

CARMIL2 is a novel and direct molecular connection between vimentin filaments and actin assembly during cell migration and invadopodia formation. Through two distinct domains, CARMIL2 localizes to vimentin filaments and regulates actin assembly. The biochemical activities of both domains are necessary for cell migration and invasion.
Abstract
Cancer cell migration requires the regulation of actin networks at protrusions associated with invadopodia and other leading edges. Carcinomas become invasive after undergoing an epithelial–mesenchymal transition characterized by the appearance of vimentin filaments. While vimentin expression correlates with cell migration, the molecular connections between vimentin- and actin-based membrane protrusions are not understood. We report here that CARMIL2 (capping protein, Arp2/3, myosin-I linker 2) provides such a molecular link. CARMIL2 localizes to vimentin, regulates actin capping protein (CP), and binds to membranes. CARMIL2 is necessary for invadopodia formation, as well as cell polarity, lamellipodial assembly, membrane ruffling, macropinocytosis, and collective cell migration. Using point mutants and chimeras with defined biochemical and cellular properties, we discovered that localization to vimentin and CP binding are both essential for the function of CARMIL2 in cells. On the basis of these results, we propose a model in which dynamic vimentin filaments target CARMIL2 to critical membrane-associated locations, where CARMIL2 regulates CP, and thus actin assembly, to create cell protrusions.
INTRODUCTION
Invasion of body tissues by metastatic tumor cells is the main cause of death in patients with cancer (Weigelt et al., 2005). Cell migration and invasion require assembly of actin-based structures, such as invadopodia. Within invadopodia and at the leading edge of migrating cells in general, actin filaments polymerize at their barbed ends, which are oriented toward the plasma membrane. The addition of actin subunits onto barbed ends provides the force that generates protrusion of the plasma membrane. Regulation of the number of free barbed ends is crucial for controlling actin networks during cell migration and invasion (Pollard and Cooper, 2009).
Invadopodia and protrusions at the leading edges of migrating cells are based on assemblies of branching networks of short actin filaments interspersed with bundles of longer unbranched filaments (Cooper and Sept, 2008; Nurnberg et al., 2011). A key regulator of actin filament barbed ends is capping protein (CP). For branched networks that are induced to form by the Arp2/3 complex, CP contributes to network assembly and architecture by capping older filaments, which restricts filament length and confines polymerization to new filaments near the membrane (Cooper and Sept, 2008; Pollard and Cooper, 2009; Pollard and Borisy, 2003; Nurnberg et al., 2011). Loss of CP in cells leads to loss of Arp2/3-based lamella and lamellipodia, increased numbers of filopodia, and decreased cell migration (Mejillano et al., 2004). Therefore insight into the regulation of CP is critical for understanding actin-based motility.
CARMIL (CP, Arp2/3, myosin-I linker) family proteins are large (∼1400 aa) multidomain polypeptides that directly bind and regulate CP (Edwards et al., 2014). CARMILs contain a CP-binding region (CBR) that interacts with CP via two motifs in tandem: the CP-interaction (CPI) motif and the CARMIL-specific interaction (CSI) motif (Hernandez-Valladares et al., 2010). CARMIL proteins have similar domain architectures (Figure 1A), consisting of an N-terminal noncanonical pleckstrin homology (PH) domain, a leucine-rich repeat (LRR) domain, a helical homodimerization domain (HD), and an extended intrinsically disordered region that contains the CBR and a proline-rich domain (PRD), which interacts with SH3 domains of class-I myosins (Zwolak et al., 2013; Edwards et al., 2014).
FIGURE 1:
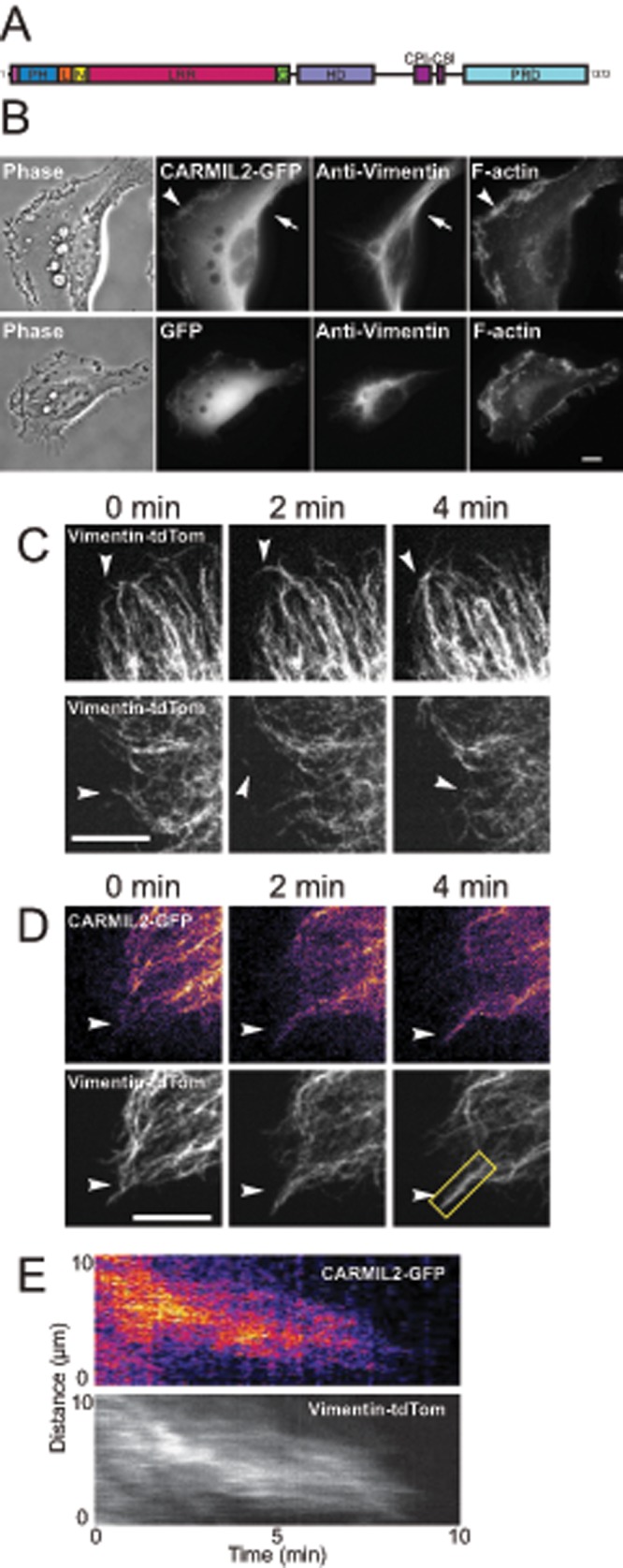
CARMIL2 localizes to dynamic vimentin filaments at the leading edge of migrating cells. (A) Domain map of CARMIL2. (B) Localization of expressed CARMIL2-GFP, compared with vimentin and F-actin, in the top panels. Arrow, example of colocalization of CARMIL2-GFP with vimentin; arrowhead: example of CARMIL2-GFP localization in leading-edge ruffles. Bottom panels show a negative control with expressed GFP. (C) Dynamics of expressed vimentin-tdTomato in frames from live-cell movies (Supplemental Movies S1 and S2). Arrowheads, examples of dynamic vimentin filaments at the leading edge. (D) Colocalization of CARMIL2-GFP with dynamic vimentin-tdTomato filaments at the leading edge (arrowheads). Frames from a live-cell movie (Supplemental Movie S3). Scale bars: 10 μm. (E) Kymograph of vimentin filament boxed in yellow from panel D.
Vertebrates have three genes encoding highly conserved CARMIL isoforms that have distinct cellular functions (Liang et al., 2009; Edwards et al., 2014). CARMIL1 regulates CP at the leading edges of migrating cells, playing critical roles in lamellipodia formation, ruffling, and macropinocytosis (Edwards et al., 2013). For CARMIL2 and CARMIL3, much less is known. CARMIL2 was identified as a transcript down-regulated in the affected skin of psoriasis patients (Matsuzaka et al., 2004). Widely expressed in human tissues (Matsuzaka et al., 2004), CARMIL2 has been implicated in costimulation pathways leading to T-cell activation (Liang et al., 2013). Cultured HT1080 fibrosarcoma cells lacking CARMIL2 show impaired migration in wound-healing assays, diminished lamellipodial ruffling, decreased macropinocytosis, and a striking cell polarity phenotype characterized by multiple leading edges that pull the cell in different directions (Liang et al., 2009). In addition, CARMIL2 was found to localize to vimentin intermediate filaments (Liang et al., 2009), the significance of which has not been addressed.
The connection of CARMIL2 to vimentin is intriguing, because vimentin appears to play a critical role in actin-based cell migration. First, vimentin expression is a hallmark of the epithelial–mesenchymal transition in metastatic carcinomas, correlating with the migration and invasion potential of cancer cells (Gilles et al., 1999; Eckes et al., 2000; Savagner, 2010; Rogel et al., 2011; Satelli and Li, 2011; Menko et al., 2014). Second, vimentin filaments have been functionally linked to processes that require coordination of the actin network during cell migration and invasion (Schoumacher et al., 2010; Helfand et al., 2011; Sakamoto et al., 2013; Sutoh Yoneyama et al., 2014; Havel et al., 2015). However, the mechanisms by which vimentin influences actin-based cell motility remain poorly understood. We reasoned that CARMIL2 was a strong candidate to connect the vimentin and actin filament systems, because CARMIL2 localizes to vimentin in cells and can regulate actin polymerization through its interaction with CP.
