

The controlled permeabilization of lysosomes and vacuoles may represent an ancient manner of programmed cell death. It is shown that TORC1 is required for lysosomal membrane permeabilization and death of yeast cells that have been exposed to antifungals, and that a novel FYVE-domain protein regulates TORC1 signaling in these conditions.
Abstract
Lysosomal membrane permeabilization (LMP) is a poorly understood regulator of programmed cell death that involves leakage of luminal lysosomal or vacuolar hydrolases into the cytoplasm. In Saccharomyces cerevisiae, LMP can be induced by antifungals and endoplasmic reticulum stressors when calcineurin also has been inactivated. A genome-wide screen revealed Pib2, a relative of LAPF/phafin1 that regulates LMP in mammals, as a pro-LMP protein in yeast. Pib2 associated with vacuolar and endosomal limiting membranes in unstressed cells in a manner that depended on its FYVE domain and on phosphatidylinositol 3-phosphate (PI(3)P) biosynthesis. Genetic experiments suggest that Pib2 stimulates the activity of TORC1, a vacuole-associated protein kinase that is sensitive to rapamycin, in a pathway parallel to the Ragulator/EGO complex containing the GTPases Gtr1 and Gtr2. A hyperactivating mutation in the catalytic subunit of TORC1 restored LMP to the gtr1∆ and pib2∆ mutants and also prevented the synthetic lethality of the double mutants. These findings show novel roles of PI(3)P and Pib2 in the regulation of TORC1, which in turn promoted LMP and nonapoptotic death of stressed cells. Rapamycin prevented the death of the pathogenic yeast Candida albicans during exposure to fluconazole plus a calcineurin inhibitor, suggesting that TORC1 broadly promotes sensitivity to fungistats in yeasts.
INTRODUCTION
Lysosomal membrane permeabilization (LMP) has been implicated as an important trigger and amplifier of apoptosis and a crucial component of both developmental and stress-induced necrosis in diverse species of animals (Boya and Kroemer, 2008; Mrschtik and Ryan, 2015) and plants (van Doorn, 2011; Hatsugai et al., 2015). In mammals, involution of the mammary gland upon cessation of nursing relies on LMP and necrosis rather than apoptosis (Kreuzaler et al., 2011). In the fruit fly Drosophila melanogaster, normal germ cell development relies on LMP (Peterson and McCall, 2013; Yacobi-Sharon et al., 2013). In the nematode Caenorhabditis elegans, neurons that have been stressed by hypoxia or hyperactive ion channels undergo LMP-dependent necrosis-like cell death in a process that involves cytosolic Ca2+, calpain proteases, the lysosomal membrane V-ATPase, and release of lysosomal cathepsin proteases and acids (Syntichaki et al., 2002, 2005). A similar calpain–cathepsin cascade triggers LMP in hippocampal CA1 neurons of primates during oxidative stress (Yamashima and Oikawa, 2009). In filamentous fungi, LMP may drive the vacuole-dependent programmed cell deaths that occur very rapidly upon mating of incompatible hyphal cells (Glass and Dementhon, 2006; Pinan-Lucarre et al., 2007). LMP also occurs naturally in cells of the yeast Saccharomyces cerevisiae undergoing the developmental stage of sporulation, as part of the autolytic process that supplies essential nutrients to developing spores (Eastwood et al., 2013). Though the occurrence of LMP is apparently widespread among eukaryotes, the underlying molecular mechanisms that regulate these phenomena are poorly understood.
LMP may also be responsible for the fungicidal synergism of combining the azole class of fungistatic compounds (e.g., fluconazole, miconazole) commonly used to treat a wide array of fungal infections in humans with inhibitors of calcineurin (e.g., FK506, cyclosporin A; Fox and Heitman, 2002; Steinbach et al., 2007; Liu et al., 2015). All the azoles target Erg11 (lanosterol desaturase), an essential enzyme in the endoplasmic reticulum (ER) that is required for cell proliferation but not required for cell survival. Azoles alone slow proliferation rates but do not kill fungal cells, but coadministration of azoles with calcineurin inhibitors causes dramatic loss of viability (i.e., a decline in colony-forming units). In most fungi, inhibitors of calcineurin alone or even knockout of calcineurin-encoding genes have little effect on growth rate in vitro, though virulence in vivo can be attenuated in some cases. The activation of calcineurin by Ca2+ and calmodulin therefore represents a protective response in many fungal species from the cidal effects of azole-class drugs.
The regulatory mechanism responsible for fungistat-induced and calcineurin-repressed LMP has been investigated most thoroughly in S. cerevisiae. Because this yeast is naturally resistant to most azole-class fungistats, the fungistatic compounds tunicamycin and dithiothreitol (DTT; inhibitors of Alg7 and Ero1 in the ER, which are required for protein N-glycosylation and protein disulfide bonding) were utilized instead and were shown to become fungicidal in the presence of calcineurin inhibitors (FK506 and cyclosporin A). Tunicamycin and DTT cause ER stress and activation of the UPR signaling pathway, but the Ire1 sensor/transducer of the UPR was not involved in either the activation of calcineurin or the suppression of cell death (Bonilla et al., 2002). Instead, calcineurin became activated through the activation of a stress-activated MAP kinase cascade (Bonilla and Cunningham, 2003), induction of K+ transporters (Stefan and Cunningham, 2013; Stefan et al., 2013), and activation of a high-affinity Ca2+ influx system that elevated cytosolic Ca2+ and activated calmodulin (Muller et al., 2001; Martin et al., 2011). In addition to calcineurin, the Ca2+/calmodulin-dependent protein kinase Cmk2 was found to weakly suppress cell death (Dudgeon et al., 2008), similar to the cell death caused by high mating pheromones (Zhang et al., 2006). A genome-wide screen for mutants that failed to die when exposed to ER stressors plus calcineurin inhibitors identified the V-ATPase, which directly pumps H+ and acidifies lysosomes and vacuoles, as necessary for LMP that precedes each cell death (Kim et al., 2012). The V-ATPase had been shown previously to promote LMP and necrosis-like deaths of degenerating neurons in C. elegans (Syntichaki et al., 2005) and of involuting mammary glands (Arnandis et al., 2012). V-ATPase inhibitors blocked early stages of the response to tunicamycin, such as the fission of large vacuoles into many smaller ones, but were ineffective at blocking the later calcineurin-sensitive stages that lead to LMP and cell death (Kim et al., 2012). Though required for the early phase of LMP regulation, the V-ATPase is not likely a relevant target of calcineurin in these conditions. The substrates of calcineurin involved in LMP remain to be identified, as do the protein kinases responsible for their initial phosphorylation.
In this paper, we report the identification of the TORC1 protein kinase as a critical promoter of LMP and cell death in S. cerevisiae cells exposed to ER stressors and calcineurin inhibitors. TORC1 and its homologues in animals are multisubunit enzymes that associate peripherally with the limiting membranes of lysosomes and vacuoles and are the direct targets of the immunosuppressive drug rapamycin (Loewith and Hall, 2011). TORC1 is well known as a master regulator of cell growth and proliferation, integrating a variety of nutrient and stress conditions and coordinating ribosome synthesis and function in translation. Mammalian TORC1 receives inputs from the V-ATPase, the RAG family of small GTPases, the Rheb family of small GTPases, and other regulators, which themselves relay information on cellular amino acid availability, energy resources, and stresses to TORC1 (Sengupta et al., 2010). Mammalian TORC1 has both prodeath and prosurvival activities in different cell types and circumstances (Wang et al., 2009). In S. cerevisiae, TORC1 activity depends partly on the RAG-family GTPases (Gtr1, Gtr2) and their membrane-tethering subunits (Ego1, Ego3), which together form the EGO complex that directly binds the vacuolar membrane and subunits of TORC1 (Loewith and Hall, 2011). S. cerevisiae mutants lacking any subunit of the EGO complex have reduced TORC1 signaling and are hypersensitive to rapamycin and other inhibitors of TORC1 kinase activity such as caffeine (Reinke et al., 2006; Wanke et al., 2008). LMP, and subsequent death, of cells exposed to ER stressors and calcineurin inhibitors is shown below to be strongly delayed upon the loss of the EGO complex or the addition of rapamycin.
One of the few proteins found to promote LMP in mammalian cell lines is LAPF/phafin1 (Chen et al., 2005), which associates with lysosomal membranes through interactions with its conserved FYVE domain and a short C-terminal tail motif (Li et al., 2007). Overexpression of LAPF/phafin1 additionally promoted autophagy (Lin et al., 2012), a process that is normally repressed by TORC1 signaling. An uncharacterized relative of LAPF/phafin1 in S. cerevisiae, termed Pib2, is shown in our study to utilize its FYVE domain and tail motif to promote LMP and death of stressed calcineurin-deficient cells through the stimulation of TORC1 signaling. These findings reveal a novel input to TORC1 signaling that involves PI(3)P interaction with FYVE domains of Pib2-family proteins. They also reveal a novel output of TORC1 signaling in S. cerevisiae and in C. albicans to promote LMP and nonapoptotic cell death responsible for conversion of fungistats into fungicides.
RESULTS
Pib2, a FYVE-domain protein related to LAPF/phafin1, promotes LMP and cell death
A recent study of more than 4800 viable gene-knockout mutants of S. cerevisiae revealed the multisubunit V-ATPase as necessary for LMP and the subsequent death of cells that were stressed with either tunicamycin or DTT in the presence of calcineurin inhibitors (Kim et al., 2012). Several additional vacuole-associated proteins were found to have roles in this process, and we focused on Pib2 first, because of its similarity to human LAPF/phafin1, which was previously shown to promote LMP and caspase-independent cell death in cell lines exposed to tumor necrosis factor α (Chen et al., 2005). Pib2 has not been studied extensively to date and was named originally for the presence of a putative PI(3)P-binding FYVE domain near the C-terminus (Shin et al., 2001). Clear orthologues of Pib2 were found encoded in the genomes of nearly all species of fungi using PSI-BLAST searches (see Materials and Methods). Alignment of Pib2 sequences from multiple fungal species revealed only two universally conserved motifs: the FYVE domain near the C-termini and a 13-amino-acid “tail motif” at the extreme C-termini, which were separated by a nonconserved spacer segment (see Figure 1A). Though the N-terminal portions of these proteins were extremely variable and difficult to align, several other weakly conserved motifs (designated A to E motifs) could be discerned only among the yeasts or the ascomycetes (summarized in Figure 1A). Of the 27 human proteins listed in the SMART database as containing FYVE domains, only LAPF/phafin1 contains a FYVE domain near the C-terminus and a conserved tail motif at the C-terminus. Multiple sequence alignments of LAPF/phafin1 orthologues from jawed vertebrates indicate similarity between its tail motif and that of the Pib2 family, except that it is extended by two amino acid residues (Figure 1A). Vertebrates and most invertebrates also express a close paralogue of LAPF/phafin1 termed EAPF/phafin2, which appears to lack the entire tail motif but retains the FYVE domain as well as a strongly conserved pleckstrin homology (PH) domain at the N-terminus (Chen et al., 2005). In spite of the N-terminal differences, Pib2 and its orthologues in fungi seem to be the closest relatives of LAPF/phafin1 proteins in vertebrates.
FIGURE 1:

Domain architecture and subcellular localization of Pib2. (A) Schematic diagrams of Pib2 from S. cerevisiae, other budding yeasts (C. albicans, Yarrowia lipolytica), fission yeast (Schizosaccharomyces pombe), a mushroom (Lichtheimia hyalospora), plus EAPF/phafin1 and LAPF/phafin2 (Homo sapiens), indicating the approximate positions of partially conserved motifs (A–E) and the universally conserved FYVE domain and tail motif (T). Gray bars indicate segments of poor conservation. Sequence logos of tail motifs from Pib2- and LAPF/phafin1 proteins are shown. (B) Fluorescence micrographs of wild-type and vps34∆ mutant strains of S. cerevisiae expressing GFP or GFP-Pib2 fusion protein (with and without the FYVE domain) after 30 min of staining with FM4-64 to reveal vacuolar and endosomal membranes.
