

The molecular basis by which prions arise spontaneously is poorly understood. The present data point toward oxidative protein damage as one of the triggers of de novo prion formation. Autophagy functions to clear oxidatively damaged proteins before their conversion to the prion form.
Abstract
Prions are self-propagating, infectious proteins that underlie several neurodegenerative diseases. The molecular basis underlying their sporadic formation is poorly understood. We show that autophagy protects against de novo formation of [PSI+], which is the prion form of the yeast Sup35 translation termination factor. Autophagy is a cellular degradation system, and preventing autophagy by mutating its core components elevates the frequency of spontaneous [PSI+] formation. Conversely, increasing autophagic flux by treating cells with the polyamine spermidine suppresses prion formation in mutants that normally show a high frequency of de novo prion formation. Autophagy also protects against the de novo formation of another prion, namely the Rnq1/[PIN+] prion, which is not related in sequence to the Sup35/[PSI+] prion. We show that growth under anaerobic conditions in the absence of molecular oxygen abrogates Sup35 protein damage and suppresses the high frequency of [PSI+] formation in an autophagy mutant. Autophagy therefore normally functions to remove oxidatively damaged Sup35, which accumulates in cells grown under aerobic conditions, but in the absence of autophagy, damaged/misfolded Sup35 undergoes structural transitions favoring its conversion to the propagatable [PSI+] form.
INTRODUCTION
Prions are infectious agents arising from misfolded proteins. They cause transmissible spongiform encephalopathies (TSEs) typified by Creutzfeldt–Jakob disease in humans and bovine spongiform encephalopathy in cattle. Conversion of the normal prion protein (PrP) into its infectious PrPSc confirmation underlies the pathogenesis of TSEs (Collinge and Clarke, 2007). This protein-only mechanism of infectivity can also explain the unusual genetic behavior of several prions found in the yeast Saccharomyces cerevisiae (Wickner, 1994; Alberti et al., 2009). [PIN+] and [PSI+] are the best-studied yeast prions and are formed from the Rnq1 and Sup35 proteins, respectively (Wickner, 1994; Derkatch et al., 1997). [PSI+] is the altered conformation of the Sup35 protein, which normally functions as a translation termination factor during protein synthesis. The de novo formation of [PSI+] is enhanced by the presence of the [PIN+] prion (Derkatch et al., 1996, 2001; Osherovich and Weissman, 2001), which is the altered form of the Rnq1 protein, whose native protein function is unknown (Treusch and Lindquist, 2012).
Yeast Sup35 normally functions in translation termination in its soluble form but is sequestered away from this function in its amyloid or aggregated form (Wickner, 1994). Recently, aggregated Sup35 has been shown to retain its translation termination function, and alterations in amyloid heterogeneity have been shown to underlie changes in Sup35 protein–only phenotypes (Pezza et al., 2014). The amyloid state is a highly structured, insoluble fibrillar deposit, consisting of many repeats of the same protein. This type of aggregation is central to the pathology of many neurodegenerative diseases, including Alzheimer’s, Parkinson’s, Huntington’s, and prion diseases. Although the exact point at which these disease-related proteins are toxic is debatable, it is well accepted that the process of amyloid formation is generally detrimental to human health. Fungal and mammalian prions form de novo, but the mechanism is poorly understood in molecular terms. An initial alternative conformational state might be instigated by spontaneous misfolding event(s) that might be triggered by mutation, mistranslation, environmental stresses, and/or disruption of the chaperone network (DeMarco and Daggett, 2005). Hence any defense systems that can eliminate these initially misfolded species might prevent conversion to the amyloid or disease-causing form of the protein.
Studies in yeast cells have identified intricate protein quality control systems in which insoluble proteins are partitioned into defined sites in the cell; amyloid and amorphous aggregates are believed to be processed separately (Sontag et al., 2014). The ubiquitin proteasome system (UPS) is the main proteolytic system that subsequently degrades misfolded and damaged proteins or proteins that are no longer required in cells. Proteins destined for degradation by the proteasome are tagged with ubiquitin, and previous studies showed that alterations in the ubiquitin system affect prion formation (Allen et al., 2007). In addition, a number of genes that affect the UPS have been identified in an unbiased genome-wide screen for factors that modify the frequency of [PSI+] induction (Tyedmers et al., 2008). This suggests that the UPS normally functions to prevent conversion of misfolded Sup35 into its transmissible amyloid form. However, ubiquitinated Sup35 has not been directly detected in yeast, and it is unclear whether the proteasome plays a direct role in suppressing [PSI+] prion formation (Allen et al., 2007).
Autophagy is the cellular proteolytic system that degrades organelles and clears protein aggregates via vacuolar/lysosomal degradation (Parzych and Klionsky, 2014). During autophagy, an elongated isolation membrane sequesters cell material for degradation, forming a double-membrane-bound vesicle called the autophagosome. Fusion of the autophagosome with vacuoles/lysosomes introduces acidic hydrolases, which degrade the contained proteins and organelles. The progress of the autophagy pathway is regulated by the products of autophagy-related genes (ATG genes). Approximately 35 ATG genes have been identified in yeast, and several mammalian homologues have been functionally characterized. A number of possible links between autophagy and protein aggregation diseases have been described. For example, a link between autophagy and prion disease was first suggested by the observance of autophagic vacuoles in neurons from a scrapie hamster model (Boellaard et al., 1991). Suppression of basal autophagy in mice causes neurodegenerative diseases, suggesting that autophagy plays a role in the clearance of misfolded proteins (Hara et al., 2006; Komatsu et al., 2006), and amyloidogenic aggregates such as those formed by α-synuclein and huntingtin have been identified as substrates of autophagy (Webb et al., 2003; Ravikumar et al., 2004; Iwata et al., 2005). In addition, a correlation has been observed between pharmacological interventions that induce autophagy and enhanced cellular degradation of prions in prion-infected neuronal cell models (Heiseke et al., 2009). All of this suggests that autophagy plays a protective role against prion toxicity. However, the molecular details of how autophagy might promote prion clearance/degradation are unclear, and it is unknown whether a defective autophagy pathway might promote increased prion formation/accumulation.
In the present study, we analyze the role of autophagy in protecting against de novo [PSI+] prion formation in yeast. We show that the frequency of [PSI+] and [PIN+] prion formation is elevated in mutants deficient in autophagy. Conversely, induction of autophagy acts to protect against de novo prion formation. We show that oxidatively damaged Sup35 accumulates in autophagy mutants and suggest that autophagy normally functions to remove such oxidatively damaged Sup35, preventing spontaneous prion formation. In agreement with this idea, we show that growth under anaerobic conditions suppresses the de novo formation of [PSI+] in an autophagy mutant.
RESULTS
[PSI+] prions are formed in autophagy mutants
A representative range of autophagy mutants was constructed to determine whether Sup35 aggregation is elevated in the absence of autophagy. Deletion mutants were made in a [PIN+][psi−] version of yeast strain 74D-694, which is commonly used for prion studies (Chernoff et al., 1995). Mutants were constructed in a [PIN+][psi−] strain background with defects in the core autophagy machinery, including the Atg1 kinase complex (atg1), the PI3K complex (atg14), the Atg9 cycling complex (atg9), the Atg8 ubiquitin-like conjugation system (atg8, atg4), and the Atg12 ubiquitin-like conjugation system (atg12, atg7). Mutants were also constructed lacking a vacuolar lipase (atg15), a receptor protein for the cytoplasm-to-vacuole targeting (Cvt) pathway (atg19), an adapter protein required for cargo loading in pexophagy (atg11), and a mitochondrial cargo receptor required in mitophagy (atg32).
Sup35 aggregate formation was visualized using a SUP35NM–green fluorescent protein (GFP) fusion construct under the control of the copper-regulatable CUP1 promoter (Patino et al., 1995). After short-term induction of this construct, diffuse cytoplasmic fluorescence is observed in [psi−] cells, whereas coalescence of newly made Sup35NM-GFP with preexisting Sup35 aggregates in [PSI+] cells allows the detection of [PSI+] foci. [PIN+][psi−] strains were grown for 16 h and SUP35NM-GFP induced with copper for 1 h to visualize Sup35 aggregation. As expected, diffuse cytoplasmic fluorescence was observed in the control [psi−] strain (Figure 1A). Similar diffuse cytoplasmic Sup35 fluorescence was detected in the atg11 and atg32 mutants. In contrast, many large Sup35 puncta were detected in all of the remaining atg mutants (Figure 1A). Quantification of aggregate formation revealed that ∼2–5% of mutant cells examined contained visible fluorescent foci after 16 h of growth (Figure 1B).