In this study, we investigated the physiological relevance of localization to vimentin and binding to CP with respect to the role of CARMIL2 in cell migration and invadopodia formation. We observed CARMIL2 localization to dynamic vimentin filaments at the leading edges of cells, identified the vimentin-localizing region of CARMIL2, and characterized the biochemical interaction between CARMIL2 and CP. Most important, using expression of specific mutants and chimeras, we found that both localization to vimentin and the ability to bind CP are completely necessary for function, including invadopodia formation as assayed by matrix degradation.
RESULTS
CARMIL2 localizes to dynamic vimentin filaments at leading edges
We examined the mechanism and significance of the colocalization of CARMIL2 with vimentin and hypothesized that CARMIL2 links vimentin filaments with actin assembly at the membrane. We then asked whether CARMIL2 localizes to dynamic vimentin filaments at the edges of cells, where actin assembly is dynamic. First, using expressed CARMIL2–green fluorescent protein (GFP), we confirmed colocalization of CARMIL2 with the bulk of vimentin filaments in the central cytoplasm, accompanied by localization of CARMIL2 in leading-edge ruffles (Figure 1B). Next we imaged individual vimentin filaments at the edge of migrating HT1080 cells, using expressed vimentin-tdTomato. We found that vimentin formed highly dynamic, thin, filamentous structures in close proximity to the plasma membrane (Figure 1C and Supplemental Movies S1 and S2), consistent with findings in other motile cells (Helfand et al., 2011; Sakamoto et al., 2013; Havel et al., 2015). We observed motile filaments of vimentin at the leading edge, extending toward and retracting away from the membrane.
To determine whether CARMIL2 localized to these dynamic vimentin filaments, we coexpressed CARMIL2-GFP with vimentin-tdTomato. In live-cell movies of single migrating cells, CARMIL2-GFP colocalized along the length of dynamic vimentin filaments at the leading edge (Figure 1D and Supplemental Movie S3). Heat-map pseudocoloring was used to show enrichment of CARMIL2-GFP on vimentin filaments. To investigate the timing of vimentin-filament retraction and extension with respect to CARMIL2 colocalization along filaments, we created kymographs, drawing lines along vimentin filaments (Figure 1E). Vimentin and CARMIL2 dynamics were coincident in time, consistent with CARMIL2 bound to vimentin filaments that extend and retract by trafficking along microtubules via microtubule motors, a model previously described by others (Gyoeva and Gelfand, 1991; Prahlad et al., 1998; Helfand et al., 2002).
CARMIL2-LRR is necessary and sufficient for localization to vimentin
To create probes for testing the functional significance of localization to vimentin by CARMIL2, we first identified regions of CARMIL2 necessary and sufficient for localization to vimentin. We used GFP fusions of CARMIL2 truncations and chimeras of CARMIL2 with CARMIL1, with truncation and fusion sites determined from secondary structure predictors (PSIPRED) and the CARMIL1 crystal structure (Zwolak et al., 2013; Figure 2A). First, we observed that the N-terminal half of CARMIL2 (N-Term) localized with vimentin, but the C-terminal half (C-Term) did not. Instead, the C-Term of CARMIL2 localized to leading-edge membranes, including ruffles (Figure 2B). Dividing the N-Term further, we found that localization to vimentin was seen with the LRR domain and not with the PH domain (Figure 2B). Smaller fragments were not tested, because they were not expected to be stable, based on previous biochemical studies (Zwolak et al., 2013).
FIGURE 2:

LRR domain of CARMIL2 is necessary and sufficient for localization to vimentin. (A) Domain map of CARMIL2 indicating CARMIL2 truncations and CARMIL1/CARMIL2 chimeras, along with summary of localization to vimentin results. (B) Localization of CARMIL-GFP fusion proteins compared with vimentin-tdTomato. (C) Manders overlap coefficients for expressed CARMIL2-GFP fusion proteins and vimentin-tdTomato. Number of data points as follows: GFP, 2; CARMIL2, 4; N-Term, 5; C-Term, 4; PH, 4; LRR, 3; PH1/LRR2/C-Term1, 5; PH2/LRR1/C-Term2, 4; PH1/LRR2, 5; PH2/LRR1, 5; PH1/LRRN1/LRRC2, 4; PH1/LRRN2/LRRC2, 4; PH2/LRRN2/LRRC1, 5; PH2/LRRN1/LRRC2, 5. Statistical significance as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001. (D) Initial frame of movies (Supplemental Movies S4–S9) comparing localization of expressed CARMIL-GFP fusion proteins with vimentin-tdTomato. Scale bars: 10 μm.
Next we constructed chimeras between CARMIL1 and CARMIL2, interchanging the PH domains, the LRR domains, and the C-Terms of each protein. Splice sites were determined using sequence alignments and the CARMIL1 crystal structure to avoid disrupting secondary structures. Chimeras were created and cloned into GFP-fusion expression vectors using Gibson assembly (Gibson, 2011). First, we tested chimeras composed of domains from the N-Term of the protein. The PH domain of CARMIL1 fused to the LRR domain of CARMIL2 (PH1/LRR2) localized to vimentin, while the converse construct, PH2/LRR1, did not. Next we tested chimeras consisting of full-length protein. A chimera composed of the PH domain of CARMIL1 (PH1) with the LRR domain of CARMIL2 (LRR2) and the C-Term of CARMIL1 (C-Term1), PH1/LRR2/C-Term1, localized to vimentin filaments, whereas the converse chimera, PH2/LRR1/C-Term2, localized to the leading-edge membrane, including ruffles (Figure 2B). We conclude that information in the LRR domain of CARMIL2 is necessary and sufficient for localization with vimentin in the context of full-length CARMIL or the N-Term of CARMIL.
We further divided the LRR domain, which consists of 16 LRRs. The LRR domain has a highly conserved region in the eighth repeat, on the ascending loop between the β-strand and α-helix (Zwolak et al., 2013). Using this location as a splice site, we observed that chimeras with the first eight LRRs of CARMIL2 (LRRN2) and the second eight LRRs of CARMIL1 (LRRC1), expressed in the context of the N-Term of CARMIL, localized to vimentin; however, the degree of localization to vimentin for these split constructs was not as strong as for those chimeras with all 16 LRRs from CARMIL2. Thus the first eight LRRs are more important for localization to vimentin than the second eight LRRs, but the second eight repeats do make a noticeable contribution.
To quantify colocalization with vimentin, we calculated the Manders overlap coefficients for the CARMIL2 truncations and chimeras (Figure 2C). In support of the qualitative observations, the values revealed that truncations and chimeras containing the LRR domain of CARMIL2 overlapped significantly more with vimentin than constructs that did not. Chimeras with the first eight LRRs displayed intermediate levels of overlap with vimentin filaments, also consistent with the qualitative observations.
To extend the analysis, we collected dual-label movies of living migrating cells and examined localization patterns for full-length CARMIL-GFP chimeras with vimentin-tdTomato (Figure 2D and Supplemental Movies S4–S9), using heat-map pseudocoloring of CARMIL2-GFP. Wild-type CARMIL2-GFP was observed on dynamic vimentin filaments, as noted above. Localization to vimentin was found with the PH1/LRR2/C-Term1 chimera but not the PH2/LRR1/C-Term2 chimera, which localized to leading-edge membranes and ruffles. The N-Term of CARMIL2 localized with vimentin, and the C-Term localized to leading-edge membrane ruffles. Thus the movie analysis of living cells confirmed the previous single-image results, in every respect.
CARMIL2 mutant unable to bind CP
Human CARMIL2, like all vertebrate CARMILs, contains a domain called the CP-binding region (CBR), which includes two tandem CP-binding motifs: CPI and CSI (Figure 1A; Hernandez-Valladares et al., 2010; Edwards et al., 2014). For CARMIL1, biochemical studies showed that the CBR region is sufficient to bind CP and decrease the actin-capping activity of CP (Yang et al., 2005; Fujiwara et al., 2010; Hernandez-Valladares et al., 2010; Zwolak et al., 2010; Kim et al., 2012; Edwards et al., 2013). This ability had not been tested for any CARMIL2 isoform, so we performed the relevant assays here for human CARMIL2, using the putative CBR, consisting of Pro-961 to Arg-1072.
First, we tested the ability of CARMIL2-CBR to inhibit actin capping by CP in a pyrene-actin–seeded polymerization assay. In this assay, CP inhibits polymerization of actin by binding to the barbed ends of growing filaments. Inhibition by CP was relieved with increasing concentrations of CARMIL2-CBR (Figure 3A). Kinetic modeling with a simple 1:1 mechanism for CBR binding to CP (reaction 3, Materials and Methods section) fitted the data well, yielding an apparent Kd of 2.9 ± 0.4 nM. Second, we tested the ability of CARMIL2-CBR to uncap actin filaments that are capped by CP, another known property of CARMIL1 (Yang et al., 2005; Hernandez-Valladares et al., 2010; Kim et al., 2012; Edwards et al., 2013). We modified the pyrene-actin–seeded polymerization assay by adding CARMIL2-CBR after, not before, the actin barbed ends were capped by CP. The rate of actin polymerization increased rapidly, consistent with uncapping (Figure 3B). We conclude that CARMIL2 binds and inhibits CP, with a level of activity similar to that observed previously for CARMIL1 (Kim et al., 2012).