For testing whether Pib2 associates with PI(3)P in the membranes of endosomes and vacuoles, a green fluorescent protein (GFP)-Pib2 fusion protein was expressed at high levels from a methionine-repressible promoter in wild type and vps34∆ mutants, which lack the sole enzyme capable of synthesizing PI(3)P (Schu et al., 1993). GFP-Pib2 clearly associated with the limiting membranes of large vacuoles and smaller endosome-like structures and was often enriched at sites of contact between these organelles (Figure 1B). The dye FM4-64 was similarly localized but not enriched at these contact sites after endocytosis (Figure 1B, bottom panels). GFP-Pib2 relocalized to the cytoplasm in vps34∆ similar to GFP alone and a derivative of GFP-Pib2 that specifically lacked the FYVE domain (Figure 1B). Therefore Pib2 localized to PI(3)P-rich intracellular membranes in S. cerevisiae similar to LAPF/phafin1-family proteins in animal cells.
The plasmid expressing GFP-Pib2 was able to fully restore high rates of cell death when introduced into pib2∆ mutants in partially repressing conditions (100% methionine added), but this complementation declined unexpectedly as GFP-Pib2 became overexpressed by lowering methionine to 12.5% (Figure 2A). In this experiment, cell death was measured 8 h after exposure of cells to tunicamycin plus FK506, an inhibitor of calcineurin, in standard synthetic complete (SC) medium. Interestingly, overexpression of GFP-Pib2 in wild-type cells also slowed the rate of cell death to levels approaching those of pib2∆ cells (Figure 2A). Therefore low levels of GFP-Pib2 complemented the pib2∆ mutant, and high levels produced a dominant-negative effect in wild-type cells.
FIGURE 2:
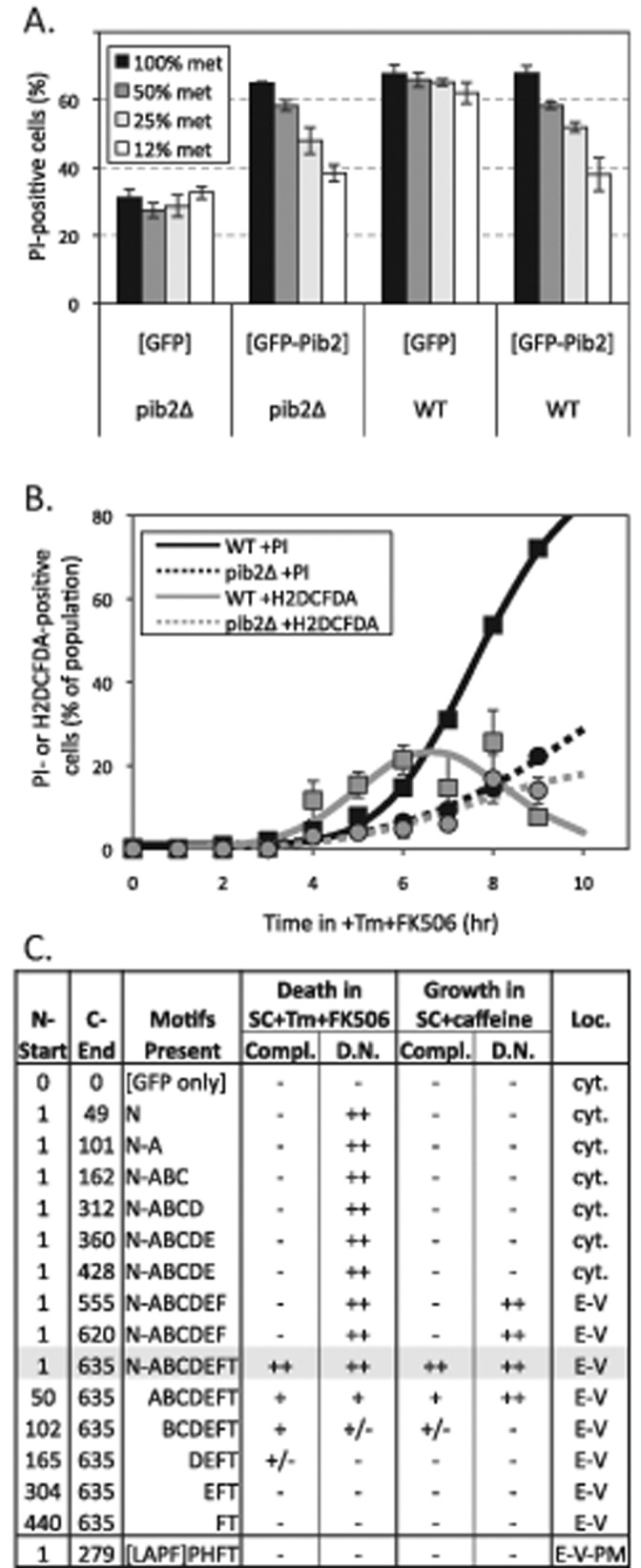
Complementation and dominant-negative activities of GFP-Pib2 and truncated derivatives. (A) Wild-type and pib2∆ mutant strains bearing plasmids that express GFP or GFP-Pib2 from a methionine-repressible promoter were grown in SC-leucine medium at different levels of methionine. Cells were then exposed to 2 μg/ml tunicamycin and 1 μg/ml FK506 for 8 h, stained with PI, and analyzed by flow cytometry. Bars indicate averages of three replicate cultures (± SD). (B) Wild-type (squares, solid curves) and pib2∆ mutant (circles, broken curves) cultures were exposed to tunicamycin and FK506 for the indicated times, then stained with PI and analyzed by flow cytometry (black symbols) or stained with dihydro-DCFDA and analyzed microscopically (gray symbols) at the indicated times of incubation at 30°C. The average data from three replicate cultures were fitted to standard sigmoid equations (black curves) or the difference between two standard sigmoid equations (gray curves) using nonlinear regression as described in Materials and Methods. (C) A series of truncated GFP-Pib2 fusion proteins were expressed at low levels (100% met) in pib2∆ cells or at high levels (12% met) in wild-type cells and then assayed for cell death (as in Figure 2A), for hypersensitivity to caffeine (see Materials and Methods), and for subcellular localization (as in Figure 1B). The degrees of complementation in the pib2∆ mutant (compl.) and of dominant-negative activity in the wild-type strain (D.N.) were summarized on a scale of none (−) to full (++). Predominant localization to the cytoplasm (cyt.) or endosomal-vacuolar membranes (E-V) is indicated.
The slower rates of cell death in pib2∆ mutants and in strains overexpressing GFP-Pib2 could be attributable to either delayed onset of LMP or longer survival of post-LMP cells. To distinguish these possibilities, we stained cells with dihydro-DCFDA at different times after exposure to tunicamycin plus FK506. Dihydro-DCFDA specifically stains the cytoplasm of post-LMP cells that have not yet died (Kim et al., 2012), and therefore the frequency of positive-staining cells in the population rises, peaks, and declines as the individual cells undergo LMP and cell death. Cultures of wild-type cells exhibited such transient staining with dihydro-DCFDA, which was best fitted by a mathematical model in which the median cell remained alive for ∼1.1 h following LMP (Figure 2B, solid curves). The pib2∆ mutant culture exhibited slower cell death and lower frequencies of staining with dihydro-DCFDA than the wild-type culture (Figure 2B, broken curves), which rules out the possibility that these cells underwent LMP at wild-type rates but survived longer in the post-LMP state. Overexpressed GFP-Pib2 in wild-type cells also exhibited lower staining with dihydro-DCFDA (unpublished data). Thus endogenous Pib2 promoted the onset of LMP, and overexpressed Pib2 delayed the onset of LMP, without any obvious effect on post-LMP survival times. This also suggests that endpoint measurements of cell death using propidium iodide (PI) serve as an excellent proxy for earlier LMP events that are transient and difficult to measure directly.
For determination of which conserved elements of Pib2 are important for its functions, several derivatives of the GFP-Pib2 fusion protein with N- and C-terminal truncations were expressed at low levels in pib2∆ mutants (100% methionine) and at high levels in the wild-type parent strain (12% methionine). Then the levels of complementation and dominant-negative activity were assessed after exposure to tunicamycin plus FK506, as in Figure 2A. Removal of the tail motif completely destroyed complementation in pib2∆ mutants, and removal of N-terminal motifs progressively diminished complementation (Figure 2C). Overexpression of the truncated derivatives in pib2∆ mutants did not improve complementation (unpublished data), and potential dominant-negative effects could not be measured because of the low rate of cell death existing in pib2∆ mutants. In wild-type cells, dominant-negative effects of overexpressed GFP-Pib2 truncations were clearly evident in all derivatives containing the poorly conserved N-terminal segment. Thus the N-terminal 49 amino acids of Pib2 were necessary and sufficient for most of the dominant-negative activity of overexpressed Pib2 in this test. Because Pib2 and all of the tested derivatives were unable to increase the rate of cell death in wild-type cells, Pib2 is not likely to function as a dose-dependent toxin that directly induces LMP and subsequent cell death. Instead, the dominant-negative activity of Pib2 and several of its derivatives is more consistent with a scaffold-like function wherein Pib2 could facilitate interactions between additional cellular factors when expressed at low levels and interfere with those interactions when overexpressed.
High-throughput screens have revealed that pib2∆ mutants grow more poorly than wild-type strains in culture media supplemented with caffeine (Hoepfner et al., 2014). We confirmed 3.6-fold caffeine hypersensitivity of pib2∆ mutants (IC50 = 1.15 mM) relative to wild type (IC50 = 4.17 mM) in YPD medium (see Materials and Methods), and exploited this growth phenotype to reanalyze the functionalities of the truncated GFP-Pib2 derivatives described above. When expressed at low levels, only the full-length GFP-Pib2 protein fully reversed the caffeine hypersensitivity of the pib2∆ mutants, and the N- and C-terminal truncated derivatives failed to complement caffeine hypersensitivity in a pattern that closely resembled the complementation results obtained from the cell death assays (Figure 2C). Complementation by the truncated derivatives was not improved by overexpression, and indeed several truncations (two lacking just the tail motif and one lacking the N-terminal segment) strongly diminished caffeine resistance of the pib2∆ mutants by an additional 2.1- to 4.6-fold (unpublished data). These same truncated derivatives also exhibited dominant-negative activity in the caffeine tolerance assays when overexpressed in wild-type cells, as did full-length GFP-Pib2 (Figure 2C). Therefore motifs A through FYVE conferred dominant-negative activity in both the caffeine tolerance and the cell death assays, whereas the A motif alone conferred dominant-negative activity in just the cell death assay. Because human LAPF/phafin1 lacks motifs A through E, it was not expected to exhibit dominant-negative or complementation activities when expressed in S. cerevisiae, and indeed it was inert, though it still localized to vacuolar and endosomal membranes (Figure 2C). The general concordance of the two complementation tests suggests that promotion of caffeine resistance and LMP are both manifestations of a single output of Pib2 that requires multiple conserved motifs, including the conserved tail motif.
