

The role of mitochondrial dynamics in cellular responses to ionizing radiation (IR) is still largely unknown. This study demonstrates that IR triggers Drp1-dependent mitochondrial fission and that Drp1 inhibition attenuates radiation-induced mitotic catastrophe, suggesting that Drp1 is involved in determining the fate of cells after irradiation.
Abstract
Accumulating evidence suggests that mitochondrial dynamics is crucial for the maintenance of cellular quality control and function in response to various stresses. However, the role of mitochondrial dynamics in cellular responses to ionizing radiation (IR) is still largely unknown. In this study, we provide evidence that IR triggers mitochondrial fission mediated by the mitochondrial fission protein dynamin-related protein 1 (Drp1). We also show IR-induced mitotic catastrophe (MC), which is a type of cell death associated with defective mitosis, and aberrant centrosome amplification in mouse embryonic fibroblasts (MEFs). These are attenuated by genetic or pharmacological inhibition of Drp1. Whereas radiation-induced aberrant centrosome amplification and MC are suppressed by the inhibition of Plk1 and CDK2 in wild-type MEFs, the inhibition of these kinases is ineffective in Drp1-deficient MEFs. Furthermore, the cyclin B1 level after irradiation is significantly higher throughout the time course in Drp1-deficient MEFs than in wild-type MEFs, implying that Drp1 is involved in the regulation of cyclin B1 level. These findings strongly suggest that Drp1 plays an important role in determining the fate of cells after irradiation via the regulation of mitochondrial dynamics.
INTRODUCTION
Mitochondria are vital organelles for the survival of eukaryotic cells through the production of cellular energy. In addition, they play essential roles in many cellular activities, including cellular ion homeostasis, heat production, and the execution of apoptosis (Nunnari and Suomalainen, 2012). As such, they contribute significantly to the preservation of cellular quality and function. Therefore it is necessary for the host cells to be equipped with a mechanism to maintain mitochondrial function and to ensure their integrity.
Mitochondria are dynamic organelles that constantly change shape as a result of a balance between fusion and fission. Correct regulation of the mitochondrial fission–fusion equilibrium, called mitochondrial dynamics, is essential for cellular homeostasis (Detmer and Chan, 2007; Youle and van der Bliek, 2012). Previous studies showed the basic molecular mechanisms involved in the morphological dynamics of mitochondria. Fusion relies on the inner membrane protein optic atrophy 1 and the outer membrane proteins mitofusin 1 and 2. On the other hand, fission requires the translocation of dynamin-related protein 1 (Drp1) from the cytosol to mitochondria, where it presumably docks on mitochondrial fission 1 protein, its adapter protein on the outer membrane (Hoppins et al., 2007). Similar to the endocytosis motor dynamin, Drp1 is believed to polymerize into ring- or spiral-shaped superstructures that constrict and eventually cleave mitochondria via a GTP hydrolysis–dependent mechanism (Hoppins et al., 2007). Cell culture studies demonstrated that excessive mitochondrial fission is associated with apoptosis, neuronal dysfunction, and cell death (Frank et al., 2001; Karbowski and Youle, 2003; Barsoum et al., 2006; Cheung et al., 2007). Drp1 inhibition by expression of a Drp1 dominant-negative mutant or by RNA interference leads to increased length and interconnectivity of mitochondria, thereby inhibiting the fission process and preventing cell death (Frank et al., 2001; Jagasia et al., 2005). Recently an infant patient with a dominant-negative Drp1 allele and mice lacking Drp1 were found to present a wide range of developmental abnormalities (Waterham et al., 2007; Ishihara et al., 2009). In addition, Drp1-mediated mitochondrial fission is also suggested to be involved in cell proliferation and to be essential for the proper distribution of mitochondria into daughter cells (Taguchi et al., 2007; Ishihara et al., 2009; Rehman et al., 2012). These findings collectively suggest a critical role of Drp1-regulated mitochondrial fission in the maintenance of cellular and organismal functions.
Ionizing radiation (IR) elicits a variety of responses in eukaryotic cells, including DNA repair, reversible or irreversible cell cycle arrest, and activation of stress signaling (Jackson and Bartek, 2009; Eriksson and Stigbrand, 2010). All of these cellular responses ultimately influence the fate of irradiated cells. As one of the cellular radioresponses, IR alters mitochondrial functions. Several studies showed that IR leads to alterations in mitochondrial respiration, elevation of mitochondrial mass, and increase in reactive oxygen species (ROS) production (Leach et al., 2001; Nugent et al., 2007; Ogura et al., 2009). Recently we investigated the effect of IR on mitochondrial function and found that IR increased mitochondrial ROS production, mitochondrial membrane potential, mitochondrial respiration, and mitochondrial ATP production (Yamamori et al., 2012). IR changes mitochondrial gene expression profiles that are related to cell survival (Kulkarni et al., 2010). Moreover, mitochondria have been reported as the primary target for radiation-induced apoptosis (Taneja et al., 2001). Furthermore, mitochondria are also suggested to be involved in radiation-induced intracellular and intercellular signaling (Gong et al., 1998; Zhou et al., 2008; Rajendran et al., 2011). These findings indicate that mitochondria are likely to be a major target of IR and to play a critical role in cellular radioresponses.
Only a limited number of studies have reported the role of mitochondrial dynamics in cellular radioresponses. Kobashigawa et al. (2011) showed that γ-irradiation promotes Drp1-dependent mitochondrial fission and mitochondrial ROS production in immortalized fibroblasts. Cytoplasmic irradiation with α-particles was also found to cause mitochondrial fission, accompanied by a reduction in respiratory-chain enzyme activities (Zhang et al., 2013). These data indicate that IR stimulates mitochondrial fission, thereby altering mitochondrial functions. However, it remains elusive how IR affects mitochondrial dynamics and the fate of irradiated cells. Here we provide evidence that IR triggers mitochondrial fission regulated by Drp1 and that its inhibition attenuates radiation-induced mitotic catastrophe (MC), which is a form of cell death associated with aberrant mitosis. Our findings imply that Drp1-dependent mitochondrial fission plays an important role in determining the fate of cells after irradiation.
RESULTS
IR triggers mitochondrial fission
To determine whether x-irradiation affects mitochondrial morphology, we performed microscopic analysis of wild-type mouse embryonic fibroblasts (WT MEFs) with or without irradiation using a fluorescent dye, MitoTracker Green FM, and confocal laser scanning microscopy. As shown in Figure 1, A and B, mitochondria in untreated cells exhibited highly networked and tubular shapes. In contrast, exposure to IR (10 Gy, 12 h) led to an increase in the number of mitochondria with fragmented and granular forms (Figure 1, C and D). To quantitatively evaluate the mitochondrial morphologies, we classified them into four categories (highly connected, tubular, intermediate, and fragmented) by manually analyzing the microscopic images (Supplemental Figure S1). Quantitative analysis revealed that IR led to the gradual increase of fragmented mitochondria and the reduction of fused (highly connected plus tubular) mitochondria up to 12 h postirradiation (Figure 1E and Supplemental Figure S2). At 24 h postirradiation, the proportion of cells with fragmented mitochondria was ∼30%, whereas that of cells with fused mitochondria increased. Furthermore, we found an increase in mitochondrial fragmentation when cells were irradiated at 5 Gy and higher and a significant decrease in fused mitochondria at 10 Gy (Figure 1F and Supplemental Figure S2). These results demonstrate that IR triggers mitochondrial fission.
FIGURE 1:

IR triggers mitochondrial fission. After x-irradiation at 10 Gy, WT MEFs were incubated for 12 h. Mitochondria were stained with 300 nM MitoTracker Green FM and analyzed by confocal laser scanning microscopy. (A) Representative confocal image of mitochondria in nonirradiated cells. (B) Magnified image of the boxed region in A. (C) Representative confocal image of mitochondria in irradiated cells. (D) Magnified image of the boxed region in C. (E, F) Quantitative analysis of mitochondrial morphologies. Microscopic analyses were conducted at the indicated times after x-irradiation at 10 Gy (E) or at 12 h after x-irradiation at the indicated doses (F). Quantitative image analysis of mitochondrial morphologies was performed as described in Materials and Methods. Data are expressed as means ± SD of three experiments.
