

Abstract
Organosulfates are important secondary organic aerosol (SOA) components and good tracers for aerosol heterogeneous reactions. However, the knowledge of their spatial distribution, formation conditions, and environmental impact is limited. In this study, we report two organosulfates, an isoprene-derived isoprene epoxydiols (IEPOX) (2,3-epoxy-2-methyl-1,4-butanediol) sulfate and a glycolic acid (GA) sulfate, measured using the NOAA Particle Analysis Laser Mass Spectrometer (PALMS) on board the NASA DC8 aircraft over the continental U.S. during the Deep Convective Clouds and Chemistry Experiment (DC3) and the Studies of Emissions and Atmospheric Composition, Clouds, and Climate Coupling by Regional Surveys (SEAC4RS). During these campaigns, IEPOX sulfate was estimated to account for 1.4% of submicron aerosol mass (or 2.2% of organic aerosol mass) on average near the ground in the southeast U.S., with lower concentrations in the western U.S. (0.2–0.4%) and at high altitudes (<0.2%). Compared to IEPOX sulfate, GA sulfate was more uniformly distributed, accounting for about 0.5% aerosol mass on average, and may be more abundant globally. A number of other organosulfates were detected; none were as abundant as these two. Ambient measurements confirmed that IEPOX sulfate is formed from isoprene oxidation and is a tracer for isoprene SOA formation. The organic precursors of GA sulfate may include glycolic acid and likely have both biogenic and anthropogenic sources. Higher aerosol acidity as measured by PALMS and relative humidity tend to promote IEPOX sulfate formation, and aerosol acidity largely drives in situ GA sulfate formation at high altitudes. This study suggests that the formation of aerosol organosulfates depends not only on the appropriate organic precursors but also on emissions of anthropogenic sulfur dioxide (SO2), which contributes to aerosol acidity.
Key Points
IEPOX sulfate is an isoprene SOA tracer at acidic and low NO conditions
Glycolic acid sulfate may be more abundant than IEPOX sulfate globally
SO2 impacts IEPOX sulfate by increasing aerosol acidity and water uptake
Keywords: organosulfate, IEPOX sulfate, glycolic acid sulfate, free troposphere aerosols, aerosol acidity, relative humidity
1 Introduction
Atmospheric aerosols affect climate forcing by directly absorbing and scattering sunlight and acting as cloud condensation nuclei (CCN) to initiate cloud formation [Charlson et al., 1992; Scott et al., 2014]. Aerosols are also atmospheric pollutants because they are harmful to human health, especially the respiratory and cardiovascular systems [Pope et al., 2002], and they decrease atmosphere visibility. Secondary organic aerosol (SOA) accounts for a significant fraction of organic aerosol (OA) mass [Murphy et al., 2006; Zhang et al., 2007a]. Due to our limited knowledge of SOA formation, the modeled SOA mass often has discrepancies with observations [Heald et al., 2005; Volkamer et al., 2006]. The formation mechanism, properties, and fates of SOA must be characterized to evaluate their environmental impact [Hallquist et al., 2009].
Organosulfates are important secondary aerosol components. They are formed by reactions between organic material and sulfate, which are the main chemical components of atmospheric aerosols [Murphy et al., 2006; Zhang et al., 2007a]. Organosulfates have been detected in ambient aerosols [Iinuma et al., 2007; Surratt et al., 2007, 2008; Gomez-Gonzalez et al., 2008; Froyd et al., 2010; Chan et al., 2010; Hatch et al., 2011; Olson et al., 2011; Zhang et al., 2012; Worton et al., 2013; Shalamzari et al., 2013] and cloud water [Pratt et al., 2013] and are estimated to comprise up to 5–10% of OA mass over the continental U.S. [Tolocka and Turpin, 2012]. Organosulfates are thought to be good tracers for heterogeneous aerosol phase chemistry and SOA formation since the known formation mechanisms involve reactive uptake of gas phase organic species onto aerosol [Surratt et al., 2010; McNeill et al., 2012; Zhang et al., 2012]. Organosulfates are polar, hydrophilic, and low-volatility SOA compounds, which may help nanoparticle growth [Smith et al., 2008; Yli-Juuti et al., 2013] and increase their potential to become CCN. Therefore, investigation of organosulfate abundance, distributions, sources, formation mechanisms, and fates is an important step to improve our knowledge of SOA.
One of the most well studied and abundant aerosol organosulfates is isoprene epoxydiols (IEPOX) (2,3-epoxy-2-methyl-1,4-butanediol) sulfate (C5H11SO7−) (chemical structure shown in Figure1a). IEPOX sulfate is one of the most abundant individual organic molecules in aerosols (e.g., 1–2% of carbon mass in the southeast U.S.) [Chan et al., 2010; Lin et al., 2013]. IEPOX sulfate was discovered to be a key intermediate in SOA formation from isoprene, the largest nonmethane carbon source [Surratt et al., 2010; Paulot et al., 2009b]. Uptake of IEPOX by acid-catalyzed ring opening of epoxydiol, followed by addition of inorganic sulfate, is known to form IEPOX sulfate [Darer et al., 2011; Eddingsaas et al., 2010; Surratt et al., 2010; Paulot et al., 2009b]. Organosulfates have also been proposed to form by reactive uptake of unsaturated compounds into the particle phase and reaction with the sulfate radical [Rudzinski et al., 2009; Nozière et al., 2010; Schindelka et al., 2013]. The vertical profiles of IEPOX sulfate were measured by the Particle Analysis by Laser Mass Spectrometry (PALMS) instrument during previous airborne campaigns [Froyd et al., 2010]. In those studies IEPOX sulfate accounted for about 2–3% of aerosol mass in the southeast U.S. and higher fractions in the tropical-free troposphere. IEPOX sulfate temporal profiles measured by aerosol time-of-flight mass spectrometry (ATOFMS) in Atlanta [Hatch et al., 2011] have also been reported. The IEPOX sulfate mass loading over the southeast U.S. has been estimated to be 10–60 ng/m3 by a model study [Pye et al., 2013].
Figure 1.
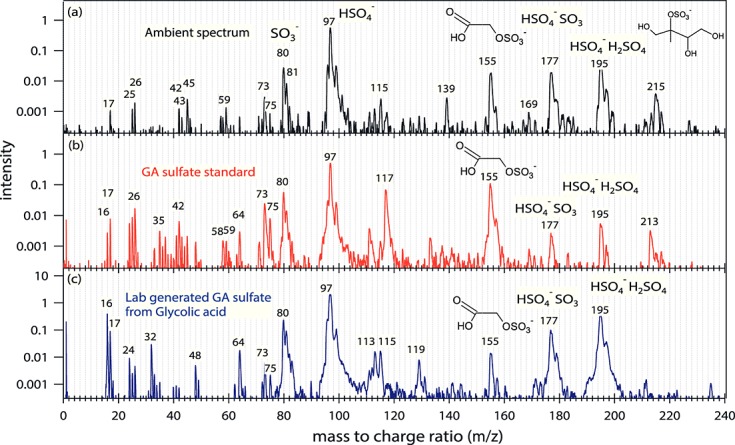
(a) Mass spectra of an ambient particle containing GA sulfate signal, (b) a laboratory particle generated from GA sulfate standard, and (c) a particle generated from a mixed solution of glycolic acid, NH4HSO4, and H2SO4.
A few other organosulfates have been quantified. For example, an organosulfate derived from 2-methyl-3 buten-2-ol, an important biogenic volatile organic emitted from pine trees, was measured to account for 0.25% of OA mass in the Manitou Forest Observatory in Colorado [Zhang et al., 2012]. The aromatic organosulfate benzyl sulfate, thought to form from anthropogenic emissions, was measured in Lahore, Pakistan, and found to account for a very small mass fraction of OA (2 ppm) but might be a useful tracer [Kundu et al., 2013].
Glycolic acid (GA) sulfate (C2H3SO6−) (chemical structure shown in Figure1a) is another potentially important organosulfate. Like IEPOX sulfate, the proposed formation mechanism of GA sulfate is the reaction of a gas phase organic precursor with acidic aerosol sulfate, although the formation mechanism remains to be proven. GA sulfate has been detected in ambient aerosols [Olson et al., 2011; Surratt et al., 2008] and in SOA generated by isoprene oxidation in chamber studies [Surratt et al., 2008]. GA sulfate can also form from the particle phase reaction of methyl vinyl ketone, a first generation oxidation product of isoprene, with the sulfate radical through a pathway similar to that proposed for hydroxyacetone sulfate formation [Schindelka et al., 2013], although it is unclear how important this pathway is under ambient conditions. GA sulfate formed from isoprene oxidation and other sources might account for significant aerosol mass. Ambient GA sulfate was measured to be 1.9–11.3 ng/m3 from filters collected on the ground in California, Ohio, and Mexico [Olson et al., 2011]. C2H3SO6− was also observed as a gas phase ion and may be the only gas-phase observation of an organosulfate [Ehn et al., 2010]. However, the spatial distribution of GA sulfate measurements is limited. The sources, formation mechanisms, and atmospheric importance of these species are unclear.
This study reports measurements of IEPOX sulfate C5H11SO7− and the less studied GA sulfate C2H3SO6−. Measurements were made with the NOAA PALMS instrument on board the NASA DC-8 airplane over the continental U.S. during the Deep Convective Clouds and Chemistry Experiment (DC3) in May and June 2012 and the Studies of Emissions and Atmospheric Composition, Clouds, and Climate Coupling by Regional Surveys (SEAC4RS) in August and September 2013. The potential sources and formation conditions of the two organosulfates in the atmosphere are also discussed.