Pib2 regulates TORC1 signaling independent of the EGO complex
No specific functions of Pib2 or any of its orthologues have been reported to date. To determine whether Pib2 functions are similar to any other proteins in S. cerevisiae, we downloaded two large data sets of chemical interaction profiles (Hoepfner et al., 2014) and genetic interaction profiles (Costanzo et al., 2010) that were collected for thousands of gene-knockout mutants and performed hierarchical clustering of all the tested mutants. In the data set of chemical interactions, pib2∆ mutants clustered tightly with several mutants that lack nonessential regulators or subunits of TORC1 (gtr1∆, gtr2∆, ego1∆, ego3∆, lst4∆, lst7∆, tco89∆, and tor1∆). All members of this group exhibit strong hypersensitivities to a cluster of chemicals that include rapamycin, caffeine, and related molecules, and consequently their profiles of chemical interactions are all strongly correlated with one another (Figure 3A). In the data set of genetic interactions, pib2∆ mutants again grouped with the TORC1-deficient mutants (gtr1∆, gtr2∆, ego1∆, ego3∆, lst4∆, and lst7∆), and all the mutants in this group correlated strongly with one another in their profiles of genetic interaction (Figure 3B). These independent studies therefore implicate Pib2 as a potential regulator or subunit of TORC1 that had not been recognized previously.
FIGURE 3:
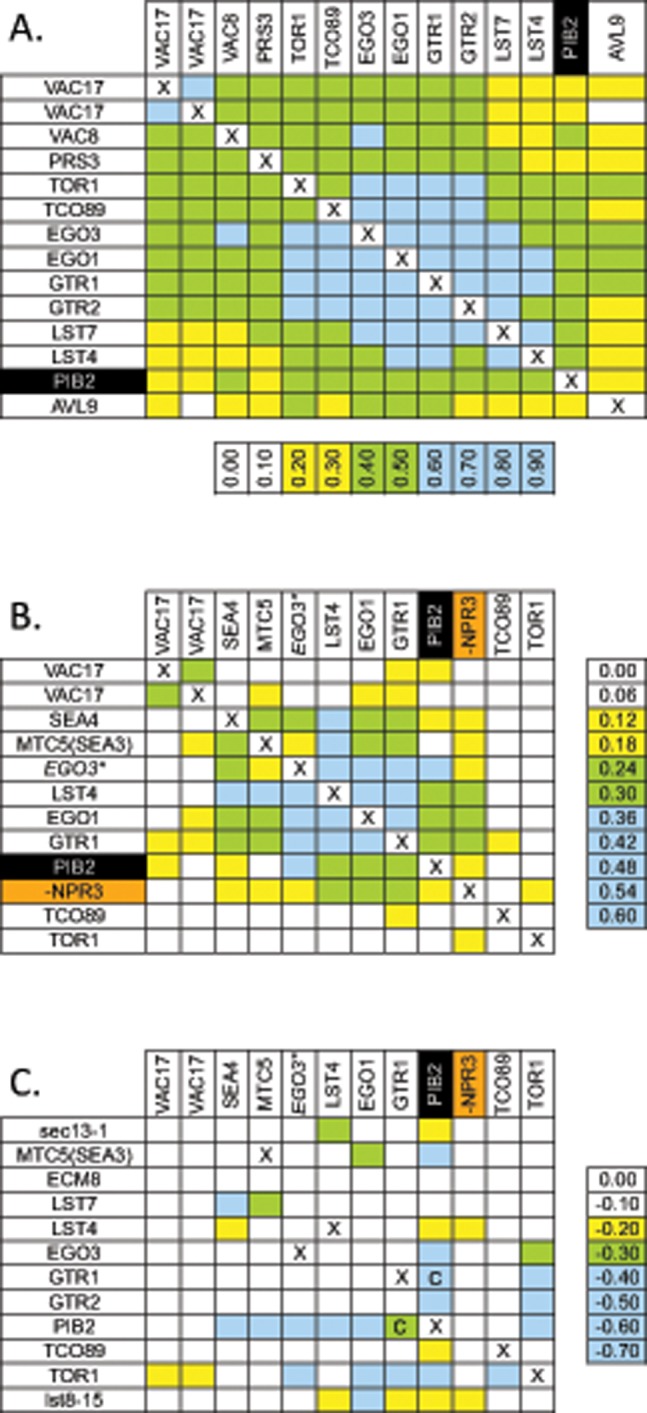
pib2∆ mutants cluster with mutants deficient in TORC1 signaling. Data sets of chemical interactions (Hoepfner et al., 2014) and genetic interactions (Costanzo et al., 2010) were subjected to hierarchical clustering (see Materials and Methods), and the clusters containing pib2∆ mutants were visualized by calculating Pearson correlation coefficients between gene profiles (A and B). Raw scores for genetic interactions are also shown (C), and all tables were colored according to the indicated scale. The mutant lacking NPR3, a negative regulator of TORC1, exhibited anticorrelation with pib2∆ and was manually moved into the cluster along with inverting the sign of correlation coefficient (B) and genetic interactions (C). The strain labeled EGO3* was labeled ECM8 in the original studies, but this mutant failed to complement an ego3∆ mutant and was renamed (see Materials and Methods). Mutant strains that could not be constructed or compared are indicated (X). Mutants for which synthetic lethality was confirmed by tetrad analysis are indicated (C).
If Pib2 functions upstream of TORC1 as a positive regulator, the pib2∆ knockout mutation may exhibit strong fitness defects when combined with knockout mutations in genes encoding other upstream regulators, such as the EGO complex (composed of Gtr1, Gtr2, Ego1, and Ego3, and stimulated by Lst4 and Lst7). Remarkably, double mutants carrying a pib2∆ mutation and either a gtr1∆, gtr2∆, ego1∆, ego3∆, lst4∆, lst7∆, or tco89∆ mutation each exhibited synthetic lethality or strong fitness defects, unlike the double mutants lacking two subunits of the EGO complex or its regulators (illustrated in Figure 3C). Tetrad analysis of a sporulated gtr1∆/+ pib2∆/+ double heterozygous diploid strain confirmed synthetic lethality of all gtr1∆ pib2∆ double mutant ascospores except when they inherited a plasmid bearing a functional GTR1 or PIB2 gene (unpublished data). All of the viable gtr1∆ pib2∆ [GTR1 URA3] strains failed to grow in media containing 5’-FOA (5-fluoroorotic acid) that counterselects for the plasmid (see Materials and Methods), again confirming synthetic lethality of gtr1∆ pib2∆ double mutants. Importantly, viability of the gtr1∆ pib2∆ double mutant was fully restored by a single nucleotide substitution within the TOR1 gene (TOR1-I1954S) that we recovered as a spontaneous mutation that reversed caffeine hypersensitivity of a gtr1∆ mutant strain (see Materials and Methods). The TOR1-I1954S mutation also reversed caffeine hypersensitivity of pib2∆ mutants and even increased caffeine resistance of wild-type cells (unpublished data), similar to a previously characterized TOR1-I1954T mutation that causes hyperactivation of TORC1 (Reinke et al., 2006). The rescue of gtr1∆ pib2∆ synthetic lethality by TOR1-I1954S in normal growth conditions strongly suggests that Pib2 and EGO complexes function independently and additively as upstream activators of TORC1 signaling, even in normal growth conditions.
Diminished rates of cell death were also observed in gtr1∆ mutants (Figure 4A) and in three other mutants lacking EGO complex subunits (unpublished data). Introduction of TOR1-I1954S into the gtr1∆ mutant background and the pib2∆ mutant background strongly increased the rates of cell death up to wild-type levels (Figure 4A). Even the gtr1∆ pib2∆ TOR1-I1954S triple-mutant strain exhibited a wild-type rate of cell death (Figure 4A, blue squares), showing that neither Pib2 nor the EGO complex were required for LMP and cell death as long as TORC1 signaling was sufficient. However, the hyperactive TOR1-I1954S mutation in an otherwise wild-type strain background did not accelerate the rate of cell death beyond the wild-type level (Figure 4A, white vs. black circles). On the other hand, introduction of npr2∆ mutation, which causes TORC1 hyperactivation by eliminating inhibitors of the EGO complex (Wu and Tu, 2011) and produces a genetic interaction profile that is anticorrelated with gtr1∆ mutants (Figure 3B), increased the rates of cell death in both pib2∆ mutant and the wild-type backgrounds but not in the gtr1∆ mutant background (Figure 4B). Therefore the onset of LMP and the subsequent death of stressed cells correlated closely with overall levels of TORC1 signaling, which were diminished in pib2∆ and gtr1∆ and enhanced in npr2∆ mutants.
FIGURE 4:
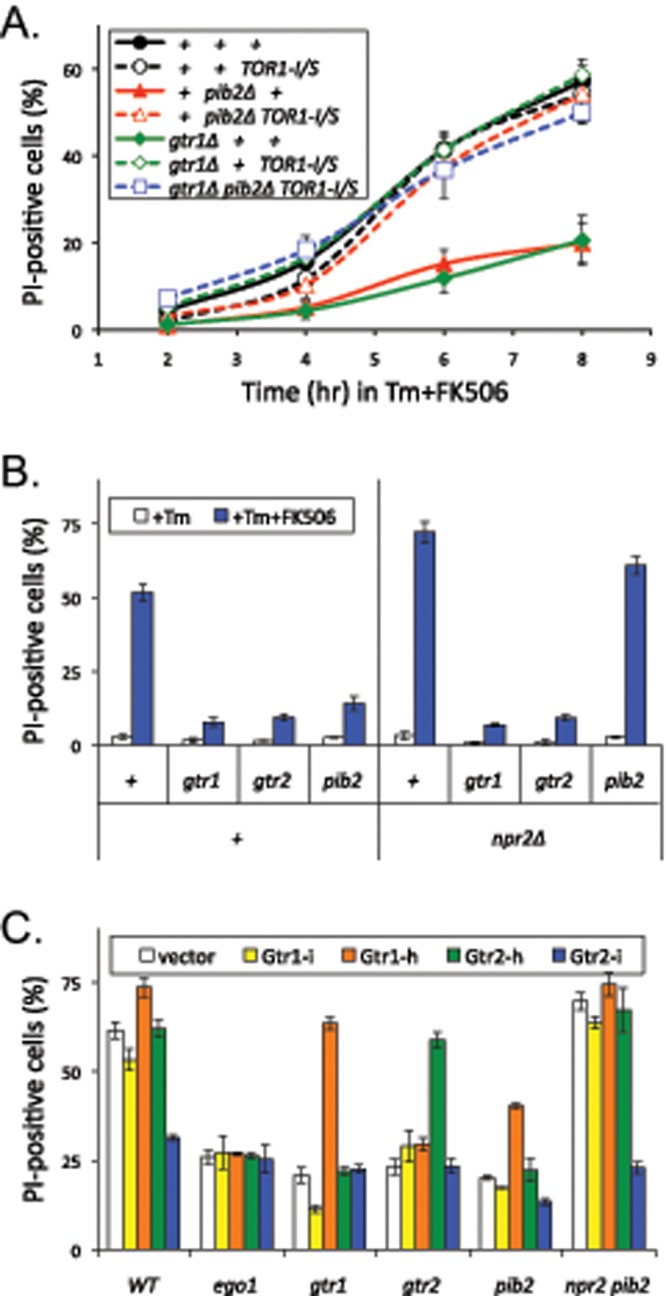
Pib2 stimulates TORC1 signaling independent of the Ragulator/EGO complex. Strains bearing pib2∆, gtr1∆, gtr2∆, or ego1∆ knockout mutations (∆ symbol sometimes omitted) and also bearing either the TOR1-I1954S point mutation (A), the npr2∆ mutation (B), or plasmids expressing hyperactive (-h) and inactive (-i) variants of Gtr1 or Gtr2 (C) were grown in SC or SC-uracil medium, exposed to tunicamycin plus FK506 for 8 h, stained with PI, and analyzed by flow cytometry. The percentages of PI-positive (dead) cells were calculated from the average of three replicate cultures (± SD).