IR stimulates Drp1 translocation
Previous evidence showed that Drp1 is essential for mitochondrial fission and that its translocation from the cytosol to mitochondria triggers mitochondrial fission (Hoppins et al., 2007). Therefore we examined whether IR causes mitochondrial translocation of Drp1 by performing subcellular fractionation and Western blotting. We found that Drp1 accumulated in mitochondria after IR, peaking at 8–12 h postirradiation and declining thereafter (Figure 2A). Dose–response analysis revealed that mitochondrial translocation of Drp1 emerged after 5-Gy irradiation and became significant when cells were irradiated at 10 Gy (Figure 2B). These data suggest that IR stimulates mitochondrial translocation of Drp1, leading to mitochondrial fission.
FIGURE 2:
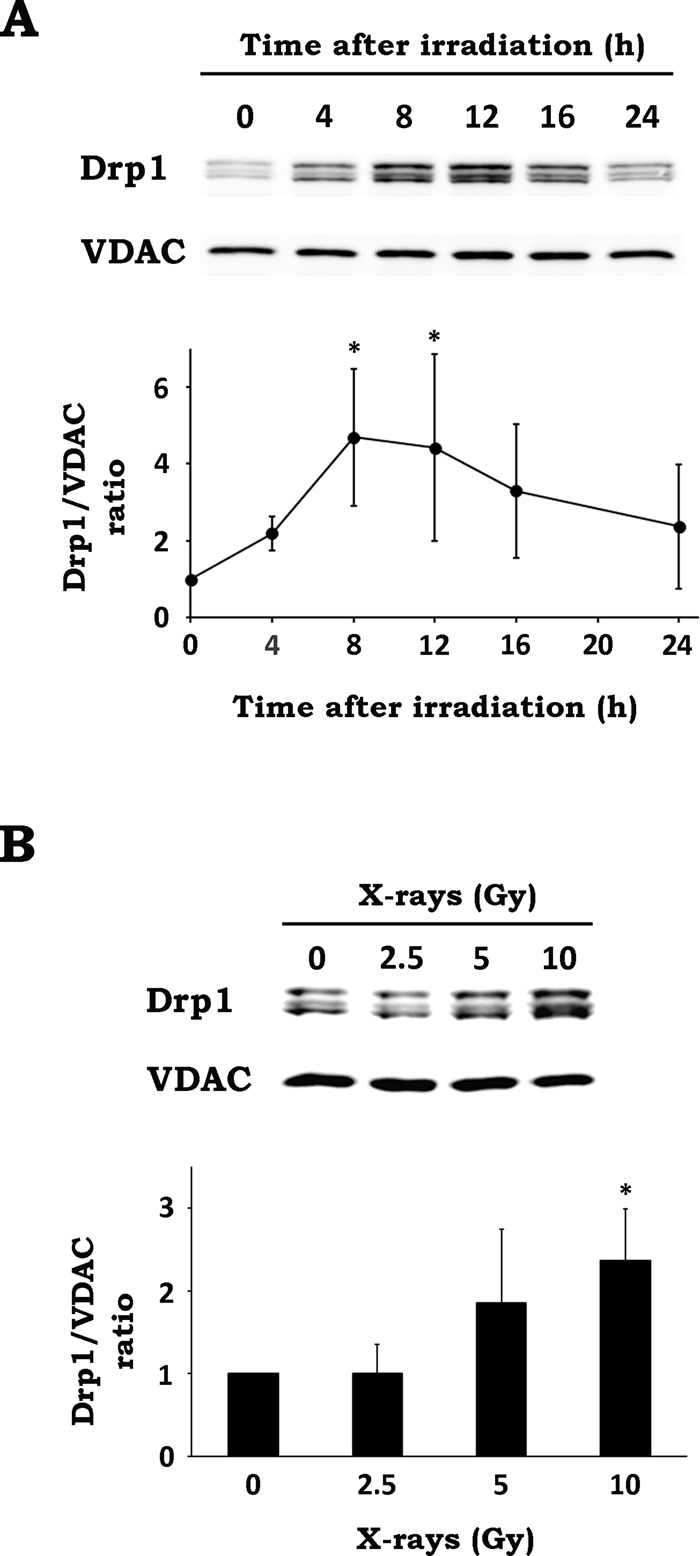
IR stimulates Drp1 translocation. (A) After x-irradiation at 10 Gy, WT MEF cells were cultured for the indicated times. After incubation, organelle fractions containing mitochondria were isolated and analyzed by Western blotting. Top, representative blots of Drp1 and VDAC. Bottom, time-course analysis of Drp1 translocation. The intensities of Drp1 bands were normalized to those of VDAC bands. Data are expressed as means ± SD of five experiments. *p < 0.05 vs. 0 h (Dunnett test). (B) After x-irradiation at the indicated doses, WT MEF cells were cultured for 12 h. After incubation, organelle fractions containing mitochondria were isolated and analyzed by Western blotting. Top, representative blots of Drp1 and VDAC. Bottom, dose–response analysis of Drp1 translocation. Data are expressed as means ± SD of four experiments. *p < 0.05 vs. 0 Gy (Dunnett test).
Drp1 inhibition reduces radiation-induced mitochondrial fission
Because IR was demonstrated to stimulate Drp1 translocation, we next investigated whether Drp1 is involved in radiation-induced mitochondrial fission. To explore the role of Drp1, we used two lines of immortalized MEFs, one derived from wild-type mice (WT MEFs) and the other from Drp1-deficient mice (KO MEFs; Ishihara et al., 2009). We evaluated the mitochondrial morphology in both cells with or without x-irradiation. In the resting state, mitochondria in KO MEFs were more fused than those in WT MEFs (Figure 3, A and B, and Supplemental Figure S3A). Whereas IR caused significant increase in fragmented mitochondria and decrease in fused mitochondria in WT MEFs, the effect of IR on mitochondrial morphology in KO MEFs was less prominent (Figure 3, A and B, and Supplemental Figure S3A), indicating that Drp1 is involved in mitochondrial fission after IR. To examine further the involvement of Drp1, we next evaluated the effect of a Drp1 inhibitor, Mdivi-1 (Cassidy-Stone et al., 2008). After treatment of WT MEFs with Mdivi-1, we analyzed mitochondrial morphology. In the resting state, Mdivi-1 treatment resulted in an increase of the number of cells with highly connected mitochondria without changing the overall ratio of fused mitochondria (Figure 3, C and D, and Supplemental Figure S3B). Furthermore, mitochondrial fission after irradiation was clearly attenuated by Mdivi-1, exhibiting similar mitochondrial morphology with or without irradiation. We also found that overexpression of Drp1 dominant-negative mutant (Drp1 K38A) in WT MEFs attenuated radiation-induced mitochondrial fission (Supplemental Figure S4, A and B). Taken together, these results show that Drp1 inhibition reduces radiation-induced mitochondrial fission, suggesting the involvement of Drp1 in this process.
FIGURE 3:

Inhibition of Drp1 diminishes radiation-induced mitochondrial fission. (A, B) WT and KO MEFs were irradiated at 10 Gy of x-rays and incubated for 12 h. (A) Representative confocal images of mitochondria in MEFs with or without x-irradiation. IR, ionizing radiation. (B) Quantitative analysis of mitochondrial morphologies in WT and KO MEFs. (C, D) After x-irradiation at 10 Gy in the presence or absence of 25 μM Mdivi-1, WT MEFs were incubated for 12 h. (C) Representative confocal images of mitochondria in WT MEFs in the presence or absence of Mdivi-1 with or without x-irradiation. (D) Quantitative analysis of mitochondrial morphologies in WT MEFs in the presence or absence of Mdivi-1. Quantitative image analysis of mitochondrial morphologies was performed as described in Materials and Methods. Data are expressed as means ± SD of three experiments.
Drp1 inhibition attenuates radiation-induced mitotic catastrophe
MC is a form of cell death associated with aberrant mitosis, owing to uncoordinated or improper M-phase progression, and is considered the major cell death mechanism after IR, especially in apoptosis-impaired tumor cells (Vakifahmetoglu et al., 2008; Eriksson and Stigbrand, 2010; Lauber et al., 2012). To investigate whether Drp1-dependent mitochondrial fission participates in determining the fate of irradiated cells, we next examined the effect of Drp1 inhibition on radiation-induced MC. After x-irradiation, we stained cell nuclei and scored the cells with features of aberrant mitotic nuclei such as micronuclei, multilobular nuclei, and fragmented nuclei as cells undergoing MC (Figure 4A). As shown in Figure 4B, MC was significantly increased at 18 h and later after irradiation. MC was stimulated when cells were exposed to x-rays at 2.5 Gy and higher, whereas it was clearly reduced in Drp1-null cells compared with WT cells (Figure 4C), suggesting the involvement of Drp1 in this process. Similarly, MC after irradiation was reduced by Mdivi-1 treatment (Figure 4D), as well as by expressing Drp1 K38A in WT MEFs (Supplemental Figure S4C), supporting the previous data. Because Mdivi-1 treatment exhibited no effect on the radiation-induced MC in Drp1-deficient cells (Supplemental Figure S5A), this corroborated that Mdivi-1 reduced MC by inhibiting Drp1. Furthermore, we investigated the effect of Drp1 deficiency on cellular survival after IR by clonogenic survival assay. As shown in Figure 4E, KO MEFs were less sensitive to IR and showed higher survival than WT MEFs. These findings strongly suggested that Drp1 is involved in determining the fate of cells after irradiation via the induction of MC.