2 Methods
The NOAA PALMS instrument measures the chemical composition of individual particles using a laser evaporation and ionization technique [Murphy and Thomson, 1995]. Particles with a diameter larger than about 200 nm can be sized by two continuous laser beams (405 nm) and ionized by a pulsed excimer laser (193 nm). The ions are extracted and detected by a time of flight mass spectrometer [Murphy, 2007]. The PALMS instrument detects m/z > 500 with unit mass resolution. However, fragmentation can limit the detection of large molecules. The PALMS instrument is able to detect inorganic (e.g., potassium, sulfate, and metals) and organic compounds in single particles. Most of the organic compounds are fragmented due to excimer laser ionization. However, Froyd et al. [2010] found that IEPOX sulfate is not fully fragmented and can be detected as C5H11SO7− by PALMS at m/z 215. PALMS measured the aerosol chemical composition on board the NASA DC8 airplane during DC3 and SEAC4RS over the continental U.S. from about 400 m to 12 km in altitude. The organosulfate aerosols were sampled by a forward facing solid diffuser inlet based on the University of Hawaii design [McNaughton et al., 2007] for 90% and 84% of the data in DC3 and SEAC4RS and a High Cross-flow Aerosol Sampler designed by Dr. Suresh Dhaniyala at Clarkson University as an experimental inlet for the remaining data. The solid diffuser inlet sampled up to 2.8 µm at unit efficiency and with a 0.5 s inlet residence time. The experimental inlet sampled particles up to at least 1 µm at unit efficiency and with a 1.4 s inlet residence time. Because almost all organosulfate signals (>95%) were detected in submicron particles, both inlets have the same unit sampling efficiency for organosulfates. The sampling tubing temperature was about 20°C. Additional measurements are reported for flight campaigns over the Alaskan Arctic (ARCPAC, Aerosol, Radiation, and Cloud Processes affecting Arctic Climate) in 2008 and over Central America and nearby oceans: Pre-AVE (Pre-Aura Validation Experiment ) in 2004, CR-AVE (Costa Rica Aura Validation Experiment) in 2006, and TC4 (Tropical Composition, Cloud, and Climate Coupling) in 2007.
A peak at m/z 155 was the most intense organosulfate signal on average detected by PALMS during DC3 and SEAC4RS. The ratio of the isotopic signals at m/z 157 to m/z 155 of 0.04 is consistent with the presence of sulfur in the molecule (Figure1a). An accurate mass analysis [Froyd et al., 2010] of m/z 155 for the DC3 and SEAC4RS data indicates that the empirical formula is C2H3SO6−. The structure of ambient C2H3SO6− was investigated by Galloway et al. [2009] who suggested that ambient C2H3SO6− is likely GA sulfate since the mass and elution time of ambient C2H3SO6− is the same as that of a glycolic acid sulfate standard C2H3O2SO4−. Since PALMS cannot distinguish between isomeric compounds, we adopt the structural identification by Galloway et al. [2009] for this work. No signal at m/z 155 was observed for many spectra that contain IEPOX sulfate at m/z 215 in the field nor when the larger organosulfate BEPOX (2,3-epoxy-1,4-butanediol, C4H8O2) sulfate was sampled in the lab [Froyd et al., 2010], suggesting that C2H3SO6− is not formed from fragmentation of a larger organosulfate.
IEPOX sulfate was quantified using the calibration described by Froyd et al. [2010]. A synthesized GA sulfate standard [Olson et al., 2011] was used to calibrate PALMS signals at m/z 155. The mass spectrum of the GA sulfate standard is shown in Figure1b. Due to the high variability of laser vaporization and ionization, quantitative PALMS measurements are challenging. The ion signal intensity depends on the excimer laser power and the ablation position, both of which vary from particle to particle. Therefore, the average of the relative signal intensity at m/z 155, which is the signal intensity at m/z 155 normalized by the total ion intensity, was used to quantify the compounds instead of the individual absolute signal intensity. Aerosols generated from 13 solutions containing different known mass fractions of GA sulfate in the range from 0.3% to 2% were delivered to the PALMS and the corresponding average relative signals of m/z 155 at medium (1.7 mJ or 1.7 × 109 W/cm2) and low (0.84 mJ or 8.4 × 108 W/cm2) excimer laser power were recorded. The chemical compounds besides GA sulfate in the solutions were sulfate, ammonium, and an oxidized organic (succinic acid). They were used to mimic the general composition of the atmospheric particles in which organosulfates were detected. The mass ratios of organic to inorganic constituents were about 0.1, and the detailed chemical compositions of the solutions are provided in Table S1 in the supporting information. The ionization efficiencies of PALMS do not depend strongly on the relative concentrations of the organic to inorganic constituents as long as metal cations are absent. The calibration was also extrapolated to high excimer laser power (2.8 mJ or 2.8 × 109 W/cm2). For the simulated aerosol the uncertainty in organosulfate ionization efficiency due to the aerosol matrix was smaller than the variation of laser power. Figure2 shows the calibration plot. The relative signal intensity at m/z 155 as a function of aerosol GA sulfate mass fraction is used to determine the sensitivity to GA sulfate and convert the PALMS m/z 155 signal to GA sulfate aerosol mass fraction. Due to the variation of laser power pulses and the inhomogeneous chemical composition of individual aerosols, the calibration has significant uncertainty denoted as the error bars. PALMS is about 3 times more sensitive to GA sulfate than IEPOX sulfate. Increased laser power reduces the organosulfate parent peaks because of increased fragmentation. The effect is less pronounced for GA sulfate than for IEPOX sulfate. This may be due to less fragmentation of the smaller GA sulfate compound. Because different laser powers are optimal for different aerosol species, PALMS was operated alternately at medium and low laser power during DC3 and at high and low laser power during SEAC4RS.
Figure 2.
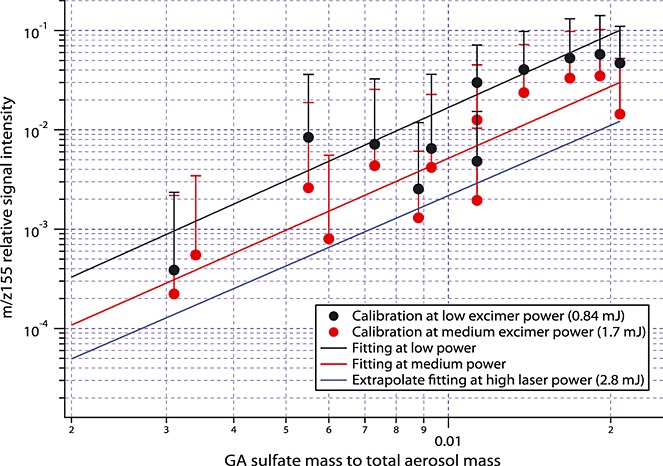
Average PALMS relative signals at m/z 155 (dots) for different GA sulfate mass fractions at low and medium PALMS laser power. The fits at low power (black line) and medium power (red line) and the extrapolated fit at high power (blue line) are used to convert PALMS relative signals at m/z 155 in ambient air to GA sulfate mass fractions.
Other organosulfate signals observed during DC3 and SEAC4RS were much smaller. For example, an organosulfate signal at m/z 169 may be lactic acid sulfate [Olson et al., 2011]. A signal at m/z 169 was first observed by electrospray ionization mass spectrometry (ESI-MS) in isoprene oxidation studies with methylglyoxal as the proposed precursor [Surratt et al., 2007]. The signal at m/z 169 was 1.5% of the signal at m/z 155 on average by PALMS. The vertical distribution of the signals at m/z 169 was similar to that of GA sulfate. Other potential organosulfate species at m/z 139, 141, 153, 183, and 199 had signal intensities of about 9%, 7%, 0.3%, 0.1%, and 0.1% of GA sulfate signals, respectively. Except for m/z 141, the above organosulfate ions were derived mostly from isoprene oxidation chemistry [Surratt et al., 2007]. We estimated they constitute a very small aerosol mass assuming that their sensitivities are between IEPOX sulfate and GA sulfate. Such small peaks were often below the detection limit so we make no further attempt to quantify them in this study. However, these organosulfates may account for a significant mass fraction if their sensitivities are orders of magnitude lower. Offline analysis of organosulfates by liquid chromatography and ESI-MS and availability of additional organosulfate standards are required to address this important question.
Aerosol acidity or neutralization can be estimated from the PALMS signal at m/z 195, which corresponds to a sulfuric acid cluster HSO4− · H2SO4. Acidic particles form this peak as well as HSO4−, whereas neutralized particles favor formation of only HSO4− ions. Particles in the stratosphere and in volcanic plumes are known to be more acidic and consequently have higher HSO4− · H2SO4 signals compared to tropospheric particles [Murphy et al., 2007; Carn et al., 2011]. The [HSO4− · H2SO4]/([HSO4−] + [HSO4− · H2SO4]) signals measured in the field generally increased as particle-into-liquid sampler [NH4+] to [SO42−] ratios decreased [Froyd et al., 2009, 2010]. Lab experiments also found that the [HSO4− · H2SO4]/[HSO4−] signals from a similar Aerosol Time-of-Flight Mass spectrometer (ATOFMS) increased with decreased aerosol neutralization [Yao et al., 2011]. This acidity indicator signal also varies with laser power. Due to the different laser powers used during these two campaigns, the acidity response ion HSO4− · H2SO4 was biased high (more acidic) during DC3 and biased low (less acidic) during SEAC4RS. The aerosol pH values were also estimated from the thermodynamic model extended aerosol inorganic model (E-AIM) [Clegg et al., 1998; Wexler and Clegg, 2002; Zhang et al., 2007b] for DC3 and SEAC4RS. Hennigan et al. [2014] and Guo et al. [2014] evaluated the aerosol acidity estimated from thermodynamic models ISORROPIA and E-AIM with and without gas phase inputs. Submicron aerosol sulfate (SO42−), ammonium (NH4+), and nitrate (NO3−) from a High-Resolution Aerosol Mass Spectrometer (HR-AMS), gas phase nitric acid (HNO3) from the California Institute of Technology (Caltech) chemical ionization mass spectrometer (CIMS), ambient relative humidity, temperature, and pressure were used as inputs. The impact of organic compounds on pH values was not considered. The predicted gas phase HNO3 levels agreed well with the measurements. The predicted pH values generally anticorrelated with the PALMS acidity signal, as expected (Figures3c, 3d, 3g, and 3h). Since the pH predictions from the thermodynamic model probably become less accurate as sulfate approaches complete neutralization, and since gas phase NH3 measurements were not available to constrain the model, the PALMS acidity signal was used as the main aerosol acidity indicator.
Figure 3.