The ability of an npr2∆ mutation to increase death of pib2∆ mutants but not gtr1∆ mutants is consistent with the prevailing model, in which Npr2 functions as a negative regulator of the EGO complex (Wu and Tu, 2011) independent of Pib2. To further establish the independence of the EGO complex and Pib2, we tested whether hyperactive variants of Gtr1 and Gtr2 (Gao and Kaiser, 2006) would increase death of pib2∆ mutants when expressed from plasmids (Figure 4C). Hyperactive Gtr1-h significantly increased the rate of death in pib2∆ and gtr1∆ mutants but not in gtr2∆ or ego1∆ mutants that lack other components of the EGO complex. Hyperactive Gtr2-h increased the rate of death in gtr2∆ mutants but not in gtr1∆ or ego1∆ or pib2∆ mutants, suggesting it was less efficient in activating the EGO complex than hyperactive Gtr1-h. Inactive Gtr2-i did not complement the gtr2∆ mutant and exhibited dominant-negative effects on the rate of cell death in pib2∆ and especially in npr2∆ pib2∆ double mutants, which lack the Pib2 input into TORC1 and therefore rely more heavily on the EGO complex (Figure 4C). Thus the EGO complex and its upstream regulators all interacted as expected from earlier studies and regulated cell death through TORC1 independent of Pib2.
To determine whether Pib2 affects basal signaling through TORC1, we studied the phosphorylation status of several TORC1 effectors by Western blot analysis of whole-cell lysates. Phosphorylation of ribosomal protein S6 (Rps6) dramatically declines upon addition of rapamycin to growing cells (Gonzalez et al., 2015). Using antibodies that specifically recognize phospho-Rps6, we confirmed that rapamycin causes a large and rapid decline of Rps6 phosphorylation in growing wild-type cells relative to loading control (Pgk1; Figure 5A). However, the gtr1∆ and pib2∆ mutants exhibited little or no decrease in phospho-Rps6 levels during exponential growth (Figure 5A). Introduction of the npr2∆ mutation into these strains did not detectably increase phospho-Rps6 levels, but did diminish the effects of rapamycin when Gtr1 and Pib2 were functioning, and not in the gtr1∆ and pib2∆ mutant strains. On the other hand, the Par32 phosphoprotein, which undergoes large shifts in mobility on SDS–PAGE in response to rapamycin (Huber et al., 2009), shifted significantly in gtr1∆ and pib2∆ mutants relative to wild type (Figure 5B), suggesting partial deficiency of TORC1 signaling in both of the mutants. The simplest hypothesis consistent with all these findings is that Pib2 and the EGO complex independently stimulate basal TORC1-dependent signaling in growing S. cerevisiae cells.
FIGURE 5:

Altered phosphorylation of TORC1-sensitive proteins in pib2∆ mutants. (A) Wild type, pib2∆, and gtr1∆ mutants with and without additional npr2∆ knockout mutation were grown to log phase in SC medium and exposed to 200 ng/ml rapamycin for 1 h. Proteins were extracted, separated by SDS–PAGE, and analyzed by Western blotting using antibodies specific for phospo-Rps6 and Pgk1 as a loading control. The same strains bearing a Par32-HA expression plasmid were grown in SC-uracil medium (B) or SC-uracil minus all other nonessential amino acids to diminish basal TORC1 signaling (C), exposed to rapamycin as above, and analyzed by Western blotting using anti-HA monoclonal antibodies.
TORC1-sensitive processes respond to ER stress
Does TORC1 signaling increase in response to ER stresses or calcineurin signaling? To answer this question, we monitored the TORC1-sensitive changes in phosphorylation of Rps6 and Par32 at several different times following exposure to tunicamycin or tunicamycin plus FK506. Within the first 2–3 h of exposure to either drug, the phosphorylation status of both proteins was not significantly altered in either wild-type cells or in pib2∆ cells, though the mutant cells exhibited reduced TORC1 activity (Figure 6), as observed earlier. Even an ire1∆ mutant, which lacks a functional UPR signaling pathway and is hypersensitive to tunicamycin, exhibited no apparent change in the TORC1-sensitive phosphoproteins within the first 2 h (Figure 6). Thus TORC1 signaling appeared unresponsive to ER stress and calcineurin signaling within this time frame.
FIGURE 6:

Tunicamycin and FK506 have little impact on TORC1 signaling. Wild-type, pib2∆ mutant, and ire1∆ mutant strains were grown to log phase in SC medium, exposed to tunicamycin (Tm), rapamycin (Rap), and calcineurin inhibitor (FK506) as indicated. Aliquots of each culture were removed before or 0.25, 0.5, 1, 2, or 4 h after drug addition and extracted rapidly, and total cell protein was analyzed by SDS–PAGE and Western blotting using antibodies that recognize phosopho-Rps6, total Rps6, Pib2, Pgk1, and the HA epitope of Par32-HA, which was expressed from plasmid pMS034.
Interestingly, total Rps6 protein levels declined dramatically in all three strains after 3–4 h of exposure to tunicamycin (Figure 6). Such a decline was not observed for Pgk1, which served as a control for gel loading. In spite of this decline in total Rps6 abundance, the residual Rps6 remaining at 4 h was completely shifted to a faster-migrating species that probably represented the dephosphorylated form, as it was not detected by anti phospho-Rps6 antibodies. In the presence of FK506, the decline in total Rps6 was unaltered, but the residual protein in wild-type and ire1∆ mutant cells was found in both phosphorylated and dephosphorylated forms. No residual phospho-Rps6 was detected in the pib2∆ mutant. These findings may indicate that a decline in TORC1 signaling occurs after several hours of exposure to tunicamycin and that calcineurin signaling may hasten the decline. However, this conclusion was not substantiated when Par32 was used as the reporter of TORC1 signaling (Figure 6).
Finally, we examined Pib2 mobility under these same conditions to determine whether this protein responds to ER stress, calcineurin, or TORC1. In both wild type and ire1∆ mutants, Pib2 shifted from a slightly slower and more diffuse band to a faster and sharper band after 1 h of exposure to rapamycin (Figure 6, right lanes), suggesting that phosphorylation of Pib2 is also responsive to TORC1, as observed previously (MacGurn et al., 2011). Similar mobility changes were observed during the first 2 h of exposure to tunicamycin or tunicamycin plus FK506. Thus Pib2 appeared to become modified in response to ER stress, and the modification was potentially a result of diminished TORC1 signaling. In any case, we obtained no evidence for increased TORC1 signaling at any time during the response to ER stress.
Vacuole fragmentation during ER stress requires TORC1
ER stress induces slow vacuole fragmentation without leakage of contents, which arises as a consequence of diminished vacuole–vacuole fusion events in the face of ongoing fission events (Baars et al., 2007). Interestingly, GFP-Pib2 became concentrated near the sites of vacuole–vacuole contact as indicated by staining with FM4-64 (Figure 7A). For investigation of the potential roles of Pib2 and TORC1 signaling in this process, FM4-64–stained cells were exposed to tunicamycin in the presence and absence of rapamycin and then monitored over time. While rapamycin greatly slowed the rate of vacuole fragmentation in wild-type cells (Figure 7B, left), the pib2∆ and gtr1∆ mutants were indistinguishable from wild-type cells in the response to tunicamycin (right). These findings suggest that low levels of TORC1 signaling may be sufficient for normal vacuole fission in these conditions. A potent inhibitor of the V-ATPase concanamycin C also blocked vacuole fragmentation in wild-type cells during ER stress (Baars et al., 2007). If the V-ATPase were required only for TORC1 activation, strains with hyperactive TORC1 signaling would still undergo vacuole fragmentation in the presence of V-ATPase inhibitor. In contrast to this hypothesis, concanamycin C efficiently inhibited vacuole fragmentation and cell death in both TOR1-I1974S and npr2∆ mutant strains (unpublished data). Therefore the V-ATPase and TORC1 both played important and potentially independent roles in the vacuole fragmentation that occurs in response to ER stress.
FIGURE 7:

Vacuole fragmentation during ER stress depends on TORC1 signaling but not Pib2 or Gtr1. (A) Wild-type cells expressing GFP-Pib2 and prestained with FM4-64 were exposed to tunicamycin or vehicle control for 2 h and imaged by fluorescence microscopy. (B) Wild-type, pib2∆ mutant, and gtr1∆ mutant strains were prestained with FM4-64, exposed to tunicamycin for 2 h (right) or up to 2 h (left), and imaged by fluorescence microscopy, and then 200 cells were scored as having either 1 or 2 (large) vacuoles or >3 vacuoles. Data points and bars represent the average of three replicate cultures (± SD).
TORC1 acts at early stages in the pathway leading to LMP and cell death
For determination of whether TORC1 activity promotes steps in addition to vacuole fragmentation that lead to LMP and cell death, rapamycin was added together with a calcineurin inhibitor at different times after exposure of wild-type cells to tunicamycin. To avoid possible interference between rapamycin and FK506, which both utilize the same FKBP-12 receptor protein to mediate their inhibition of their targets, we used cyclosporine A as the inhibitor of calcineurin. Interestingly, rapamycin strongly delayed cell death when added at the same time as tunicamycin but completely failed to slow cell death when added 2 h after tunicamycin exposure, when vacuoles had already fragmented in nearly all cells (Figure 8A). This finding suggests that TORC1 activity, like V-ATPase activity, performs its prodeath functions in the early stages of the response to ER stress and is not required for the calcineurin-sensitive stages that occur later and lead to LMP and cell death. As shown earlier, TORC1 loses activity after several hours of tunicamycin exposure (Figure 6).
FIGURE 8:
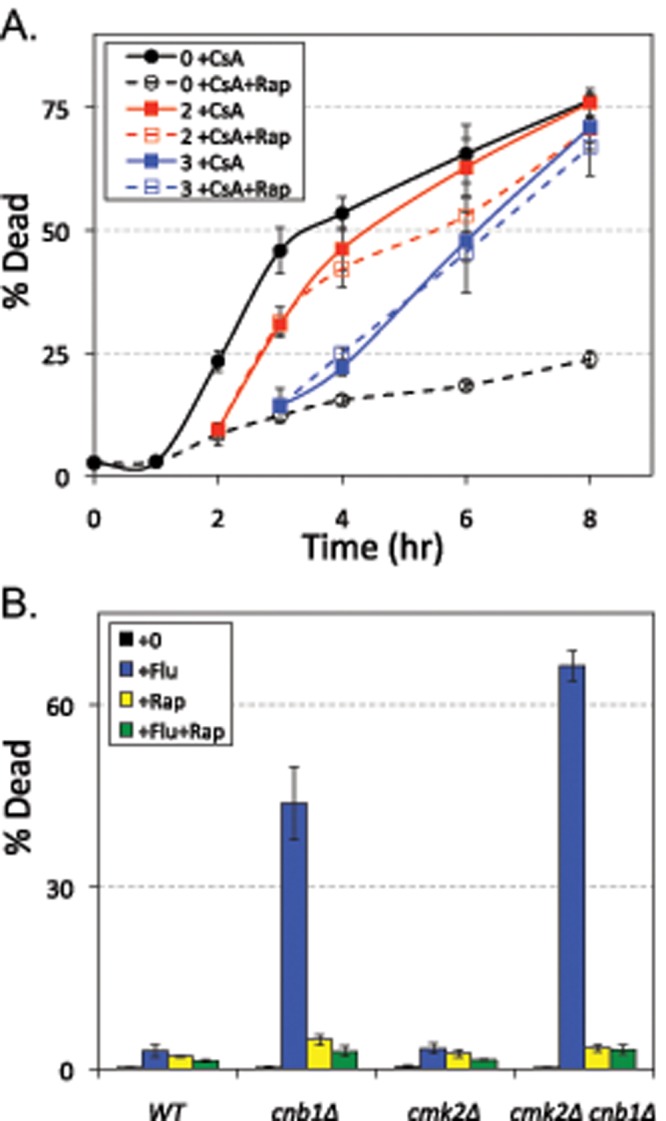
TORC1 signaling promotes fungicidal synergism in S. cerevisiae and C. albicans. (A) The wild-type strain of S. cerevisiae was grown to log phase in SC medium, exposed to tunicamycin beginning at 0 h, and then either calcineurin inhibitor (CsA) or calcineurin plus TORC1 inhibitors (CsA+Rap) were added 0, 2, or 4 h later (black, red, or blue lines). At the indicated times, the cultures were sampled and stained with PI, and the fraction of dead cells was determined by flow cytometry. The average (± SD) of three replicate cultures is shown. (B) Wild type, cnb1∆ mutant, cmk2∆ mutant, and cmk2∆ cnb1∆ double mutants of C. albicans were grown to log phase in SC medium; exposed to fluconazole (+Flu), rapamycin (+Rap), or both (+Flu+Rap) for 8 h; and then stained with PI and analyzed by flow cytometry for the fraction of dead cells in the population. Bars represent the averages (± SD) of three replicate cultures.