FIGURE 1:
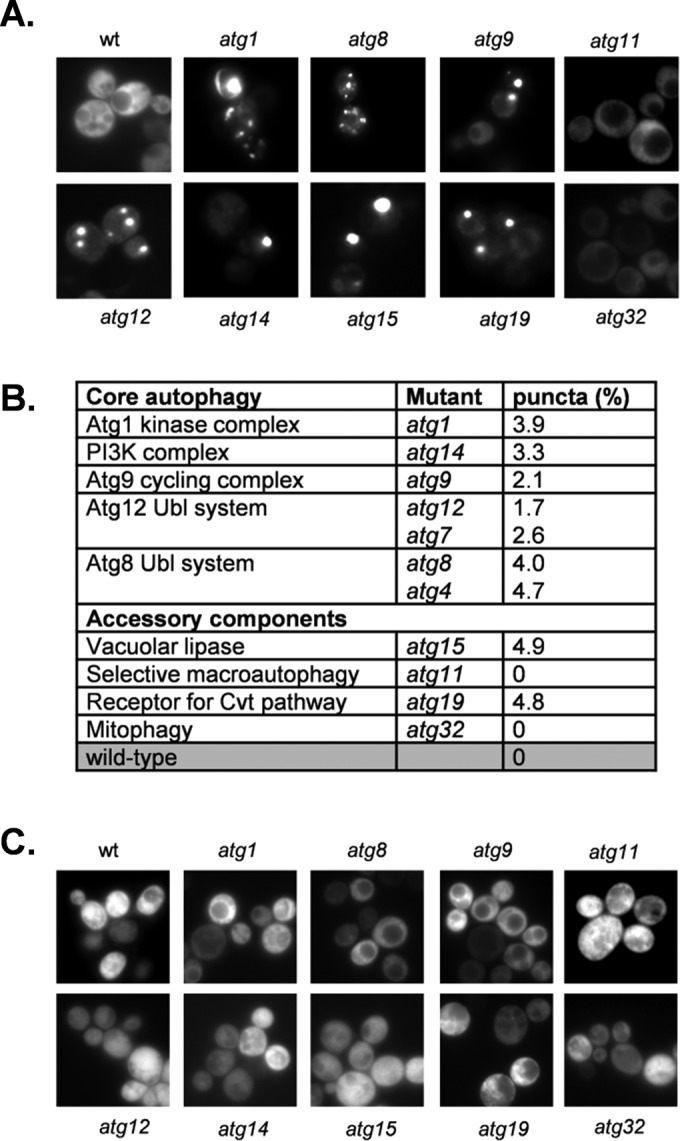
[PSI+] prions are formed in autophagy mutants. (A) Representative fluorescence micrographs for the wild-type and indicated autophagy mutant strains containing the Sup35NM-GFP plasmid. The Sup35NM-GFP plasmid was induced for 1 h using copper before visualizing aggregate formation after 16 h of growth. (B) The aggregation frequency was calculated in the indicated autophagy mutants as a percentage of the number of cells containing fluorescent foci out of ∼300 cells counted. (C) Representative fluorescence micrographs for strains cured with GdnHCl.
One well-defined genetic criterion for a yeast prion is its reversible curability (Wickner, 1994). This is commonly tested using guanidine hydrochloride (GdnHCl), which blocks the propagation of yeast prions by inhibiting the key ATPase activity of Hsp104, a molecular chaperone that is absolutely required for yeast prion propagation (Ferreira et al., 2001; Jung and Masison, 2001). Curing autophagy mutants with GdnHCl resulted in diffuse cytoplasmic Sup35-GFP fluorescence, with no detectable foci, indicating that these puncta may represent [PSI+] prion formation rather than amorphous Sup35 protein aggregates (Figure 1C)
Increased frequency of de novo [PSI+] and [PIN+] prion formation in autophagy mutants
Strain 74D-694 contains the ade1-14 mutant allele, which confers adenine auxotrophy due to the presence of a premature UGA stop codon in the ADE1 gene. Thus [psi−] ade1-14 cells are unable to grow in the absence of exogenous adenine and accumulate an intermediate in the adenine biosynthetic pathway that causes the colonies to be red. Suppression of the ade1-14 mutation in [PSI+] cells allows growth in the absence of adenine, giving rise to white or pink colonies. Spontaneous white/pink Ade+ colonies were isolated from atg1, atg8, and atg19 mutants (Figure 2A). The Ade+ phenotype was eliminated by growth in the presence of GdnHCl, giving rise to red Ade− colonies, confirming the de novo formation of [PSI+] in these cells (Figure 2A). Semidenaturing detergent agarose gel electrophoresis (SDD-AGE) was used to provide further evidence that autophagy mutants form [PSI+] prions. [PSI+] prions form SDS-resistant, high–molecular weight aggregates that can be detected using SDD-AGE (Kryndushkin et al., 2003). Such Sup35 aggregates were detected in a control [PSI+] strain (Figure 2B). Growth of this strain in the presence of GdnHCl shifted Sup35 back to its monomeric size due to the requirement for Hsp104 to propagate [PSI+] prion formation. Similar SDS-resistant, high–molecular weight aggregates were detected in the [PSI+] versions of the atg1, atg4, atg8, and atg15 mutants, which were also curable by growth in the presence of GdnHCl (Figure 2B).
FIGURE 2:
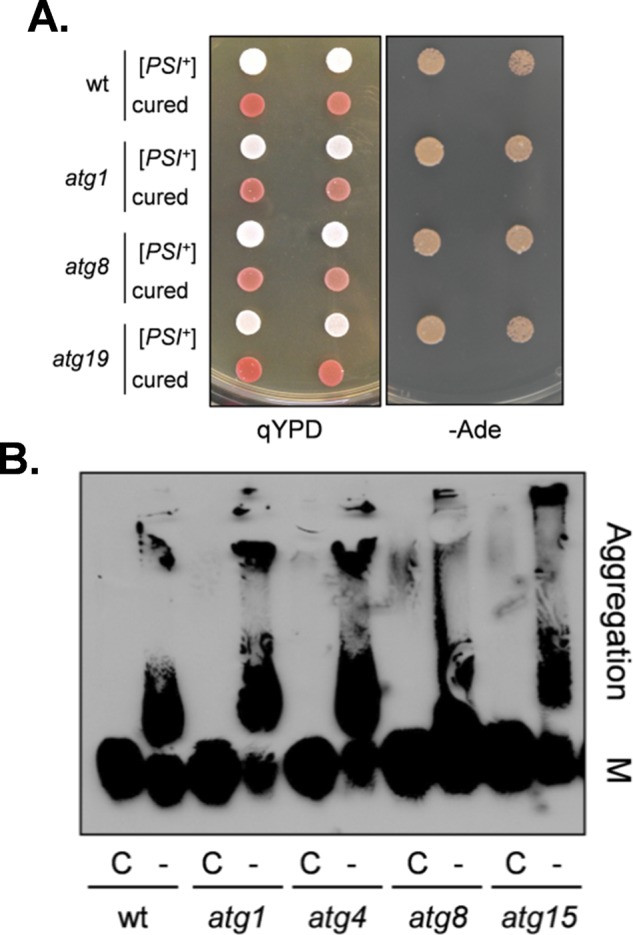
The [PSI+] prion is formed in autophagy mutants. (A) [PSI+] prion formation was visualized in the wild-type (74D-694) and atg1, atg8, and atg19 mutant strains by pink/white colony formation and growth on minimal medium in the absence of adenine. Curing with GdnHCl gives rise to red Ade− colonies, confirming the de novo formation of [PSI+] in these cells. (B) Cell extracts were prepared from exponentially growing cells and analyzed by SDD-AGE. SDS-resistant Sup35 aggregates were detected in atg1, atg4, atg8, and atg15 mutant strains. Aggregate and monomer (M) forms are indicated.
To quantify [PSI+] formation, we used a plasmid with a ura3-14 allele containing the ade1-14 nonsense mutation engineered into the wild-type URA3 gene (Manogaran et al., 2006). The ura3-14 allele allows [PSI+] prion formation to be scored by growth on media lacking uracil, indicative of decreased translational termination efficiency in [PSI+] cells. We used this assay rather than scoring [PSI+] formation by suppression of the ade1-14 nonsense mutation and growth on media lacking adenine to avoid any possible complications arising from adenine metabolism in autophagy mutants. Formation of the red pigment in adenine mutants arises due to its accumulation in vacuoles (Chaudhuri et al., 1997), and we reasoned that autophagy mutants might affect vacuolar function. [PSI+] formation was differentiated from nuclear gene mutations that give rise to uracil prototrophy by their irreversible elimination in GdnHCl. Using this assay, we estimated the frequency of de novo [PSI+] prion formation in a control [PIN+][psi−] strain to be ∼1 × 10−5 (Figure 3A), comparable to previously reported frequencies (Lund and Cox, 1981; Lancaster et al., 2010). Loss of ATG1, ATG8, or ATG19 caused a modest increase in the frequency of de novo [PSI+] formation of approximately twofold to threefold. Given that increased cellular concentration of Sup35 can promote [PSI+] prion formation, we examined Sup35 protein levels in autophagy mutants (Figure 3B). This analysis confirmed that similar levels of Sup5 are present in a wild-type and atg1, atg8, and atg19 mutant strains, ruling out any effects on Sup35 protein concentration.