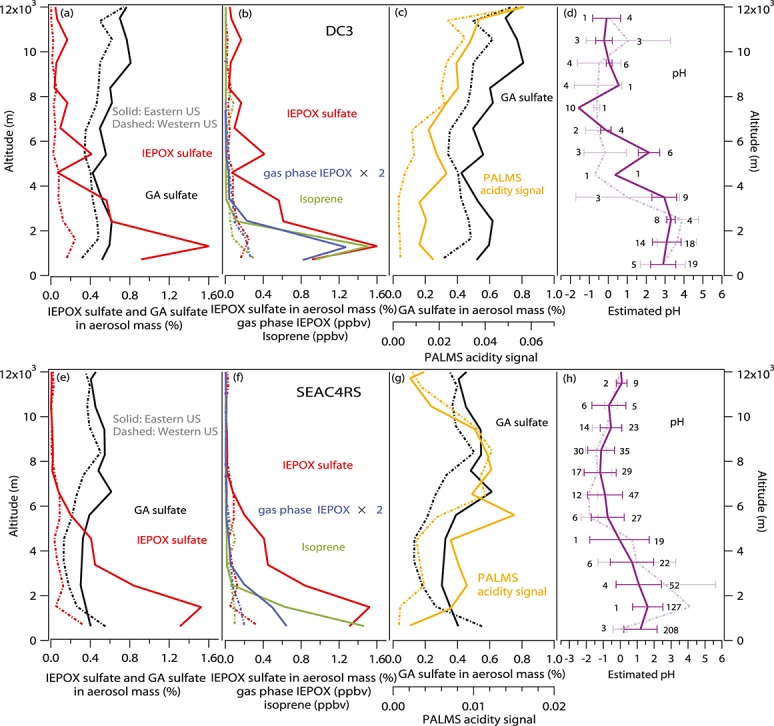
Average vertical profiles of measured (a) IEPOX sulfate (red) and GA sulfate (black) in aerosol mass (%), (b) isoprene (green), gas phase IEPOX (blue), (c) and PALMS acidity signal (orange), and (d) estimated pH (purple) in the eastern (solid) and western (dashed) U.S. during DC3 and (e–h) SEAC4RS. Longitude 96°W is used to separate eastern U.S. data from western U.S. data. The error bars in the estimated pH panels represent ±1 standard deviation of the 5 min average data. The data points in each bin for eastern and western U.S. are on the right and left of the error bars, respectively.
To investigate the potential formation mechanisms and precursors of GA sulfate, laboratory experiments were carried out to synthesize the organosulfate at m/z 155 from both glyoxal and glycolic acid. Aerosols were nebulized from a (0.4M: 0.2M: 0.2M) mixture of glyoxal, ammonium bisulfate (NH4HSO4), and sulfuric acid (H2SO4) or a (0.2M: 0.2M: 0–0.2M) mixture of glycolic acid, NH4HSO4, and H2SO4. Some solutions were exposed to UV light (254 nm) from a standard mercury pen lamp to initiate photochemistry for a range of time periods (Table1). A differential mobility analyzer was used to select the sizes representative of ambient particles containing GA sulfate. The size-selected aerosols were sampled by PALMS. Table1 shows the lab experiment cases, number fraction of acidic aerosols containing m/z 155, relative m/z 155 signals in acidic aerosols, and size distribution of the aerosols detected by PALMS. Figure1c shows the mass spectrum of a particle generated from a solution of glycolic acid, NH4HSO4, and H2SO4.
Table 1.
Laboratory Experiments on GA Sulfate Formation and Precursors
| Experiment Casesa | Add H2O2 | Reaction Time Before Making Aerosols | UV Radiation | Acidic Aerosols | Number Fraction of Generated Aerosols is Acidic | Number Fraction of Acid Aerosols Containing m/z 155 | Relative Peak Area at m/z 155 in Acidic Particles | Acid Particle Mode Diameters (µm) |
|---|---|---|---|---|---|---|---|---|
| A. Glyoxal + NH4HSO4 + H2SO4 | no | <1 h | no | yes | 0.17 | 0 | 0 | 0.5 |
| B. Glyoxal + NH4HSO4 + H2SO4 | no | 4 days | no | yes | 0.47 | 0.01 | 2 × 10−4 | 0.6 (82%) and 1.8 (18%) |
| C. Glyoxal + NH4HSO4 + H2SO4 | no | <6 h | 5 h | 0.54 | 0.03 | 3 × 10−4 | 0.6 (80%) and 2.0 (20%) | |
| D. Glyoxal + NH4HSO4 + H2SO4 | no | 6 days | 5 h | yes | 0.68 | 0.11 | 3.5 × 10−4 | 1 (23%) and 2.3 (78%) |
| E. Glycolic acid + NH4HSO4 + H2SO4 | no | 1 day | no | yes | 0.90 | 0.28 | 1.5 × 10−3 | 0.6 (44%) and 2.0 (56%) |
| F. Glycolic acid + NH4HSO4 + H2SO4 | yes | New (<2 h) | 75 min | yes | 0.69 | 0.53 | 6 × 10−3 | 0.6 (10%) and 2.0 (90%) |
| G. Glycolic acid + (NH4)2SO4 + H2SO4 | yes | New (<2 h) | 40 min | Less acidic pH = 3 | 0.1 | 0.02 | 8 × 10−4 | distributed between 0.5 and 2.0 |
| H. Glycolic acid + NH4HSO4 + H2SO4 | yes | 1 day | 75 min | yes | 0.91 | 0.41 | 3 × 10−3 | 1.6 |
The solutions were exposed to lab light in the daytime after they were made.
Other trace gas and aerosol concentrations were measured by a suite of instruments on board the NASA DC8 airplane during DC3 and SEAC4RS. Isoprene was measured by both proton transfer reaction mass spectrometry (PTR-MS, University of Innsbruck) [Hansel et al., 1999] and by whole air sampling (WAS; University of California Irvine) followed by laboratory analysis using gas chromatography [Colman et al., 2001]. PTR-MS measurements have a higher spatial and temporal resolution compared to WAS data. Isoprene measurements by PTR-MS do, however, suffer from furan interference in biomass burning (BB) plumes [Christian et al., 2004]. In BB-impacted air masses (identified by elevated acetonitrile levels), WAS data were used for the analysis. Two gas phase isoprene oxidation products, hydroxy hydroperoxides (ISOPOOH) and dihydroxyepoxides (IEPOX), were measured by the Caltech CIMS [Paulot et al., 2009a; St. Clair et al., 2010]. NO and O3 were measured by a NOAA chemiluminescence instrument [Ryerson et al., 2001]. The aerosol mass of organic material, sulfate, and ammonium was measured by a University of Colorado Aerodyne HR-AMS [DeCarlo et al., 2006] and is reported under standard temperature and pressure conditions (1 atm and 273 K). Submicron total aerosol mass was derived from the total submicron aerosol volume measured by the Ultra-High Sensitivity Aerosol Spectrometer and the laser aerosol spectrometer aerosol sizing instruments operated by the NASA LARGE group [Ziemba et al., 2013] with the assumption of an average aerosol density of 1.4 g/cm3. The submicron total aerosol mass was used to multiply the PALMS organosulfate mass fraction products to get to the absolute organosulfate mass loading.
3 Results and Discussion
3.1 Vertical Distribution of IEPOX Sulfate and GA Sulfate Over the Continental U.S
The average vertical aerosol mass fractions of IEPOX sulfate and GA sulfate in the eastern U.S. and western U.S. during DC3 and SEAC4RS are shown in Figures3a and 3e, respectively. Flights during the DC3 campaign targeted continental convective inflow and high altitude outflow. Most of the SEAC4RS flights targeted a wide range of tropospheric environments. SEAC4RS campaign data were excluded for marine flights and for the flight on 2 September that sampled convective outflow, where the vertical profile was similar to DC3 with enhanced IEPOX sulfate at high altitudes. IEPOX sulfate is estimated to account for 1.3% and 0.2% of submicron aerosol mass on average near the ground in the eastern and western U.S., respectively, during DC3, and 1.4% and 0.2% during SEAC4RS. Near the ground is defined as from the lowest GPS altitudes sampled by DC-8 (about 400 m) to 1500 m since the typical daytime boundary layer height was about 1–2 km. The IEPOX sulfate mass fraction decreased in the upper troposphere and lower stratosphere (to < 0.2% of submicron aerosol mass). GA sulfate accounted for 0.6% and 0.3% of submicron aerosol mass near the ground and increased to 0.7% and 0.6% of submicron aerosol mass in the upper troposphere in the eastern and western U.S., respectively, during DC3, compared to 0.4% and 0.6% near the ground and 0.6% and 0.4% in the upper troposphere in the eastern and western U.S. during SEAC4RS. Total IEPOX sulfate mass accounted for 2.2% and 0.7% of total organic mass measured by AMS near the ground in the eastern and western U.S. and for 5.0% and 1.9% of total sulfate mass measured by AMS on average in these two campaigns. Total GA sulfate mass accounted for 0.9% and 0.7% of total organic mass measured by AMS near the ground in the eastern and western U.S. and for 2.0% and 2.5% of total sulfate mass measured by AMS on average in these two campaigns. Most of the eastern U.S. flights were over the southeast U.S. There were several low-altitude flight segments over the “isoprene volcano” region in the Ozark Mountains of Arkansas and Missouri during SEAC4RS. Although isoprene observed in the “isoprene volcano” region was significantly enhanced, there was no enhancement of IEPOX sulfate observed compared to average levels in the eastern U.S.
The IEPOX sulfate mass fraction was highest near the ground in the eastern U.S. where the isoprene emissions were most intense. Compared to IEPOX sulfate, the vertical profiles of GA sulfate mass fraction were nearly constant with altitude and peaked in the upper troposphere, suggesting that GA precursors are more widely distributed and probably longer lived than the IEPOX-derived sulfate. Accordingly, GA sulfate may account for significant aerosol mass throughout the global troposphere. The average IEPOX sulfate mass fraction of 1.4% of total submicron aerosol mass or of 2.2% of total OA mass in the southeast U.S. from 400 m to 1500 m was close to the aircraft measurements in 2004 by Froyd et al. [2010] (3% of submicron aerosol mass at low altitude) and the ground measurements by Lin et al. [2013] (1–2% of OA mass). The estimated mass loading of GA sulfate of 20 ng/m3 at low altitudes was near the upper limit of GA sulfate detected from filter measurements collected from several ground sites [Olson et al., 2011]. To our knowledge, these are the first-online and the first-airborne measurements of GA sulfate reported.