FIGURE 3:
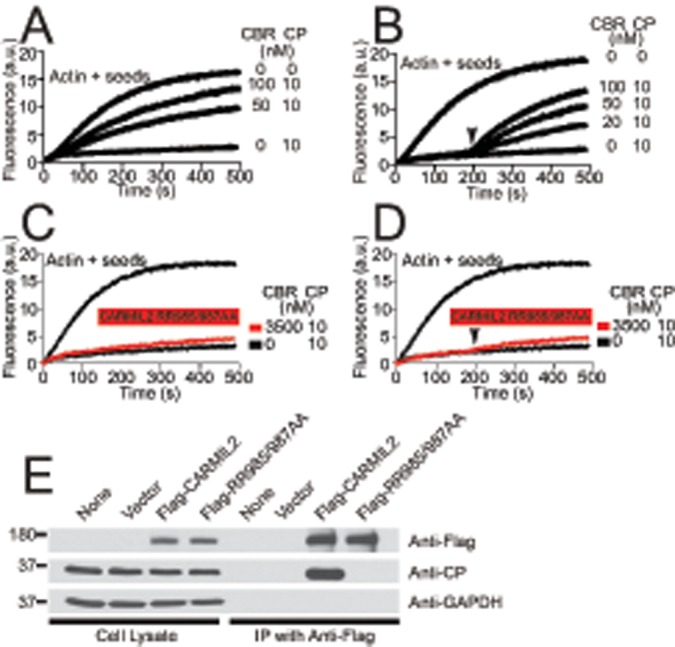
CARMIL2 mutant RR985/987AA loses ability to bind CP. (A) Inhibition of capping activity by CBR fragment of wild-type CARMIL2 in pyrene-actin–seeded polymerization assay. CBR and CP were mixed before time zero. (B) Reversal of capping by CBR fragment of wild-type CARMIL2 in a similar assay. CP was added at time zero, and CARMIL2-CBR was added at 200 s (arrowhead). (C) CARMIL2 RR985/987AA mutant fails to inhibit capping activity and (D) fails to reverse capping, in experiments similar to those in A and B. (E) Full-length CARMIL2 RR985/987AA mutant does not interact with CP in whole-cell lysates. FLAG-tagged full-length CARMIL2, wild-type, and RR985/987AA mutant, was precipitated from whole-cell lysates. Precipitates were probed with anti-CP, anti-FLAG, and anti-GAPDH.
Next we created a CARMIL2 mutant defective in binding CP, by changing two amino acid residues. CARMIL2 contains a CPI motif, defined as LxHxTxxRPK(x)6P (Bruck et al., 2006), and cocrystal structures of CARMIL1-CBR with CP show several close contacts between the CPI motif and CP (Hernandez-Valladares et al., 2010). In previous work with CARMIL1, we found that changing two basic residues, K987 and R989, to alanine led to a nearly complete loss of binding (Edwards et al., 2013). We mutated the analogous residues of CARMIL2-CBR, R985 and R987, to alanine. Repeating the pyrene-actin–seeded polymerization assays, we found that the mutant had little to no ability to interact with CP, either in the assay for inhibition of capping (Figure 3C) or the assay for uncapping (Figure 3D).
These assays were performed with the CBR fragment, using purified proteins in vitro. In a complementary approach, we tested the ability of full-length CARMIL2, wild-type and mutant, to interact with CP in HT1080 cells by immunoprecipitation. We found that CP coprecipitated with FLAG-tagged wild-type CARMIL2. In contrast, CP did not coprecipitate with the RR985/987AA mutant form of FLAG-CARMIL2 (Figure 3E). On the basis of these results, we conclude that the interaction of CARMIL2 with CP was essentially lost in the RR985/987AA mutant.
The physiological significance of CARMIL2 interaction with vimentin and CP
We used the CARMIL2/CARMIL1 chimeras and the CARMIL2 RR985/987AA mutant to test the physiological significance of localization to vimentin and CP binding for the function of CARMIL2. We asked whether expression of the chimeras or the mutant was able to rescue traits that result from loss of CARMIL2 (Liang et al., 2009). To deplete endogenous CARMIL2, we used a lentiviral system to deliver short hairpin RNA (shRNA) targeting CARMIL2. A second lentivirus was used to introduce shRNA-resistant versions of GFP fusions of the chimeras and mutants. Immunoblots confirmed that endogenous CARMIL2 was depleted and that the expression levels of the rescue constructs were appropriate (Figure 4A).
FIGURE 4:

Localization to vimentin and CP-binding ability of CARMIL2 are necessary for lamellipodial ruffling, macropinocytosis, and cell polarity. (A) Expression of CARMIL-GFP mutants and chimeras to test for rescue of loss of CARMIL2. Immunoblots of whole-cell lysates probed with anti-CARMIL2, anti-GFP, and anti-GAPDH. (B) Cell polarity phenotype revealed in images. First frame of 1-h movies (Supplemental Movies S10–S16) of migrating cells plated sparsely. PH2/LRR1/C-Term2 chimera and RR985/987AA CP-binding mutant fail to rescue effects of depletion of CARMIL2; however, rescue is provided by wild-type CARMIL2 and the PH1/LRR2/C-Term1 chimera. Scale bar: 10 μm. (C) Quantification of cell polarity from the images based on calculation of circularity, as described in Materials and Methods, for CARMIL2-depletion and expression-rescue cells (n = 30). *, p < 0.0001. Box-and-whisker format showing median, interquartile range, and the extremes. (D) Quantification of macropinocytosis based on counting macropinosomes in CARMIL2-depletion and expression-rescue cells (n = 30). Error bars are SEM. *, p < 0.0001. (E) Persistence of individually migrating cells (n = 30). Error bars are SEM. (F) Distance traveled of individually migrating cells (n = 30). Error bars are SEM. (G) Mean-squared displacement of individually migrating cells (n = 30). Error bars are SEM. (H) Assembly of the lamellipodial actin network, but not the vimentin network, at the cell edge depends on ability of CARMIL2 to localize to vimentin and to bind CP. CARMIL2-depleted and expression-rescue cells were stained with anti-vimentin, anti-CP, or fluorescent phalloidin. Arrowheads, CP; arrows, F-actin in lamellipodia. Scale bar: 10 μm.
We first examined the cell polarity, lamellipodial assembly, ruffling, and macropinocytosis defects resulting from loss of CARMIL2 (Liang et al., 2009). Phase-contrast movies of individual, randomly migrating CARMIL2-depleted and expression-rescue cells were collected (Figure 4B and Supplemental Movies S10–S16). Uninfected cells and cells infected with a scramble-sequence shRNA served as negative controls. In the movies, CARMIL2-depleted cells exhibited a polarity defect, with multiple protrusions pulling the cell in many directions (Figure 4B), as previously described (Liang et al., 2009). The leading edges of CARMIL2-depleted cells were smooth and flat, lacking the prominent lamellipodia and membrane ruffles seen in the controls. In addition, the depleted cells formed few macropinosomes, consistent with the loss of ruffles, which mediate macropinosome formation (Kerr and Teasdale, 2009).
We quantitated the cell polarity defect by calculating the circularity of the cell (Figure 4C), defined as the area of the cell divided by the area of a circle with the same perimeter as the cell (see Materials and Methods). We quantitated macropinocytosis by counting the number of macropinosomes per cell (Figure 4D). Loss of CARMIL2 led to statistically significant moderate decreases in the value of circularity and large decreases in the number of macropinosomes (Figure 4, C and D).
To test for rescue by expression, we first confirmed that all of the traits of the CARMIL2-depletion phenotype could be rescued by expression of shRNA-resistant wild-type CARMIL2, demonstrating that the effects of the shRNA were specific for CARMIL2, without off-target effects (Figure 4, B–D).
We found that expression of the CARMIL chimera PH2/LRR1/C-Term2, which does not localize to vimentin, did not rescue any of the traits of the loss-of-function phenotype (Figure 4, B–D). However, expression of the converse chimera, PH1/LRR2/C-Term1, which does localize to vimentin, was able to provide full rescue (Figure 4, B–D). Thus localization to vimentin is necessary for the function of CARMIL2 in cells. The results with the converse chimera show that providing CARMIL1 with the ability to localize to vimentin (by changing the LRR domain) is sufficient to allow CARMIL1 to substitute for CARMIL2. Thus the LRR of CARMIL2 has a specific and essential function, while the functions of the other domains are similar for CARMIL1 and CARMIL2.
Expression of the CP-binding mutant did not rescue any of the traits caused by loss of CARMIL2 (Figure 4, B–D). Thus the ability of CARMIL2 to bind CP is also completely necessary for its function.