FIGURE 4:

Drp1 inhibition attenuates radiation-induced MC and increases cellular survival after x-irradiation. (A) Cells undergoing MC after x-irradiation. After WT MEFs were x-irradiated at 10 Gy and incubated for 24 h, cell nuclei were stained with 300 nM DAPI. Representative images of the cells with features of aberrant mitotic nuclei such as micronuclei, multilobular nuclei, and fragmented nuclei. (B) Time course of MC induction after irradiation. WT MEFs were cultured for the indicated times after x-irradiation at 10 Gy. After incubation, nuclear morphology of the irradiated cells was analyzed. At least 200 cells were counted, and the percentage of cells undergoing MC was determined. Data are expressed as means ± SD of three experiments. *p < 0.05 and **p < 0.01 vs. 0 h (Dunnett test). (C) Effect of Drp1 deficiency on radiation-induced MC. After x-irradiation at the indicated doses, WT (black) and KO (white) MEFs were cultured for 24 h. MC was scored as described. Data are expressed as means ± SD of three experiments. *p < 0.05 and **p < 0.01 vs. WT (Student’s t test). (D) Effect of Mdivi-1 on radiation-induced MC. After x-irradiation at the indicated doses in the presence of 0 (black), 5 (white), or 25 (gray) μM Mdivi-1, WT MEFs were cultured for 24 h. MC was scored as described. Data are expressed as means ± SD of three experiments. **p < 0.01 vs. 0 μM Mdivi-1 (Student’s t test). (E) Effect of Drp1 deficiency on clonogenic survival after x-irradiation. After x-irradiation at the indicated doses, WT (black) and KO (white) MEFs were cultured for 7 d. Colonies containing >50 cells were scored as surviving cells. Survival fractions were calculated as described in Materials and Methods. Data are expressed as means ± SD of three experiments. *p < 0.05 vs. WT (Student’s t test).
The apoptosis pathway is dispensable for radiation-induced mitotic catastrophe
It has been shown that Drp1-mediated mitochondrial fission plays a role in the activation of apoptosis signaling and the execution of apoptosis (Li et al., 2010; Montessuit et al., 2010; Zhao et al., 2014). In addition, activation of the apoptosis-signaling pathway via caspase-2 is involved in the induction of MC (Castedo et al., 2004a, b). These findings prompted us to examine whether the apoptosis-signaling pathway is involved in radiation-induced MC. When we tested the effect of a pan-caspase inhibitor Z-VAD-FMK, it showed no influence on MC level after irradiation (Figure 5A). In addition, we could scarcely detect the release of cytochrome c and Smac from the mitochondria to the cytosol up to 24 h after irradiation in WT MEFs (Figure 5B). Furthermore, IR resulted in an increase of cellular ATP content not only in WT MEFs but also in KO MEFs, which were previously shown to be oligomycin sensitive (Yamamori et al., 2012; Supplemental Figure S6). These results imply that mitochondrial function is not compromised after irradiation regardless of the presence or absence of Drp1. These results demonstrate that IR does not activate the intrinsic apoptosis pathway initiated by the cytosolic release of mitochondrial proteins and the concomitant impairment of mitochondrial function and that radiation-induced MC is independent of apoptosis signaling.
FIGURE 5:

The apoptosis pathway is not involved in radiation-induced MC. (A) Effect of a caspase inhibitor on radiation-induced MC. WT MEFs were x-irradiated at 10 Gy in the presence of pan-caspase inhibitor Z-VAD-FMK at 0 (black), 5 (white), or 20 (gray) μM. After 24 h of incubation, MC was scored as described in Materials and Methods. Data are expressed as means ± SD of three experiments. n.s., not significant. (B) Cytosolic release of mitochondrial proteins after x-irradiation. After x-irradiation at 10 Gy, WT MEFs were incubated for the indicated times. Cytosol and organelle fractions were collected, and subcellular distribution of cytochrome c (cyt c) and Smac was analyzed by Western blotting. Treatment with etoposide (Eto; 50 μM, 8 h) was used as positive control. GAPDH and VDAC were loading controls for the cytosol and organelle fraction, respectively. Representative blots of three experiments.
Drp1 inhibition prevents aberrant centrosome amplification after IR
Whereas a normal cell contains one or two centrosomes, a cell that underwent MC presents an atypical number of centrosomes (Dodson et al., 2007; Rello-Varona et al., 2010). This abnormality in centrosome number is mainly due to a dysfunction in the centrosome duplication system or other defects in the dividing cells that result in the acquisition of additional centrosomes (Brinkley, 2001). When we conducted a microscopic analysis of cellular centrosomes by immunostaining with an anti–γ-tubulin antibody, >95% of cells contained one or two centrosomes under nonirradiated conditions (Figure 6, A and B). In contrast, after irradiation, we observed cells carrying more than two centrosomes. They also presented an abnormal nuclear morphology (Figure 6A). The proportion of cells with more than two centrosomes was found to increase 15 h after irradiation and accumulated up to 24 h postirradiation (Figure 6B), suggesting that IR triggered aberrant centrosome amplification. To examine whether Drp1 inhibition affects this abnormality, we evaluated the number of centrosomes in WT and Drp1-deficient MEFs with or without x-irradiation. Whereas WT and KO MEFs showed no difference in the proportion of the cells with more than two centrosomes when not irradiated, irradiated KO MEFs exhibited a significantly reduced abnormality in the centrosome number compared with irradiated WT MEFs (Figure 6C). In addition, Drp1 inhibition by Mdivi-1 also suppressed the abnormality in centrosome number after irradiation in a concentration-dependent manner (Figure 6D). Because Mdivi-1 treatment showed no effect on the radiation-induced abnormality in centrosome number in Drp1-deficient cells (Supplemental Figure S5B), this corroborated that Mdivi-1 attenuated radiation-induced centrosome abnormality by inhibiting Drp1. These findings indicate that Drp1 is involved in the aberrant centrosome amplification after IR.
FIGURE 6:

Drp1 inhibition prevents aberrant centrosome amplification after IR. (A) x-irradiation triggers aberrant centrosome amplification. After WT MEFs were x-irradiated at 10 Gy and incubated for 24 h, γ-tubulin and cell nuclei were stained. Representative images of cells without irradiation (IR(−)) and after X-irradiation (IR(+)). (B) Time-course change in the number of centrosomes after irradiation. WT MEFs were cultured for the indicated times after x-irradiation at 10 Gy. The number of centrosomes in a cell was measured by counting the γ-tubulin foci per cell. Data are expressed as means ± SD of three experiments. *p < 0.05 vs. 0 h (Dunnett test). (C) Effect of Drp1 deficiency on radiation-induced aberrant centrosome amplification. After x-irradiation at 10 Gy, WT (black) and KO (white) MEFs were cultured for 24 h. The number of centrosomes in a cell was scored as described. Data are expressed as means ± SD of three experiments. *p < 0.05 vs. WT (Student’s t test). (D) Effect of Mdivi-1 on radiation-induced aberrant centrosome amplification. After x-irradiation at 10 Gy in the presence of 0 (black), 5 (white), or 25 (gray) μM Mdivi-1, WT MEFs were cultured for 24 h. The number of centrosomes in a cell was scored as described. Data are expressed as means ± SD of three experiments. **p < 0.01 vs. 0 μM Mdivi-1 (Student’s t test).