TORC1 promotes fungicidal actions of fluconazole in calcineurin-deficient C. albicans
In many pathogenic yeasts and fungi, calcineurin inhibitors convert fungistats such as fluconazole into fungicides, possibly by inducing VMP. To test whether TORC1 promotes death in these circumstances, we exposed cultures of calcineurin-deficient (cnb1∆ homozygous mutant) C. albicans to fluconazole in the presence and absence of rapamycin and analyzed them for cell death using PI stain. Fluconazole strongly killed the cnb1∆ mutants in comparison with the isogenic wild-type parent strain, and rapamycin completely blocked this effect (Figure 8B). Similar results were obtained using tunicamycin as the stressor instead of fluconazole (unpublished data). In S. cerevisiae cells, a Ca2+/calmodulin-dependent protein kinase (Cmk2) plays a secondary role in suppression of LMP and cell death that can be visualized only in calcineurin-deficient cells (Dudgeon et al., 2008). A secondary role of Cmk2 in suppression of cell death was also observed in C. albicans cells exposed to mating pheromones (Alby et al., 2010). Similar to S. cerevisiae strains, the cmk2∆ mutant behaved like wild type, and the cmk2∆ cnb1∆ double mutant exhibited slightly higher levels of cell death than the cnb1∆ single mutant after exposure to fluconazole, and those cell deaths were fully blocked by rapamycin (Figure 8B). Therefore TORC1 played important prodeath roles in calcineurin-deficient C. albicans and S. cerevisiae cells alike when the cells were stressed by either synthetic (e.g., fluconazole) or natural (e.g., tunicamycin) fungistats.
DISCUSSION
The molecular mechanism by which calcineurin prevents death of fungal cells that have been exposed to fungistatic drugs is not understood, but it seems to be conserved across diverse fungal phyla (Fox and Heitman, 2002; Steinbach et al., 2007; Liu et al., 2015). Early studies in S. cerevisiae ruled out the known targets of calcineurin as strong regulators of cell death (Dudgeon et al., 2008), and later a genome-wide screen revealed the V-ATPase and leakage of luminal material from vacuoles into the cytoplasm as critical components of the pathway leading to cell death (Kim et al., 2012). Calcineurin also promotes survival of S. cerevisiae cells that have been stressed with aggregated forms of human α-synuclein, which is a primary determinant of neurodegeneration in Parkinson's disease (Caraveo et al., 2014). In other models of neurodegeneration and in mammary gland involution, the V-ATPase and LMP have been implicated (Syntichaki et al., 2005; Arnandis et al., 2012). These findings hint at possible conservation between fungi and animals of a mechanism that controls and regulates LMP. In this study, we extended this comparison by focusing on a protein (Pib2) that is related to a protein (LAPF/phafin1) implicated as an inducer of LMP in human L929 cells (Chen et al., 2005). We show that Pib2 increases TORC1 signaling and that full TORC1 signaling is required for LMP and cell death in both S. cerevisiae and C. albicans caused by the combination of fungistats and calcineurin inhibitors.
Lysosomes and vacuoles naturally undergo fission and fusion reactions without leakage of luminal content (Starai et al., 2007). In response to tunicamycin and other fungistats, vacuole fusion appears to be selectively inhibited, and due to ongoing fission, vacuoles become highly fragmented in appearance within the first 2 h (Kim et al., 2012). This fragmented state persists for several hours with little cell death in the population. Though calcineurin inhibitors did not alter kinetics of vacuole fragmentation, the fragmented vacuoles appeared to fuse again about the same time as luminal molecules were released into the cytoplasm (Kim et al., 2012). Pib2 associated with the limiting membranes of vacuoles and endosomes in unstressed cells and concentrated at the sites of vacuole–vacuole contact in the stressed cells, in a manner that depended on its FYVE domain and PI(3)P synthesis in these membranes. Therefore Pib2 is well positioned to mediate LMP, membrane fusion, or other phenomena that could lead to death of calcineurin-deficient cells. However, overexpression of Pib2 or its truncated derivatives in wild-type cells did not accelerate the rate of cell death in any circumstances tested and, upon exposure to tunicamycin plus FK506, interfered with the process in a dominant-negative manner that is more typical of scaffolds than of a pore-forming protein such as Bax.
The data presented here show that Pib2 naturally stimulates TORC1 signaling via a pathway that is independent of the EGO complex and its upstream regulators. Though pib2∆ mutants exhibit chemical and genetic interaction profiles that are correlated with gtr1∆ and other mutants deficient in the EGO complex, Pib2 had not been associated previously with TORC1 signaling. Synthetic lethality between pib2∆ and gtr1∆ mutations may have obscured the association. Like mutants deficient in the EGO complex, pib2∆ mutants are viable and exhibit phenotypes indicative of diminished TORC1 signaling (hypersensitivity to rapamycin and caffeine, hypo-phosphorylation of Rps6, hyperphosphorylation of Par32, and diminished rates of cell death), at least some of which can be reversed by a hyperactivating I1954S substitution in Tor1 that is very similar to a previously characterized hyperactivating I1954T substitution (Reinke et al., 2006). Thus Pib2 and EGO complexes seem to activate TORC1 independently and additively, with full TORC1 activity being necessary for LMP and cell death in the conditions studied here. A second EGO complex–independent pathway of TORC1 activation was recently identified as responsive to glutamine feeding (Stracka et al., 2014). It will be interesting to determine whether that regulatory pathway involves Pib2 or some other factor, whether other proteins function with Pib2 in the regulation of TORC1, and whether Pib2 communicates the concentration of some nutrient or metabolite to TORC1.
This simple model becomes more complicated, however, when additional findings are also considered. First, Pib2 interacted with all four subunits of the EGO complex in the protein-fragment complementation assay (PCA) format of a yeast two-hybrid screen (Tarassov et al., 2008). We independently confirmed interactions between Pib2 and Gtr1, Ego1, and the Kog1 subunit of TORC1 using a split-ubiquitin yeast two-hybrid system (Binda et al., 2009; unpublished data), but we could not confirm physical interactions in coimmunoprecipitation experiments. While Pib2 stimulated TORC1 signaling, it may also inhibit TORC1 signaling by inhibiting functions of the EGO complex. This hypothesis could explain why truncated nonstimulatory derivatives of Pib2 exhibited strong caffeine hypersensitivity when overexpressed in pib2∆ and wild-type cells. Second, phosphorylation of Pib2 changed in response to rapamycin (MacGurn et al., 2011; Figure 6), suggesting that Pib2 may be an effector or feedback regulator of TORC1. The nonessential Tco89 regulatory subunit of TORC1 also undergoes rapid changes in phosphorylation in response to rapamycin (Huber et al., 2009; Oliveira et al., 2015) that are of unknown significance. More research is necessary to fully understand the complete set of interactions between Pib2 and TORC1. The mechanism by which the EGO complex regulates TORC1 will be easier to determine in the pib2∆ mutant background, which lacks the alternative and potentially confounding source of inputs into TORC1.
Recently phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2) in vacuolar membranes was found to be important for activation of TORC1 signaling in S. cerevisiae through interactions with the essential subunit Kog1 and certain effectors such as Sch9 (Jin et al., 2014). Raptor, the mammalian homologue of Kog1, also bound PI(3,5)P2 (Bridges et al., 2012). Because PI(3)P, the immediate precursor of PI(3,5)P2, and the FYVE domain of Pib2 were necessary for recruitment of the protein to the vacuolar membrane, it is tempting to speculate that TORC1 signaling also responds to PI(3)P. Though vps34∆ mutants exhibit hypersensitivity to rapamycin and caffeine (Banuelos et al., 2010; Bridges et al., 2012), this mutant lacks both PI(3)P and PI(3,5)P2. In S. cerevisiae, two distinct PI(3) kinases are thought to exist that share several subunits (Vps34, Vps15, Atg6) and also contain a unique subunit for complex I (Atg14) and for complex II (Vps38), which promote autophagy and vesicle trafficking, respectively (Kihara et al., 2001). A vps38∆ mutant exhibited a 67% decrease in PI(3)P accumulation and weak hypersensitivity to rapamycin (Bridges et al., 2012), but these mutants died at wild-type rates when exposed to tunicamycin plus FK506 (Kim et al., 2012). Interestingly, atg14∆ mutants were resistant to rapamycin and exhibited diminished rates of cell death (Kim et al., 2012), potentially exposing a role for autophagy in the regulation of LMP. However, atg6∆ and many other autophagy-deficient mutants of S. cerevisiae exhibited near wild-type rates of cell death (Kim et al., 2012). Thus the PI(3)P synthesized by complex I may be more important for promoting cell death, while the PI(3)P synthesized by complex II may be more important for TORC1 activation in yeasts. The situation may be even more complicated in mammalian cells, in which TORC1 phosphorylates Atg14 and inhibits PI(3)P synthesis by complex I (Yuan et al., 2013).
Currently there is no direct evidence for LAPF/phafin1 regulation of TORC1 in animals. LAPF/phafin1 requires its FYVE domain for localization to lysosomal and endosomal limiting membranes, but it differs from Pib2 in that lysosomal localization also required the tail motif (Lin et al., 2012). In HEK293T cells, overexpressed LAPF/phafin1 caused enlargement of endocytic vesicles and enhanced endocytosis and autophagy (Lin et al., 2012). Though these effects were not observed upon overexpression of EAPF/phafin2, which lacks the tail motif, overexpression of the tail-less homologue in Drosophila (termed Rush) produced similar effects and appeared to modulate endocytosis through interactions with regulators of Rab7 GTPase activity (Gailite et al., 2012). These effects on endocytosis may be unique properties of the phafins in animals, as these proteins all contain well-conserved pleckstrin homology (PH) domains at their N-termini that are not present in the fungal homologues of Pib2. Likewise, there is no direct evidence for a role of Pib2 in endocytosis outside of its role in regulating TORC1 signaling. Therefore the fungal and animal families of proteins containing FYVE domains and tail motifs may not be descended from a common ancestor, or they may function divergently in the fungal and animal kingdoms.