To quantify single-cell migration, we calculated the persistence, distance traveled, and mean-square displacement of randomly migrating individual cells. We found no differences between untreated, CARMIL2-depleted, or any of the rescue conditions (Figure 4, E–G). From these results, we concluded that loss of CARMIL2 has no effect on single-cell migration in the absence of a directional cue.
Next, to investigate the hierarchy of the molecular interactions, we examined the vimentin and lamellipodial actin networks in the depletion and expression-rescue cells by staining cells with anti-vimentin, anti-CP, and fluorescent phalloidin. The vimentin filament networks were similar in all cases (Figure 4H). In contrast, disruptions of the lamellipodial actin network were observed when CARMIL2 function was not present. Anti-CP staining, which in control cells was prominent at the leading edge and in puncta surrounding macropinosomes, was greatly diminished at those locations in cells lacking CARMIL2 (Figure 4H). The CP-staining pattern was rescued by expression of shRNA-resistant wild-type CARMIL2 and the PH1/LRR2/C-Term1 chimera, whereas the staining pattern of the PH2/LRR1/C-Term2 chimera and the RR985/987AA CP-binding mutant resembled CARMIL2-depleted cells. Examination of the F-actin network with fluorescent phalloidin yielded similar results (Figure 4H). CARMIL2-depleted cells showed decreased phalloidin staining, and this was rescued by expression of the PH1/LRR2/C-Term1 chimera but not the PH2/LRR1/C-Term2 chimera or the RR985/987AA CP-binding mutant. On the basis of these results, we conclude that the abilities of CARMIL2 both to localize to vimentin and to bind CP are necessary for lamellipodial actin assembly, membrane ruffling, macropinocytosis, and cell polarity.
Collective cell migration depends on localization to vimentin but not CP-binding ability
We investigated the importance of the individual biochemical and cellular functions of CARMIL2 for the process of cell migration in wound healing. Cell migration defects in wound-healing assays have been described for cells lacking CARMIL2 (Liang et al., 2009). Here we first confirmed that CARMIL2-depleted cells migrate slowly, compared with control cells, when they fill a scratch wound in a tissue-culture monolayer (Figure 5A). The rate of movement of the edge of the monolayer was decreased by almost half (Figure 5B).
FIGURE 5:
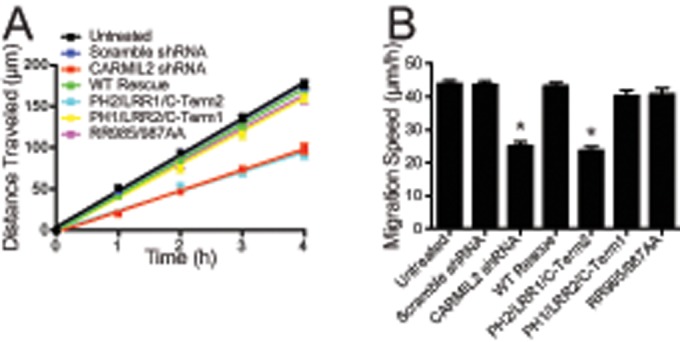
Cell migration in wound-healing assay depends on the ability of CARMIL2 to localize to vimentin but not its ability to bind CP. (A) Distance traveled from edge of wound by CARMIL2-depleted and expression-rescue cells over time. Error bars on plotted points are SEM. (B) Average migration speed of CARMIL2-depleted and expression-rescue cells during wound-healing assays. Data are the mean of nine repetitions of each experiment collected on the same day; error bars are SEM. *, p < 0.0001.
We found that expression of wild-type CARMIL2 and the PH1/LRR2/C-Term1 chimera rescued the migration defect completely (Figure 5, A and B); however, expression of the PH2/LRR1/C-Term2 chimera had no effect. Thus the ability of CARMIL2 to interact with vimentin is necessary for the function of CARMIL2 in cell migration in wound healing.
In a surprising contrast, expression of the CP-binding mutant RR985/987AA rescued the cell migration defect completely (Figure 5, A and B), which was not the case for all the other loss-of-function traits discussed above, including cell polarity, lamellipodial assembly, ruffling, and macropinocytosis. Thus the absence of lamellipodia and ruffling in the CP-binding mutant cells had no effect on the rate of cell migration, indicating that these prominent dynamic features of the leading edge are not important for cell migration in the context of wound healing. This conclusion is consistent with other studies of cells with impaired lamellipodial assembly created by other perturbations (Gupton et al., 2005; Suraneni et al., 2012; Wu et al., 2012; Edwards et al., 2013).
Together these results lead us to two conclusions. First, the localization to vimentin function of CARMIL2, but not the ability to bind CP, is necessary for collective cell migration. Second, lamellipodia and ruffles are dispensable for cell migration in the wound-healing setting.
Invadopodia formation and matrix degradation
Vimentin filaments have been proposed to play a role in the formation of invadopodia (Schoumacher et al., 2010; Sutoh Yoneyama et al., 2014). Invadopodia are filled with dynamic actin filament networks nucleated by the Arp2/3 complex (Buccione et al., 2004; Lorenz et al., 2004; Yamaguchi et al., 2005; Weaver, 2006). We hypothesized that CARMIL2 might serve as a molecular link connecting vimentin to actin assembly in invadopodia. To assay invadopodia formation, we performed matrix-degradation assays by imaging the loss of fluorescent matrix under HT1080 cells. In control cells, sites of matrix degradation were found in association with bright puncta of F-actin, which are the defining hallmarks of invadopodia (Figure 6A; Chen, 1989). In contrast, sites of matrix degradation were absent in CARMIL2-depleted cells, based on qualitative observations of images and a quantitative method of image analysis that involved converting the fluorescent-matrix image to a binary format (Figure 6B; see Materials and Methods). The matrix-degradation phenotype was rescued by expression of wild-type CARMIL2 and the PH1/LRR2/C-Term1 chimera, but not the PH2/LRR1/C-Term2 chimera or the RR985/987AA CP-binding mutant. On the basis of these results, we conclude that the ability of cells to degrade matrix depends on CARMIL2’s ability to interact with vimentin and bind CP.
FIGURE 6:
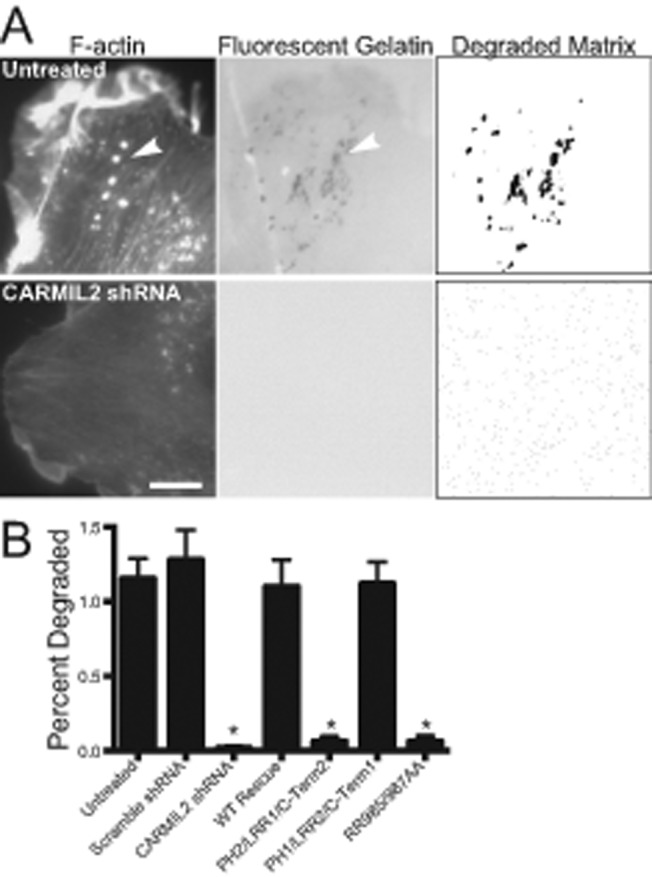
Invadopodia formation and degradation of underlying matrix depends on abilities of CARMIL2 to localize to vimentin and to bind CP. (A) Uninfected cells and CARMIL2-depleted cells plated on fluorescent gelatin. Invadopodia (arrowheads) are identified with fluorescent phalloidin staining. Sites of matrix degradation are identified as dark areas in the fluorescent gelatin image. For quantification, the fluorescent gelatin image was converted with a threshold set at 3 SDs below mean pixel intensity. Scale bar: 10 μm. (B) Quantification of matrix degradation. Mean percent of cell area with matrix degradation in threshold images for CARMIL2-depletion and expression-rescue cells. Error bars are SEM. n = 20 cells. *, p < 0.0001.
DISCUSSION
In this study, we report the discovery of a novel molecular connection between vimentin intermediate filaments and lamellipodial actin dynamics. First, we found that CARMIL2 localizes to dynamic vimentin filaments at the leading edges of migrating cells, mediated by its LRR domain. We showed that CARMIL2 binds and inhibits CP, similar to other CARMILs. Most important, we created mutants and chimeras with specific functional properties, which demonstrate that both localization to vimentin and the CP-binding ability of CARMIL2 are necessary for the function of CARMIL2 in invadopodia formation and cell migration, hallmarks of malignant cancers.