Aberrant centrosome amplification and MC after IR are dependent on Polo-like kinase 1 and cyclin-dependent kinase 2
The centrosome duplication cycle consists of four major steps—centriole disengagement, centrosome duplication, centrosome maturation, and centrosome separation. During this cycle, Polo-like kinase 1 (Plk1) and cyclin-dependent kinase 2 (CDK2) are essential for centrosome maturation and centrosome duplication, respectively (Lane and Nigg, 1996; Meraldi et al., 1999). Therefore we believed that these kinases might be involved in the Drp1-dependent abnormality in centrosome numbers and MC after IR. To test this hypothesis, we first examined whether Plk1 and CDK2 are activated in response to IR. The activation of Plk1 and CDK2 was evaluated by measuring their phosphorylation at the activation sites (T210 on Plk1, T160 on CDK2). As shown in Figure 7A, IR stimulated the phosphorylation of both kinases, peaking at 12 h after irradiation, indicating their activation. In addition, it was also noticeable that the expression level of total Plk1 and the Plk1 T210 phosphorylation were slightly higher in WT MEFs than KOs, whereas the CDK2 phosphorylation profiles were almost equal in both cells. To determine whether these two enzymes are involved in the Drp1-dependent abnormality in centrosome numbers and MC after IR, we next examined the effects of pharmacological inhibitors for these kinases. WT and KO MEFs were irradiated in the presence of vehicle, a Plk1 inhibitor, BI2536, or a CDK2 inhibitor, NU6140. After 24-h incubation, we performed immunostaining with anti–γ-tubulin antibody and nuclear staining with 4′,6-diamidino-2-phenylindole (DAPI), followed by microscopic analysis. As shown in Figure 7B, ∼50% of WT MEFs contained more than two centrosomes after IR. BI2536 or NU6140 treatment significantly reduced this number. In contrast, KO MEFs exhibited a much lower level of radiation-induced abnormality in centrosome number after IR (∼15%), which was not affected by the drug treatments (Figure 7B). We also examined the effect of these inhibitors on the radiation-induced MC in WT and KO MEFs (Figure 7C). In WT MEFs, the radiation-induced MC was greatly diminished by BI2536 or NU6140 treatment. In contrast, a lower induction of MC was observed in KO MEFs after irradiation, and the BI2536 and NU6140 treatments had no effect on it. Furthermore, we found that the BI2536 and NU6140 treatments attenuated the post-IR phosphorylation of histone H3 at Ser-10, which serves as a mitosis marker, in WT MEFs (Supplemental Figure S7), indicating that these drugs affected the mitosis progression in irradiated cells. These results suggest that Plk1 and CDK2 are required for radiation-induced aberrant centrosome amplification and subsequent MC mediated by Drp1.
FIGURE 7:
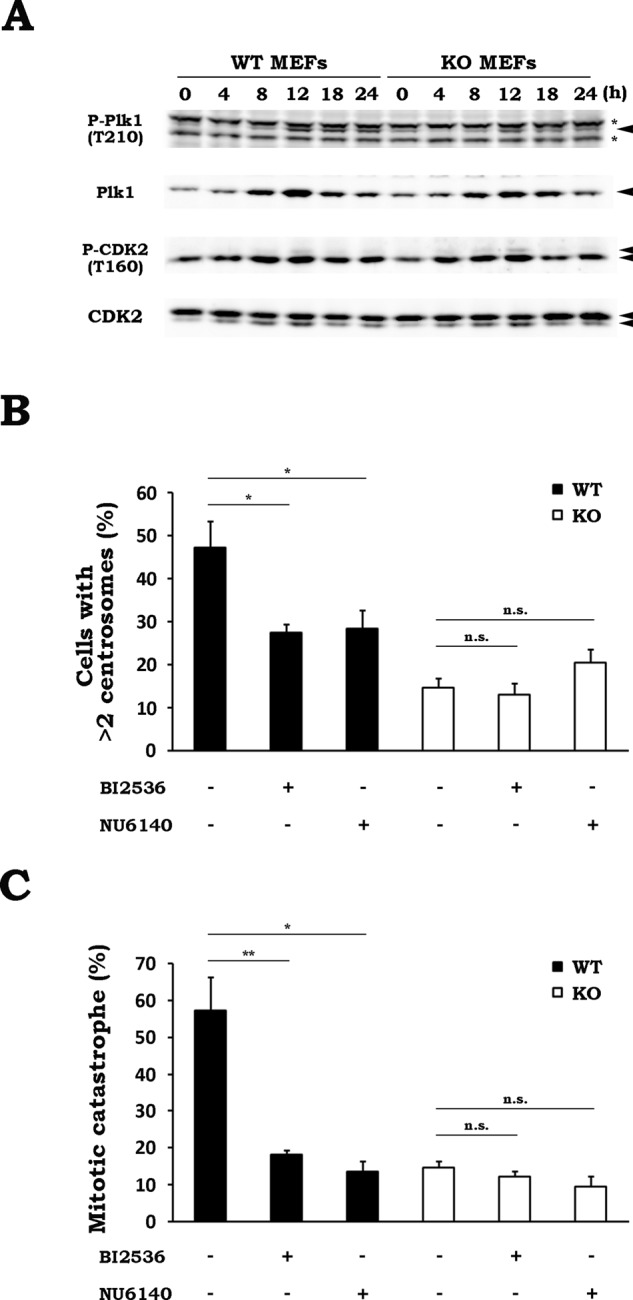
Aberrant centrosome amplification and MC after IR are dependent on Plk1 and CDK2. (A) Phosphorylation profiles of Plk1 and CDK2 after IR in WT and KO MEFs. After WT or KO MEFs were x-irradiated at 10 Gy, the cells were collected at the indicated times. The phosphorylation and expression of the indicated proteins were analyzed by Western blotting. Representative blots of three experiments. Asterisks denote nonspecific bands. (B) Effect of kinase inhibitors on radiation-induced aberrant centrosome amplification. WT (black) or KO (white) MEFs were x-irradiated at 10 Gy in the presence or absence of 100 nM BI2536 (a Plk1 inhibitor) or 2 μM NU6140 (a CDK2 inhibitor). After incubation for 24 h, the number of centrosomes in a cell was measured by counting the γ-tubulin foci per cell. Data are expressed as means ± SD of three experiments. *p < 0.05 (Student’s t test). n.s., not significant. (C) Effect of kinase inhibitors on radiation-induced MC. WT (black) or KO (white) MEFs were x-irradiated at 10 Gy in the presence or absence of 100 nM BI2536 or 2 μM NU6140. After incubation for 24 h, the percentage of cells undergoing MC was determined by monitoring the nuclear morphology of the irradiated cells. Data are expressed as means ± SD of three experiments. *p < 0.05, **p < 0.01 (Student’s t test). n.s., not significant.
Drp1 deficiency elevates cyclin B1 level after irradiation
In response to DNA damage by various stimuli, including IR, eukaryotic cells trigger cell cycle arrest via the activation of DNA damage checkpoints and repair the damage (Iliakis et al., 2003). However, when the cell cycle arrest is prolonged due to excessive DNA damage, it restarts prematurely, carrying unrepaired damages. This premature reentry in mitosis was suggested to be involved in the events associated with defective or deregulated mitosis (Dodson et al., 2007; Huang et al., 2008). For successful mitosis, it is essential to execute the synthesis and degradation of mitotic regulators in a timely manner (Hunt et al., 1992; Descombes and Nigg, 1998). Therefore we speculated that Drp1 affects the expression of mitotic regulators after IR, thereby affecting the radiation-induced defective mitosis. To test this hypothesis, we evaluated the expression levels of three mitotic regulators (cyclin A, E, and B1) and the phosphorylation levels of two proteins (histones H2AX and H3) in WT and KO MEFs (Figure 8A). In WT MEFs, the increase in histone H2AX phosphorylation (γ-H2AX) was first observed at 4 h after irradiation and was elevated until 18 h postirradiation. This was similarly observed in KO MEFs, suggesting that there was no difference in levels of IR-triggered DNA double-strand breaks (DSBs) throughout the time course in both cells. The histone H3 phosphorylation disappeared at 4 h after IR, reappeared at 12 h, and remained elevated until 18 h in WT MEFs. This pattern was similarly observed in KO MEFs, indicating that, after temporal cell cycle arrest after irradiation, reentry in mitosis occurred no later than 12 h postirradiation in both cells. These findings suggest that WT and KO MEFs possess a similar level of DSB up to 18 h after irradiation and undergo mitosis reentry, carrying unrepaired DSBs. Cyclin B1 increased after IR, peaked at 12 h postirradiation, and declined thereafter to the basal level. The cyclin B1 level was also elevated after irradiation in KO MEFs, but to a much greater extent than in WT MEFs. Furthermore, although the IR-induced cyclin B1 level in KO MEFs peaked at 12 h postirradiation and decreased thereafter in a similar manner to that of WT MEFs, it did not reach the basal level in KO MEFs. Quantitative analysis of the cyclin B1 level confirmed the clear difference in cyclin B1 abundance after IR between WT and KO MEFs, with greater increase, higher peak level, and weaker postpeak decline in KO MEFs than with WT MEFs (Figure 8B). The checkpoint-dependent fluctuation of cyclin B1 level is essentially under the control of the ubiquitin/proteasome system, and its proper regulation is essential for mitosis to be correct (van Leuken et al., 2008). Thus our data suggest that Drp1 is involved in the regulation of cyclin B1 level, presumably by influencing its degradation through the ubiquitin/proteasome system.