How does TORC1 promote LMP and death of stressed cells lacking calcineurin function, and thereby contribute to cidal effects of fungistats? TORC1 signaling stimulates expression of ribosomes and the initiation stages of translation, the expression and trafficking of amino acid permeases, the down-regulation of autophagy, and the modulation of several metabolic and regulatory cascades (Loewith and Hall, 2011). At present, it is unclear how such diverse outputs could contribute to LMP and cell death in these circumstances. If the rapamycin-induced up-regulation of autophagy were somehow protective, autophagy-deficient mutants would be expected to exhibit higher rates of cell death than wild type. Of the 30 different autophagy-deficient (atg) mutants tested in our original genome-wide screen, only the atg15∆ mutants exhibited elevated rates of cell death. The atg15∆ mutants lack a lipase in the lumen of vacuoles that is responsible for degrading luminal vesicles and is not directly involved in autophagic uptake of cellular material into vacuoles (Epple et al., 2001; Teter et al., 2001). We report here another role of TORC1 signaling during the response to stress: the stimulation of vacuole fission or inhibition of vacuole fusion. Though gtr1∆ and pib2∆ mutants exhibited roughly wild-type rates of vacuole fragmentation during exposure to tunicamycin, rapamycin strongly inhibited vacuole fragmentation in stressed cells. Residual TORC1 signaling in gtr1∆ and pib2∆ cells was therefore sufficient to promote vacuole fragmentation but not for eventual cell death. TORC1 signaling has previously been shown to promote vacuole fission in vitro and in live S. cerevisiae cells responding to salt stress (Michaillat et al., 2012). Using several readouts of TORC1 signaling, we found no evidence that tunicamycin stimulates TORC1 signaling and evidence that TORC1 signaling slowly declines to undetectable levels after about 2 h of stress, and even faster in gtr1∆ and pib2∆ mutants. The ability of rapamycin to block cell death also declined to undetectable levels by 2 h of stress (Figure 8). All of these findings together suggest that TORC1 signaling is necessary only in the early stages of the response to stress, priming the cells for later calcineurin-sensitive stages that lead to vacuole fusion, LMP, and cell death.
While TORC1 activity promoted LMP and cell death in stressed yeasts, it had the opposite effect on necrosis-like death of degenerating neurons in the nematode C. elegans, potentially through the down-regulation of autophagy (Toth et al., 2007). This model of neurodegeneration is induced by hyperactive ion channels that are expressed in the dying neurons and has been shown previously to depend on elevation of cytosolic Ca2+, calpain activation, V-ATPase activity, and release of lysosomal cathepsins into the cytoplasm, and therefore is a well-established model of LMP in animals (Boya and Kroemer, 2008; Yamashima and Oikawa, 2009; Mrschtik and Ryan, 2015). On the other hand, TORC1 can also promote cell death in other models of neurodegeneration, usually through induction of apoptosis (Wang et al., 2009). LMP regulatory mechanisms in animals are thus complicated by the variable types of cellular stresses, the diversity of cell types, and the emerging cross-talk between LMP, autophagy, and apoptosis. More research on LMP control mechanisms in fungi and animals may provide insights into controlling fungal infections and neurodegenerative disease.
MATERIALS AND METHODS
Yeast strains, culture media, and reagents
The yeast strains used in this study were generated from the wild-type parent BY4741 and BY4742 strain backgrounds using standard gene-knockout procedures (Longtine et al., 1998) and/or crosses (see Table 1). The primers used for gene knockouts are available upon request, and all knockouts were confirmed by PCR analysis using flanking oligonucleotides. Yeast strains were cultured in rich yeast–peptone–dextrose (YPD) medium, SC medium containing all 20 amino acids plus adenine and uracil, or SC medium lacking a nutrient for plasmid selection. For some experiments, the concentration of methionine was varied from 100% (100 μg/ml) to 12%.
TABLE 1:
Yeast strains and plasmids used in this study. Continued
| Strain name | Genotype | Source | Parent | F primera | R primera |
|---|---|---|---|---|---|
| S. cerevisiae strains | |||||
| BY4741 | MATa his3∆1 leu2∆2 met15∆0 ura3∆0 | Giaever et al., 2002 | |||
| BY4742 | MATα his3∆1 leu2∆2 lys2∆0 ura3∆0 | Giaever et al., 2002 | |||
| K1259 | MATa ire1::G418 | Bonilla et al., 2002 | BY4741 | ||
| AK018 | MATa pib2::NAT | This study | BY4741 | Pib2 KO F11 | Pib2 KO R11 |
| AK025 | MATa npr2::HIS3 | This study | BY4741 | Npr2 KO F1 | Npr2 KO R0 |
| AK060 | MATa gtr1::NAT | This study | BY4741 | Gtr1 KO F11 | Gtr1 KO R11 |
| AK061 | MATa gtr2::NAT | This study | BY4741 | Gtr2 KO F11 | Gtr2 KO R11 |
| AK062 | MATa ego1::NAT | This study | BY4741 | Ego1 KO F11 | Ego1 KO R11 |
| AK063 | MATa ego3::NAT | This study | BY4741 | Ego3 KO F11 | Ego3 KO R11 |
| AK109 | MATa iml1::HIS3 | This study | BY4741 | Iml1 KO F11 | Iml1 KO R11 |
| AK069 | MATa npr2::HIS3 gtr1::NAT | This study | AK060 | Npr2 KO F1 | Npr2 KO R0 |
| AK070 | MATa npr2::HIS3 gtr2::NAT | This study | AK061 | Npr2 KO F1 | Npr2 KO R0 |
| AK071 | MATa iml1::HIS3 gtr1::NAT | This study | AK060 | Iml1 KO F11 | Iml1 KO R11 |
| AK072 | MATa iml1::HIS3 gtr2::NAT | This study | AK061 | Iml1 KO F11 | Iml1 KO R11 |
| AK105 | MATα pib2::G418 | This study | BY4742 | Pib2 KO F11 | Pib2 KO R11 |
| AK101 | MATα gtr1::NAT | This Study | BY4742 | Gtr1 KO F11 | Gtr1 KO R11 |
| AK060-b03 | MATa tor1-I1954S gtr1::NAT | This study | AK060 | ||
| AK102 | MATα tor1-I1954S gtr1::NAT | This study | AK105/060-b03 | ||
| AK104 | MATα tor1-I1954S | This study | AK105/060-b03 | ||
| AK106 | MATα tor1-I1954S pib2::G418 | This study | AK105/060-b03 | ||
| AK107 | MATα tor1-I1954S gtr1::NAT pib2::G418 | This study | AK105/060-b03 | ||
| C. albicans strains | |||||
| RBY1182 | a/α leu2/leu2 his1/his1 | Alby et al., 2010 | |||
| DSY797 | cnb1::HIS1/cnb1::LEU2 | Alby et al., 2010 | |||
| KAY483 | cnb1::HIS1/cnb1::LEU2 cmk2∆/cmk2::SAT1 | Alby et al., 2010 | |||
| KAY481 | cmk2::SAT1/cmk2::SAT1 | Alby et al., 2010 | |||
| Plasmids | |||||
| pSM214 | [MET]-GFP AmpR CEN LEU2 | Mehta et al., 2009 | p415MET25 | ||
| pAK009 | [MET]-GFP-Pib2 | This study | pSM214 | Pib2-F-SpeI | Pib2-Stop-XhoI |
| pAK010 | [MET]-GFP-Pib2(1-49) | This study | pSM214 | Pib2-F-SpeI | Pib2+147R |
| pAK011 | [MET]-GFP-Pib2(1-101) | This study | pSM214 | Pib2-F-SpeI | Pib2+303R |
| pAK012 | [MET]-GFP-Pib2(1-162) | This study | pSM214 | Pib2-F-SpeI | PIB2+488R |
| pAK013 | [MET]-GFP-Pib2(1-312) | This study | pSM214 | Pib2-F-SpeI | PIB2+938R |
| pAK014 | [MET]-GFP-Pib2(1-360) | This study | pSM214 | Pib2-F-SpeI | Pib2 +1080R |
| pAK015 | [MET]-GFP-Pib2(1-428) | This study | pSM214 | Pib2-F-SpeI | PIB2+1284R |
| pAK016 | [MET]-GFP-Pib2(1-555) | This study | pSM214 | Pib2-F-SpeI | Pib2 +1665R |
| pAK017 | [MET]-GFP-Pib2(1-620) | This study | pSM214 | Pib2-F-SpeI | Pib2 +1860R |
| pAK018 | [MET]-GFP-Pib2(50-635) | This study | pSM214 | Pib2+148F | Pib2-Stop-XhoI |
| pAK019 | [MET]-GFP-Pib2(102-635) | This study | pSM214 | Pib2+304F | Pib2-Stop-XhoI |
| TABLE 1: Yeast strains and plasmids used in this study. | |||||
| pAK020 | [MET]-GFP-Pib2(165-635) | This study | pSM214 | PIB2+493F | Pib2-Stop-XhoI |
| pAK021 | [MET]-GFP-Pib2(304-635) | This study | pSM214 | PIB2+910F | Pib2-Stop-XhoI |
| pAK022 | [MET]-GFP-Pib2(440-635) | This study | pSM214 | PIB2+1318F | Pib2-Stop-XhoI |
| pAK023 | [MET]-GFP-Pib2(-∆FYVE) | This study | pSM214 | Pib2 1582-FYVEF | Pib2 1326-FYVER |
| pCK294 | Gtr1-i (-S20L) CEN URA3 | Gao and Kaiser, 2006 | |||
| pCK295 | Gtr1-h (-Q65L) CEN URA3 | Gao and Kaiser, 2006 | |||
| pCK296 | Gtr2-h (-S23L) CEN URA3 | Gao and Kaiser, 2006 | |||
| pCK297 | Gtr2-i (-Q66L) CEN URA3 | Gao and Kaiser, 2006 | |||
| pMS034 | Par32-HA CEN URA3 | Huber et al., 2009 | |||
aSequences of forward and reverse PCR primers are available upon request.
S. cerevisiae strains bearing TORC1 hyperactivating mutations were obtained as follows. First, independent colonies of the caffeine-sensitive gtr1∆ mutant strain were grown in YPD medium and plated on YPD agar medium containing 8 mM caffeine. One caffeine-resistant colony (suppressor) was picked from each parent colony, purified, and then tested for restoration of wild-type rates of cell death when exposed to tunicamycin plus FK506 in SC medium. Of the 29 suppressors (80% of total) that passed this test, four were found to confer caffeine resistance in a dominant manner (in heterozygous diploids), and the remainder were less dominant or recessive. One dominant suppressor and four recessive suppressors were subjected to whole-genome resequencing at the Next Generation Sequencing Center (Johns Hopkins Medical Institutions), and in each case, only one single nucleotide substitution was found, four of which altered the sequence of TOR1 (resulting in Tor1-I1954S [dominant], -A1927E, -A1957V, -L2271V), and one of which altered the sequence of KOG1 (resulting in Kog1-S1511L). The gtr1∆ TOR1-I1954S double-mutant strain was crossed to a pib2∆ mutant, resulting in pib2∆ TOR1-I1954S double mutants and viable gtr1∆ pib2∆ TOR1-I1954S triple mutants, in addition to the single mutants. The TOR1-I1954S mutant behaved similar to the hyperactive TOR1-I1954T mutant isolated previously as a caffeine-resistant mutant (Reinke et al., 2006).
The plasmids used in this study were generated using standard cloning procedures. Plasmid pAK018 was generated by PCR amplification of PIB2 codon sequences from genomic DNA using primers Pib2-F-SpeI and Pib2-Stop-XhoI. The product was digested with SpeI and XhoI and ligated into pSM214 (Mehta et al., 2009) containing an in-frame N-terminal GFP under the methionine-repressible MET25 promoter. All truncations of Pib2 were generated by PCR amplification of the desired segment from pAK018 using the specified primers. The products were then digested with SpeI and XhoI and ligated into SM214. pAK023, which contains GFP-Pib2 lacking a functional FYVE domain, was generated by two separate PCRs of the N-terminus and C-terminus. The N-terminus, containing all sequence N-terminal of the FYVE domain (1–1326 base pairs), was digested with SpeI and XmaI, and the C-terminus, containing all sequence C-terminal of the FYVE domain (1581–1908 base pairs), was digested with XmaI and XhoI. The two pieces were ligated simultaneously into pSM214.