CARMIL family proteins have similar domain architectures, with a high degree of sequence similarity throughout (Liang et al., 2009; Zwolak et al., 2013). However, the three vertebrate isoforms possess highly conserved differences that distinguish one from another (Edwards et al., 2014), suggesting that they have different functions. Our previous work revealed that CARMIL1 and CARMIL2 have different localizations and functions in cultured human cells. Most important, expression of one was not able to rescue phenotypes resulting from loss of the other (Liang et al., 2009).
CARMIL2 is known to colocalize with vimentin filaments (Liang et al., 2009). However, the physiological significance and mechanism of the interaction was not known. In addition, the extent of colocalization was not examined, especially whether CARMIL2 colocalized with dynamic vimentin filaments at the cell edge. Vimentin filaments at the leading edge are particularly interesting because of evidence linking vimentin to processes of membrane-associated actin assembly, including the formation of cell protrusions, notably invadopodia, which are features of cancer cells invading tissue (Schoumacher et al., 2010; Helfand et al., 2011; Sakamoto et al., 2013; Sutoh Yoneyama et al., 2014; Havel et al., 2015).
To investigate the mechanism and significance of the vimentin–CARMIL2 interaction, we first identified the LRR domain of CARMIL2 as necessary and sufficient for localization to vimentin. LRR domains of other proteins, including NOD2 and NLRP3, also interact with vimentin (Stevens et al., 2013; dos Santos et al., 2015), but we found no obvious sequence or structural similarities between those LRR domains and the LRR domain of CARMIL2. To test the physiological significance of CARMIL2’s localization to vimentin, we created chimeras with specific loss or gain of vimentin interaction by exchanging LRR domains between CARMIL1 and CARMIL2. Our results show that the ability to interact with vimentin is completely necessary for CARMIL2 to function and that addition of this ability to CARMIL1 is sufficient to allow CARMIL1 to function in place of CARMIL2.
Loss of CARMIL2 causes a number of phenotypes (Liang et al., 2009). We found that PH2/LRR1/C-Term2, the chimera that does not localize to vimentin, was unable to rescue defects in lamellipodia, ruffling and macropinocytosis; these membrane-associated phenomena depend on one another and on actin assembly (Chhabra and Higgs, 2007; Kerr and Teasdale, 2009). This chimera was also unable to rescue the distinctive cell polarity defect displayed by CARMIL2-depleted cells, which resembles the polarity defects of cells lacking myosin-IIB, Cdc42, or PAK1 (Nobes and Hall, 1999; Sells et al., 1999; Lo et al., 2004; Cau and Hall, 2005; Liang et al., 2009; Parrini et al., 2009).
We also found that localization to vimentin is important for CARMIL2’s role in collective cell migration, based on wound-healing assays, and for formation of invadopodia, based on matrix-degradation assays. Vimentin expression is a hallmark of the epithelial–mesenchymal transition of carcinomas, correlating with the potential for cancer cells to migrate and invade (Gilles et al., 1999; Eckes et al., 2000; Savagner, 2010; Rogel et al., 2011; Satelli and Li, 2011; Menko et al., 2014), so CARMIL2 may be a critical link that connects vimentin to invasion and migration of cancer cells.
In contrast to collective cell migration, random migration of individual cells is not affected by loss of CARMIL2. Collective cell migration depends on guidance of leader cells and cooperation of follower cells. This is mediated by localization of key proteins, such as Rac1, to the leading edge and transduction of migrational cues through cell–cell junctions, respectively (Friedl, 2004; Dumortier et al., 2012; Collins and Nelson, 2015; Haeger et al., 2015; Yamaguchi et al., 2015). CARMIL2 depletion does not disrupt the ability of cells to maintain contact during wound healing, and the overall levels of Rac1 activation are not decreased in CARMIL2-depleted cells (Liang et al., 2009). However, the localization of activated Rac1 in CARMIL2-depleted cells may be impaired during wound healing. If so, then localization of active Rac1 would depend on the ability of CARMIL2 to localize to vimentin but not bind CP. Elucidating the differences in the role of CARMIL2 between random and directed migration requires further study.
One striking result was that replacement of the LRR domain of CARMIL1 with the vimentin-localizing LRR domain of CARMIL2 was sufficient to rescue CARMIL2 phenotypes. Thus the other domains of CARMIL1, including the noncanonical PH domain, the CBR, the basic and hydrophobic membrane-binding domain (Brzeska et al., 2010), and the PRD, have sufficient functional overlap to replace the corresponding domains of CARMIL2. Only the LRR domain has a specific function unique to CARMIL2. Of note, the chimera of CARMIL2 with the CARMIL1 LRR (PH2/LRR1/C-Term2) localizes to the leading edge but is not able to rescue the lamellipodial defects of CARMIL2-depleted cells. This observation suggests that the key function of CARMIL2 is to provide a functional connection between vimentin filaments and membrane-associated actin during cell migration and invasion.
We investigated the physiological significance of the ability of CARMIL2 to bind and inhibit CP, which has been established for CARMIL1 (Edwards et al., 2013). We found that the apparent affinity for CARMIL2 binding to CP was similar to that of CARMIL1 (Hernandez-Valladares et al., 2010; Kim et al., 2012), consistent with the sequence similarities and the cocrystal structures. In addition, we found a mutant form of CARMIL2 with a nearly complete loss of the ability to bind CP was unable to rescue the loss of lamellipodial ruffling, macropinocytosis, cell polarity, and invadopodia-mediated matrix degradation. Thus the ability to bind CP is necessary for the function of CARMIL2 in cells, as is the case for CARMIL1 (Edwards et al., 2013). This result supports the hypothesis that CP binding is one critical function, held in common by CARMIL1 and CARMIL2, that is targeted to vimentin filaments by CARMIL2.
On the other hand, the CARMIL2 CP-binding mutant was able to rescue the wound-healing cell migration defect of CARMIL2-depleted cells. These cells lack lamellipodia, so this result provides additional evidence that lamellipodia are dispensable for cell migration in certain contexts (Gupton et al., 2005; Suraneni et al., 2012; Wu et al., 2012). A similar result was obtained for CARMIL1 (Edwards et al., 2013); therefore another function of CARMIL1 and CARMIL2 must be necessary for cell migration in wound healing. That function may be binding to membranes, which is mediated by a basic and hydrophobic membrane-binding domain, as described by us in a separate study (Lanier et al., 2015).
The interaction of CARMIL2-CBR with CP decreased, but did not eliminate, the ability of CP to bind barbed ends, as is the case for CPI-motif proteins in general (Hernandez-Valladares et al., 2010; Kim et al., 2012; Edwards et al., 2014). Thus CARMIL2 may have a role in targeting CP to sites of actin assembly and in modulating its capping activity, bringing it down to a level appropriate for a physiological setting (Yang et al., 2005; Uruno et al., 2006). Consistent with this view, a recent study from our lab revealed that the interaction of CP with a CPI-motif protein is necessary for the function of CP in cells (Edwards et al., 2015). In that study, expression of CP with point mutations that ablate its interaction with the CPI motif caused loss-of-function phenotypes in dominant-negative and expression-rescue experiments. One model for how CP may be activated by a CPI motif is that CP may be constitutively inhibited by the protein V1/myotrophin and that CPI-motif proteins relieve that inhibition by an allosteric mechanism (Fujiwara et al., 2014).
On the basis of these results, we propose a model of CARMIL2 function in which vimentin filaments target CARMIL2 and its domains, including the CBR, basic and hydrophobic membrane-binding domain, PH domain, and PRD, to critical membrane-associated sites for lamellipodia formation, cell migration, and invadopodia-mediated matrix degradation. (Figure 7). The properties of this novel multifunctional multidomain protein provide new insight into the molecular mechanisms that underlie cancer cell invasion and migration.
FIGURE 7:
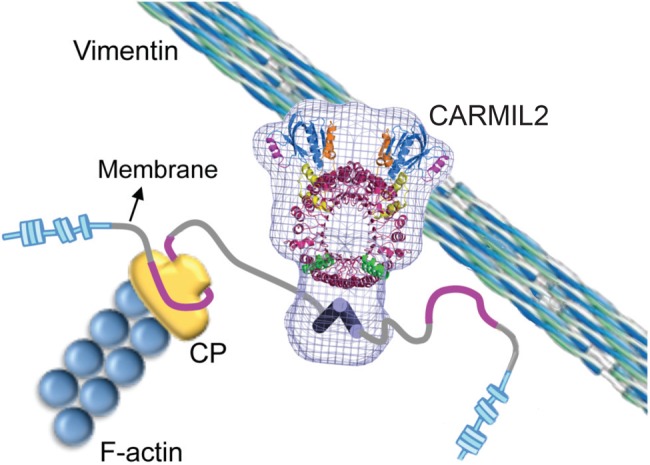
Model of CARMIL2 function in cells. CARMIL2 interacts with dynamic vimentin filaments at the leading edge of cells through its LRR domain. Interaction with vimentin is necessary for targeting CARMIL2 and its domains, such as the CBR and basic and hydrophobic membrane-binding domain, to functional membrane-associated locations for lamellipodia formation, cell migration, and invadopodia formation.
