

Abstract
There are numerous studies that demonstrate the anti-neoplastic activity of phosphatidylinositol 3-kinase (PI3K) inhibitors and the mechanisms of inducing autophagy in cancer cells. The new anticancer drug puquitinib mesylate (XC-302) is a molecular-targeted drug, which suppresses the activity of PI3K directly. However, it remains unclear whether XC-302 can develop an antitumor effect by inducing autophagy in naso-pharyngeal cancer cells. The MTT assay was used to study the anti-proliferative effects of XC-302. Subsequently, autophagy was determined by monodansylcadaverine (MDC) staining, punctate localization of green fluorescent protein (GFP)-light chain 3 (LC3), LC3 protein blotting and electron microscopy. The expression levels of beclin 1, p62, protein kinase B (AKT), phospho (p)-AKT, mechanistic target of rapamycin (mTOR) and p-mTOR in XC-302-induced autophagy were detected. Autophagy inhibition was assayed by 3-methyladenine (3-MA) or small interfering RNA (siRNA) silencing of beclin 1. XC-302 inhibited the viability of CNE-2 in a dose-dependent manner and the IC50 of 72 h was 5.2 µmol/l. After cells were exposed to XC-302 for 24 h, MDC-labeled autophagolysosomes were evident in CNE-2 cells by fluorescence microscope. Autophagosomes and autolysosomes were identified by transmission electron microscopy. Following transfection with GFP-LC3, XC-302 induced a significant accumulation of GFP-LC3, as monitored by a confocal microscope, which was reduced by 3-MA. XC-302 induced the formation of LC3-II, increased beclin 1 levels and decreased the expression of p62. Additionally, the expression levels of p-AKT and p-mTOR were reduced with the elevation of XC-302. Knockdown of beclin 1 with siRNA or co-treatment with 3-MA enhanced significantly the survival of CNE-2 and promoted the ability of clone formation. XC-302 also induced apoptosis in CNE-2, and when autophagy was inhibited by 3-MA, the apoptosis rate was decreased. The present data provides the evidence that XC-302 can induce autophagy in CNE-2, which promotes the program of cell death and inhibits the PI3K/AKT/mTOR signaling pathway. Furthermore, XC-302 also promoted apoptosis in CNE-2 cells, which could be reduced when autophagy was suppressed, meaning that autophagy may interact with apoptosis to induce cell death.
Keywords: puquitinib mesylate, phosphatidylinositol 3-kinase inhibitors, inhibitor, autophagy, apoptosis
Introduction
The aberrant activation of the phosphatidylinositol 3-kinase (PI3K) signaling pathway is an essential component of cellular processes that are critical for malignant tumor development, such as growth, proliferation and invasion (1,2). The small molecule inhibitors, including PI3K itself and associated pathway proteins, have been investigated as potential targets for new anticancer drugs (3). Puquitinib mesylate (XC-302) is a novel multiple-target agent that has been developed independently by Xinchang Pharmacy Corp. (Zhejiang Medicine Co., Ltd., Zhejiang, China). The ability to immediately suppress the activity of PI3K (subtype IA, IC50 against PI3K isoforms: p110α=766.6 nM, p110β=699.4 nM, p110δ=2.8 nM and p110γ=89.7 nM) in vitro has been reported in a preclinical study. It also can apparently inhibit the protein kinase B (AKT) phosphorylation mediated by epidermal growth factor receptor, the activity of receptor tyrosine kinase (kinase insert domain receptor and platelet-derived growth factor receptor β) and the formation of the vascular endothelial cells in the lumen. Preliminary studies have reported that XC-302 has clear antitumor efficacy in xenograft nude mice and in vitro, including cells of colon cancer, lung cancer and lymphoma (all with the IC50 in a range of 0.5–2.0 µM). In addition, XC-302 shows low toxicity in toxicological experiments. It is also rapid to absorb and has a high-level absolute bioavailability, which have been indicated by the preclinical pharmacokinetics tests. The phase I clinical trials also exhibit notable safety, tolerability and efficiency of XC-302 in advanced solid tumors and hematological malignancies (data are unpublished information from Xinchang Pharmacy Corp.).
Epidemiology studies show that nasopharyngeal carcinoma (NPC) is a type of familiar head and neck cancer in Southern China and Southeast Asia (4). Currently, irradiation alone or chemotherapy combined with radiotherapy is the major treatment on the basis of the disease stage (5). Due to the marked breakthroughs in diagnosis and treatment (such as intensity-modulated radiation therapy), the majority of early-stage patients could be cured (6). Even in patients with locally advanced stage disease, the 5-year local control rate is more than 90% (7). However, there remain a portion of locally advanced and numerous metastasis NPC patients who fail to achieve long-term disease control and succumb from the disease when it begins to progress out of control. One of the reasons for the poor prognosis may be treatment resistant (8). Therefore, evolving novel antitumor drugs research and development is a notable prospect for oncotherapy. Preclinical studies demonstrated that the PI3K/mechanistic target of rapamycin (mTOR) signaling pathway is commonly activated in numerous cancers and inhibition of this pathway could increase radiosensitivity, including NPC (9,10), suggesting that the PI3K/mTOR pathway may be a promising target in NPC (11). A previous study has shown that BEZ235, a dual PI3K/mTOR inhibitor, has a therapeutic potential and can reverse resistance to cisplatin in NPC (10). Another study reported that GSK2126458 and PKI-587, which are also dual PI3K/mTOR inhibitors, suppress tumor progression and increase radiosensitivity in NPC (9).