FIGURE 8:
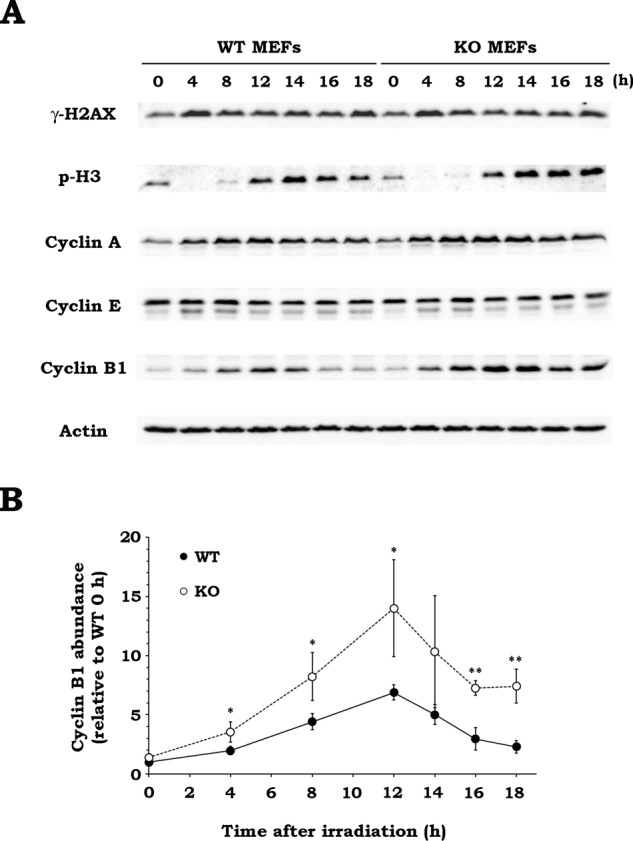
Drp1 deficiency elevates cyclin B1 expression after irradiation. (A) The effect of Drp1 deficiency on protein expression profiles after irradiation. After WT or KO MEFs were x-irradiated at 10 Gy, the cells were collected at the indicated times. The expression of the indicated proteins was analyzed by Western blotting. Representative blots of three experiments. Actin was used as loading control. p-H3, phospho–histone H3. (B) Time-course change of cyclin B1 abundance after irradiation in WT and KO MEFs. WT (black) or KO (white) MEFs were x-irradiated at 10 Gy, and the expression of cyclin B1 was analyzed by Western blotting. The abundance of cyclin B1 in both types of MEFs at the indicated times was quantified relative to that in WT MEFs at 0 h. Data are expressed as means ± SD of three experiments. *p < 0.05, **p < 0.01 vs. WT MEFs (Student’s t test).
DISCUSSION
In the present study, we showed that x-irradiation causes Drp1-dependent mitochondrial fission. Previous studies demonstrated that γ- and α-irradiation also induce mitochondrial fission via Drp1 (Kobashigawa et al., 2011; Zhang et al., 2013), suggesting that mitochondrial fission is a general radioresponse in mammalian cells. However, the mechanism by which IR activates mitochondrial fission via Drp1 remains largely unknown. Drp1 undergoes various posttranslational modifications (PTMs), including phosphorylation, ubiquitination, SUMOylation, and S-nitrosylation, modulating its activity (Otera et al., 2013). Although the underlying mechanism has not been investigated, it is possible that IR induces as-yet- unidentified PTMs on Drp1, altering the property of Drp1 and leading to mitochondrial fission. Thus further studies are required to elucidate what types of PTMs are stimulated by IR and how they affect Drp1 activity and mitochondrial fission.
Our results show that Drp1 is involved in the radiation-induced cell death evaluated by MC and clonogenicity after IR. In fact, both MC and clonogenicity were significantly reduced by Drp1 inhibition (Figure 4). It is well documented that genotoxic stresses, including IR, induce cell death through MC in tumor cells (Vakifahmetoglu et al., 2008; Eriksson and Stigbrand, 2010; Lauber et al., 2012). Although defective or deregulated mitosis is believed to be responsible for the emergence of MC (Portugal et al., 2010; Vitale et al., 2011), its precise mechanism has not been determined. In this study, the pan-caspase inhibitor did not affect radiation-induced MC, and IR did not cause the release of cytochrome c and Smac from the mitochondria to the cytosol, which activates the intrinsic apoptosis pathway (Figure 5). In contrast, IR resulted in aberrant centrosome amplification, which was diminished by Drp1 inhibition (Figure 6). These findings imply that IR induces MC not via the activation of the apoptosis pathway, but via the aberrant amplification of centrosomes and that Drp1 is involved in the latter process. Previous studies indicated that IR, MC, and aberrant centrosome amplification are interrelated. IR was shown to trigger MC, as well as aberrant centrosome amplification (Dodson et al., 2007). In mitotic cells, overreplicated centrosomes lead to the formation of multipolar spindles, resulting in abnormal chromosomal division or failure of cytokinesis, each of which is associated with the induction of MC (Sato et al., 2000; Pihan, 2013). Thus it was indicated that aberrant centrosome amplification mediates MC after IR.
In this study, we showed that radiation-induced aberrant centrosome amplification and MC were significantly suppressed by inhibition of Plk1 and CDK2 in WT MEFs (Figure 7). This suggests the involvement of these kinases in radiation-induced defective/deregulated mitosis. Plk1 and CDK2 are widely considered indispensable for the progression of M and G1/S phase, respectively (Iliakis et al., 2003). Moreover, they are also essential for centrosome maturation and centrosome duplication (lane and Nigg, 1996; Meraldi et al., 1999). Prosser et al. (2012) reported that Plk1 and CDK2 play important roles in aberrant centrosome amplification induced by CDK1 inhibition. CDK2 activation by checkpoint kinase 1 is also involved in aberrant centrosome amplification after γ-irradiation (Bourke et al., 2010). Furthermore, Inanç et al. (2010) demonstrated that Plk1 inhibition attenuates aberrant centrosome amplification after γ-irradiation. These observations are in line with our data, strongly suggesting that these kinases are critical for the radiation-induced aberrant centrosome amplification.
Ubiquitin-dependent destruction of mitotic regulators by the proteasome is crucial for the regulation of cell cycle progression, as well as for the genotoxic stress response. Anaphase-promoting complex/cyclosome (APC/C) is a ubiquitin ligase that initiates the metaphase–anaphase transition and mitotic exit by targeting proteins such as securin and cyclin B1 (Pines, 2011). Normally, cells exposed to genotoxic stress activate cell cycle checkpoints and halt the progression of the cell cycle until DNA damage is resolved (Iliakis et al., 2003). However, in this study, we observed a significant decrease in cyclin B1 level, which indicates APC/C activation, from 12 to 18 h postirradiation in WT MEFs, although γ-H2AX level, which is an index of DSBs, remained elevated during the same period of time (Figure 8A). This finding suggests that the cells aborted the cell cycle checkpoints prematurely and resumed progression into mitosis. This assumption was supported by the time-course change of phospho–histone H3 level in WT MEFs after IR, indicating reentry into mitosis by no later than 12 h postirradiation. It has been shown that during microtubule poison–induced mitotic arrest, the spindle assembly checkpoint (SAC) prevents cell cycle progression by keeping APC/C inactive (Schnerch et al., 2012). However, the SAC does not arrest cells permanently, and escape from mitosis eventually takes place in the presence of the unsatisfied SAC (Brito and Rieder, 2006). This process is known as “mitotic slippage,” in which cells exit mitosis without chromosome segregation or cell division. Slow but steady degradation of cyclin B1 by APC/C in the presence of an active SAC is a biochemical hallmark of mitotic slippage and drives a cell out of mitosis (Brito and Rieder, 2006; Blagosklonny, 2007). Of note, mitotic slippage has been associated with the execution of MC (Vitale et al., 2011). Previous studies reported that IR, similar to microtubule poisons, elicits SAC activation (Dodson et al., 2007), as well as mitotic slippage (Suzuki et al., 2012). Given these data and the continuous decay of cyclin B1 from 12 to 18 h after irradiation observed especially in WT MEFs (Figure 8A), it is possible that IR triggers mitotic slippage via cyclin B1 degradation, resulting in the defective mitotic exit and consequent MC.