FIGURE 3:
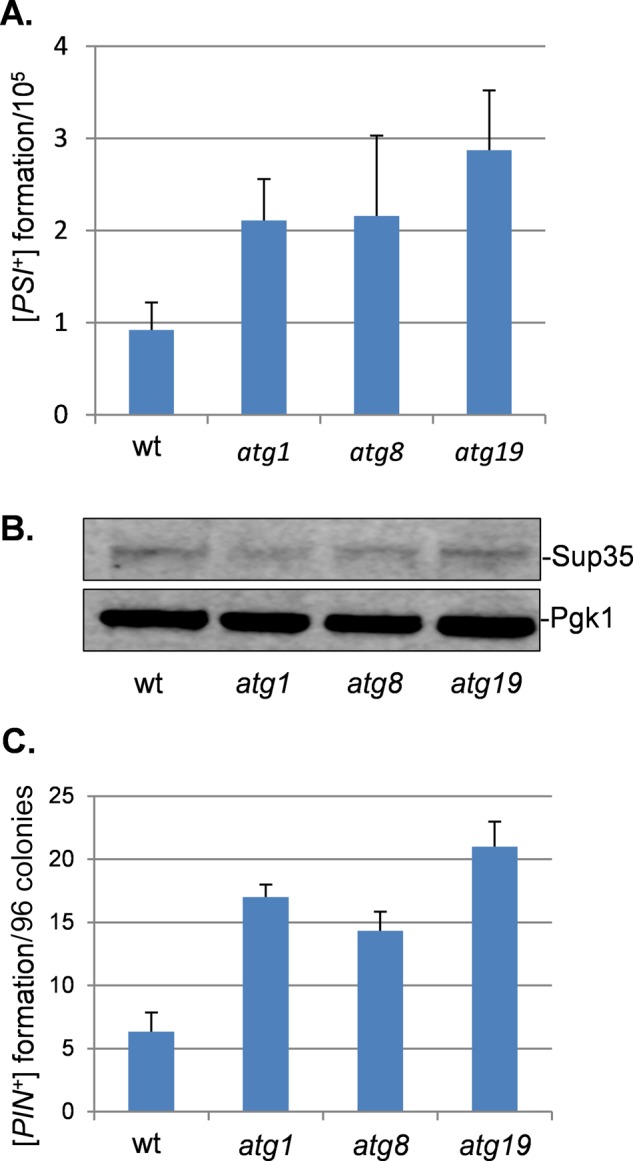
Increased frequency of de novo [PSI+] and [PIN+] prion formation in autophagy mutants. (A) [PSI+] prion formation was quantified in the wild-type and atg1, atg8, and atg19 mutant strains using an engineered ura3-14 allele, which contains the ade1-14 nonsense mutation inserted into the wild-type URA3 gene (Manogaran et al., 2006). [PSI+] prion formation was scored by growth on medium lacking uracil, indicative of decreased translational termination efficiency. [PSI+] formation was differentiated from nuclear gene mutations that give rise to uracil prototrophy by their irreversible elimination in GdnHCl. Data shown are the means of at least three independent biological repeat experiments expressed as the number of colonies per 105 viable cells. Error bars denote the SD. (B) Western blot analysis showing Sup35 protein levels in wild-type and atg1, atg8, and atg19 mutant strains. Blots were probed with α-Pgk1 as a loading control. (C) [PIN+] prion formation was quantified in wild-type and atg1, atg8, and atg19 mutant strains and is expressed as the number of [PIN+] colonies formed per 96 colonies examined. Data shown are the means of at least three independent biological experiments; error bars denote the SD.
Given the increased frequency of [PSI+] prion formation in autophagy mutants, we examined the de novo formation of another yeast prion, namely the Rnq1/[PIN+] prion, which is not related in sequence to the Sup35/[PSI+] prion. The de novo formation of [PIN+] prions was detected in ∼6% of control [pin−] cells (Figure 3C), comparable to previous measurements (Sideri et al., 2011). The frequency of [PIN+] prion formation was elevated by approximately twofold to threefold in atg1, atg8, and atg19 mutants (Figure 3C). Taken together, these data indicate that an increased frequency of de novo prion formation occurs in mutants defective in the core autophagy machinery, suggesting that active autophagy is required to suppress prion formation during normal growth conditions.
The frequency of induced [PSI+] formation is increased in an autophagy mutant
[PSI+] prion formation can be induced by the overexpression of Sup35 in [PIN+] strains since the excess Sup35 increases the possibility for prion seed formation (Wickner, 1994). Strains were initially grown overnight, before SUP35NM-GFP was induced with copper to promote [PSI+] prion formation. We found that overexpression of Sup35NM-GFP in a [PIN+][psi−] control strain resulted in detectable protein aggregates after 18 h of expression induced by copper addition, and aggregates were detected in ∼4.7% of cells by 24 h (Figure 4A), similar to previous reports (Mathur et al., 2010). Overexpression of SUP35NM-GFP in [PIN+][psi−] cells facilitates the detection of ring- and ribbon-like aggregates that are believed to be characteristic of de novo prion formation. These structures can be found in the cell periphery or surrounding the vacuole and mature into an infectious prion state, detected as large, dot-like aggregates (Ganusova et al., 2006). Ring and ribbon-like aggregates characteristic of the de novo formation of [PSI+] could be detected in 0.6% of control cells by 18 h (Figure 4A). When the same experiment was repeated in an atg1 mutant, increased aggregation was detected after overnight growth and induction of SUP35NM-GFP for 1 h, as expected from Figure 1. Sup35 aggregation continued to increase in the atg1 mutant, with 13.6% of cells examined containing visible aggregates after 24 h of SUP35NM-GFP expression (Figure 4A). Ring- and ribbon-like aggregates characteristic of the de novo formation of [PSI+] could also be detected within 12 h. Thus autophagy mutants appear to show an increased frequency of both spontaneous and induced [PSI+] prion formation.
FIGURE 4:

Induction of [PSI+] prion formation in autophagy mutants. (A) Fluorescence micrographs for [PIN+] [psi−] versions of the wild-type and atg1 mutant strains containing the Sup35NM-GFP plasmid induced with copper for the indicated times. Top rows, representative images in which puncta formation was first detected in the wild-type (18 h) and atg1 (1 h) strains. Bottom rows, representative images in which rod- and ribbon-like aggregates, indicative of de novo prion formation, were detected in the wild-type (18 h) and atg1 (12 h) strains. The percentage of cells containing visible puncta or rod- and ribbon-like aggregates is shown for each strain from an average of 300 cells counted. (B) Western blot analysis of the wild-type and atg1 mutant strains after induction of Sup35NM-GFP for 1, 8, 18, or 24 h. Blots were probed with αSup35 or α-Pgk1 as a loading control. (C) Fluorescence micrographs for cured versions of the wild-type and atg1 mutant strains after induction of Sup35NM-GFP for 2 or 24 h. Representative images in which puncta were only detected in atg1 mutant cells (3.6%) after 24-h induction. (D) [PSI+] prion formation was quantified in the wild-type and atg1 mutant strains containing the Sup35NM-GFP plasmid after 0 , 1, and 24 h of copper induction. [PSI+] formation was quantified using the ade1-14 mutant allele by growth on medium lacking adenine and differentiated from nuclear SUPX gene mutations by their irreversible elimination in GdnHC1. Data shown are the means of at least three independent biological repeat experiments expressed as the number of colonies per 104 viable cells. Error bars denote the SD. (E) Sup35 toxicity was examined in [psi−] or [PSI+] versions of the indicated strains containing SUP35 under the control of a GAL inducible promoter. Strains were initially grown overnight in raffinose-containing medium before dilution (A600 = 1, 0.1, 0.01, 0.001) and spotting onto agar plates containing galactose or glucose. Overexpression of Sup35 inhibits growth in the [PSI+] versions of autophagy mutants compared with [psi−] versions.
Sup35 Western blot analysis was used to rule out any differences in SUP35NM-GFP induction in the atg1 mutant compared with the wild-type strain (Figure 4B). This analysis showed that a similar profile of increased SUP35NM-GFP was detected in both the wild-type and atg1 mutant strains. Strains were cured with GdnHCl before SUP35NM-GFP overexpression to determine the requirement for Hsp104 for induced puncta formation. No puncta were detected in the cured wild-type strain after 2 or 24 h induction of SUP35NM-GFP (Figure 4C). Similarly, no puncta were detected in the atg1 mutant after 2 h of induction of SUP35NM-GFP, but 3.6% of atg1 mutant cells contained puncta after 24 h of induction (Figure 4C). These data confirm that the induced aggregate formation in the wild type and the atg1 mutant is largely [PIN+] dependent.