3.2 IEPOX Sulfate and Its Precursors
The IEPOX sulfate vertical profiles had a similar pattern to those of isoprene and its gas phase oxidation product IEPOX (Figures3b and 3f). Average isoprene mixing ratios were similar during DC3 and SEAC4RS in the eastern U.S. The gas phase IEPOX (and ISOPOOH) was significantly lower during SEAC4RS, which was probably due to higher NO concentration observed in the southeast U.S. during SEAC4RS than DC3. Considering lower levels of IEPOX (523 pptv in DC3 versus 285 pptv in SEAC4RS on average) during SEAC4RS, a similar IEPOX sulfate mass fraction in both campaigns, and twice higher submicron aerosol mass loading during SEAC4RS near the ground in the southeast U.S., production of IEPOX sulfate from gas phase IEPOX seemed to be much more efficient during SEAC4RS. The lower pH near the ground estimated during SEAC4RS may contribute to the more efficient IEPOX sulfate formation. Another possibility is that reactive uptake of IEPOX formed other species such as 2-methyltetrols and C5 alkene triols [Surratt et al., 2010] in particle phase under the DC3 conditions. Further study is needed to explore the reasons for the higher IEPOX sulfate production efficiency during SEAC4RS.
The vertical profiles of IEPOX sulfate and its precursors may provide clues about the lifetime of IEPOX sulfate, which has not been investigated. Generally, isoprene, gas phase IEPOX, and aerosol phase IEPOX sulfate dropped to insignificant levels above 3.5 km, 4 km, and 8 km, respectively. The chemical lifetime of isoprene is about 1–3 h depending on hydroxyl radicals (OH) concentrations [Paulot et al., 2009a]. The lifetime of gas phase IEPOX due to OH oxidation is about 3–28 h [Jacobs et al., 2013; Bates et al., 2014], and loss to aerosol uptake varies from about 1 h to days depending on the aerosol acidity [Surratt et al., 2010; Gaston et al., 2014]. The IEPOX sulfate vertical profile suggests that the lifetime of IEPOX sulfate is longer than those of isoprene and gas phase IEPOX. Higher IEPOX sulfate mass fractions in the upper troposphere observed during DC3 compared to SEAC4RS may be due to shortened vertical transport time of either IEPOX sulfate or isoprene from convection because DC3 flights were designed to target convection, and SEAC4RS mostly sampled nonconvectively active air masses. The observed vertical profiles of isoprene, IEPOX, and IEPOX sulfate and the relevant HOx and NO levels may help modelers to estimate the IEPOX reactive uptake rates under ambient conditions that can be compared with rates measured in the laboratory at different conditions (e.g., aerosol acidity).
Two case studies of IEPOX sulfate and isoprene measurements are shown in Figure4. The case study on the 11 June 2012 flight to the southeast U.S. during DC3 (Figures4a and 4b) further confirms that IEPOX sulfate is formed from isoprene. IEPOX sulfate mass fraction generally tracks the concentrations of isoprene and gas phase IEPOX. The consistent low levels of isoprene (<200 pptv), IEPOX (<200 pptv), and IEPOX sulfate (<0.3% mass fraction) at low altitudes near 10:25 P.M. UTC are due to flying over the low vegetation and low isoprene emission area between Arkansas and Mississippi (Figure4a). This indicates that isoprene is required in IEPOX sulfate formation and is consistent with the IEPOX sulfate formation from oxidation of isoprene as proposed by Surratt et al. [2010] and Paulot et al. [2009a]. This also indicates that IEPOX sulfate formation can be isoprene-limited in low-leaf areas in the southeast U.S. Meanwhile, the levels of IEPOX sulfate generally followed the OA mass concentrations. This may indicate that SOA mass formed from biogenic VOCs oxidation contributed substantially to OA mass in the southeast U.S. and that IEPOX sulfate can be used as a tracer for SOA formed from isoprene oxidation. Figures4c and 4d demonstrate a case study near Northern California and southern Oregon on 6 August 2013 where there were significant isoprene and gas phase IEPOX concentrations, but no IEPOX sulfate was detected. In the enhanced isoprene periods, the aerosols were mostly neutralized, and the airplane was sampling biomass burning plumes. A similar case (not shown) with high isoprene and no detectable IEPOX sulfate was found on the 26 August 2013 flight. In general, little IEPOX sulfate was observed in biomass burning plumes because the aerosols were neutralized. This indicates that either IEPOX does not effectively react with neutralized aerosols or the reactive uptake forms other SOA products (e.g., 2-methyltetrols and C5 alkene triols) not IEPOX sulfate [Surratt et al., 2010].
Figure 4.

(a) A low-altitude flight leg on 11 June 2012 over the southeast U.S. during DC3 color coded and sized with airborne PTR-MS isoprene measurements plotted over an isoprene emission inventory BEIS3.13 map [Pierce et al., 1998]. (b) The corresponding time series of flight altitude (black), isoprene (light green), IEPOX (blue), IEPOX sulfate (red), organic aerosol mass (dark green), and PALMS acidity signal (orange). (c) A low-altitude flight leg on 6 August 2013 over North California and south Oregon during SEAC4RS color coded and sized with isoprene measurements by airborne whole air sampler, and (d) the corresponding time series plot.
In addition to the appropriate organic and sulfate precursors, the formation of IEPOX sulfate from isoprene is modulated by sulfate aerosol acidity and gas phase NO levels. Figures5a and 5b show IEPOX sulfate mass fraction versus isoprene mixing ratios when the aerosols were acidic (PALMS acidity signal > 0.002) (red) and near neutralized (PALMS acidity signal < 0.002) (black). Isoprene concentrations in Figure5 are from PTR-MS with a detection limit of 6 pptv (2σ for 5 min data) and were filtered for acetonitrile less than 200 pptv to exclude biomass burning plumes. More IEPOX sulfate was formed on average at the same isoprene levels when aerosols were acidic compared to near neutralized. The same data are plotted for low (<100 pptv) and high NO (>200 pptv) levels in Figures5c and 5d. Generally, higher IEPOX sulfate mass fractions were observed at low NO conditions for both field campaigns, which is consistent with efficient formation of gas phase IEPOX from isoprene oxidation under low NO conditions. The significant IEPOX sulfate present at high NO conditions (even > 500 pptv) during SEAC4RS may be due to transport or formation of IEPOX from isoprene hydroxynitrate oxidation [Jacobs et al., 2014], which probably plays a small role. Aerosol SO42− concentrations are known to be important for IEPOX sulfate formation, and NH4+ concentrations may also be important [Nguyen et al., 2014]. The average IEPOX sulfate mass fraction increased with aerosol SO42− or NH4+ at low levels (SO42− < 2 or NH4 + < 1 µg/m3) but not at higher mass loadings. This indicates that the aerosol SO42− and NH4+ levels are important but may not be limiting factors of IEPOX sulfate formation in most ambient conditions. It is worth noting that almost all IEPOX sulfate and GA sulfate signals were observed in particles classified as sulfate-organic mixtures.
Figure 5.
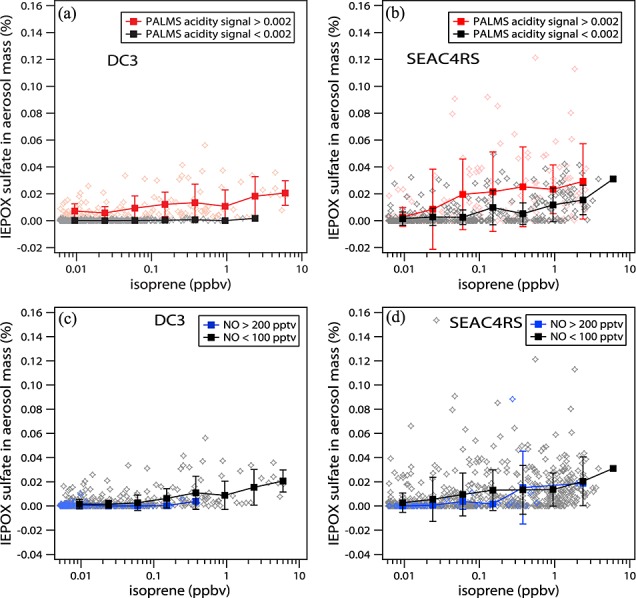
IEPOX sulfate mass fraction versus isoprene levels plotted as 5 min averages for (a) acidic aerosols (PALMS acidity signal > 0.002) (orange stars) and (b) near neutralized aerosols (PALMS acidity signal <0.002) (gray stars). IEPOX sulfate mass fraction versus isoprene levels at high NO (>200 pptv) (blue stars) and low NO (<100 pptv) (gray stars) during (c) DC3 and (d) SEAC4RS. The red, black, and blue squares are the averages and error bars represent ±1 standard deviation at different isoprene levels in logarithm scale.
3.3 Aerosol Acidity
The PALMS acidity signal has undergone previous validation as a qualitative indicator of sulfate acidity [Murphy et al., 2007; Froyd et al., 2009, 2010; Carn et al., 2011; Yao et al., 2011]. Our data show that under some conditions aerosol acidity as measured by PALMS is important in both ambient IEPOX sulfate and GA sulfate formation. At low altitudes (<1000 m) in the eastern U.S. where isoprene is more abundant, the IEPOX sulfate and GA sulfate signals were well correlated with PALMS aerosol acidity signal [HSO4− · H2SO4]/([HSO4−] + [HSO4− · H2SO4]) during DC3 and SEAC4RS when IEPOX sulfate and GA sulfate signals were above their respective detection limits (see Figure6). This correlation indicates that higher acidity tends to promote formation of IEPOX sulfate and GA sulfate near the ground when the organic precursors are abundant. This provides field evidence for an important role of aerosol acidity in ambient IEPOX sulfate formation. The importance of acidity agrees with both the acid-catalyzed epoxydiol ring opening formation mechanism [Surratt et al., 2010] and the sulfate radical initiated organosulfate formation because efficient formation of sulfate radicals also requires acidity [Schindelka et al., 2013]. At comparable isoprene concentrations, higher levels of IEPOX sulfate were generally observed in particles with higher acidity (see Figures5a and 5b), also demonstrating the important role of acidity in IEPOX sulfate formation. Conversely, a correlation was not observed with aerosol sulfate near the ground in the southeastern U.S. In a recent study at a rural ground site in southeastern U.S., Xu et al. [2014] reported a correlation of biogenic OA mass with aerosol sulfate but not with aerosol acidity. This apparent discrepancy is at least partially explained by the inherent differences in the measured aerosol properties. PALMS gives a direct measure of two organosulfate species that comprise 1–2% of aerosol mass, whereas Xu et al. [2014] used Aerodyne AMS bulk measurements to extract a biogenic OA factor representing 10–30% of aerosol mass. However, other factors besides aerosol acidity such as gas phase IEPOX partitioning, aerosol water content, and subsequent neutralization during particle aging may affect IEPOX sulfate concentrations and the apparent connection to aerosol acidity. Accordingly, the correlation with acidity was not generally observed above the boundary layer. The important role of relative humidity in IEPOX sulfate formation is discussed in section 3.4.