In addition to the reduced mitochondrial fission, we found the following effects of Drp1 inhibition on cellular radioresponses: 1) Drp1 inhibition prevented the induction of aberrant centrosome amplification and MC; 2) aberrant centrosome amplification and MC in Drp1-deficient cells were insensitive to the inhibition of Plk1 and CDK2; and 3) Drp1 deficiency resulted in the accumulation of cyclin B1 after IR. These findings suggest that Drp1 inhibition impairs APC/C-dependent cyclin B1 destruction, leading to the attenuation of radiation-induced aberrant centrosome amplification and MC, which normally occurs via the activation of Plk1- and CDK2-mediated signaling pathways in Drp1-expressing cells. Because DSB level, evaluated by γ-H2AX, was not influenced by Drp1 inhibition (Figure 8A), the effects of Drp1 inhibition are unlikely to be due to the suppression of the DNA damage repair capacity by Drp1 inhibition. Previous studies reported that Drp1 inhibition hampers cell cycle progression by delaying G2/M progression (Marsboom et al., 2012; Qian et al., 2012), potentially accounting for the reduced induction of defective mitosis in KO MEFs. However, we found that the cell cycle profiles, judged by cellular DNA content, after IR were quite similar in WT and KO MEFs (unpublished data), indicating otherwise. From these observations, it is possible that there is a Drp1-dependent regulatory mechanism of APC/C, controlling the degradation of its substrates, including cyclin B1. If the aforementioned mechanism behind radiation-induced MC is correct, we speculate that Drp1 inhibition prevents it by disturbing APC/C activity via a currently unknown mechanism. Of interest, it has been shown that Drp1 siRNA attenuates Cdh1 expression (Qian et al., 2012), which serves as a coactivator of APC/C, and that Cdh1 depletion elevates cyclin B1 level (Engelbert et al., 2008). These findings imply that the increase in cyclin B1 level in Drp1-deficient cells could result from the Drp1-dependent down-regulation of Cdh1. On the other hand, it has been shown that APC/CCdh1 mediates the proteasomal degradation of Drp1 (Horn et al., 2011), suggesting that there might be a feedback mechanism between Drp1 and Cdh1 to regulate each other’s protein level.
In summary, the present study demonstrates that IR triggers Drp1-dependent mitochondrial fission and that Drp1 inhibition attenuates radiation-induced MC, suggesting that Drp1 is involved in determining the fate of cells after irradiation by controlling mitochondrial shape. Our findings imply that mitochondrial dynamics plays an important role in cellular radioresponses and that interfering with mitochondrial dynamics could be an approach to altering cellular radiosensitivity and the efficacy of radiotherapy.
MATERIALS AND METHODS
Reagents
MitoTracker Green FM, DAPI, and Alexa Fluor 488– phalloidin were purchased from Life Technologies (Carlsbad, CA). Digitonin and etoposide were from Wako Pure Chemical Industries (Osaka, Japan). Mdivi-1 was from Enzo Life Sciences (Farmingdale, NY). Z-VAD-FMK was purchased from Peptide Institute (Osaka, Japan). NU6140 and BI2536 were from Santa Cruz Biotechnology (Santa Cruz, CA). The following antibodies were used for Western blotting and immunostaining: anti-Drp1, anti–cytochrome c (BD Biosciences, Billerica, MA), anti–γ-tubulin (Sigma-Aldrich, St. Louis, MO), anti-Smac (ProSci, Poway, CA), anti–phospho-Plk1 (Thr-210), anti-Plk1 (Abcam, Cambridge, United Kingdom), anti–phospho-CDK2 (Thr-160), anti-γ-H2AX, anti–phospho-histone H3 (Ser-10; Cell Signaling Technology, Beverly, MA), Alexa Fluor 488 anti-mouse immunoglobulin G (Life Technologies), anti–voltage-dependent anion-selective channel protein 1 (VDAC1), anti–glyceraldehyde 3-phosphate dehydrogenase (GAPDH), anti-CDK2, anti–cyclin A, anti–cyclin E, anti–cyclin B1, anti-actin, and horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz Biotechnology). The Western Lightning Plus-ECL chemiluminescence detection kit was purchased from PerkinElmer-Cetus (Boston, MA).
Cell culture and x-irradiation
The SV40-immortalized MEFs derived from Drp1-deficient mice (KO MEFs) and its control wild-type cells (WT MEFs) were kindly provided by Masatoshi Nomura (Kyushu University, Fukuoka, Japan; Ishihara et al., 2009) and were maintained in DMEM (Life Technologies) containing 10% (vol/vol) fetal bovine serum (Biowest, Nuaille, France) at 37°C in 5% CO2. The x-irradiation was performed at room temperature using a Shimadzu PANTAK HF-320 x-ray generator (Shimadzu; Kyoto, Japan) or an X-RAD iR-225 x-ray irradiator (Precision X-Ray, North Branford, CT) with a dose rate of 2.54 Gy/min at 200 kVp and 20 mA with a 1.0-mm aluminum filter and 1.37 Gy/min at 200 kVp and 15 mA with a 1.0-mm aluminum filter, respectively. Where necessary, cells were treated with drugs for 2 h before x-irradiation. After irradiation, the medium was replaced with fresh growth medium containing drugs, and the cells were cultured for analysis.
Mitochondrial morphology
Cells were seeded on 35-mm glass-bottomed dishes (AGC Techno Glass, Shizuoka, Japan) and cultured overnight. At the times indicated after x-irradiation, the cells were incubated with serum-free medium containing 300 nM MitoTracker Green FM for 30 min at 37°C. After two washes with serum-free medium, fresh growth medium was supplied. Fluorescence images of live cells were obtained using an LSM700 confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany) at 37°C in 5% CO2. To quantify mitochondrial morphology, a cell was judged to have fragmented mitochondria if >75% of the mitochondria visible in the cell were punctate or circular and fused if <25% of the mitochondria were punctate or circular. The fraction of fused mitochondria was subdivided into highly connected and tubular, depending on the length of mitochondrial filaments (“highly connected” mitochondria appear more elongated than “tubular” mitochondria). If cells presented mitochondria with mixed morphologies, we classified them as intermediate. Representative images of the different mitochondrial morphologies in each category are shown in Supplemental Figure S1. In one experiment, >50 cells/condition were randomly chosen and classified as described, and the percentage of each category was calculated. Experiments were repeated three times.
Crude subcellular fractionation
At the indicated times after irradiation, cells were collected and counted. After 1 × 106 cells were transferred to a 1.5-ml tube and centrifuged at 3000 × g for 1 min at 4°C, the pellets were suspended with 50 μl of plasma membrane permeabilization buffer (400 μg/ml digitonin, 137 mM NaCl, 8.1 mM Na2HPO4, 80 mM KCl, and 1.47 mM KH2PO4) and incubated for 5 min on ice. After centrifugation at 800 × g for 5 min at 4°C, the supernatants were collected (cytosol fraction). The remaining pellets were suspended with lysis buffer (50 mM Tris-HCl, pH 7.5, 1% [vol/vol] Triton X-100, 5% [vol/vol] glycerol, 5 mM EDTA, and 150 mM NaCl) and incubated for 15 min on ice. After centrifugation at 20,000 × g for 15 min at 4°C, the supernatants were collected (organelle fraction). For Western blotting analysis, 3× Laemmli’s sample buffer (0.1875 M Tris-HCl, pH 6.8, 15% [vol/vol] β-mercaptoethanol, 6% [wt/vol] SDS, 30% [vol/vol] glycerol, and 0.006% [wt/vol] bromophenol blue) was added to the collected supernatants, and the samples were boiled for 3 min.