The feasibility of medicines in anticancer treatments is assessed by their capacity to induce cell death (12). Three major modalities of cell death have been defined, including apoptosis (type I), autophagy (type II), which has been regarded as 'programmed cell death', and necrosis (type III), which has been considered as accidental and unregulated (13). There are limited studies that have observed that autophagy can be induced by drugs in NPC, including drugs targeted for the PI3K/AKT/mTOR pathway (14,15). As a consequence, XC-302 may not exhibit an antitumor effect by inducing autophagy in NPC cells. In addition, we hypothesize that the present study could provide an initial basis of XC-302 for subsequent clinical application.
Materials and methods
Cell cultures and reagents
The human NPC cell line, CNE-2 (poorly differentiated), was supplied from State Key Laboratory of Oncology (Sun Yat-sen University Cancer Center, Guangzhou, China). The cell lines were maintained in RPMI-1640 with 10% fetal bovine serum (FBS). Cells in the logarithmic phase were used in the experiments. The puquitinib mesylate (XC-302; chemical structure shown in Fig. 1) was obtained from Xinchang Pharmacy Corp. and dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO, USA) at a storage concentration of 40 mM.
Figure 1.

The structural formula of XC-302. C17H15N7•2CH3SO3H•2H2O; Molecular weight: 545.59.
Cell proliferation assay
In brief, tumor cells (3×103/100 µl/well) were plated in 96-well plates and incubated for 24 h. Culture medium containing gradient concentrations of XC-302 (16–0.125 µM) were added to each well, and 0.1% DMSO was set as the negative control. MTT (5.0 mg/l; 20 µl) was added after the cells were incubated for 68 h, followed by the addition of 150 µl DMSO into each well to dissolve the dark blue crystals. The absorbance (A) was measured at wavelengths of 570 and 630 nm, respectively (Model-550; Bio-Rad, Hercules, CA, USA). Finally, the cell viability value was calculated using the following formula: (Asample - Ablank)/(Acontrol - Ablank) × 100%. All the experiments were executed independently at least three times in triplicate.
Monodansylcadaverine (MDC) staining
The MDC assay was used to label preliminary autophagosomes (16). Cells grown on 6-well plates were treated with XC-302 (0.5–8 µM) for 24 h, incubated with 0.05 mM MDC for 30 min at 37°C and washed with phosphate-buffered saline (PBS). Fluorescence images were captured by an Olympus IX71 fluorescence microscope (Olympus, Center Valley, PA, USA).
Transmission electron microscopy (TEM)
The presence of autophagosomes in TEM is the standard for detecting autophagy. Cells grown on 6-well plates were treated with XC-302 (4 µM) and 24 h later were trypsinized and washed with PBS prior to fixing in fixative buffer. Subsequently, cells were collected by centrifugation at 500 × g for 10 min, suspended and incubated for 2 h in 2.5% glutaraldehyde. Following fixation, cell samples were treated with 2% osmium tetroxide in 0.1 M sodium cacodylate buffer, dehydrated through a graded series of acetone and embedded in resin. Finally, the samples were sliced into 65-nm sections, which were processed for TEM (Hitachi electron microscope H-600; Hitachi, Ltd., Toyko, Japan).
Transfection of green fluorescent protein (GFP)-light chain 3 (LC3) expression vector
The GFP-LC3 expression vector was generously provided by Professor X.F. Zhu (State Key Laboratory of Oncology). Following transfection of the GFP-LC3 expression vector with Lipofectamine 2000 (Invitrogen) for 24 h, following the manufacturer's protocol, cells were divided into groups based as follows: i) Negative control with 0.1% DMSO (4 µM); ii) XC-302 (4 and 8 µM); iii) 3-MA (5 mM); and iv) 3-MA (5 mM) + XC-302 (4 and 8 µM). Following the indicated treatment, the localization of GFP was directly observed with a laser scanning confocal microscope.