Figure 6.
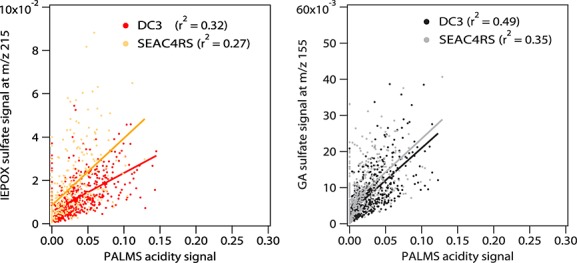
(a) Scatterplots of PALMS IEPOX sulfate relative signal at m/z 215 versus PALMS acidity signal at low altitudes (<1000 m) in the eastern U.S. during DC3 (red) and SEAC4RS (orange). (b) The scatterplot of PALMS GA sulfate relative signal at m/z 155 versus PALMS acidity signal at low altitudes (<1000 m) in the eastern U.S. during DC3 (black) and SEAC4RS (gray). Each point represents one single particle.
The anticorrelation between GA sulfate and aerosol pH vertical profiles and the correlation between GA sulfate and PALMS acidity signal vertical profiles (Figures3c, 3d, 3g, and 3h) showed that acidity also plays an important role in GA sulfate formation at high altitudes. The formation of IEPOX sulfate in the upper troposphere may be limited by the amount of gas phase IEPOX rather than acidity.
3.4 The Impact of Relative Humidity
Ambient relative humidity determines aerosol liquid water content, which affects the partitioning of gas phase compounds to aerosols, aerosol pH, and ionic strength [Zhang et al., 2007b]. IEPOX sulfate and GA sulfate had opposite trends with relative humidity (RH). Most (95%) of the data collected during these two aircraft campaign were during the daytime so that the RH trends were likely not due to the daytime and nighttime RH differences. The IEPOX sulfate mass fraction generally increased as RH increased up to 60%–80% (Figure7). The same trend was observed if IEPOX sulfate mass fraction was normalized by isoprene mixing ratios or if only low-altitude data were plotted (not shown). This may indicate that aerosol liquid water promotes IEPOX partitioning and suggest that uptake can be an important step in IEPOX sulfate formation. Conversely, the GA sulfate mass fraction generally decreased with increasing RH (Figure7). This may indicate that more concentrated or more acidic aerosols promote formation of GA sulfate at low RH levels [McNeill et al., 2012] and that partitioning of organic precursors may not limit GA sulfate formation in ambient air.
Figure 7.
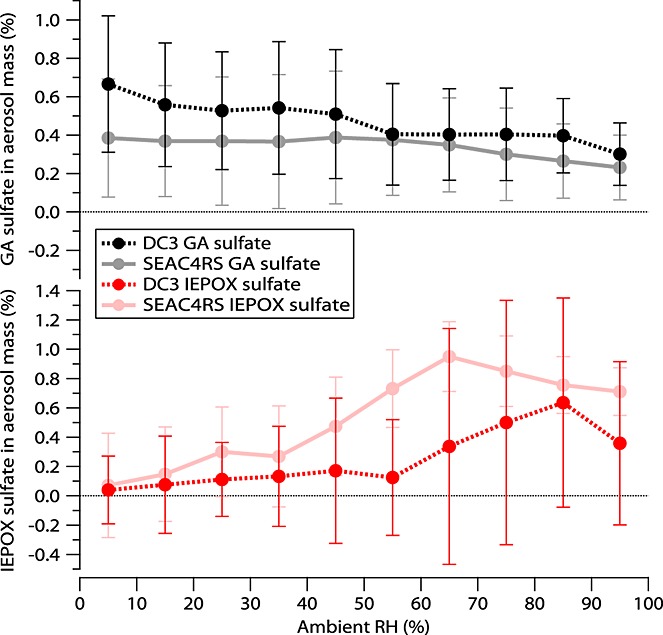
GA sulfate (black for DC3 and gray for SEAC4RS) and IEPOX sulfate (red for DC3 and pink for SEAC4RS) mass fractions from PALMS binned to different relative humidity. Error bars represent ±1 standard deviation.
3.5 GA Sulfate in the Lower Stratosphere and Evidence for In Situ Formation
In stratospheric air ([O3]/[CO] > 2.3), the GA sulfate mass fraction decreased as O3 increased (Figure8a). This indicates that stratospheric GA sulfate or its gas phase precursors likely come from the troposphere and were diluted or removed via physical and chemical processes as they traveled further into the stratosphere. The GA sulfate mass fraction (Figures3 and 8a) reached a maximum (∼1%) near the tropopause because this region contains both highly acidic aerosols and more abundant organic precursors than the lower stratosphere. Particles that originate in the stratosphere or tropopause region are clearly identified by their high sulfate content and high acidity [Murphy et al., 2007]. The number fraction of such stratospheric origin aerosols decreased dramatically (Figure8b, red) at lower altitudes. However, when examining just these stratospheric origin particles, the number fraction containing GA sulfate increased from 16% to 45% when they mixed into the troposphere (Figure8b, black). This indicates that the maximum GA sulfate concentration near the tropopause is likely formed in situ when gas phase organic precursors from lower altitudes encounter highly acidic particles from the stratosphere.
Figure 8.
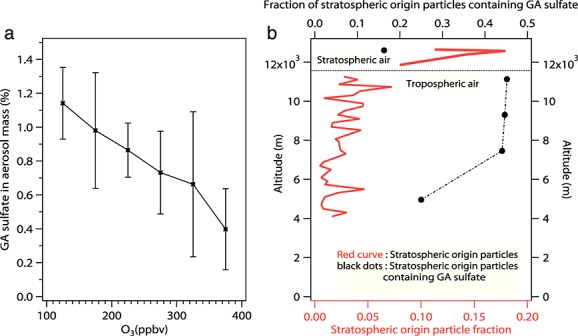
Organosulfates in the stratosphere. (a) GA sulfate aerosol mass fraction binned at different O3 levels in stratospheric air. Error bars represent ±1 standard deviation. No significant IEPOX sulfate was detected during DC3 in stratospheric air. (b) Number fraction of particles originating from stratosphere (red) and the number fraction of this type of particles containing GA sulfate (black) are plotted for both lower stratosphere and upper troposphere regions. Stratospheric air is defined as ([O3]/[CO] > 2.3). Since SEAC4RS did not sample stratospheric air, only DC3 data are shown.
3.6 Investigation of Potential GA Sulfate Organic Precursors
Ambient measurements provide some clues about the sources of GA sulfate. Although GA sulfate had a different spatial distribution from IEPOX sulfate, GA sulfate had a correlation (r2 = 0.34 for SEAC4RS; r2 = 0.33 for DC3 and SEAC4RS together) with IEPOX sulfate at low altitudes (below 1000 m) (Figure9). Moreover, aerosols containing high IEPOX sulfate always had significant GA sulfate, but aerosols with significant GA sulfate may or may not contain IEPOX sulfate. This pattern probably indicates that the organic precursors of GA sulfate are more diverse than IEPOX sulfate, which is formed only from isoprene.
Figure 9.
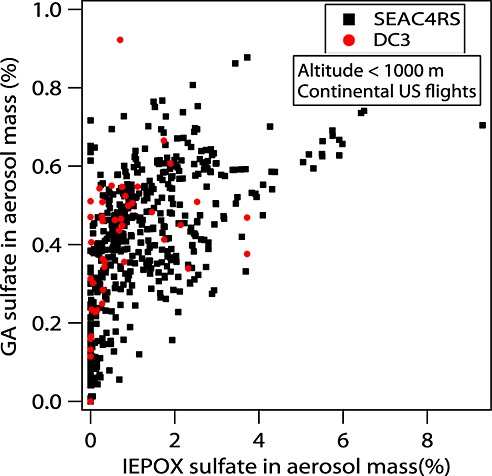
Scatterplot of GA sulfate mass fraction versus IEPOX sulfate mass fraction at low altitudes (<1000 m) for continental U.S. flights for DC3 (red) and SEAC4RS (black). Each data point represents 5 min average PALMS data.
Lab experiments were performed to investigate some potential organic precursors and formation mechanisms of GA sulfate (see Table1). Glyoxal was proposed to be a potential organic precursor of GA sulfate under UV irradiation [Galloway et al., 2009]. Aerosols generated from a mixture of glyoxal, ammonium bisulfate, and sulfuric acid were delivered to PALMS to investigate the formation of GA sulfate. No signal at m/z 155 was detected when the solution containing glyoxal was newly made and not exposed to UV radiation. The relative signal at m/z 155 increased to ∼3 × 10−4 when the solution was a few days old or exposed to UV radiation. This indicates that GA sulfate can form in a glyoxal/ammonium bisulfate/sulfuric acid solution with low efficiency and that UV radiation can accelerate the reaction. Aerosols generated from a solution containing glycolic acid instead of glyoxal had higher relative signals (1.5–6 × 10−3) at m/z 155 even when the solution was newly made and not exposed to UV radiation. Lowering the solution acidity (pH > 3) significantly decreased the relative signals of GA sulfate (8 × 10−4). The laboratory experiments suggest that the organosulfate at m/z 155 is formed from a glycolic acid solution more efficiently than from a glyoxal solution, and acidity largely promotes formation of GA sulfate. Although glyoxal in aerosol can be converted to glycolate by a disproportionation reaction [Fratzke and Reilly, 1986] and thereby provides a possible route to GA sulfate formation, the disproportionation is negligibly slow in acidic solutions.