MATERIALS AND METHODS
Antibodies and reagents
Reagents and materials were from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA), unless stated otherwise. To detect CARMIL2 protein, we produced a rabbit polyclonal antibody (pAb) against a peptide consisting of amino acid residues 665–678 (HPTRARPRPRRQHH). To detect vimentin by immunostaining, we used mouse monoclonal antibody (mAb) 3B4 (Millipore, Billerica, MA). Other antibodies were as follows: mouse mAb anti-Flag M2 and horseradish peroxidase–conjugated secondary antibodies (Sigma-Aldrich); mouse mAb anti-CP 2A3 (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA); rabbit pAb anti-GFP 6C5 and mouse mAb anti-GAPDH (Abcam, Cambridge, MA); and Alexa Fluor–conjugated secondary antibodies (Life Technologies, Carlsbad, CA). To stain for F-actin, we used Alexa Fluor–conjugated phalloidin (Sigma-Aldrich).
Cell culture, transfection, knockdown, and rescue of CARMIL2
Plasmids are listed in Supplemental Table S1. Human CARMIL2b cDNA (NCBI: FJ026014) and CARMIL1a (NCBI: FJ009082) were the starting materials for all subcloning and chimera construction. GFP fusions were created by subcloning CARMIL2 and fragments into pAcGFP1-C1 (Clontech, Mountain View, CA) at BglII and HindIII sites. CARMIL chimeras were created using Gibson assembly methods (Gibson, 2011). For fragments and chimeras, truncation and splice sites were chosen based on PSIPRED and alignments with the mouse CARMIL1 structure of Zwolak et al. (2013). Adam Zwolak and Roberto Dominguez of the University of Pennsylvania provided critical advice on selecting sites to produce stable fragments. For chimeras, domain boundaries were as follows: PH1 1-147; PH2 1-148; LRR1 148-684; LRR2 149-687; C-Term1 685-1371; C-Term2 688-1372. Vimentin (NCBI: NM_003380) was subcloned into tdTomato-N1 at the BglII and HindIII sites.
Human HT1080 cells (American Type Culture Collection, Manassas, VA) were grown in DMEM (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (Sigma-Aldrich) at 37°C with 5% CO2. Cells were transfected using Transit-LT1 (Mirus, Madison, WI).
For depletion of endogenous CARMIL2, an shRNA construct targeting CARMIL2, in the lentiviral vector pFLRu-FH-GFP, was used as previously described (Liang et al., 2009). The shRNA targeting sequence was GCAAAGATGGCGAGATCAAG, with CAGTCGCGTTTGCGACTGG as a nontargeting scrambled control. For expression rescue, shRNA-resistant CARMIL2 constructs were subcloned into pBOB-GFP for lentiviral-based expression as described (Mooren et al., 2014). Resistance to shRNA was created with three codon-silent nucleotide changes (lowercase): GCAAgGAcGGgGAGATCAAG, using QuikChange mutagenesis (Stratagene, La Jolla, CA). The CARMIL2 RR985/987AA CP-binding mutant was created using QuikChange site-directed mutagenesis (Stratagene).
Immunofluorescence, live-cell imaging, and image analysis
For immunofluorescence, HT1080 cells grown on glass coverslips coated with 30 μg/ml fibronectin (Sigma-Aldrich) were fixed 24 h posttransfection. For staining with fluorescent phalloidin or antibodies, cells were generally fixed in 2.5% paraformaldehyde and 0.25% glutaraldehyde in PIPES buffer (125 mM NaCl, 5 mM KCl, 1.25 mM NaH2PO4, 0.4 mM KH2PO4, 1 mM MgCl2, 5.5 mM glucose, 0.5 mM ethylene glycol tetraacetic acid, 20 mM 1,4-piperazinediethanesulfonic acid, pH 7.1) at 37°C for 10 min, then quenched with 1 mg/ml NaBH4 in Dulbecco’s phosphate-buffered saline (pH 7.1). Cells were permeabilized with 0.1% Triton X-100 at room temperature for 10 min. Different fixation protocols were used for vimentin (ice-cold MeOH at −20°C for 10 min) and CP (GlyoFixx at 37°C for 10 min; Thermo-Scientific, Kalamazoo, MI). Immunostaining was performed with the primary and secondary antibodies listed above. Cells were imaged using 60×/1.4 numerical aperture (NA) and 100×/1.4 NA objectives on an Olympus IX70 inverted microscope (Olympus, Melville, NY). Wide-field images were collected with a CoolSnap HQ camera (Photometrics, Woburn, MA), and spinning-disk confocal images were collected with an Orca Flash 4.0 camera (Hamamatsu, Bridgewater, NJ). Images were collected and initially processed with Micromanager (Edelstein et al., 2010) and ImageJ.
For live-cell fluorescence movies, cells were grown on glass-bottom culture dishes (MatTek, Ashland, MA) coated with 30 μg/ml fibronectin (Sigma-Aldrich). Cells were maintained at 37°C with 5.0% CO2. Spinning-disk confocal or wide-field fluorescence images were captured every 6 s for up to 10 min using a 100×/1.4 NA objective. Cells were imaged 24 h posttransfection. Heat-map pseudocoloring was performed in ImageJ using the Fire look-up table.
For phase-contrast movies, cells were handled in a similar manner. Images of migrating single cells (n = 30) were acquired every 60 s for 1 h using a 60×/1.4 NA phase-contrast objective on an Olympus IX70 inverted microscope. Cells were imaged 72 h postinfection with lentivirus. To avoid observer bias in selecting cells for movie analysis, we imaged the first 30 isolated cells encountered when systemically surveying the disk in a grid pattern.
To quantitate polarity, we calculated circularity as (areacell × 4π)/(perimetercell2) (Thurston et al., 1988). ImageJ was used to measure the area and perimeter of single cells from initial frames of time-lapse movies (n = 30). Macropinosomes were counted as phase-bright vesicles in the initial frame of phase-contrast time-lapse movies (n = 30) of single cells. For calculating mean-squared displacement, distance traveled, and persistence, displacements of individual cell nuclei were tracked frame by frame. For quantitation of colocalization, Manders overlap coefficients were calculated using ImageJ from images of cells coexpressing GFP-CARMIL2 constructs and vimentin-tdTomato. Kymographs along vimentin filaments were generated using ImageJ with a 5-pixel line width.
Coimmunoprecipitations and immunoblots
Immunoprecipitation with anti-FLAG M2 affinity beads (Sigma-Aldrich) was performed according to the manufacturer’s instructions. The beads were washed, and precipitated protein was eluted with 3X-FLAG peptide. Supernatant was boiled with SDS-loading buffer and analyzed by SDS–PAGE and immunoblotting.
Immunoblots were performed with the primary and secondary antibodies listed above. Immunoblots were developed with SuperSignal West Pico Chemiluminescent substrate (Thermo-Scientific) and exposed to autoradiography film.
Protein expression and purification
The CBR fragments of human CARMIL2b Pro-961–Arg-1072 (pBJ 1843) were amplified from cDNA by PCR and cloned into pGEX-6P-3 (GE Healthcare, Piscataway, NJ). Complete DNA sequencing of the insert and junctions verified the identity and integrity of the plasmids. The mutant CARMIL2-CBR RR985/987AA was created using QuikChange site-directed mutagenesis (Stratagene). GST-fusion proteins were expressed in BL21 (DE3) Escherichia coli and purified with glutathione Fast-Flow Sepharose resin (GE Healthcare). Cultures were grown and induced with isopropyl-β-d-thiogalactoside at 23°C. After elution from the glutathione resin, GST-CBR was mixed with PreScission protease (GE Healthcare). The mixture was dialyzed into S-Sepharose buffer A (10 mM Tris, pH 8.0, 10 mM KCl, 0.1 mM EDTA, 0.5 mM dithiothreitol [DTT], 1 mM NaN3) overnight, applied to an S-Sepharose column, and eluted with a KCl gradient (10–700 mM). For storage, CBR was dialyzed into 10 mM Tris (pH 8.0), 40 mM KCl, 0.1 mM EDTA, 0.5 mM DTT, and 1 mM NaN3 and kept on wet ice. The concentration of CBR was calculated from A280, based on predicted extinction coefficients, and confirmed by SDS–PAGE with Coomassie blue staining.
Actin polymerization assays
Actin was purified from rabbit skeletal muscle as previously described (Wear et al., 2003). Pyrene–actin polymerization assays, including inhibition and uncapping of by CARMIL, were performed as described (Wear et al., 2003; Kim et al., 2012). The actin concentration was 1.5 μM, with 7% pyrene label. For capping assays, 10 nM CP was added to a mixture of pyrene-labeled actin and spectrin–actin seeds at time zero. To assay for reversal of capping, pyrene–actin was polymerized from seeds in the presence of 10 nM CP. After 200 s, the CBR fragment of CARMIL was added, and polymerization was followed for 300 s. Apparent binding-affinity constants between CP and CBR were determined by fitting the data to polymerization rate equations with Berkeley Madonna (www.berkeleymadonna.com), based on the following mechanism:
In these reactions, A is actin monomer, Nb is free barbed end, CP is capping protein, CPNb is capped barbed end, and CBR is CBR-CARMIL. These equations assume that a capped barbed end, CPNb, can neither add nor lose actin subunits and that the complex of CP with CARMIL, CPCBR, cannot interact with a barbed end. For the elongation rate constants in Reaction 1, k+ was 11.6 μM−1s−1 and k- was 1.4 s−1 (Pollard, 1986). The rate constants for capping in Reaction 2 were determined by fitting the experimental data for seeded actin polymerization with a series of concentrations of CP. The rate constants for CP binding to CARMIL in Reaction 3 were determined by fitting a set of experimental data produced by addition of CARMIL at various concentrations to 10 nM CP.