The induction of [PSI+] prion formation was quantified using the ade1-14 mutant allele, which confers adenine auxotrophy and is differentiated from nuclear SUPX gene mutations by its irreversible elimination in guanidine hydrochloride (Tuite et al., 1981). The frequency of [PSI+] formation was ∼10-fold higher in a wild-type strain containing the Sup35-GFP plasmid before copper induction compared with a nontransformed strain (compare Figures 4D and 3A). This presumably reflects increased basal levels of Sup35 expression from the Sup35-GFP plasmid. Before copper induction, the frequency of [PSI+] formation was approximately threefold higher in the atg1 mutant than with the wild-type strain (Figure 4D). This is very similar to the difference observed between wild-type and atg1 mutant strains using the ura3-14 assay (Figure 3A). [PSI+] formation was strongly induced in response to copper addition. This induction was stronger in the atg1 mutant than with the wild-type strain, confirming that autophagy suppresses induced [PSI+] prion formation (Figure 4D).
Overexpression of Sup35 in a [PSI+] background can be toxic due to increased Sup35 aggregation titrating Sup35 away from its normal function in translation termination (Derkatch et al., 1996; Allen et al., 2007; Vishveshwara et al., 2009). We examined whether autophagy is required to protect against this toxicity. Sup35 was overexpressed under the control of the GAL1 promoter in [PIN+][PSI+] versions of the wild-type and atg1, atg8, and atg19 mutant strains. Sup35 overexpression was more toxic in the autophagy mutants than with the wild-type strain (Figure 4E). This toxicity depended on the [PSI+] status of the cells, since it was largely abrogated in [PIN+][psi−] mutants.
Inducing autophagy protects against de novo [PSI+] prion formation
Because loss of autophagy results in an increased frequency of prion formation, we examined whether inducing autophagy could protect against de novo [PSI+] prion formation. This is difficult to examine in a wild-type strain, given the rare occurrence of spontaneous [PSI+] prion formation. We therefore used mutants that are known to have an increased frequency of de novo [PSI+] prion formation. This included a mutant lacking the Tsa1 and Tsa2 peroxiredoxins (Sideri et al., 2010). Peroxiredoxins are important cellular antioxidants, and oxidative damage to Sup35 is believed to trigger the formation of [PSI+] prions in this mutant (Sideri et al., 2011). A number of factors that modify the frequency of [PSI+] induction were identified in an unbiased genome-wide screen (Tyedmers et al., 2008). We used one such mutant identified in this screen that is deleted for PPQ1 (SAL6), encoding a protein phosphatase of unknown function. Ppq1 (Sal6) was originally identified as a mutant that increases the efficiency of translational suppressors, including nonsense suppressors (Vincent et al., 1994). Rather than affecting prion formation, this mutant presumably allows the detection of [PSI+] strains that are normally too weak to detect. Using the ura3-14 allele plasmid assay, we found the frequency of de novo [PSI+] prion formation to be elevated by approximately fourfold in a [PIN+][psi−] tsa1 tsa2 mutant and by 20-fold in a [PIN+][psi−] ppq1 mutant (Figure 5A). Autophagy can be stimulated by a number of pharmacological agents including the naturally occurring polyamine spermidine (Eisenberg et al., 2009; Morselli et al., 2011). Growth of the tsa1 tsa2 and ppq1 mutants in the presence of 4 mM spermidine significantly reduced the elevated frequency of de novo [PSI+] prion formation normally observed in these mutants but not in an atg1 mutant (Figure 5A).
FIGURE 5:
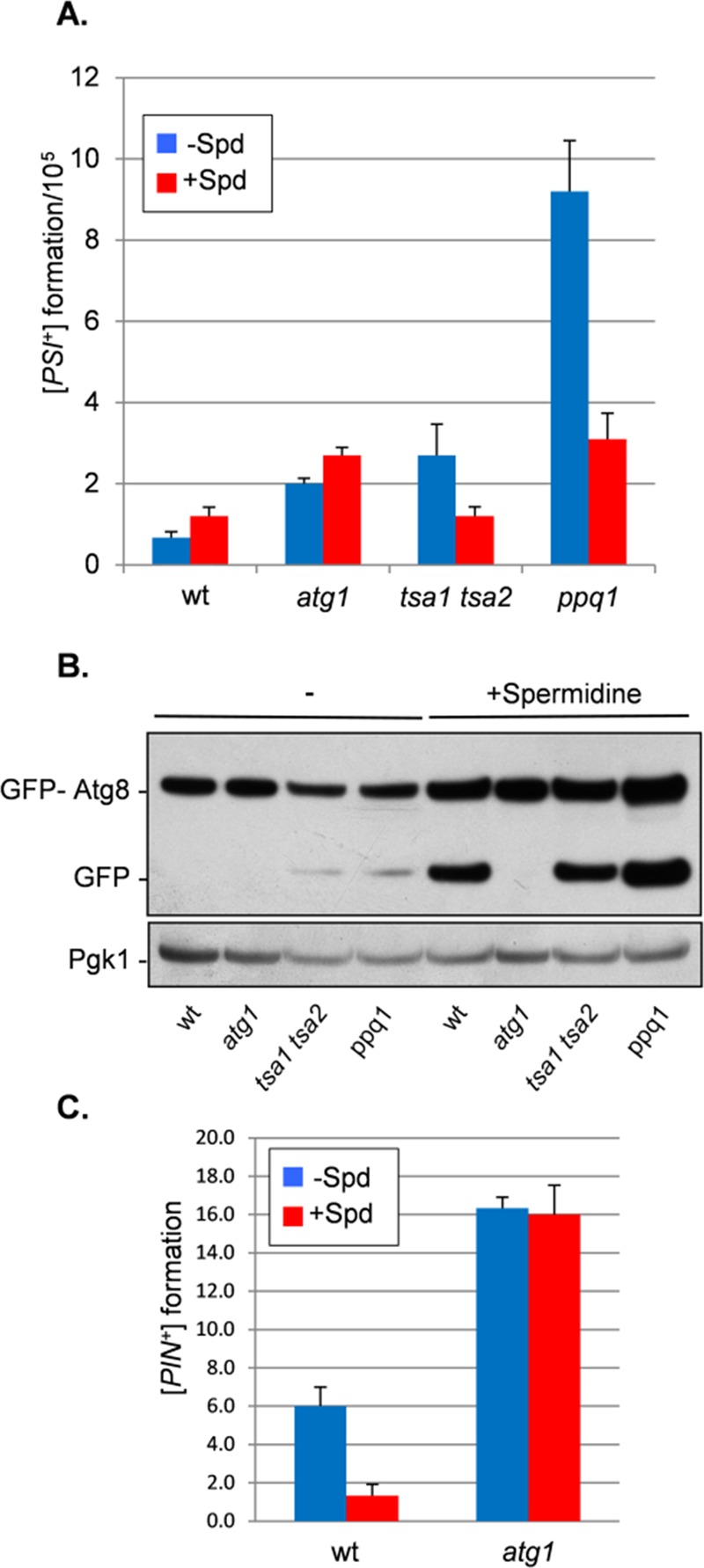
Inducing autophagy with spermidine protects against de novo [PSI+] and [PIN+] prion formation. (A) [PSI+] prion formation was quantified in wild-type and tsa1 tsa2 and ppq1 mutant strains using an engineered ura3-14 allele as described for Figure 3A. Autophagy was induced by growing cells in the presence of 4 mM spermidine (+ Spd). (B) Autophagic flux was monitored in cells expressing GFP-Atg8. Growth in the presence of spermidine induced autophagy in wild-type and tsa1 tsa2 and ppq1 mutant strains as detected by the appearance of free GFP indicative of autophagic flux. No free GFP was detected in an atg1 mutant. (C) [PIN+] prion formation was quantified in wild-type and atg1 mutant cells as described for Figure 3C. Autophagy was induced by growing cells in the presence of 4 mM spermidine (+ Spd).
A GFP-Atg8 construct was used as a control to confirm that spermidine induces autophagy in these mutants (Noda et al., 1995). This assay follows the autophagy-dependent proteolytic liberation of GFP from GFP-Atg8, which is indicative of autophagic flux. Free GFP was detected in the wild-type and tsa1 tsa2 and ppq1 mutant strains after spermidine treatment but not in the atg1 mutant, which is defective in autophagy (Figure 5B). Of interest, low levels of free GFP were detected in the tsa1 tsa2 and ppq1 mutants in the absence of spermidine, suggesting that these mutants already have elevated basal levels of autophagy. Because de novo [PIN+] formation occurs at a relatively high rate, we examined whether inducing autophagy with spermidine also protects against [PIN+] formation. This analysis showed that spermidine reduces de novo [PIN+] formation from ∼6% to <1% (Figure 5C). Spermidine treatment did not affect the elevated frequency of de novo [PIN+] formation observed in an atg1 mutant.