Glycolic acid, a potential important precursor of GA sulfate identified in lab experiments, has both biogenic and anthropogenic sources [Warneck, 2005]. Methyl vinyl ketone, another potential gas phase precursor of GA sulfate [Schindelka et al., 2013], has isoprene as a biogenic source. Glycolic acid can form from oxidation of glycolaldehyde (CHOCH2OH) [Perri et al., 2009; Warneck, 2005] and acetic acid (CH3COOH) [Tan et al., 2012], both of which come from biogenic and anthropogenic sources [e.g., de Gouw et al., 2009; Goldstein et al., 1996; Chebbi and Carlier, 1996; Paulot et al., 2011]. Glycolaldehyde is water soluble and can undergo reactive uptake on aerosols (Heff. = 107 M/atm) [Nguyen et al., 2013]. In the aqueous phase, the oxidation of glycolaldehyde by OH may be more efficient than that of acetic acid [Lim et al., 2005]. The reaction of glycolaldehyde in the aerosol phase forms glycolic acid on a time scale of minutes [Ortiz-Montalvo et al., 2012].The lifetime of glycolaldehyde due to photolysis and reaction with OH is relatively long (1 day and > 2.5 days) [Bacher et al., 2001]. Glycolaldehyde is the second generation product from isoprene oxidation by OH, and about 10–30% of isoprene oxidation yields glycolaldehyde at high NO conditions [Bates et al., 2014; Galloway et al., 2011; Paulot et al., 2009a] but only 3% yields glycolaldehyde at low NO conditions [Bates et al., 2014]. Glycolaldehyde can also form from oxidation of anthropogenic emissions of ethene [Spaulding et al., 2003; Warneck, 2005]. Although the organic precursors of GA sulfate in the ambient are not well known, glycolic acid and its precursors (such as glycoaldehyde, acetic acid, ethene, and isoprene) are likely precursors according to known photooxidation pathways, lab experiments, and ambient measurements. However, no clear correlation was found in the aircraft data between potential GA sulfate gas phase organic precursors (glycoaldehyde or ethene) and aerosol phase GA sulfate mass fraction. This is not completely unexpected because GA sulfate vertical profiles seem to correlate with aerosol acidity, and gas phase GA sulfate organic precursors may not be the limiting factor of GA sulfate formation in most of the troposphere. Compared to the precursor of IEPOX sulfate, the precursors for GA sulfate have more diverse emission sources and longer photochemical lifetimes. These probably contribute to a much wider spatial distribution of GA sulfate in the troposphere.
3.7 GA Sulfate Measurements in the Tropics and Arctic
The IEPOX sulfate measurements in the tropics and eastern U.S. were summarized by Froyd et al. [2010]. GA sulfate and PALMS acidity signal measurements in the tropics during the CR-AVE, PRE-AVE, and TC4 field studies and in the Arctic during the ARCPAC field study are shown in Figure10. The GA sulfate vertical profiles in these campaigns also generally correlated with the PALMS acidity signal. The highest mass fraction of GA sulfate in the upper troposphere in CR-AVE was likely driven by the very acidic aerosols at those altitudes. Lower aerosol acidity during ARCPAC (3–7 km) also contributed to the lower GA sulfate mass fraction. GA sulfate was significantly lower near the ground in the Arctic compared to midlatitudes (e.g., DC3) even with similar aerosol acidity to DC3. This is consistent with the notion that GA sulfate originates from biogenic and anthropogenic sources, both of which are much weaker in the Arctic region (Alaska and the Arctic Oceans). The measurements of GA sulfate in the Arctic, midlatitudes, and tropics demonstrate that GA sulfate is widespread in locations that have acidic aerosols and significant biogenic and anthropogenic emissions or outflow.
Figure 10.
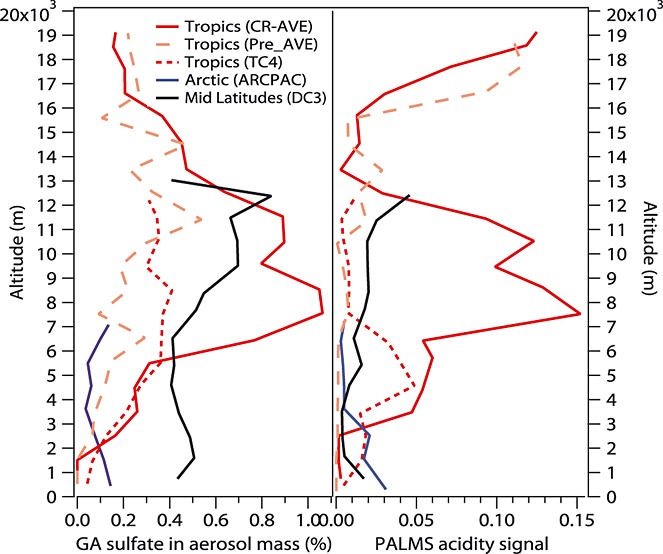
Vertical profiles of GA sulfate aerosol mass fraction and the PALMS acidity signal during previous field campaigns in the Central American tropics (CR-AVE, Pre-AVE, and TC4) (red), in the Arctic (ARCPAC) (blue), and in the midlatitudes (DC3) (black).
4 Conclusions
IEPOX sulfate and GA sulfate were quantified in aerosols sampled over a wide variety of tropospheric and lower stratospheric environments. IEPOX sulfate was most abundant near the ground in the southeast U.S. where it was estimated to account for about 1.4% of submicron aerosol mass. The measurements confirm that IEPOX sulfate can be used as a tracer of SOA formed primarily from low-NO isoprene oxidation chemistry in regions of acidic aerosols such as southeast U.S. GA sulfate was more spatially uniform and was estimated to account for about 0.5% of submicron aerosol mass on average from near the ground to the upper troposphere and lower stratosphere. Considering its spatial distribution, GA sulfate may be even more abundant than IEPOX sulfate globally. The wide spatial distribution of GA sulfate is consistent with multiple organic precursors of both anthropogenic and biogenic origin. This study provides field evidence of the importance of acidity in formation of both organosulfates under some conditions. Aerosol acidity is measured by the PALMS acidity signal. Higher relative humidity also promotes formation of IEPOX sulfate. The different RH dependence of IEPOX sulfate and GA sulfate may indicate that the rate-limiting steps in their ambient formation are different. The in situ formation of GA sulfate in the upper troposphere and lower stratosphere provides evidence for aerosol chemical reactions at high altitudes and low RH and for the presence of gas phase oxygenated organic compounds in the upper troposphere. Other potential organosulfate species have concentrations a factor of >10 lower than IEPOX sulfate and GA sulfate, assuming similar detection sensitivities. Although the total organosulfate burden is limited to a few percent of aerosol mass over the continental U.S., organosulfates are good tracers for SOA formation. The emission of SO2, which contributes to aerosol acidity and water uptake, likely promotes the formation of SOA compounds such as IEPOX sulfate. Regions with both elevated VOCs and SO2, such as Asian pollution plumes or continental convective outflow in the upper troposphere, may produce significantly more aerosol mass via these mechanisms.
Acknowledgments
The majority of the study is supported by the NASA grant NNH12AT29I from the Upper Atmosphere Research Program, Radiation Sciences Program, and Tropospheric Chemistry Program and by NOAA base funding. The GA sulfate standard is based upon work supported by the National Science Foundation under grant CHE-1213723. IEPOX and ISOPOOH measurements were supported by NASA NNX12AC06G. PTR-MS measurements were supported by BMVIT/FFG-ALR (Austrian Space Applications Programme, ASAP), the NASA Postdoctoral Program (NPP), and the National Institute of Aerospace (NIA). NO and O3 measurements were supported by NASA grant NNH12AT30I. AMS measurements were supported by NASA NNX12AC03G, NSF AGS-1243354, and NOAA NA13OAR4310063. We thank Barbara Ervens at NOAA and University of Colorado, Boulder for helpful discussion. We also would like to thank all the NASA DC8 crew for their assistance to integrate, maintain, and deintegrate the instrument on the airplane. The data are publicly available at NASA data achieve https://www-air.larc.nasa.gov/cgi-bin/ArcView/dc3-seac4rs and http://www-air.larc.nasa.gov/missions/seac4rs/index.html. The analysis results are available upon requested from jin.liao@noaa.gov and karl.froyd@noaa.gov.