Stocks of tunicamycin (Sigma-Aldrich, St. Louis, MO), concanamycin C (Santa Cruz Biotechnology, Dallas, TX), and FK506 (Astrellas Pharma, Chu¯o¯, Tokyo, Japan) were dissolved in dimethyl sulfoxide (DMSO) at 1, 1, and 1 mg/ml concentrations, respectively, and stored at −20°C. Fluconazole (Sigma-Aldrich) was dissolved in ethanol to 50 mg/ml and stored at −20°C. Caffeine was freshly dissolved into SC or YPD medium at various concentrations and sterilized by filtration. Propidium iodide (Sigma-Aldrich) was dissolved in phosphate-buffered saline at a concentration of 1 μg/ml; dihydro-DCFDA (Sigma-Aldrich) and FM4-64 (Invitrogen, Carlsbad, CA) were dissolved in DMSO at concentrations of 1 and 1 mg/ml, respectively.
Caffeine-sensitivity assays
S. cerevisiae strains were grown overnight to stationary phase in SC medium or SC-leucine medium (to select for plasmids) containing 100–12% methionine, then diluted 500-fold into the same medium containing serial dilutions of caffeine, ranging from 25 to 0.1 mM. After 24 h of incubation at 30°C in flat-bottom 96-well dishes, the optical densities at 650 nm were measured. The concentration of caffeine causing 50% inhibition of growth (IC50) was determined by nonlinear regression of the data to the standard four-parameter sigmoid equation, and the IC50 determinations from replicate experiments with each mutant strain were averaged.
Cell-staining methods
Log-phase S. cerevisiae cells growing in SC medium were exposed to tunicamycin (2 μg/ml), fluconazole (25 μg/ml), rapamycin (0.2 μg/ml), caffeine (various concentrations), concanamycin C (3 μM), FK506 (1 μg/ml), or combinations thereof for various lengths of time; pelleted; and stained for 15 min at room temperature in the same medium containing freshly prepared PI (0.25 μg/ml) and/or dihydro-DCFDA (1 μg/ml). Stained cells were imaged using an LSM 510 META confocal microscope (Zeiss) with appropriate filter sets, photographed, and counted manually after appropriate binning. In some experiments, log-phase cells were prestained with FM4-64 (10 μg/ml) for 15 min at 30°C, washed, and incubated for an additional 15 min at 30°C to allow complete endocytosis to the vacuole before addition of stressors and imaging (Vida and Emr, 1995). Most often, 2000 live and dead cells in each culture were counted after PI staining using a BD FACSArray instrument, as described previously (Dudgeon et al., 2008; Kim et al., 2012). For generating the black curves in Figure 2B, the measured frequencies of PI-stained cells in the cultures were fitted to the standard sigmoid equation (= min + (max − min)/(1 + (MLS/t)slope); where min and max were pegged at 1 and 100% and median lifespan and slope factor were solved). For generating the gray curves in Figure 2B, the measured frequencies of dihydro-DCFDA–stained cells in the cultures were fitted to a standard sigmoid equation (as above) minus the best-fit sigmoid equation obtained from the PI-stained cells in that culture.
Western blotting
Cultures were grown to log phase at 30°C in SC medium and adjusted to the same density. Samples were treated with FK506 (1 μg/ml), tunicamycin (2 μg/ml), or rapamycin (0.2 μg/ml) for various lengths of time at 30°C before processing. Processing involved centrifuging 800 μl of cells at 4°C, lysis of cells with 10% ice-cold trichloroacetic acid, extraction of proteins with SDS sample buffer at 37°C, then SDS–PAGE, and Western blotting, as described previously (Mehta et al., 2009). Blots were probed with anti-hemagglutinin (HA) monoclonal antibodies at 1:10,000 dilution (Covance, Princeton, NJ), anti-phospho S6 (Ser-235/236) monoclonal antibodies at 1:5000 dilution (Cell Signaling, Danvers, MA), anti-RPS6 polyclonal antibodies at 1:5000 dilution (Abcam, Cambridge, United Kingdom), or anti-Pib2 antibodies (kind gift of Chris Burd, Yale University). Protein standards were probed with anti-PGK1 antibodies at 1:10,000 (Abcam).
Computational methods
For identification of orthologues of Pib2, standard protein PSI-BLAST searches of nonredundant protein sequences were restricted to fungi (taxid:4751; Altschul et al., 1997). The hits were collected and subjected to multiple sequence alignment using the CLUSTAL W (Thompson et al., 1994) with MegAlign software, and segments of strong sequence conservation were identified among the sequences (labeled A–E, FYVE, and T in Figure 1). True orthologues of Pib2 could not be identified readily in metazoa because of strong conservation of FYVE domains in several different protein families. We therefore utilized the SMART database (Schultz et al., 2000) to identify all 27 human proteins bearing FYVE domains and to focus attention on those proteins for which the FYVE domain was located near the C-terminus and a C-terminal sequence motif was conserved in orthologues from other species. Only LAPF/phafin1 and its orthologues contained a tail motif separated from the FYVE domain by a poorly conserved spacer sequence, similar to Pib2 and its orthologues. Sequence logos that summarize the degree of conservation at each amino acid position were generated from multiple sequence alignments of Pib2 and LAPF/phafin1 orthologues using WebLogo (Crooks et al., 2004). Hierarchical clustering of chemical and genetic interaction profiles was performed using Cluster 3.0 using the correlation (uncentered) setting (Eisen et al., 1998). After clustering was complete, the tables of genetic and chemical interactions were reordered, and new tables containing the Pearson correlation coefficients between each gene and every other gene were generated. Portions of these tables are shown in Figure 3, A and B, and the complete tables are available upon request.
While studying the clusters containing pib2∆ mutants, we noticed that an ecm8∆ mutant coclustered with pib2∆ and other TORC1-deficient strains when it was used as a query strain but not as an array strain. An independently generated ecm8∆ mutant strain behaved like wild-type parents in similar assays (unpublished data). Because the ECM8 gene is located adjacent to the EGO3 gene, we tested whether the ecm8∆ query strain harbors an unexpected ego3 mutation by performing complementation tests. The ecm8∆ array strain failed to complement the ego3∆ query strain in assays of cell death and caffeine hypersensitivity, and therefore the ECM8 gene was relabeled as EGO3* in Figure 3.
Acknowledgments
The authors are particularly indebted to Stephen Helliwell (Novartis), who shared unpublished data on Pib2 localization and pib2∆ phenotypes; Chris Burd (Yale University), who shared unpublished results and provided anti-Pib2 polyclonal antibodies; and Michael Costanzo and Brenda Andrews (University of Toronto), who shared unpublished SGA data that supplemented Figure 3. Robbie Loewith (University of Geneva), Claudio De Virgilio (University of Fribourg), and Chris Kaiser (Massachusetts Institute of Technology) supplied helpful plasmids. Richard Bennett (Brown University) supplied C. albicans strains. We thank Hyemin Kim, Nathan Snyder, and other members of the Cunningham lab for their advice on the research. This research was generously supported by grants from the National Institutes of Health (T32-GM007231 to the CMDB graduate training program at Johns Hopkins University; R21-AI115016 to K.W.C.).
Abbreviations used:
- Cmk2
Ca2+/calmodulin-dependent protein kinase
- DCFDA
2′,7′ –dichlorofluorescin diacetate
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- HA
hemagglutinin
- LMP
lysosomal membrane permeabilization
- PH
pleckstrin homology
- PI(3)P
phosphatidylinositol 3-phosphate
- PI(3,5)P2
phosphatidylinositol 3,5-bisphosphate
- Rps6
ribosomal protein S6
- SC
synthetic complete
- UPR
unfolded protein response
- YPD
yeast–peptone–dextrose.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-08-0581) on October 28, 2015.
REFERENCES
- Alby K, Schaefer D, Sherwood RK, Jones SK, Jr, Bennett RJ. Identification of a cell death pathway in Candida albicans during the response to pheromone. Eukaryot Cell. 2010;9:1690–1701. doi: 10.1128/EC.00155-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnandis T, Ferrer-Vicens I, Garcia-Trevijano ER, Miralles VJ, Garcia C, Torres L, Vina JR, Zaragoza R. Calpains mediate epithelial-cell death during mammary gland involution: mitochondria and lysosomal destabilization. Cell Death Differ. 2012;19:1536–1548. doi: 10.1038/cdd.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baars TL, Petri S, Peters C, Mayer A. Role of the V-ATPase in regulation of the vacuolar fission-fusion equilibrium. Mol Biol Cell. 2007;18:3873–3882. doi: 10.1091/mbc.E07-03-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banuelos MG, Moreno DE, Olson DK, Nguyen Q, Ricarte F, Aguilera-Sandoval CR, Gharakhanian E. Genomic analysis of severe hypersensitivity to hygromycin B reveals linkage to vacuolar defects and new vacuolar gene functions in Saccharomyces cerevisiae. Curr Genet. 2010;56:121–137. doi: 10.1007/s00294-009-0285-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda M, Peli-Gulli MP, Bonfils G, Panchaud N, Urban J, Sturgill TW, Loewith R, De Virgilio C. The Vam6 GEF controls TORC1 by activating the EGO complex. Mol Cell. 2009;35:563–573. doi: 10.1016/j.molcel.2009.06.033. [DOI] [PubMed] [Google Scholar]
- Bonilla M, Cunningham KW. Mitogen-activated protein kinase stimulation of Ca2+ signaling is required for survival of endoplasmic reticulum stress in yeast. Mol Biol Cell. 2003;14:4296–4305. doi: 10.1091/mbc.E03-02-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla M, Nastase KK, Cunningham KW. Essential role of calcineurin in response to endoplasmic reticulum stress. EMBO J. 2002;21:2343–2353. doi: 10.1093/emboj/21.10.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27:6434–6451. doi: 10.1038/onc.2008.310. [DOI] [PubMed] [Google Scholar]
- Bridges D, Fisher K, Zolov SN, Xiong T, Inoki K, Weisman LS, Saltiel AR. Rab5 proteins regulate activation and localization of target of rapamycin complex 1. J Biol Chem. 2012;287:20913–20921. doi: 10.1074/jbc.M111.334060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraveo G, Auluck PK, Whitesell L, Chung CY, Baru V, Mosharov EV, Yan X, Ben-Johny M, Soste M, Picotti P, et al. Calcineurin determines toxic versus beneficial responses to alpha-synuclein. Proc Natl Acad Sci USA. 2014;111:E3544–E3552. doi: 10.1073/pnas.1413201111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Li N, Chen T, Han Y, Li C, Wang Y, He W, Zhang L, Wan T, Cao X. The lysosome-associated apoptosis-inducing protein containing the pleckstrin homology (PH) and FYVE domains (LAPF), representative of a novel family of PH and FYVE domain-containing proteins, induces caspase-independent apoptosis via the lysosomal-mitochondrial pathway. J Biol Chem. 2005;280:40985–40995. doi: 10.1074/jbc.M502190200. [DOI] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudgeon DD, Zhang N, Ositelu OO, Kim H, Cunningham KW. Nonapoptotic death of Saccharomyces cerevisiae cells that is stimulated by Hsp90 and inhibited by calcineurin and Cmk2 in response to endoplasmic reticulum stresses. Eukaryot Cell. 2008;7:2037–2051. doi: 10.1128/EC.00291-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastwood MD, Cheung SW, Meneghini MD. Programmed nuclear destruction in yeast: self-eating by vacuolar lysis. Autophagy. 2013;9:263–265. doi: 10.4161/auto.22881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epple UD, Suriapranata I, Eskelinen EL, Thumm M. Aut5/Cvt17p, a putative lipase essential for disintegration of autophagic bodies inside the vacuole. J Bacteriol. 2001;183:5942–5955. doi: 10.1128/JB.183.20.5942-5955.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox DS, Heitman J. Good fungi gone bad: the corruption of calcineurin. Bioessays. 2002;24:894–903. doi: 10.1002/bies.10157. [DOI] [PubMed] [Google Scholar]