Sup35 protein oxidation is increased in autophagy mutants
To begin to address the mechanism by which prions form spontaneously in autophagy mutants, we examined whether the localization of [PSI+] foci is altered in an autophagy mutant. Induced [PSI+] prion formation is believed to proceed via targeted localization of misfolded Sup35 to the IPOD, which is formed adjacent to the vacuole (Tyedmers et al., 2010; Sontag et al., 2014). The vacuolar dye FM4-64 was used to visualize vacuolar membranes in wild-type and atg1 mutant strains (Figure 6A). This analysis revealed that similar vacuolar Sup35-GFP foci were detected in the wild-type and atg1 mutant strains after copper induction of the SUP35NM-GFP fusion (Figure 6A). We used CFP-ATG8 and CFP-ATG14 as markers of the preautophagosomal structure (PAS) in wild-type and atg1 mutant strains (Figure 6B). Similar to previous reports (Tyedmers et al., 2010), we observed that Sup35-GFP foci formed adjacent to these PAS markers. Similar Sup35-GFP foci were formed in the atg1 mutant despite the absence of PAS formation. Thus, although an atg1 mutant is deficient in macroautophagy and the recruitment of additional Atg proteins to the PAS (Parzych and Klionsky, 2014), Sup35 is still targeted to the vacuolar membrane.
FIGURE 6:
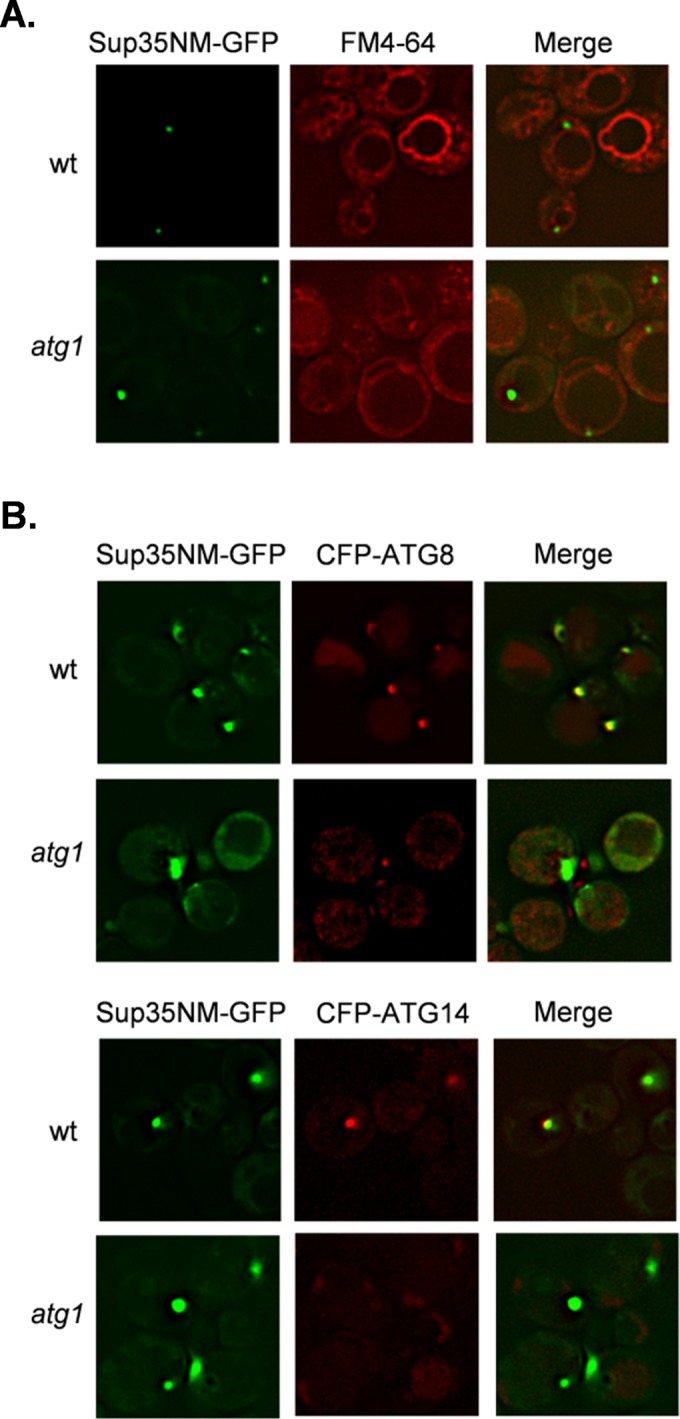
Sup35 aggregate formation occurs at similar intracellular sites in wild-type and atg1 mutant strains. (A) The vacuolar dye FM4-64 was used to visualize vacuolar membranes in wild-type and atg1 mutant strains. Similar vacuolar Sup35-GFP foci were detected in the wild-type and atg1 mutant strains after copper induction of the SUP35NM-GFP fusion construct for 1 h. (B) CFP-ATG8 and CFP-ATG14 were used as markers of PAS in wild-type and atg1 mutant strains. Strains were grown for 48 h in the presence of 4 mM spermidine to induce autophagy and SUP35NM-GFP induced with copper for 1 h. Sup35 aggregates form adjacent to PAS markers in the wild-type strain.
Given that vacuolar targeting of Sup35 appears to be unaffected in an atg1 mutant, we next examined whether oxidatively damaged Sup35 accumulates in an autophagy mutant, which might trigger [PSI+] prion formation. This is because oxidative damage to Sup35 is believed to be one possible cause of the initial misfolding event that triggers the formation of the [PSI+] prion in yeast (Sideri et al., 2011). We previously showed that loss of antioxidants results in elevated levels of Sup35 methionine oxidation and [PSI+] prion formation, suggesting that endogenous levels of reactive oxygen species (ROS) are sufficient to promote prion formation (Sideri et al., 2011). We reasoned that if autophagy acts to suppress prion formation by removing oxidatively damaged Sup35, we might detect oxidized Sup35 in an autophagy mutant. Sup35 oxidation was measured by immunoblot analysis using an antibody that recognizes methionine sulfoxide (MetO). No MetO was detected in a wild-type strain grown under nonstress conditions, but MetO was detected after exposure to 1 mM hydrogen peroxide for 1 h (Figure 7A). Formation of MetO was also detected in the tsa1 tsa2 mutant, as previously reported (Sideri et al., 2011). In addition, MetO formation was detected in an atg1 mutant in the absence of oxidative stress, suggesting that autophagy normally functions to remove oxidatively damaged Sup35 that forms during normal growth conditions and endogenous ROS exposure.
FIGURE 7:
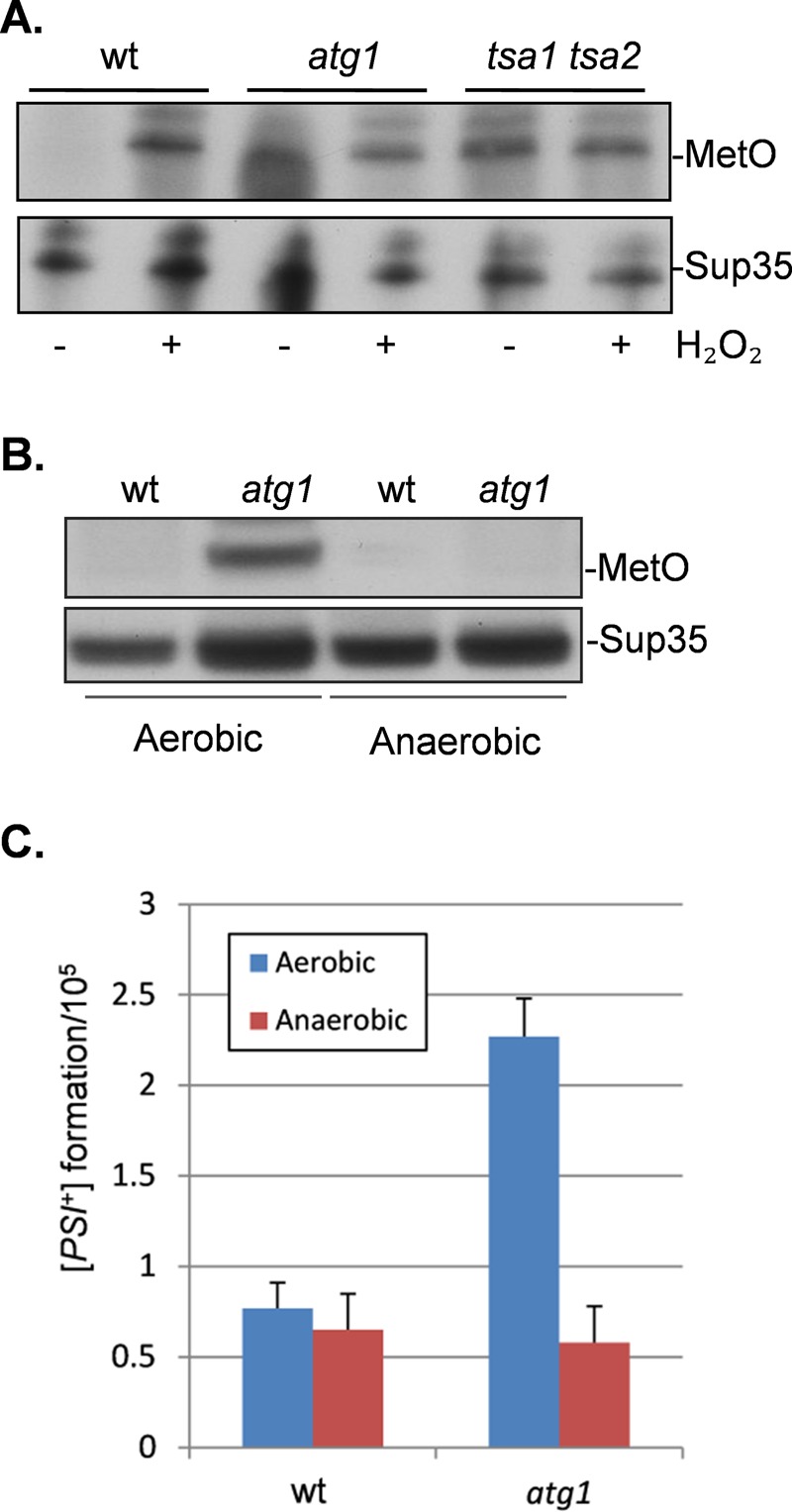
Oxidation of Sup35 in an atg1 mutant causes [PSI+] prion formation. (A) Sup35 was affinity purified using TAP chromatography from wild-type and atg1 and tsa1 tsa2 mutant strains. Western blots were probed with anti-PAP (peroxidase anti-peroxidase) to confirm that similar amounts of Sup35 were purified from each strain. Sup35 oxidation was detected using antibodies that recognize MetO. Strains were treated with 1 mM hydrogen peroxide where indicated. (B) Sup35 methionine oxidation was detected in wild-type and atg1 mutant strains grown under aerobic vs. anaerobic conditions. Western blots were probed with anti-Sup35 to confirm that similar amounts of Sup35 were purified from each strain. (C) [PSI+] prion formation was quantified in wild-type and atg1 mutant strains after growth under aerobic or anaerobic conditions.