Supporting Information
Table S1
References
- Bacher C, Tyndall GS. Orlando JJ. The atmospheric chemistry of glycolaldehyde. J. Atmos. Chem. 2001;39(2):171–189. doi: 10.1023/A:1010689706869. [Google Scholar]
- Bates KH, Crounse JD, St. Clair JM, Bennett NB, Nguyen TB, Seinfeld JH, Stoltz BM. Wennberg PO. Gas phase production and loss of isoprene epoxydiols. J. Phys. Chem. A. 2014;118(7):1238–1246. doi: 10.1021/jp4107958. doi: 10.1021/jp4107958. [DOI] [PubMed] [Google Scholar]
- Carn SA, et al. In situ measurements of tropospheric volcanic plumes in Ecuador and Colombia during TC4. J. Geophys. Res. 2011;116 D00J24, doi: 10.1029/2010JD014718. [Google Scholar]
- Chan MN, et al. Characterization and quantification of isoprene-derived epoxydiols in ambient aerosol in the southeastern United States. Environ. Sci. Technol. 2010;44(12):4590–4596. doi: 10.1021/es100596b. doi: 10.1021/Es100596b. [DOI] [PubMed] [Google Scholar]
- Charlson RJ, Schwartz SE, Hales JM, Cess RD, Coakley JA, Hansen JE. Hofmann DJ. Climate forcing by anthropogenic aerosols. Science. 1992;255(5043):423–430. doi: 10.1126/science.255.5043.423. doi: 10.1126/science.255.5043.423. [DOI] [PubMed] [Google Scholar]
- Chebbi A. Carlier P. Carboxylic acids in the troposphere, occurrence, sources, and sinks: A review. Atmos. Environ. 1996;30(24):4233–4249. doi: 10.1016/1352-2310(96)00102-1. [Google Scholar]
- Christian TJ, Kleiss B, Yokelson RJ, Holzinger R, Crutzen PJ, Hao WM, Shirai T. Blake DR. Comprehensive laboratory measurements of biomass-burning emissions: 2. First intercomparison of open-path FTIR, PTR-MS, and GC- MS/FID/ECD. J. Geophys. Res. 2004;109 D02311, doi: 10.1029/2003JD003874. [Google Scholar]
- Clegg SL, Brimblecombe P. Wexler AS. Thermodynamic model of the system H+−NH4+−SO42–NO3–H2O at tropospheric temperatures. J. Phys. Chem. A. 1998;102(12):2137–2154. [Google Scholar]
- Colman JJ, Swanson AL, Meinardi S, Sive BC, Blake DR. Rowland FS. Description of the analysis of a wide range of volatile organic compounds in whole air samples collected during PEM-tropics A and B. Anal. Chem. 2001;73(15):3723–3731. doi: 10.1021/ac010027g. doi: 10.1021/Ac010027g. [DOI] [PubMed] [Google Scholar]
- Darer AI, Cole-Filipiak NC, O’Connor AE. Elrod MJ. Formation and stability of atmospherically relevant isoprene-derived organosulfates and organonitrates. Environ. Sci. Technol. 2011;45(5):1895–1902. doi: 10.1021/es103797z. doi: 10.1021/Es103797z. [DOI] [PubMed] [Google Scholar]
- DeCarlo PF, et al. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 2006;78(24):8281–8289. doi: 10.1021/ac061249n. doi: 10.1021/Ac061249n. [DOI] [PubMed] [Google Scholar]
- De Gouw JA, et al. Airborne measurements of ethene from industrial sources using laser photo-acoustic spectroscopy. Environ. Sci. Technol. 2009;43(7):2437–2442. doi: 10.1021/es802701a. doi: 10.1021/Es802701a. [DOI] [PubMed] [Google Scholar]
- Eddingsaas NC, VanderVelde DG. Wennberg PO. Kinetics and products of the acid-catalyzed ring-opening of atmospherically relevant butyl epoxy alcohols. J. Phys. Chem. A. 2010;114(31):8106–8113. doi: 10.1021/jp103907c. doi: 10.1021/Jp103907c. [DOI] [PubMed] [Google Scholar]
- Ehn M, et al. Composition and temporal behavior of ambient ions in the boreal forest. Atmos. Chem. Phys. 2010;10(17):8513–8530. doi: 10.5194/acp-10-8513-2010. [Google Scholar]
- Fratzke AR. Reilly PJ. Kinetic-analysis of the disproportionation of aqueous glyoxal. Int. J. Chem. Kinet. 1986;18(7):757–773. doi: 10.1002/kin.550180704. [Google Scholar]
- Froyd KD, Murphy DM, Sanford TJ, Thomson DS, Wilson JC, Pfister L. Lait L. Aerosol composition of the tropical upper troposphere. Atmos. Chem. Phys. 2009;9(13):4363–4385. [Google Scholar]
- Froyd KD, Murphy SM, Murphy DM, de Gouw JA, Eddingsaas NC. Wennberg PO. Contribution of isoprene-derived organosulfates to free tropospheric aerosol mass. Proc. Natl. Acad. Sci. U.S.A. 2010;107(50):21,360–21,365. doi: 10.1073/pnas.1012561107. doi: 10.1073/pnas.1012561107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway MM, Chhabra PS, Chan AWH, Surratt JD, Flagan RC, Seinfeld JH. Keutsch FN. Glyoxal uptake on ammonium sulphate seed aerosol: Reaction products and reversibility of uptake under dark and irradiated conditions. Atmos. Chem. Phys. 2009;9(10):3331–3345. [Google Scholar]
- Galloway MM, Huisman AJ, Yee LD, Chan AWH, Loza CL, Seinfeld JH. Keutsch FN. Yields of oxidized volatile organic compounds during the OH radical initiated oxidation of isoprene, methyl vinyl ketone, and methacrolein under high-NOx conditions. Atmos. Chem. Phys. 2011;11(21):10,779–10,790. doi: 10.5194/acp-11-10779-2011. [Google Scholar]
- Gaston GJ, Riedel TP, Zhang Z, Gold A, Surratt JD. Thornton JA. Reactive uptake of an isoprene-derived epoxydiol to submicron aerosol particles. Environ. Sci. Technol. 2014;48(19):11,178–11,186. doi: 10.1021/es5034266. doi: 10.1021/es5034266. [DOI] [PubMed] [Google Scholar]
- Goldstein AH, Fan SM, Goulden ML, Munger JW. Wofsy SC. Emissions of ethene, propene, and 1-butene by a midlatitude forest. J. Geophys. Res. 1996;101(D4):9149–9157. doi: 10.1029/96JD00334. [Google Scholar]
- Gomez-Gonzalez Y, et al. Characterization of organosulfates from the photooxidation of isoprene and unsaturated fatty acids in ambient aerosol using liquid chromatography/(−) electrospray ionization mass spectrometry. J. Mass Spectrom. 2008;43(3):371–382. doi: 10.1002/jms.1329. doi: 10.1002/Jms.1329. [DOI] [PubMed] [Google Scholar]
- Guo H, et al. Particle water and pH in the southeastern United States. Atmos. Chem. Phys. Discuss. 2014;14:27,143–27,193. doi: 10.5194/acpd-14-27143-2014. [Google Scholar]
- Hallquist M, et al. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Atmos. Chem. Phys. 2009;9(14):5155–5236. [Google Scholar]
- Hansel A, Jordan A, Warneke C, Holzinger R, Wisthaler A. Lindinger W. Proton-transfer-reaction mass spectrometry (PTR-MS): On-line monitoring of volatile organic compounds at volume mixing ratios of a few pptv. Plasma Sources Sci. Technol. 1999;8(2):332–336. doi: 10.1088/0963-0252/8/2/314. [Google Scholar]
- Hatch LE, Creamean JM, Ault AP, Surratt JD, Chan MN, Seinfeld JH, Edgerton ES, Su YX. Prather KA. Measurements of isoprene-derived organosulfates in ambient aerosols by aerosol time-of-flight mass spectrometry—Part 2: Temporal variability and formation mechanisms. Environ. Sci. Technol. 2011;45(20):8648–8655. doi: 10.1021/es2011836. doi: 10.1021/Es2011836. [DOI] [PubMed] [Google Scholar]
- Heald CL, Jacob DJ, Park RJ, Russell LM, Huebert BJ, Seinfeld JH, Liao H. Weber RJ. A large organic aerosol source in the free troposphere missing from current models. Geophys. Res. Lett. 2005;32 L18809, doi: 10.1029/2005GL023831. [Google Scholar]
- Hennigan CJ, Izumi J, Sullivan AP, Weber RJ. Nenes A. A critical evaluation of proxy methods used to estimate the acidity of atmospheric particles. Atmos. Chem. Phys. Discuss. 2014;14:27,579–27,618. doi: 10.5194/acpd-14-27579-2014. [Google Scholar]
- Iinuma Y, Muller C, Berndt T, Boge O, Claeys M. Herrmann H. Evidence for the existence of organosulfates from beta-pinene ozonolysis in ambient secondary organic aerosol. Environ. Sci. Technol. 2007;41(19):6678–6683. doi: 10.1021/es070938t. doi: 10.1021/Es070938t. [DOI] [PubMed] [Google Scholar]
- Jacobs MI, Darer AI. Elrod MJ. Rate constants and products of the OH reaction with isoprene-derived epoxides. Environ. Sci. Technol. 2013;47(22):12,868–12,876. doi: 10.1021/es403340g. doi: 10.1021/Es403340g. [DOI] [PubMed] [Google Scholar]
- Jacobs MI, Burke WJ. Elrod MJ. Kinetics of the reactions of isoprene-derived hydroxynitrates: Gas phase epoxide formation and solution phase hydrolysis. Atmos. Chem. Phys. 2014;14:8933–8946. doi: 10.5194/acp-14-8933-2014. [Google Scholar]
- Kundu S, Quraishi TA, Yu G, Suarez C, Keutsch FN. Stone EA. Evidence and quantitation of aromatic organosulfates in ambient aerosols in Lahore, Pakistan. Atmos. Chem. Phys. 2013;13(9):4865–4875. doi: 10.5194/acp-13-4865-2013. [Google Scholar]
- Lim HJ, Carlton AG. Turpin BJ. Isoprene forms secondary organic aerosol through cloud processing: Model simulations. Environ. Sci. Technol. 2005;39(12):4441–4446. doi: 10.1021/es048039h. doi: 10.1021/Es048039h. [DOI] [PubMed] [Google Scholar]
- Lin YH, Knipping EM, Edgerton ES, Shaw SL. Surratt JD. Investigating the influences of SO2 and NH3 levels on isoprene-derived secondary organic aerosol formation using conditional sampling approaches. Atmos. Chem. Phys. 2013;13(16):8457–8470. doi: 10.5194/acp-13-8457-2013. [Google Scholar]
- McNaughton CS, et al. Results from the DC-8 Inlet Characterization Experiment (DICE): Airborne versus surface sampling of mineral dust and sea salt aerosols. Aerosol Sci. Technol. 2007;41(2):136–159. doi: 10.1080/02786820601118406. [Google Scholar]
- McNeill VF, Woo JL, Kim DD, Schwier AN, Wannell NJ, Sumner AJ. Barakat JM. Aqueous-phase secondary organic aerosol and organosulfate formation in atmospheric aerosols: A modeling study. Environ. Sci. Technol. 2012;46(15):8075–8081. doi: 10.1021/es3002986. doi: 10.1021/Es3002986. [DOI] [PubMed] [Google Scholar]
- Murphy DM. The design of single particle laser mass spectrometers. Mass Spectrom. Rev. 2007;26(2):150–165. doi: 10.1002/mas.20113. doi: 10.1002/Mas.20113. [DOI] [PubMed] [Google Scholar]
- Murphy DM. Thomson DS. Laser ionization mass-spectroscopy of single aerosol-particles. Aerosol Sci. Technol. 1995;22(3):237–249. doi: 10.1080/02786829408959743. [Google Scholar]
- Murphy DM, Cziczo DJ, Froyd KD, Hudson PK, Matthew BM, Middlebrook AM, Peltier RE, Sullivan A, Thomson DS. Weber RJ. Single-particle mass spectrometry of tropospheric aerosol particles. J. Geophys. Res. 2006;111 D23S32, doi: 10.1029/2006JD007340. [Google Scholar]