Cell migration in a wound-healing model
HT1080 cells were infected with lentivirus for endogenous CARMIL2 depletion and expression rescue of mutants and chimeras. Infected cells were grown to a confluent monolayer on glass-bottom cell culture dishes (MatTek) coated with 30 μg/ml fibronectin (Sigma-Aldrich). At 72 h postinfection, a wound was scratched with a pipette tip. Measurements were made every hour for 4 h using an ocular micrometer.
Invadopodia matrix-degradation assay
Coverslips coated with fluorescent gelatin (Life Technologies) were prepared as previously described (Artym et al., 2009). HT1080 cells were infected with lentivirus for depletion of CARMIL2 and expression rescue as above. At 72 h postinfection, cells were plated onto fluorescent gelatin–coated coverslips. Cells were allowed to degrade the matrix for 3 h before fixation, permeabilization, and phalloidin staining.
To quantitate matrix degradation, we created a binary image from the fluorescent gelatin image by imposing a threshold at three SDs below the mean pixel intensity. Total cell area and area of degradation were measured from phalloidin-stained images and binary images, respectively, using ImageJ. To avoid counting stray pixels in the area of degradation, we constrained the analysis to areas greater than 10 pixels.
Statistical analysis
We performed Student’s t test on population values to determine whether means differed by statistically significant amounts. Data analysis and graphical representations were done using Prism 6 (GraphPad, La Jolla, CA).
Supplementary Material
Acknowledgments
We thank Roberto Dominguez and Adam Zwolak of the University of Pennsylvania for their advice with this project. We also thank our lab colleagues for their comments and assistance, specifically Yun Liang for providing plasmids and Jinmei Li for making lentivirus. This work was supported by National Institutes of Health grant GM95509 to J.A.C. M.H.L. was supported by the National Cancer Institute of the National Institutes of Health under award number F30CA171595.
Abbreviations used:
- CARMIL
capping protein
- Arp2/3
myosin-I linker
- CBR
CP-binding region
- CP
capping protein
- CPI
CP interaction
- CSI
CARMIL-specific interaction
- C-Term
C-terminal half
- DTT
dithiothreitol
- GFP
green fluorescent protein
- HD
homodimerization domain
- LRR
leucine-rich repeat
- mAb
monoclonal antibody
- NA
numerical aperture
- N-Term
N-terminal half
- pAb
polyclonal antibody
- PH
pleckstrin homology
- PRD
proline-rich domain
- PSIPRED
secondary structure predictors
- shRNA
short hairpin RNA.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-08-0552) on October 14, 2015.
REFERENCES
- Artym VV, Yamada KM, Mueller SC. ECM degradation assays for analyzing local cell invasion. Methods Mol Biol. 2009;522:211–219. doi: 10.1007/978-1-59745-413-1_15. [DOI] [PubMed] [Google Scholar]
- Bruck S, Huber TB, Ingham RJ, Kim K, Niederstrasser H, Allen PM, Pawson T, Cooper JA, Shaw AS. Identification of a novel inhibitory actin-capping protein binding motif in CD2-associated protein. J Biol Chem. 2006;281:19196–19203. doi: 10.1074/jbc.M600166200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzeska H, Guag J, Remmert K, Chacko S, Korn ED. An experimentally based computer search identifies unstructured membrane-binding sites in proteins: application to class I myosins, PAKS, and CARMIL. J Biol Chem. 2010;285:5738–5747. doi: 10.1074/jbc.M109.066910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol. 2004;5:647–657. doi: 10.1038/nrm1436. [DOI] [PubMed] [Google Scholar]
- Cau J, Hall A. Cdc42 controls the polarity of the actin and microtubule cytoskeletons through two distinct signal transduction pathways. J Cell Sci. 2005;118:2579–2587. doi: 10.1242/jcs.02385. [DOI] [PubMed] [Google Scholar]
- Chen WT. Proteolytic activity of specialized surface protrusions formed at rosette contact sites of transformed cells. J Exp Zool. 1989;251:167–185. doi: 10.1002/jez.1402510206. [DOI] [PubMed] [Google Scholar]
- Chhabra ES, Higgs HN. The many faces of actin: matching assembly factors with cellular structures. Nat Cell Biol. 2007;9:1110–1121. doi: 10.1038/ncb1007-1110. [DOI] [PubMed] [Google Scholar]
- Collins C, Nelson WJ. Running with neighbors: coordinating cell migration and cell-cell adhesion. Curr Opin Cell Biol. 2015;36:62–70. doi: 10.1016/j.ceb.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA, Sept D. New insights into mechanism and regulation of actin capping protein. Int Rev Cell Mol Biol. 2008;267:183–206. doi: 10.1016/S1937-6448(08)00604-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos G, Rogel MR, Baker MA, Troken JR, Urich D, Morales-Nebreda L, Sennello JA, Kutuzov MA, Sitikov A, Davis JM, et al. Vimentin regulates activation of the NLRP3 inflammasome. Nat Commun. 2015;6:6574. doi: 10.1038/ncomms7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumortier JG, Martin S, Meyer D, Rosa FM, David NB. Collective mesendoderm migration relies on an intrinsic directionality signal transmitted through cell contacts. Proc Natl Acad Sci USA. 2012;109:16945–16950. doi: 10.1073/pnas.1205870109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckes B, Colucci-Guyon E, Smola H, Nodder S, Babinet C, Krieg T, Martin P. Impaired wound healing in embryonic and adult mice lacking vimentin. J Cell Sci. 2000;113:2455–2462. doi: 10.1242/jcs.113.13.2455. [DOI] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N. Computer control of microscopes using μManager. Curr Protoc Mol Biol. 2010;92:14.20.1–14.20.17. doi: 10.1002/0471142727.mb1420s92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M, Liang Y, Kim T, Cooper JA. Physiological role of the interaction between CARMIL1 and capping protein. Mol Biol Cell. 2013;24:3047–3055. doi: 10.1091/mbc.E13-05-0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M, McConnell P, Schafer DA, Cooper JA. CPI motif interaction is necessary for capping protein function in cells. Nat Commun. 2015;6:8415. doi: 10.1038/ncomms9415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M, Zwolak A, Schafer DA, Sept D, Dominguez R, Cooper JA. Capping protein regulators fine-tune actin assembly dynamics. Nat Rev Mol Cell Biol. 2014;15:677–689. doi: 10.1038/nrm3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P. Prespecification and plasticity: shifting mechanisms of cell migration. Curr Opin Cell Biol. 2004;16:14–23. doi: 10.1016/j.ceb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Fujiwara I, Remmert K, Hammer JA. Direct observation of the uncapping of capping protein-capped actin filaments by CARMIL homology domain 3. J Biol Chem. 2010;285:2707–2720. doi: 10.1074/jbc.M109.031203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara I, Remmert K, Piszczek G, Hammer JA. Capping protein regulatory cycle driven by CARMIL and V-1 may promote actin network assembly at protruding edges. Proc Natl Acad Sci USA. 2014;111:E1970–E1979. doi: 10.1073/pnas.1313738111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG. Enzymatic assembly of overlapping DNA fragments. Methods Enzymol. 2011;498:349–361. doi: 10.1016/B978-0-12-385120-8.00015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles C, Polette M, Zahm JM, Tournier JM, Volders L, Foidart JM, Birembaut P. Vimentin contributes to human mammary epithelial cell migration. J Cell Sci. 1999;112:4615–4625. doi: 10.1242/jcs.112.24.4615. [DOI] [PubMed] [Google Scholar]
- Gupton SL, Anderson KL, Kole TP, Fischer RS, Ponti A, Hitchcock-DeGregori SE, Danuser G, Fowler VM, Wirtz D, Hanein D, Waterman-Storer CM. Cell migration without a lamellipodium: translation of actin dynamics into cell movement mediated by tropomyosin. J Cell Biol. 2005;168:619–631. doi: 10.1083/jcb.200406063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyoeva FK, Gelfand VI. Coalignment of vimentin intermediate filaments with microtubules depends on kinesin. Nature. 1991;353:445–448. doi: 10.1038/353445a0. [DOI] [PubMed] [Google Scholar]
- Haeger A, Wolf K, Zegers MM, Friedl P. Collective cell migration: guidance principles and hierarchies. Trends Cell Biol. 2015;25:556–566. doi: 10.1016/j.tcb.2015.06.003. [DOI] [PubMed] [Google Scholar]
- Havel LS, Kline ER, Salgueiro AM, Marcus AI. Vimentin regulates lung cancer cell adhesion through a VAV2-Rac1 pathway to control focal adhesion kinase activity. Oncogene. 2015;34:1979–1990. doi: 10.1038/onc.2014.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfand BT, Mendez MG, Murthy SN, Shumaker DK, Grin B, Mahammad S, Aebi U, Wedig T, Wu YI, Hahn KM, et al. Vimentin organization modulates the formation of lamellipodia. Mol Biol Cell. 2011;22:1274–1289. doi: 10.1091/mbc.E10-08-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfand BT, Mikami A, Vallee RB, Goldman RD. A requirement for cytoplasmic dynein and dynactin in intermediate filament network assembly and organization. J Cell Biol. 2002;157:795–806. doi: 10.1083/jcb.200202027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Valladares M, Kim T, Kannan B, Tung A, Aguda AH, Larsson M, Cooper JA, Robinson RC. Structural characterization of a capping protein interaction motif defines a family of actin filament regulators. Nat Struct Mol Biol. 2010;17:497–503. doi: 10.1038/nsmb.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr MC, Teasdale RD. Defining macropinocytosis. Traffic. 2009;10:364–371. doi: 10.1111/j.1600-0854.2009.00878.x. [DOI] [PubMed] [Google Scholar]