Given that oxidatively damaged Sup35 accumulates in an atg1 mutant, we examined whether oxidative damage underlies the increased frequency of [PSI+] prion formation in an autophagy mutant. The [PIN+][psi−] versions of the wild-type and atg1 mutant strains were grown under aerobic or anaerobic conditions in the absence of molecular oxygen. Examination of Sup35 methionine oxidation revealed that no MetO was detected in the atg1 mutant after growth under anaerobic conditions (Figure 7B). Under aerobic conditions, the frequency of [PSI+] prion formation was elevated by approximately threefold, whereas no increased [PSI+] prion formation was detected in the atg1 mutant grown under anaerobic conditions (Figure 7C). These data indicate that oxidative growth conditions are required for the increased frequency of prion formation in an autophagy mutant.
DISCUSSION
Prions form spontaneously without any underlying genetic change. The emergence of disease correlates with the appearance of PrPSc, a novel conformational form of the cellular PrP protein. This prion form replicates through a cycle of seeded polymerization and fragmentation, and it is assumed that genetic or environmental factors can trigger the conformational change in the absence of any preexisting PrPSc “seeds” (Collinge and Clarke, 2007). However, the exact mechanisms underlying the switch from a normally soluble protein to the amyloid form are poorly understood. Prions cause many neurodegenerative diseases, including Alzheimer’s, Parkinson’s, and Creutzfeldt–Jakob diseases and amyotrophic lateral sclerosis. Most cases of these human diseases are sporadic (Prusiner, 2013). It is therefore important to establish the conditions that trigger the switch to the prion or disease-causing form. Our data indicate that autophagy is required to suppress de novo formation of the yeast [PSI+] and [PIN+] prions.
Mutants lacking several different core and associated autophagy components were found to elevate prion formation. In contrast, mutants lacking ATG11 or ATG32, which are deficient in pexophagy and mitophagy, respectively, were unaffected in aggregate formation. Mutants lacking ATG19 also displayed increased spontaneous aggregate formation. This was somewhat unexpected because Atg19 is believed to function as an essential component of the cytoplasm-to-vacuole targeting pathway (Cvt) rather than in nonselective autophagy (Xie and Klionsky, 2007). However, there is some evidence for a role for Atg19 in the degradation of an ER-associated degradation substrate (Mazon et al., 2007). Atg19 is required for the efficient degradation of Pma1 in a process that also requires the UPS. It is unclear whether Sup35 may act as a substrate of the Cvt pathway or whether Sup35 aggregation and [PSI+] prion formation are indirect effects of loss of Atg19. This does not appear to arise specifically due to loss of the Cvt pathway, since Sup35 aggregation and [PSI+] prion formation are unaffected in a mutant lacking Atg11, which functions as an adapter protein required for cargo loading in pexophagy and the Cvt pathway.
The frequency of de novo [PSI+] formation is relatively low in a wild-type strain (∼1 × 10−5) and is elevated by approximately twofold to threefold in mutants lacking ATG1, ATG8, or ATG19. In contrast, Sup35NM-GFP aggregate formation was detected in ∼4–5% of cells examined for the same autophagy mutants. This suggests that not all cells with fluorescent aggregates correspond to true [PSI+] formation. It has long been known that not all SUP35-GFP aggregates will give rise to prions. For example, previous studies suggested that about half of cells with fluorescent dots will die. Some of the rest contain nonproductive (e.g., nonamyloid) aggregates (Arslan et al., 2015). Thus, whereas overexpressing SUP35-GFP provides a way to look for the general propensity to promote aggregate formation, it does not distinguish among aggregates that will be dissolved/cleared by autophagy, the ones that will kill the cell, and the ones that will give rise to [PSI+]. Our quantitative assay specifically quantifies “classical” PIN-dependent [PSI+] formation, that is, prions that are capable of propagation and strong enough to give rise to viable colonies. [PSI+] formation is strongly induced in response to copper addition, as expected. This induction is stronger in the atg1 mutant than in the wild-type strain, confirming that autophagy suppresses induced [PSI+] prion formation. Furthermore, taking the wild-type strain as an example, these experiments confirm that the frequency of true [PSI+] formation (1.2 × 10−4) is much lower than the number of cells containing visible Sup35-GFP aggregates (4.7 × 10−2).
Sup35 containing oxidized methionine (MetO) was detected in the atg1 mutant, which appears to underlie the increased frequency of [PSI+] prion formation in this mutant. Increasing evidence suggests a causal link between protein oxidation and de novo prion formation (Grant, 2015). For example, oxidized methionine residues detected in misfolded PrPSc have been proposed to facilitate the structural conversion underlying the sporadic formation of PrPSc (DeMarco and Daggett, 2005; Wolschner et al., 2009; Elmallah et al., 2013). Similarly, we showed that methionine oxidation of Sup35 in a range of yeast antioxidant mutants underlies the switch from a soluble form of the protein to the [PSI+] prion (Sideri et al., 2011; Doronina et al., 2015). Abrogating methionine oxidation in antioxidant mutants prevents [PSI+] formation, suggesting that protein oxidation may be a common mechanism underlying the aggregation of both mammalian and yeast amyloidogenic proteins (Sideri et al., 2011; Doronina et al., 2015). Similar increases in the levels of Met-SO formation were detected in tsa1 tsa2 and atg1 mutant strains and in a wild-type strain exposed to hydrogen peroxide. Note that the immunoblots used to detect Met-SO levels do not provide a quantitative measure of methionine oxidation, but the detection of oxidized methionine has been shown to correlate with elevated frequencies of [PSI+] formation (Doronina et al., 2015). We do not know why MetO formation appears to decrease in atg1 mutant cells after oxidative stress conditions. It is possible that the excess accumulation of oxidized proteins that might arise from the combination of oxidative stress and the loss of atg1 is toxic to cells, or, alternatively, other protein degradation or aggregate-clearing systems may become active under these conditions. We suggest that autophagy normally functions to remove oxidized Sup35 from cells, but in the absence of autophagy, misfolded Sup35 undergoes structural transitions favoring its conversion to the propagatable [PSI+] form. In agreement with this idea, growth under anaerobic conditions abrogated both methionine oxidation and the increased frequency of [PSI+] formation in an atg1 mutant.
Met-SO was detected in Sup35 in an atg1 mutant grown under normal conditions in the absence of any added oxidant. This suggests that endogenous ROS levels are sufficient to damage Sup35 but that autophagy normally functions to remove damaged Sup35, preventing [PSI+] formation. The frequency of prion formation was lower in the atg1 mutant than in antioxidant mutants under the same growth conditions. This presumably reflects the higher levels of endogenous ROS that are formed in the absence of antioxidants. A significant proportion of newly synthesized proteins are known to be misfolded, and this is exacerbated by conditions that promote further unfolding, such as oxidative stress (Hohn et al., 2014). These oxidized proteins are often nonfunctional and must be removed by degradation to prevent aggregate formation. The UPS and autophagy are the major routes of clearance for toxic proteins. The UPS is believed to mainly degrade short-lived proteins, whereas autophagy degrades high–molecular weight protein aggregates commonly seen in neurodegenerative disorders. Not surprisingly, therefore, dysregulation of autophagy has been implicated in the pathogenesis of neurodegenerative disorders, since aggregate-prone are eliminated more efficiently via the autophagy pathway than the UPS (Banerjee et al., 2010; Lynch-Day et al., 2012). Sup35 has been shown to be a proteasomal substrate, and proteasomal activity has been demonstrated to influence [PSI+] propagation (Kabani et al., 2014). The proteasome was found to degrade highly ordered prion protein assemblies in a manner that does not require ubiquitination. It therefore seems likely that the UPS and autophagy provide overlapping defense systems to protect against prion formation and propagation.