SDS–PAGE and Western blotting
Cells were collected and lysed with lysis buffer (50 mM Tris-HCl, pH 7.5, 1% [vol/vol] Triton X-100, 5% [vol/vol] glycerol, 5 mM EDTA, and 150 mM NaCl). After centrifugation at 18,000 × g for 15 min at 4°C, supernatants were collected. Threefold-concentrated Laemmli’s sample buffer (0.1875 M Tris-HCl, pH 6.8, 15% [vol/vol] β-mercaptoethanol, 6% [wt/vol] SDS 30% [vol/vol] glycerol, and 0.006% [wt/vol] bromophenol blue) was added to the supernatants, and the samples were boiled for 3 min. Proteins were separated by SDS–PAGE and transferred onto a nitrocellulose membrane (Advantec TOYO, Tokyo, Japan). The membrane was blocked with TBST (10 mM Tris-HCl, pH 7.4, 0.1 M NaCl, and 0.1% Tween-20) containing 5% (wt/vol) nonfat skim milk and probed with specific antibodies diluted with TBST containing 5% (wt/vol) nonfat skim milk or 5% (wt/vol) bovine serum albumin (BSA) overnight at 4°C. After probing with HRP-conjugated secondary antibodies, the bound antibodies were detected with Western Lightning Plus-ECL. Image acquisition was performed with an LAS 4000 mini image analyzer (Fujifilm, Tokyo, Japan), and image analysis was done by MultiGauge software (Fujifilm).
Analysis of MC and centrosome numbers
Cells were seeded on glass coverslips coated with collagen (Cellmatrix Type I-C; Nitta Gelatin, Osaka, Japan) and cultured overnight. After x-irradiation, the cells were fixed with 3.7% (wt/vol) paraformaldehyde/phosphate-buffered saline (PBS) for 10 min at room temperature. After permeabilization with 0.1% (vol/vol) Triton X-100/PBS for 5 min at room temperature, the cells were treated with PBS containing 1% (wt/vol) BSA for 30 min at room temperature. Subsequently they were stained with 1% (vol/vol) Alexa Fluor 488–phalloidin to visualize the cell outlines. After being washed twice with PBS, they were incubated with 300 nM DAPI for 5 min at room temperature, and coverslips were mounted with ProLong Gold antifade reagent (Life Technologies). Fluorescence microscopy analysis was performed using an Olympus BX61 microscope (Olympus, Tokyo, Japan) with a reflected light fluorescence. At least 200 cells were counted, and the cells with features of aberrant mitotic nuclei (micronuclei, multilobular nuclei, and fragmented nuclei) were scored as cells undergoing MC. For centrosomal staining, irradiated cells were fixed with ice-cold methanol for 10 min at −20°C. After permeabilization with ice-cold acetone for 1 min at −20°C, the cells were washed three times with PBS, followed by treatment with PBS containing 6% (wt/vol) goat serum for 1 h at room temperature. Subsequently they were incubated with the anti–γ-tubulin antibody (1:5000) in 3% goat serum/PBS overnight at 4°C. They were then incubated in the dark with the Alexa Fluor 488–conjugated anti-mouse secondary antibody at a 1:500 dilution in 3% goat serum/PBS for 1.5 h. After incubation, they were washed three 3 times with PBS and counterstained with 300 nM DAPI for 5 min at room temperature. The coverslips were mounted, and fluorescence microscopic images were acquired as described. The γ-tubulin foci per cell were counted manually for at least 200 cells per condition, and the percentage of the cells containing more than two foci was determined.
Clonogenic survival assay
Cells were seeded on 6-cm dishes and cultured for 6 h. The cells were then x-irradiated, and the medium was replaced with fresh growth medium. After incubation for 7 d, they were fixed with methanol and stained with Giemsa solution. Colonies containing >50 cells were scored as surviving cells. Survival fractions were calculated with a correction for the plating efficiency of the nonirradiated control.
Statistical analysis
All results are expressed as means ± SD of at least three separate experiments. Comparison of two groups was performed with Student’s t tests. For multiple comparisons, Dunnett’s test or the Tukey–Kramer test was used. The minimum level of significance was set at p < 0.05.
Supplementary Material
Acknowledgments
We are deeply grateful to Masatoshi Nomura for generously providing MEFs. This work was supported, in part, by the Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research (23780286 [T.Y.], 25861045 [H.Y.], and 24659551 [O.I.]), the Takeda Science Foundation (T.Y.), and a research grant for the Study of the Health Effects of Radiation Organized by Ministry of the Environment, Japan (O.I. and T.Y.). The sponsors had no role in study design, collection, analysis, and interpretation of data, writing of the manuscript, or decision to submit the manuscript for publication.
Abbreviations used:
- APC/C
anaphase-promoting complex/cyclosome
- BSA
bovine serum albumin
- CDK2
cyclin-dependent kinase 2
- DAPI
4′,6-diamidino-2-phenylindole
- Drp1
dynamin-related protein 1
- γ-H2AX
phosphorylated histone H2AX
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HRP
horseradish peroxidase
- IR
ionizing radiation
- MC
mitotic catastrophe
- MEF
mouse embryonic fibroblast
- PBS
phosphate-buffered saline
- Plk1
Polo-like kinase 1
- PTM
posttranslational modification
- ROS
reactive oxygen species
- SAC
spindle assembly checkpoint
- VDAC
voltage-dependent anion-selective channel protein 1.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-03-0181) on October 14, 2015.
REFERENCES
- Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Gräber S, Kovacs I, Lee WD, Waggoner J, Cui J, et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagosklonny MV. Mitotic arrest and cell fate: why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle. 2007;6:70–74. doi: 10.4161/cc.6.1.3682. [DOI] [PubMed] [Google Scholar]
- Bourke E, Brown JAL, Takeda S, Hochegger H, Morrison CG. DNA damage induces Chk1-dependent threonine-160 phosphorylation and activation of Cdk2. Oncogene. 2010;29:616–624. doi: 10.1038/onc.2009.340. [DOI] [PubMed] [Google Scholar]
- Brinkley BR. Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol. 2001;11:18–21. doi: 10.1016/s0962-8924(00)01872-9. [DOI] [PubMed] [Google Scholar]
- Brito DA, Rieder CL. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol. 2006;16:1194–1200. doi: 10.1016/j.cub.2006.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Roumier T, Valent A, Raslova H, Yakushijin K, Horne D, Feunteun J, Lenoir G, Medema R, et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene. 2004a;23:4362–4370. doi: 10.1038/sj.onc.1207572. [DOI] [PubMed] [Google Scholar]
- Castedo M, Perfettini J-L, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004b;23:2825–2837. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- Cheung ECC, McBride HM, Slack RS. Mitochondrial dynamics in the regulation of neuronal cell death. Apoptosis. 2007;12:979–992. doi: 10.1007/s10495-007-0745-5. [DOI] [PubMed] [Google Scholar]
- Descombes P, Nigg EA. The polo-like kinase Plx1 is required for M phase exit and destruction of mitotic regulators in Xenopus egg extracts. EMBO J. 1998;17:1328–1335. doi: 10.1093/emboj/17.5.1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- Dodson H, Wheatley SP, Morrison CG. Involvement of centrosome amplification in radiation-induced mitotic catastrophe. Cell Cycle. 2007;6:364–370. doi: 10.4161/cc.6.3.3834. [DOI] [PubMed] [Google Scholar]
- Engelbert D, Schnerch D, Baumgarten A, Wäsch R. The ubiquitin ligase APC(Cdh1) is required to maintain genome integrity in primary human cells. Oncogene. 2008;27:907–917. doi: 10.1038/sj.onc.1210703. [DOI] [PubMed] [Google Scholar]
- Eriksson D, Stigbrand T. Radiation-induced cell death mechanisms. Tumour Biol. 2010;31:363–372. doi: 10.1007/s13277-010-0042-8. [DOI] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- Gong B, Chen Q, Almasan A. Ionizing radiation stimulates mitochondrial gene expression and activity. Radiat Res. 1998;150:505–512. [PubMed] [Google Scholar]
- Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annu Rev Biochem. 2007;76:751–780. doi: 10.1146/annurev.biochem.76.071905.090048. [DOI] [PubMed] [Google Scholar]
- Horn SR, Thomenius MJ, Johnson ES, Freel CD, Wu JQ, Coloff JL, Yang CS, Tang W, An J, Ilkayeva OR, et al. Regulation of mitochondrial morphology by APC/CCdh1-mediated control of Drp1 stability. Mol Biol Cell. 2011;22:1207–1216. doi: 10.1091/mbc.E10-07-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Fletcher L, Beeharry N, Daniel R, Kao G, Yen TJ, Muschel RJ. Abnormal cytokinesis after X-irradiation in tumor cells that override the G2 DNA damage checkpoint. Cancer Res. 2008;68:3724–3732. doi: 10.1158/0008-5472.CAN-08-0479. [DOI] [PubMed] [Google Scholar]