- Gailite I, Egger-Adam D, Wodarz A. The phosphoinositide-associated protein Rush hour regulates endosomal trafficking in Drosophila. Mol Biol Cell. 2012;23:433–447. doi: 10.1091/mbc.E11-02-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Kaiser CA. A conserved GTPase-containing complex is required for intracellular sorting of the general amino-acid permease in yeast. Nat Cell Biol. 2006;8:657–667. doi: 10.1038/ncb1419. [DOI] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Glass NL, Dementhon K. Non-self recognition and programmed cell death in filamentous fungi. Curr Opin Microbiol. 2006;9:553–558. doi: 10.1016/j.mib.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Gonzalez A, Shimobayashi M, Eisenberg T, Merle DA, Pendl T, Hall MN, Moustafa T. TORC1 promotes phosphorylation of ribosomal protein S6 via the AGC kinase Ypk3 in Saccharomyces cerevisiae. PLoS One. 2015;10:e0120250. doi: 10.1371/journal.pone.0120250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatsugai N, Yamada K, Goto-Yamada S, Hara-Nishimura I. Vacuolar processing enzyme in plant programmed cell death. Front Plant Sci. 2015;6:234. doi: 10.3389/fpls.2015.00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoepfner D, Helliwell SB, Sadlish H, Schuierer S, Filipuzzi I, Brachat S, Bhullar B, Plikat U, Abraham Y, Altorfer M, et al. High-resolution chemical dissection of a model eukaryote reveals targets, pathways and gene functions. Microbiol Res. 2014;169:107–120. doi: 10.1016/j.micres.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Huber A, Bodenmiller B, Uotila A, Stahl M, Wanka S, Gerrits B, Aebersold R, Loewith R. Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev. 2009;23:1929–1943. doi: 10.1101/gad.532109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin N, Mao K, Jin Y, Tevzadze G, Kauffman EJ, Park S, Bridges D, Loewith R, Saltiel AR, Klionsky DJ, Weisman LS. Roles for PI(3,5)P2 in nutrient sensing through TORC1. Mol Biol Cell. 2014;25:1171–1185. doi: 10.1091/mbc.E14-01-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152:519–530. doi: 10.1083/jcb.152.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Kim A, Cunningham KW. Vacuolar H+-ATPase (V-ATPase) promotes vacuolar membrane permeabilization and nonapoptotic death in stressed yeast. J Biol Chem. 2012;287:19029–19039. doi: 10.1074/jbc.M112.363390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzaler PA, Staniszewska AD, Li W, Omidvar N, Kedjouar B, Turkson J, Poli V, Flavell RA, Clarkson RW, Watson CJ. Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol. 2011;13:303–309. doi: 10.1038/ncb2171. [DOI] [PubMed] [Google Scholar]
- Li N, Zheng Y, Chen W, Wang C, Liu X, He W, Xu H, Cao X. Adaptor protein LAPF recruits phosphorylated p53 to lysosomes and triggers lysosomal destabilization in apoptosis. Cancer Res. 2007;67:11176–11185. doi: 10.1158/0008-5472.CAN-07-2333. [DOI] [PubMed] [Google Scholar]
- Lin WJ, Yang CY, Li LL, Yi YH, Chen KW, Lin YC, Liu CC, Lin CH. Lysosomal targeting of phafin1 mediated by Rab7 induces autophagosome formation. Biochem Biophys Res Commun. 2012;417:35–42. doi: 10.1016/j.bbrc.2011.11.043. [DOI] [PubMed] [Google Scholar]
- Liu S, Hou Y, Liu W, Lu C, Wang W, Sun S. Components of the calcium-calcineurin signaling pathway in fungal cells and their potential as antifungal targets. Eukaryot Cell. 2015;14:324–334. doi: 10.1128/EC.00271-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Hall MN. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics. 2011;189:1177–1201. doi: 10.1534/genetics.111.133363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- MacGurn JA, Hsu PC, Smolka MB, Emr SD. TORC1 regulates endocytosis via Npr1-mediated phosphoinhibition of a ubiquitin ligase adaptor. Cell. 2011;147:1104–1117. doi: 10.1016/j.cell.2011.09.054. [DOI] [PubMed] [Google Scholar]
- Martin DC, Kim H, Mackin NA, Maldonado-Baez L, Evangelista CC, Beaudry VG, Dudgeon DD, Naiman DQ, Erdman SE, Cunningham KW. New regulators of a high affinity Ca2+ influx system (HACS) revealed through a genome-wide screen in yeast. J Biol Chem. 2011;286:10744–10754. doi: 10.1074/jbc.M110.177451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta S, Li H, Hogan PG, Cunningham KW. Domain architecture of the regulators of calcineurin (RCANs) and identification of a divergent RCAN in yeast. Mol Cell Biol. 2009;29:2777–2793. doi: 10.1128/MCB.01197-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaillat L, Baars TL, Mayer A. Cell-free reconstitution of vacuole membrane fragmentation reveals regulation of vacuole size and number by TORC1. Mol Biol Cell. 2012;23:881–895. doi: 10.1091/mbc.E11-08-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrschtik M, Ryan KM. Lysosomal proteins in cell death and autophagy. FEBS J. 2015;282:1858–1870. doi: 10.1111/febs.13253. [DOI] [PubMed] [Google Scholar]
- Muller EM, Locke EG, Cunningham KW. Differential regulation of two Ca2+ influx systems by pheromone signaling in Saccharomyces cerevisiae. Genetics. 2001;159:1527–1538. doi: 10.1093/genetics/159.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira AP, Ludwig C, Zampieri M, Weisser H, Aebersold R, Sauer U. Dynamic phosphoproteomics reveals TORC1-dependent regulation of yeast nucleotide and amino acid biosynthesis. Sci Signal. 2015;8:rs4. doi: 10.1126/scisignal.2005768. [DOI] [PubMed] [Google Scholar]
- Peterson JS, McCall K. Combined inhibition of autophagy and caspases fails to prevent developmental nurse cell death in the Drosophila melanogaster ovary. PLoS One. 2013;8:e76046. doi: 10.1371/journal.pone.0076046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinan-Lucarre B, Paoletti M, Clave C. Cell death by incompatibility in the fungus Podospora. Semin Cancer Biol. 2007;17:101–111. doi: 10.1016/j.semcancer.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Reinke A, Chen JC, Aronova S, Powers T. Caffeine targets TOR complex I and provides evidence for a regulatory link between the FRB and kinase domains of Tor1p. J Biol Chem. 2006;281:31616–31626. doi: 10.1074/jbc.M603107200. [DOI] [PubMed] [Google Scholar]
- Schultz J, Copley RR, Doerks T, Ponting CP, Bork P. SMART: a Web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000;28:231–234. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schu PV, Takegawa K, Fry MJ, Stack JH, Waterfield MD, Emr SD. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science. 1993;260:88–91. doi: 10.1126/science.8385367. [DOI] [PubMed] [Google Scholar]
- Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40:310–322. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin ME, Ogburn KD, Varban OA, Gilbert PM, Burd CG. FYVE domain targets Pib1p ubiquitin ligase to endosome and vacuolar membranes. J Biol Chem. 2001;276:41388–41393. doi: 10.1074/jbc.M105665200. [DOI] [PubMed] [Google Scholar]
- Starai VJ, Jun Y, Wickner W. Excess vacuolar SNAREs drive lysis and Rab bypass fusion. Proc Natl Acad Sci USA. 2007;104:13551–13558. doi: 10.1073/pnas.0704741104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan CP, Cunningham KW. Kch1 family proteins mediate essential responses to endoplasmic reticulum stresses in the yeasts Saccharomyces cerevisiae and Candida albicans. J Biol Chem. 2013;288:34861–34870. doi: 10.1074/jbc.M113.508705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan CP, Zhang N, Sokabe T, Rivetta A, Slayman CL, Montell C, Cunningham KW. Activation of an essential calcium signaling pathway in Saccharomyces cerevisiae by Kch1 and Kch2, putative low-affinity potassium transporters. Eukaryot Cell. 2013;12:204–214. doi: 10.1128/EC.00299-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach WJ, Reedy JL, Cramer RA, Jr, Perfect JR, Heitman J. Harnessing calcineurin as a novel anti-infective agent against invasive fungal infections. Nat Rev Microbiol. 2007;5:418–430. doi: 10.1038/nrmicro1680. [DOI] [PubMed] [Google Scholar]
- Stracka D, Jozefczuk S, Rudroff F, Sauer U, Hall MN. Nitrogen source activates TOR (target of rapamycin) complex 1 via glutamine and independently of Gtr/Rag proteins. J Biol Chem. 2014;289:25010–25020. doi: 10.1074/jbc.M114.574335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syntichaki P, Samara C, Tavernarakis N. The vacuolar H+-ATPase mediates intracellular acidification required for neurodegeneration in C. elegans. Curr Biol. 2005;15:1249–1254. doi: 10.1016/j.cub.2005.05.057. [DOI] [PubMed] [Google Scholar]
- Syntichaki P, Xu K, Driscoll M, Tavernarakis N. Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature. 2002;419:939–944. doi: 10.1038/nature01108. [DOI] [PubMed] [Google Scholar]
- Tarassov K, Messier V, Landry CR, Radinovic S, Serna Molina MM, Shames I, Malitskaya Y, Vogel J, Bussey H, Michnick SW. An in vivo map of the yeast protein interactome. Science. 2008;320:1465–1470. doi: 10.1126/science.1153878. [DOI] [PubMed] [Google Scholar]
- Teter SA, Eggerton KP, Scott SV, Kim J, Fischer AM, Klionsky DJ. Degradation of lipid vesicles in the yeast vacuole requires function of Cvt17, a putative lipase. J Biol Chem. 2001;276:2083–2087. doi: 10.1074/jbc.C000739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth ML, Simon P, Kovacs AL, Vellai T. Influence of autophagy genes on ion-channel-dependent neuronal degeneration in Caenorhabditis elegans. J Cell Sci. 2007;120:1134–1141. doi: 10.1242/jcs.03401. [DOI] [PubMed] [Google Scholar]
- van Doorn WG. Classes of programmed cell death in plants, compared to those in animals. J Exp Bot. 2011;62:4749–4761. doi: 10.1093/jxb/err196. [DOI] [PubMed] [Google Scholar]
- Vida TA, Emr SD. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J Cell Biol. 1995;128:779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Lao U, Edgar BA. TOR-mediated autophagy regulates cell death in Drosophila neurodegenerative disease. J Cell Biol. 2009;186:703–711. doi: 10.1083/jcb.200904090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanke V, Cameroni E, Uotila A, Piccolis M, Urban J, Loewith R, De Virgilio C. Caffeine extends yeast lifespan by targeting TORC1. Mol Microbiol. 2008;69:277–285. doi: 10.1111/j.1365-2958.2008.06292.x. [DOI] [PubMed] [Google Scholar]
- Wu X, Tu BP. Selective regulation of autophagy by the Iml1-Npr2-Npr3 complex in the absence of nitrogen starvation. Mol Biol Cell. 2011;22:4124–4133. doi: 10.1091/mbc.E11-06-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacobi-Sharon K, Namdar Y, Arama E. Alternative germ cell death pathway in Drosophila involves HtrA2/Omi, lysosomes, and a caspase-9 counterpart. Dev Cell. 2013;25:29–42. doi: 10.1016/j.devcel.2013.02.002. [DOI] [PubMed] [Google Scholar]
- Yamashima T, Oikawa S. The role of lysosomal rupture in neuronal death. Prog Neurobiol. 2009;89:343–358. doi: 10.1016/j.pneurobio.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Yuan HX, Russell RC, Guan KL. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy. 2013;9:1983–1995. doi: 10.4161/auto.26058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang NN, Dudgeon DD, Paliwal S, Levchenko A, Grote E, Cunningham KW. Multiple signaling pathways regulate yeast cell death during the response to mating pheromones. Mol Biol Cell. 2006;17:3409–3422. doi: 10.1091/mbc.E06-03-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]