- Kim T, Ravilious GE, Sept D, Cooper JA. Mechanism for CARMIL protein inhibition of heterodimeric actin-capping protein. J Biol Chem. 2012;287:15251–15262. doi: 10.1074/jbc.M112.345447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier MH, McConnell P, Cooper JA. Cell migration and invadopodia formation require a membrane-binding domain of CARMIL2. J Biol Chem. 2015:jbc.M115.676882. doi: 10.1074/jbc.M115.676882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Cucchetti M, Roncagalli R, Yokosuka T, Malzac A, Bertosio E, Imbert J, Nijman IJ, Suchanek M, Saito T, et al. The lymphoid lineage-specific actin-uncapping protein Rltpr is essential for costimulation via CD28 and the development of regulatory T cells. Nat Immunol. 2013;14:858–866. doi: 10.1038/ni.2634. [DOI] [PubMed] [Google Scholar]
- Liang Y, Niederstrasser H, Edwards M, Jackson CE, Cooper JA. Distinct roles for CARMIL isoforms in cell migration. Mol Biol Cell. 2009;20:5290–5305. doi: 10.1091/mbc.E08-10-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo CM, Buxton DB, Chua GC, Dembo M, Adelstein RS, Wang YL. Nonmuscle myosin IIb is involved in the guidance of fibroblast migration. Mol Biol Cell. 2004;15:982–989. doi: 10.1091/mbc.E03-06-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz M, Yamaguchi H, Wang Y, Singer RH, Condeelis J. Imaging sites of N-wasp activity in lamellipodia and invadopodia of carcinoma cells. Curr Biol. 2004;14:697–703. doi: 10.1016/j.cub.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Matsuzaka Y, Okamoto K, Mabuchi T, Iizuka M, Ozawa A, Oka A, Tamiya G, Kulski JK, Inoko H. Identification, expression analysis and polymorphism of a novel RLTPR gene encoding a RGD motif, tropomodulin domain and proline/leucine-rich regions. Gene. 2004;343:291–304. doi: 10.1016/j.gene.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Mejillano MR, Kojima S, Applewhite DA, Gertler FB, Svitkina TM, Borisy GG. Lamellipodial versus filopodial mode of the actin nanomachinery: pivotal role of the filament barbed end. Cell. 2004;118:363–373. doi: 10.1016/j.cell.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Menko AS, Bleaken BM, Libowitz AA, Zhang L, Stepp MA, Walker JL. A central role for vimentin in regulating repair function during healing of the lens epithelium. Mol Biol Cell. 2014;25:776–790. doi: 10.1091/mbc.E12-12-0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooren OL, Li J, Nawas J, Cooper JA. Endothelial cells use dynamic actin to facilitate lymphocyte transendothelial migration and maintain the monolayer barrier. Mol Biol Cell. 2014;25:4115–4129. doi: 10.1091/mbc.E14-05-0976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol. 1999;144:1235–1244. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurnberg A, Kitzing T, Grosse R. Nucleating actin for invasion. Nat Rev Cancer. 2011;11:177–187. doi: 10.1038/nrc3003. [DOI] [PubMed] [Google Scholar]
- Parrini MC, Camonis J, Matsuda M, de Gunzburg J. Dissecting activation of the PAK1 kinase at protrusions in living cells. J Biol Chem. 2009;284:24133–24143. doi: 10.1074/jbc.M109.015271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD. Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filaments. J Cell Biol. 1986;103:2747–2754. doi: 10.1083/jcb.103.6.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science. 2009;326:1208–1212. doi: 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahlad V, Yoon M, Moir RD, Vale RD, Goldman RD. Rapid movements of vimentin on microtubule tracks: kinesin-dependent assembly of intermediate filament networks. J Cell Biol. 1998;143:159–170. doi: 10.1083/jcb.143.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogel MR, Soni PN, Troken JR, Sitikov A, Trejo HE, Ridge KM. Vimentin is sufficient and required for wound repair and remodeling in alveolar epithelial cells. FASEB J. 2011;25:3873–3883. doi: 10.1096/fj.10-170795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto Y, Boeda B, Etienne-Manneville S. APC binds intermediate filaments and is required for their reorganization during cell migration. J Cell Biol. 2013;200:249–258. doi: 10.1083/jcb.201206010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68:3033–3046. doi: 10.1007/s00018-011-0735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savagner P. The epithelial-mesenchymal transition (EMT) phenomenon. Ann Oncol. 2010;21(suppl 7):vii89–vii92. doi: 10.1093/annonc/mdq292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoumacher M, Goldman RD, Louvard D, Vignjevic DM. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J Cell Biol. 2010;189:541–556. doi: 10.1083/jcb.200909113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sells MA, Boyd JT, Chernoff J. p21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J Cell Biol. 1999;145:837–849. doi: 10.1083/jcb.145.4.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens C, Henderson P, Nimmo ER, Soares DC, Dogan B, Simpson KW, Barrett JC, Wilson DC, Satsangi J. The intermediate filament protein, vimentin, is a regulator of NOD2 activity. Gut. 2013;62:695–707. doi: 10.1136/gutjnl-2011-301775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suraneni P, Rubinstein B, Unruh JR, Durnin M, Hanein D, Li R. The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J Cell Biol. 2012;197:239–251. doi: 10.1083/jcb.201112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutoh Yoneyama M, Hatakeyama S, Habuchi T, Inoue T, Nakamura T, Funyu T, Wiche G, Ohyama C, Tsuboi S. Vimentin intermediate filament and plectin provide a scaffold for invadopodia, facilitating cancer cell invasion and extravasation for metastasis. Eur J Cell Biol. 2014;93:157–169. doi: 10.1016/j.ejcb.2014.03.002. [DOI] [PubMed] [Google Scholar]
- Thurston G, Jaggi B, Palcic B. Measurement of cell motility and morphology with an automated microscope system. Cytometry. 1988;9:411–417. doi: 10.1002/cyto.990090502. [DOI] [PubMed] [Google Scholar]
- Uruno T, Remmert K, Hammer JA. CARMIL is a potent capping protein antagonist: identification of a conserved CARMIL domain that inhibits the activity of capping protein and uncaps capped actin filaments. J Biol Chem. 2006;281:10635–10650. doi: 10.1074/jbc.M513186200. [DOI] [PubMed] [Google Scholar]
- Wear MA, Yamashita A, Kim K, Maeda Y, Cooper JA. How capping protein binds the barbed end of the actin filament. Curr Biol. 2003;13:1531–1537. doi: 10.1016/s0960-9822(03)00559-1. [DOI] [PubMed] [Google Scholar]
- Weaver AM. Invadopodia: specialized cell structures for cancer invasion. Clin Exp Metastasis. 2006;23:97–105. doi: 10.1007/s10585-006-9014-1. [DOI] [PubMed] [Google Scholar]
- Weigelt B, Peterse JL, van’t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005;5:591–602. doi: 10.1038/nrc1670. [DOI] [PubMed] [Google Scholar]
- Wu C, Asokan SB, Berginski ME, Haynes EM, Sharpless NE, Griffith JD, Gomez SM, Bear JE. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 2012;148:973–987. doi: 10.1016/j.cell.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Lorenz M, Kempiak S, Sarmiento C, Coniglio S, Symons M, Segall J, Eddy R, Miki H, Takenawa T, Condeelis J. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–452. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi N, Mizutani T, Kawabata K, Haga H. Leader cells regulate collective cell migration via Rac activation in the downstream signaling of integrin β1 and PI3K. Sci Rep. 2015;5:7656. doi: 10.1038/srep07656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Pring M, Wear MA, Huang M, Cooper JA, Svitkina TM, Zigmond SH. Mammalian CARMIL inhibits actin filament capping by capping protein. Dev Cell. 2005;9:209–221. doi: 10.1016/j.devcel.2005.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwolak A, Uruno T, Piszczek G, Hammer JA, Tjandra N. Molecular basis for barbed end uncapping by CARMIL homology domain 3 of mouse CARMIL-1. J Biol Chem. 2010;285:29014–29026. doi: 10.1074/jbc.M110.134221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwolak A, Yang C, Feeser EA, Ostap EM, Svitkina T, Dominguez R. CARMIL leading edge localization depends on a non-canonical PH domain and dimerization. Nat Commun. 2013;4:2523. doi: 10.1038/ncomms3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.