It is difficult to model sporadic prion formation without overexpressing the corresponding protein. We used mutants that display an elevated frequency of [PSI+] prion formation without any underlying effect on Sup35 protein levels to test whether increasing autophagic flux could protect against prion formation. The elevated frequency of [PSI+] prion formation was abrogated in a tsa1 tsa2 mutant after induction of autophagy using spermidine. This does not simply reflect the role of autophagy in the turnover of oxidized proteins, since [PSI+] prion formation was also reduced in a ppq1 mutant that is not involved in the oxidative stress response. The nature of the misfolded intermediate in a ppq1 mutant is unknown but is presumably normally removed via autophagy to prevent prion formation. Enhancing autophagy has also been shown to reduce toxicity in Huntington’s disease models (Sarkar et al., 2007), suggesting that pharmacological agents that increase autophagic flux may represent a promising therapeutic route toward protecting against amyloid formation and toxicity
The role of autophagy in preventing prion formation and toxicity appears evolutionarily conserved. Previous studies established the importance of autophagy for delivery of PrPSc to lysosomes for degradation in chronically infected cells (Heiseke et al., 2010; Goold et al., 2013). Similarly, inhibitors of autophagy result in increased levels of PrPSc, and stimulating autophagy decreases PrPSc levels (Aguib et al., 2009; Heiseke et al., 2009; Goold et al., 2013; Homma et al., 2014; Joshi-Barr et al., 2014). Autophagy also appears to be stimulated in response to the de novo accumulation of prion aggregates, which may act to clear PrPSc. For example, PrP accumulates as ubiquitinated intracellular protein inclusions, which causes induction of endoplasmic reticulum chaperones, the unfolded protein response, and autophagy in a mouse model of prion disease (Joshi-Barr et al., 2014). It is interesting, therefore, that the basal levels of autophagy were also somewhat elevated in the ppq1 and tsa1 tsa2 mutants, which show an elevated frequency of [PSI+] prion formation. Yeast therefore provides a powerful genetic system to further establish and identify the protective systems that protect against the spontaneous formation of prions.
MATERIALS AND METHODS
Yeast strains and plasmids
The wild-type yeast strain 74D-694 (MATa ade1-14 ura3-52 leu2-3112 trp1-289 his3-200) was used for all experiments. Strains deleted for autophagy genes were constructed in 74D-694 using standard yeast methodology. Sup35 was tagged at its C-terminus with a tandem affinity purification (TAP) tag and was described previously (Sideri et al., 2011). Sup35 was overexpressed using an inducible GAL1-Sup35 plasmid (Josse et al., 2012). Plasmids expressing CFP-ATG8 and CFP-ATG14 were described previously (Tyedmers et al., 2010)
Growth and stress conditions
Strains were grown at 30°C with shaking at 180 rpm in rich YEPD medium (2% [wt/vol] glucose, 2% [wt/vol] bactopeptone, 1% [wt/vol] yeast extract) or minimal SD (0.67% [wt/vol] yeast nitrogen base without amino acids, 2% [wt/vol] glucose) supplemented with appropriate amino acids and bases. SGal media contained 2% (wt/vol) galactose, and SRaf media contained 2% (wt/vol) raffinose in place of glucose. Media were solidified by the addition of 2% (wt/vol) agar. Strains were cured by five rounds of growth on YEPD agar plates containing 4 mM GdnHCl. Anaerobic growth conditions were established by degassing media with nitrogen gas as previously described (Beckhouse et al., 2008).
Analyses of prion formation
A plasmid containing an engineered ura3-14 allele, which contains the ade1-14 nonsense mutation in the wild-type URA3 gene (Manogaran et al., 2006), was used to score the frequency of de novo [PSI+] prion formation as previously described. Alternatively, [PSI+] prion formation was scored by growth in absence of adenine. De novo [PIN+] formation was performed as previously described (Sideri et al., 2011). [PSI+] and [PIN+] formation was calculated based on the mean of at least three independent biological repeat experiments. De novo [PSI+] prion formation was visualized as described previously using CUP1-SUP35NM-GFP (Sideri et al., 2011). The number of cells containing Sup35 puncta was quantified from ∼300 cells counted.
Protein analysis
The analysis of Sup35 amyloid polymers by SDD-AGE was performed as described previously (Alberti et al., 2010). Sup35-TAP affinity purification and detection of methionine oxidation were performed as described previously (Sideri et al., 2011).
Autophagy analysis and induction
Autophagy was induced by growth in the presence of 4 mM spermidine (Eisenberg et al., 2009). The induction of autophagy was confirmed by examining the release of free GFP due to the proteolytic cleavage of GFP-Atg8 using a plasmid described previously (Noda et al., 1995).
Acknowledgments
S.H.S. was supported by a Wellcome Trust funded studentship. V.A.D. was supported by Biotechnology and Biological Sciences Research Council Project Grant BB/J000183/1. We thank Susan Lindquist for the CFP-ATG8 and CFP-ATG14 plasmids. The Bioimaging Facility microscopes used in this study were purchased with grants from the Biotechnology and Biological Sciences Research Council, Wellcome Trust, and University of Manchester Strategic Fund.
Abbreviations used:
- ADE
adenine
- ATG
autophagy-related gene
- Cvt
cytoplasm-to-vacuole targeting
- GAL
galactose
- GdnHCl
guanidine hydrochloride
- GFP
green fluorescent protein
- IPOD
insoluble protein deposit
- M
monomer
- MetO
oxidized methionine
- MetSO
methionine sulfoxide
- PAP
peroxidase anti-peroxidase
- PAS
preautophagosomal structure
- PrP
normal prion protein
- PrPSC
prion protein scrapie form
- ROS
reactive oxygen species
- SDD-AGE
semidenaturing detergent agarose gel electrophoresis
- Spd
spermidine
- SUPX
suppressor
- UPS
ubiquitin proteasome system
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-08-0548) on October 21, 2015.
