238. doi: 10.1161/01.cir.90.3.1225. [DOI] [PubMed] [Google Scholar]
- 51.Correia LCL, Lakatta EG, O’Connor FC, et al. Attenuated cardiovascular reserve during prolonged submaximal exercise in healthy older subjects. J Am Coll Cardiol. 2002;40:1290–1297. doi: 10.1016/s0735-1097(02)02132-0. [DOI] [PubMed] [Google Scholar]
- 52.Fleg JL, Schulman S, O’Connor F, et al. Effects of acute b-adrenergic receptor blockade on age-associated changes in cardiovascular performance during dynamic exercise. Circulation. 1994;90:2333–2341. doi: 10.1161/01.cir.90.5.2333. [DOI] [PubMed] [Google Scholar]