- Hunt T, Luca FC, Ruderman JV. The requirements for protein synthesis and degradation, and the control of destruction of cyclins A and B in the meiotic and mitotic cell cycles of the clam embryo. J Cell Biol. 1992;116:707–724. doi: 10.1083/jcb.116.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliakis G, Wang Y, Guan J, Wang H. DNA damage checkpoint control in cells exposed to ionizing radiation. Oncogene. 2003;22:5834–5847. doi: 10.1038/sj.onc.1206682. [DOI] [PubMed] [Google Scholar]
- Inanç B, Dodson H, Morrison CG. A centrosome-autonomous signal that involves centriole disengagement permits centrosome duplication in G2 phase after DNA damage. Mol Biol Cell. 2010;21:3866–3877. doi: 10.1091/mbc.E10-02-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagasia R, Grote P, Westermann B, Conradt B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature. 2005;433:754–760. doi: 10.1038/nature03316. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Youle RJ. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003;10:870–880. doi: 10.1038/sj.cdd.4401260. [DOI] [PubMed] [Google Scholar]
- Kobashigawa S, Suzuki K, Yamashita S. Ionizing radiation accelerates Drp1-dependent mitochondrial fission, which involves delayed mitochondrial reactive oxygen species production in normal human fibroblast-like cells. Biochem Biophys Res Commun. 2011;414:795–800. doi: 10.1016/j.bbrc.2011.10.006. [DOI] [PubMed] [Google Scholar]
- Kulkarni R, Marples B, Balasubramaniam M, Thomas RA, Tucker JD. Mitochondrial gene expression changes in normal and mitochondrial mutant cells after exposure to ionizing radiation. Radiat Res. 2010;173:635–644. doi: 10.1667/RR1737.1. [DOI] [PubMed] [Google Scholar]
- Lane HA, Nigg EA. Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J Cell Biol. 1996;135:1701–1713. doi: 10.1083/jcb.135.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber K, Ernst A, Orth M, Herrmann M, Belka C. Dying cell clearance and its impact on the outcome of tumor radiotherapy. Front Oncol. 2012;2:1–14. doi: 10.3389/fonc.2012.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB, Tuyle G Van. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001;61:3894–3901. [PubMed] [Google Scholar]
- Li J, Li Y, Qin D, Von Harsdorf R, Li P. Mitochondrial fission leads to Smac/DIABLO release quenched by ARC. Apoptosis. 2010;15:1187–1196. doi: 10.1007/s10495-010-0514-8. [DOI] [PubMed] [Google Scholar]
- Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J, et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res. 2012;110:1484–1497. doi: 10.1161/CIRCRESAHA.111.263848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA. Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat Cell Biol. 1999;1:88–93. doi: 10.1038/10054. [DOI] [PubMed] [Google Scholar]
- Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy-Wetzel E, Basañez G, Meda P, et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates bax oligomerization. Cell. 2010;142:889–901. doi: 10.1016/j.cell.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent SME, Mothersill CE, Seymour C, Mcclean B, Fiona M, Murphy JEJ, Lyng M, Lyng FM. Increased mitochondrial mass in cells with functionally compromised mitochondria after exposure to both direct gamma radiation and bystander factors. Radiat Res. 2007;168:134–142. doi: 10.1667/RR0769.1. [DOI] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura A, Oowada S, Kon Y, Hirayama A, Yasui H, Meike S, Kobayashi S, Kuwabara M, Inanami O. Redox regulation in radiation-induced cytochrome c release from mitochondria of human lung carcinoma A549 cells. Cancer Lett. 2009;277:64–71. doi: 10.1016/j.canlet.2008.11.021. [DOI] [PubMed] [Google Scholar]
- Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta. 2013;1833:1256–1268. doi: 10.1016/j.bbamcr.2013.02.002. [DOI] [PubMed] [Google Scholar]
- Pihan GA. Centrosome dysfunction contributes to chromosome instability, chromoanagenesis, and genome reprograming in cancer. Front Oncol. 2013;3:277. doi: 10.3389/fonc.2013.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines J. Cubism and the cell cycle: the many faces of the APC/C. Nat Rev Mol Cell Biol. 2011;12:427–438. doi: 10.1038/nrm3132. [DOI] [PubMed] [Google Scholar]
- Portugal J, Mansilla S, Bataller M. Mechanisms of drug-induced mitotic catastrophe in cancer cells. Curr Pharm Des. 2010;16:69–78. doi: 10.2174/138161210789941801. [DOI] [PubMed] [Google Scholar]
- Prosser SL, Samant MD, Baxter JE, Morrison CG, Fry AM. Oscillation of APC/C activity during cell cycle arrest promotes centrosome amplification. J Cell Sci. 2012;125:5353–5368. doi: 10.1242/jcs.106096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Choi S, Gibson GA, Watkins SC, Bakkenist CJ, Van Houten B. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J Cell Sci. 2012;125:5745–5757. doi: 10.1242/jcs.109769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran S, Harrison SH, Thomas RA, Tucker JD. The role of mitochondria in the radiation-induced bystander effect in human lymphoblastoid cells. Radiat Res. 2011;175:159–171. doi: 10.1667/rr2296.1. [DOI] [PubMed] [Google Scholar]
- Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C, Archer SL. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012;26:2175–2186. doi: 10.1096/fj.11-196543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rello-Varona S, Kepp O, Vitale I, Michaud M, Senovilla L, Jemaà M, Joza N, Galluzzi L, Castedo M, Kroemer G. An automated fluorescence videomicroscopy assay for the detection of mitotic catastrophe. Cell Death Dis. 2010;1:e25. doi: 10.1038/cddis.2010.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Mizumoto K, Nakamura M, Ueno H, Minamishima YA, Farber JL, Tanaka M. A possible role for centrosome overduplication in radiation-induced cell death. Oncogene. 2000;19:5281–5290. doi: 10.1038/sj.onc.1203902. [DOI] [PubMed] [Google Scholar]
- Schnerch D, Follo M, Krohs J, Felthaus J, Engelhardt M, Wäsch R. Monitoring APC/C activity in the presence of chromosomal misalignment in unperturbed cell populations. Cell Cycle. 2012;11:310– 321. doi: 10.4161/cc.11.2.18737. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Yamauchi M, Oka Y, Suzuki K, Yamashita S. Live-cell imaging visualizes frequent mitotic skipping during senescence-like growth arrest in mammary carcinoma cells exposed to ionizing radiation. Int J Radiat Oncol Biol Phys. 2012;83:e241–e250. doi: 10.1016/j.ijrobp.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- Taneja N, Tjalkens R, Philbert MA, Rehemtulla A. Irradiation of mitochondria initiates apoptosis in a cell free system. Oncogene. 2001;20:167–177. doi: 10.1038/sj.onc.1204054. [DOI] [PubMed] [Google Scholar]
- Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008;15:1153–1162. doi: 10.1038/cdd.2008.47. [DOI] [PubMed] [Google Scholar]
- Van Leuken R, Clijsters L, Wolthuis R. To cell cycle, swing the APC/C. Biochim Biophys Acta. 2008;1786:49–59. doi: 10.1016/j.bbcan.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385–392. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- Waterham HR, Koster J, van Roermund CWT, Mooyer PAW, Wanders RJA, Leonard J V. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736–1741. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- Yamamori T, Yasui H, Yamazumi M, Wada Y, Nakamura Y, Nakamura H, Inanami O. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic Biol Med. 2012;53:260–270. doi: 10.1016/j.freeradbiomed.2012.04.033. [DOI] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Davidson MM, Zhou H, Wang C, Walker WF, Hei TK. Cytoplasmic irradiation results in mitochondrial dysfunction and DRP1-dependent mitochondrial fission. Cancer Res. 2013;73:6700–6710. doi: 10.1158/0008-5472.CAN-13-1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y-X, Cui M, Chen S-F, Dong Q, Liu X-Y. Amelioration of ischemic mitochondrial injury and Bax-dependent outer membrane permeabilization by Mdivi-1. CNS Neurosci Ther. 2014:528–538. doi: 10.1111/cns.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Ivanov VN, Lien Y-C, Davidson M, Hei TK. Mitochondrial function and nuclear factor-kappaB-mediated signaling in radiation-induced bystander effects. Cancer Res. 2008;68:2233–2240. doi: 10.1158/0008-5472.CAN-07-5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.