REFERENCES
- Aguib Y, Heiseke A, Gilch S, Riemer C, Baier M, Schatzl HM, Ertmer A. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy. 2009;5:361–369. doi: 10.4161/auto.5.3.7662. [DOI] [PubMed] [Google Scholar]
- Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009;137:146–158. doi: 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S, Halfmann R, Lindquist S. Biochemical, cell biological, and genetic assays to analyze amyloid and prion aggregation in yeast. Methods Enzymol. 2010;470:709–734. doi: 10.1016/S0076-6879(10)70030-6. [DOI] [PubMed] [Google Scholar]
- Allen KD, Chernova TA, Tennant EP, Wilkinson KD, Chernoff YO. Effects of ubiquitin system alterations on the formation and loss of a yeast prion. J Biol Chem. 2007;282:3004–3013. doi: 10.1074/jbc.M609597200. [DOI] [PubMed] [Google Scholar]
- Arslan F, Hong JY, Kanneganti V, Park SK, Liebman SW. Heterologous aggregates promote de novo prion appearance via more than one mechanism. PLoS Genet. 2015;11:e1004814. doi: 10.1371/journal.pgen.1004814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Beal MF, Thomas B. Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends Neurosci. 2010;33:541–549. doi: 10.1016/j.tins.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckhouse AG, Grant CM, Rogers PJ, Dawes IW, Higgins VJ. The adaptive response of anaerobically grown Saccharomyces cerevisiae to hydrogen peroxide is mediated by the Yap1 and Skn7 transcription factors. FEMS Yeast Res. 2008;8:1214–1222. doi: 10.1111/j.1567-1364.2008.00439.x. [DOI] [PubMed] [Google Scholar]
- Boellaard JW, Kao M, Schlote W, Diringer H. Neuronal autophagy in experimental scrapie. Acta Neuropathol. 1991;82:225–228. doi: 10.1007/BF00294449. [DOI] [PubMed] [Google Scholar]
- Chaudhuri B, Ingavale S, Bachhawat AK. apd1+, a gene required for red pigment formation in ade6 mutants of schizosaccharomyces pombe, encodes an enzyme required for glutathione biosynthesis: a role for glutathione and a glutathione-conjugate pump. Genetics. 1997;145:75–83. doi: 10.1093/genetics/145.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+] Science. 1995;268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- DeMarco ML, Daggett V. Local environmental effects on the structure of the prion protein. C R Biol. 2005;328:847–862. doi: 10.1016/j.crvi.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: the story of [PIN+] Cell. 2001;106:171–182. doi: 10.1016/s0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics. 1997;147:507–519. doi: 10.1093/genetics/147.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics. 1996;144:1375–1386. doi: 10.1093/genetics/144.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doronina VA, Staniforth GL, Speldewinde SH, Tuite MF, Grant CM. Oxidative stress conditions increase the frequency of de novo formation of the yeast [PSI(+)] prion. Mol Microbiol. 2015;96:163–174. doi: 10.1111/mmi.12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11:1305–1314. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- Elmallah MI, Borgmeyer U, Betzel C, Redecke L. Impact of methionine oxidation as an initial event on the pathway of human prion protein conversion. Prion. 2013;7:404–411. doi: 10.4161/pri.26745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira PC, Ness F, Edwards SR, Cox BS, Tuite MF. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol Microbiol. 2001;40:1357–1369. doi: 10.1046/j.1365-2958.2001.02478.x. [DOI] [PubMed] [Google Scholar]
- Ganusova EE, Ozolins LN, Bhagat S, Newnam GP, Wegrzyn RD, Sherman MY, Chernoff YO. Modulation of prion formation, aggregation, and toxicity by the actin cytoskeleton in yeast. Mol Cell Biol. 2006;26:617–629. doi: 10.1128/MCB.26.2.617-629.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold R, McKinnon C, Rabbanian S, Collinge J, Schiavo G, Tabrizi SJ. Alternative fates of newly formed PrPSc upon prion conversion on the plasma membrane. J Cell Sci. 2013;126:3552–3562. doi: 10.1242/jcs.120477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CM. Sup35 methionine oxidation is a trigger for de novo [PSI+] prion formation. Prion. 2015;9:257–265. doi: 10.1080/19336896.2015.1065372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Heiseke A, Aguib Y, Riemer C, Baier M, Schatzl HM. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J Neurochem. 2009;109:25–34. doi: 10.1111/j.1471-4159.2009.05906.x. [DOI] [PubMed] [Google Scholar]
- Heiseke A, Aguib Y, Schatzl HM. Autophagy, prion infection and their mutual interactions. Curr Issues Mol Biol. 2010;12:87–97. [PubMed] [Google Scholar]
- Hohn A, Jung T, Grune T. Pathophysiological importance of aggregated damaged proteins. Free Radic Biol Med. 2014;71:70–89. doi: 10.1016/j.freeradbiomed.2014.02.028. [DOI] [PubMed] [Google Scholar]
- Homma T, Ishibashi D, Nakagaki T, Satoh K, Sano K, Atarashi R, Nishida N. Increased expression of p62/SQSTM1 in prion diseases and its association with pathogenic prion protein. Sci Rep. 2014;4:4504. doi: 10.1038/srep04504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- Joshi-Barr S, Bett C, Chiang WC, Trejo M, Goebel HH, Sikorska B, Liberski P, Raeber A, Lin JH, Masliah E, Sigurdson CJ. De novo prion aggregates trigger autophagy in skeletal muscle. J Virol. 2014;88:2071–2082. doi: 10.1128/JVI.02279-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josse L, Marchante R, Zenthon J, von der Haar T, Tuite MF. Probing the role of structural features of mouse PrP in yeast by expression as Sup35-PrP fusions. Prion. 2012;6:201–210. doi: 10.4161/pri.19214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G, Masison DC. Guanidine hydrochloride inhibits Hsp104 activity in vivo: a possible explanation for its effect in curing yeast prions. Curr Microbiol. 2001;43:7–10. doi: 10.1007/s002840010251. [DOI] [PubMed] [Google Scholar]
- Kabani M, Redeker V, Melki R. A role for the proteasome in the turnover of Sup35p and in [PSI(+)] prion propagation. Mol Microbiol. 2014;92:507–528. doi: 10.1111/mmi.12572. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem. 2003;278:49636–49643. doi: 10.1074/jbc.M307996200. [DOI] [PubMed] [Google Scholar]
- Lancaster AK, Bardill JP, True HL, Masel J. The spontaneous appearance rate of the yeast prion [PSI+] and its implications for the evolution of the evolvability properties of the [PSI+] system. Genetics. 2010;184:393–400. doi: 10.1534/genetics.109.110213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund PM, Cox BS. Reversion analysis of [psi-] mutations in Saccharomyces cerevisiae. Genet Res. 1981;37:173–182. doi: 10.1017/s0016672300020140. [DOI] [PubMed] [Google Scholar]
- Lynch-Day MA, Mao K, Wang K, Zhao M, Klionsky DJ. The role of autophagy in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a009357. doi: 10.1101/cshperspect.a009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manogaran AL, Kirkland KT, Liebman SW. An engineered nonsense URA3 allele provides a versatile system to detect the presence, absence and appearance of the [PSI+] prion in Saccharomyces cerevisiae. Yeast. 2006;23:141–147. doi: 10.1002/yea.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur V, Taneja V, Sun Y, Liebman SW. Analyzing the birth and propagation of two distinct prions, [PSI+] and [Het-s](y), in yeast. Mol Biol Cell. 2010;21:1449–1461. doi: 10.1091/mbc.E09-11-0927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazon MJ, Eraso P, Portillo F. Efficient degradation of misfolded mutant Pma1 by endoplasmic reticulum-associated degradation requires Atg19 and the Cvt/autophagy pathway. Mol Microbiol. 2007;63:1069–1077. doi: 10.1111/j.1365-2958.2006.05580.x. [DOI] [PubMed] [Google Scholar]
- Morselli E, Marino G, Bennetzen MV, Eisenberg T, Megalou E, Schroeder S, Cabrera S, Benit P, Rustin P, Criollo A, et al. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J Cell Biol. 2011;192:615–629. doi: 10.1083/jcb.201008167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, Matsuura A, Wada Y, Ohsumi Y. Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1995;210:126–132. doi: 10.1006/bbrc.1995.1636. [DOI] [PubMed] [Google Scholar]
- Osherovich LZ, Weissman JS. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI+] prion. Cell. 2001;106:183–194. doi: 10.1016/s0092-8674(01)00440-8. [DOI] [PubMed] [Google Scholar]
- Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20:460–473. doi: 10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 1995;273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- Pezza JA, Villali J, Sindi SS, Serio TR. Amyloid-associated activity contributes to the severity and toxicity of a prion phenotype. Nat Commun. 2014;5:4384. doi: 10.1038/ncomms5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet. 2013;47:601–623. doi: 10.1146/annurev-genet-110711-155524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, Webster JA, Lewis TA, O’Kane CJ, Schreiber SL, Rubinsztein DC. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3:331–338. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sideri TC, Koloteva-Levine N, Tuite MF, Grant CM. Methionine oxidation of Sup35 protein induces formation of the [PSI+] prion in a yeast peroxiredoxin mutant. J Biol Chem. 2011;286:38924–38931. doi: 10.1074/jbc.M111.272419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sideri TC, Stojanovski K, Tuite MF, Grant CM. Ribosome-associated peroxiredoxins suppress oxidative stress-induced de novo formation of the [PSI+] prion in yeast. Proc Natl Acad Sci USA. 2010;107:6394–6399. doi: 10.1073/pnas.1000347107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag EM, Vonk WI, Frydman J. Sorting out the trash: the spatial nature of eukaryotic protein quality control. Curr Opin Cell Biol. 2014;26C:139–146. doi: 10.1016/j.ceb.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treusch S, Lindquist S. An intrinsically disordered yeast prion arrests the cell cycle by sequestering a spindle pole body component. J Cell Biol. 2012;197:369–379. doi: 10.1083/jcb.201108146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuite MF, Mundy CR, Cox BS. Agents that cause a high frequency of genetic change from [psi+] to [psi-] in Saccharomyces cerevisiae. Genetics. 1981;98:691–711. doi: 10.1093/genetics/98.4.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyedmers J, Madariaga ML, Lindquist S. Prion switching in response to environmental stress. PLoS Biol. 2008;6:e294. doi: 10.1371/journal.pbio.0060294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyedmers J, Treusch S, Dong J, McCaffery JM, Bevis B, Lindquist S. Prion induction involves an ancient system for the sequestration of aggregated proteins and heritable changes in prion fragmentation. Proc Natl Acad Sci USA. 2010;107:8633–8638. doi: 10.1073/pnas.1003895107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent A, Newnam G, Liebman SW. The yeast translational allosuppressor, SAL6: a new member of the PP1-like phosphatase family with a long serine-rich N-terminal extension. Genetics. 1994;138:597–608. doi: 10.1093/genetics/138.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishveshwara N, Bradley ME, Liebman SW. Sequestration of essential proteins causes prion associated toxicity in yeast. Mol Microbiol. 2009;73:1101–1114. doi: 10.1111/j.1365-2958.2009.06836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264:566–5699. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- Wolschner C, Giese A, Kretzschmar HA, Huber R, Moroder L, Budisa N. Design of anti- and pro-aggregation variants to assess the effects of methionine oxidation in human prion protein. Proc Natl Acad Sci USA. 2009;106:7756–7761. doi: 10.1073/pnas.0902688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]