- Murphy DM, Cziczo DJ, Hudson PK. Thomson DS. Carbonaceous material in aerosol particles in the lower stratosphere and tropopause region. J. Geophys. Res. 2007;112 D04203, doi: 10.1029/2006JD007297. [Google Scholar]
- Nguyen TB, Coggon MM, Flagan RC. Seinfeld JH. Reactive uptake and photo-fenton oxidation of glycolaldehyde in aerosol liquid water. Environ. Sci. Technol. 2013;47(9):4307–4316. doi: 10.1021/es400538j. doi: 10.1021/Es400538j. [DOI] [PubMed] [Google Scholar]
- Nguyen TB, Coggon MM, Bates KH, Zhang X, Schwantes RH, Schilling KA, Loza CL, Flagan RC, Wennberg PO. Seinfeld JH. Organic aerosol formation from the reactive uptake of isoprene epoxydiols (IEPOX) onto non-acidified inorganic seeds. Atmos. Chem. Phys. 2014;14(7):3497–3510. doi: 10.5194/acp-14-3497-2014. [Google Scholar]
- Nozière B, Ekstrom S, Alsberg T. Holmstrom S. Radical-initiated formation of organosulfates and surfactants in atmospheric aerosols. Geophys. Res. Lett. 2010;37 L05806, doi: 10.1029/2009GL041683. [Google Scholar]
- Olson CN, Galloway MM, Yu G, Hedman CJ, Lockett MR, Yoon T, Stone EA, Smith LM. Keutsch FN. Hydroxycarboxylic acid-derived organosulfates: Synthesis, stability, and quantification in ambient aerosol. Environ. Sci. Technol. 2011;45(15):6468–6474. doi: 10.1021/es201039p. doi: 10.1021/Es201039p. [DOI] [PubMed] [Google Scholar]
- Ortiz-Montalvo DL, Lim YB, Perri MJ, Seitzinger SP. Turpin BJ. Volatility and yield of glycolaldehyde SOA formed through aqueous photochemistry and droplet evaporation. Aerosol Sci. Technol. 2012;46(9):1002–1014. doi: 10.1080/02786826.2012.686676. [Google Scholar]
- Paulot F, Crounse JD, Kjaergaard HG, Kroll JH, Seinfeld JH. Wennberg PO. Isoprene photooxidation: New insights into the production of acids and organic nitrates. Atmos. Chem. Phys. 2009a;9(4):1479–1501. [Google Scholar]
- Paulot F, Crounse JD, Kjaergaard HG, Kurten A, St. Clair JM, Seinfeld JH. Wennberg PO. Unexpected epoxide formation in the gas-phase photooxidation of isoprene. Science. 2009b;325(5941):730–733. doi: 10.1126/science.1172910. doi: 10.1126/science.1172910. [DOI] [PubMed] [Google Scholar]
- Paulot F, et al. Importance of secondary sources in the atmospheric budgets of formic and acetic acids. Atmos. Chem. Phys. 2011;11(5):1989–2013. doi: 10.5194/acp-11-1989-2011. doi: 10.5194/acp-11-1989-2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perri MJ, Seitzinger S. Turpin BJ. Secondary organic aerosol production from aqueous photooxidation of glycolaldehyde: Laboratory experiments. Atmos. Environ. 2009;43(8):1487–1497. doi: 10.1016/j.atmosenv.2008.11.037. [Google Scholar]
- Pierce T, Geron C, Bender L, Dennis R, Tonnesen G. Guenther A. Influence of increased isoprene emissions on regional ozone modeling. J. Geophys. Res. 1998;103(D19):25,611–25,629. doi: 10.1029/98JD01804. [Google Scholar]
- Pope CA, Burnett RT, Thun MJ, Calle EE, Krewski D, Ito K. Thurston GD. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. JAMA, J. Am. Med. Assoc. 2002;287(9):1132–1141. doi: 10.1001/jama.287.9.1132. doi: 10.1001/jama.287.9.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt KA, Fiddler MN, Shepson PB, Carlton AG. Surratt JD. Organosulfates in cloud water above the Ozarks’ isoprene source region. Atmos. Environ. 2013;77:231–238. doi: 10.1016/j.atmosenv.2013.05.011. [Google Scholar]
- Pye HOT, et al. Epoxide pathways improve model predictions of isoprene markers and reveal key role of acidity in aerosol formation. Environ. Sci. Technol. 2013;47(19):11,056–11,064. doi: 10.1021/es402106h. doi: 10.1021/Es402106h. [DOI] [PubMed] [Google Scholar]
- Rudzinski KJ, Gmachowski L. Kuznietsova I. Reactions of isoprene and sulphoxy radical-anions - a possible source of atmospheric organosulphites and organosulphates. Atmos. Chem. Phys. 2009;9(6):2129–2140. [Google Scholar]
- Ryerson TB, et al. Observations of ozone formation in power plant plumes and implications for ozone control strategies. Science. 2001;292(5517):719–723. doi: 10.1126/science.1058113. doi: 10.1126/science.1058113. [DOI] [PubMed] [Google Scholar]
- Schindelka J, Iinuma Y, Hoffmann D. Herrmann H. Sulfate radical-initiated formation of isoprene-derived organosulfates in atmospheric aerosols. Faraday Discuss. 2013;165:237–259. doi: 10.1039/c3fd00042g. doi: 10.1039/C3fd00042g. [DOI] [PubMed] [Google Scholar]
- Scott CE, et al. The direct and indirect radiative effects of biogenic secondary organic aerosol. Atmos. Chem. Phys. 2014;14(1):447–470. doi: 10.5194/acp-14-447-2014. [Google Scholar]
- Shalamzari MS, Ryabtsova O, Kahnt A, Vermeylen R, Herent MF, Quetin-Leclercq J, Van der Veken P, Maenhaut W. Claeys M. Mass spectrometric characterization of organosulfates related to secondary organic aerosol from isoprene. Rapid Commun. Mass Spectrom. 2013;27(7):784–794. doi: 10.1002/rcm.6511. doi: 10.1002/Rcm.6511. [DOI] [PubMed] [Google Scholar]
- Smith JN, Dunn MJ, VanReken TM, Iida K, Stolzenburg MR, McMurry PH. Huey LG. Chemical composition of atmospheric nanoparticles formed from nucleation in Tecamac, Mexico: Evidence for an important role for organic species in nanoparticle growth. Geophys. Res. Lett. 2008;35 L04808, doi: 10.1029/2007GL032523. [Google Scholar]
- Spaulding RS, Schade GW, Goldstein AH. Charles MJ. Characterization of secondary atmospheric photooxidation products: Evidence for biogenic and anthropogenic sources. J. Geophys. Res. 2003;108(D8) 4247, doi: 10.1029/2002JD002478. [Google Scholar]
- St. Clair JM, McCabe DC, Crounse JD, Steiner U. Wennberg PO. Chemical ionization tandem mass spectrometer for the in situ measurement of methyl hydrogen peroxide. Rev. Sci. Instrum. 2010;81 doi: 10.1063/1.3480552. doi: 10.1063/1.3480552. [DOI] [PubMed] [Google Scholar]
- Surratt JD, et al. Evidence for organosulfates in secondary organic aerosol. Environ. Sci. Technol. 2007;41(2):517–527. doi: 10.1021/es062081q. doi: 10.1021/Es062081q. [DOI] [PubMed] [Google Scholar]
- Surratt JD, et al. Organosulfate formation in biogenic secondary organic aerosol. J. Phys. Chem. A. 2008;112(36):8345–8378. doi: 10.1021/jp802310p. doi: 10.1021/Jp802310p. [DOI] [PubMed] [Google Scholar]
- Surratt JD, Chan AWH, Eddingsaas NC, Chan MN, Loza CL, Kwan AJ, Hersey SP, Flagan RC, Wennberg PO. Seinfeld JH. Reactive intermediates revealed in secondary organic aerosol formation from isoprene. Proc. Natl. Acad. Sci. U.S.A. 2010;107(15):6640–6645. doi: 10.1073/pnas.0911114107. doi: 10.1073/pnas.0911114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Lim YB, Altieri KE, Seitzinger SP. Turpin BJ. Mechanisms leading to oligomers and SOA through aqueous photooxidation: Insights from OH radical oxidation of acetic acid and methylglyoxal. Atmos. Chem. Phys. 2012;12(2):801–813. doi: 10.5194/acp-12-801-2012. [Google Scholar]
- Tolocka MP. Turpin B. Contribution of organosulfur compounds to organic aerosol mass. Environ. Sci. Technol. 2012;46(15):7978–7983. doi: 10.1021/es300651v. doi: 10.1021/Es300651v. [DOI] [PubMed] [Google Scholar]
- Volkamer R, Jimenez JL, San Martini F, Dzepina K, Zhang Q, Salcedo D, Molina LT, Worsnop DR. Molina MJ. Secondary organic aerosol formation from anthropogenic air pollution: Rapid and higher than expected. Geophys. Res. Lett. 2006;33 L17811, doi: 10.1029/2006GL026899. [Google Scholar]
- Warneck P. Multi-phase chemistry of C-2 and C-3 organic compounds in the marine atmosphere. J. Atmos. Chem. 2005;51(2):119–159. doi: 10.1007/s10874-005-5984-7. [Google Scholar]
- Wexler AS. Clegg SL, et al. Atmospheric aerosol models for systems including the ions H+, NH4+, Na+, SO42−, NO3−,Cl−, Br−, and H2O. J. Geophys. Res. 2002;107(D14) 4207, doi: 10.1029/2001JD000451. [Google Scholar]
- Worton DR, et al. Observational insights into aerosol formation from isoprene. Environ. Sci. Technol. 2013;47(20):11,403–11,413. doi: 10.1021/es4011064. doi: 10.1021/Es4011064. [DOI] [PubMed] [Google Scholar]
- Xu L, et al. Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States. Proc. Natl. Acad. Sci. U.S.A. 2014;112(1):37–42. doi: 10.1073/pnas.1417609112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao XH, Rehbein PJG, Lee CJ, Evans GJ, Corbin J. Jeong CH. A study on the extent of neutralization of sulphate aerosol through laboratory and field experiments using an ATOFMS and a GPIC. Atmos. Environ. 2011;45(34):6251–6256. [Google Scholar]
- Yli-Juuti T, et al. Volatility of organic aerosol: Evaporation of ammonium sulfate/succinic acid aqueous solution droplets. Environ. Sci. Technol. 2013;47(21):12,123–12,130. doi: 10.1021/es401233c. doi: 10.1021/Es401233c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HF, et al. Organosulfates as tracers for Secondary Organic Aerosol (SOA) formation from 2-methyl-3-buten-2-ol (MBO) in the atmosphere. Environ. Sci. Technol. 2012;46(17):9437–9446. doi: 10.1021/es301648z. doi: 10.1021/Es301648z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, et al. Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes. Geophys. Res. Lett. 2007a;34 L13801, doi: 10.1029/2007GL029979. [Google Scholar]
- Zhang Q, Jimenez JL, Worsnop DR. Canagaratna M. A case study of urban particle acidity and its influence on secondary organic aerosol. Environ. Sci. Technol. 2007b;41(9):3213–3219. doi: 10.1021/es061812j. doi: 10.1021/Es061812j. [DOI] [PubMed] [Google Scholar]
- Ziemba LD, et al. Airborne observations of aerosol extinction by in situ and remote-sensing techniques: Evaluation of particle hygroscopicity. Geophys. Res. Lett. 2013;40:417–422. doi: 10.1029/2012GL054428. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1