Western blot analysis
Following treatment of various concentrations of XC-302 for different times, the total protein from the CNE-2 cells was obtained by lysing in cell lysis buffer (Cell Signaling Technology, Beverly, MA, USA). The proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane. The membranes were subsequently blocked with skimmed milk (5%) and incubated overnight at 4°C with the following antibodies: Total AKT (1:1,000, #9916, rabbit anti-human), mTOR, phospho-AKT (S473; 1:2,000, #9916, rabbit anti-human), phospho-mTOR (1:1,000, #5536, rabbit anti-human), LC3 (1:1,000, #4599, rabbit anti-human), beclin 1 (1:1,000, #3495, rabbit anti-human), p62 (1:1,000, #8025, rabbit anti-human) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:3,000, #5174, rabbit anti-human), which were purchased from Cell Signaling Technology. The samples were incubated with a secondary antibody (1:4,000, #9916, anti-rabbit IgG; CST) for 1 h at room temperature and the labeled proteins were detected using chemiluminescence reagent and automatic X-ray film. Equal loading GAPDH was assessed as the internal control for western blot analysis.
Downregulation of gene expression by small interfering RNA (siRNA)
The specific siRNA oligomers against beclin 1 (forward, 5′-CAGTTTGGCACAATCAATAtt-3′) and si-control (forward, 5′-UUCUCCGAACGUGUCACGUtt-3′) were purchased from the Gene Technology Co. (Shanghai, China). The CNE-2 cells were transfected with si-beclin 1 and Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's protocol. After incubation for 12 h, the transient transfected CNE-2 were used for the following experiments.
Colony formation assay
The cells were grown for 24 h and subsequently treated for 6 h as follows: Groups without intervention of siRNA, i) negative control; ii) XC-302 (4 µM); iii) 3-MA (5 mM); and iv) 3-MA (5 mM) + XC-302 (4 µM); and groups with silencing of siRNA, i) si-control; ii) si-control cells with XC-302 (4 µM); iii) si-beclin 1; and iv) si-beclin 1 cells with XC-302 (4 µM). The cells were trypsinized and subsequently seeded in 6-well culture plates (500/well) for 2 weeks. The cells were washed and fixed with ice-cold methanol for 10 min, followed by the addition of 0.5% crystal violet solution for 10 min to observe the colony formation.
Annexin V-FITC/propidium iodide (PI) double staining
Briefly, the CNE-2 cells were treated with different concentrations of XC-302 (4 and 8 µM) and/or 5 mM 3-methyladenine (3-MA, M9281; Sigma-Aldrich) for 24 h. Following collection, the cells were stained with Annexin V-FITC and PI. The apoptotic cells were estimated using a Beckman-Gallios flow cytometer (Beckman-Gallios, Miami, FL, USA).
Statistical analysis
Whole data are exhibited as mean ± standard error of the mean of more than two independent experiments in each group. The variances between each group were calculated by Student's t-test and P<0.05 was considered to indicate a statistically significant difference.
Results
XC-302 inhibits the proliferation of CNE-2 cells by inducing autophagy and apoptosis
To assess the effects of XC-302 on NPC cell proliferation in vitro, CNE-2 cells were treated with XC-302 at different concentrations for 72 h and subsequently evaluated by the MTT assays. Exposure to XC-302 showed a dose-dependent inhibition of cell viability. MDC is known as a specific marker of autophagic vacuoles (Fig. 2A). XC-302-treated CNE-2 cells resulted in fluorescence intensity enhanced with the increasing dose of the drug (Fig. 2B). In addition to autophagy, XC-302 treatment also induced apoptosis in the CNE-2 cells. The Annexin V-FITC/PI double staining showed that cells treated with XC-302 increased the percentage of apoptosis up to 14.2% (Fig. 2C). The TEM was adopted to further examine the autophagosomes of XC-302-treated cells. Compared with the control cells, XC-302 treatment not only resulted in a rapid increase of autophagosomes (thick arrow), but also induced the formation of autolysosomes (thin arrow) in a portion of CNE-2 cells (Fig. 2D).
Figure 2.

XC-302 inhibits proliferation, as well as induces apoptosis and autophagy, in CNE-2 cell lines. (A) After 24 h treatment of XC-302 (16–0.125 µM), CNE-2 cell viability was detected by the MTT assay. (B) XC-302 treated cells were stained with monodansylcadaverine (MDC), and the clearly visible images of staining were examined by a fluorescence microscopy. (C) CNE-2 cells were treated as described for 24 h and apoptosis was analyzed by flow cytometry following Annexin V-propidium iodide staning. (D) The formation of autophagosomes in XC-302-treated cells observed under transmission electron microscopy. The whole organelles could be observed in the cells in the untreated group. The XC-302 treatment (4 µM) not only resulted in the accumulation of autophagosomes (thick arrow), but also the formation of autolysosomes (thin arrow) in a portion of CNE-2 cells (magnification, ×37,000).
XC-302 inhibits the activation of the PI3K/AKT pathway during the process of autophagy in CNE-2 cells
Microtubule-associated protein 1 LC3 is a specific marker for autophagy. It is converted from the inactive form, LC3-I, to a cleaved form (LC3-II) during autophagy, and the LC3-II protein has been frequently used for autophagy detection. In the present study, the changes of the markers in autophagy were analyzed by western blotting analysis. The LC3-II protein levels were markedly increased by XC-302 treatment. The beclin 1 level was augmented and as a degradation product, the expression of p62 was decreased (Fig. 3A and B).
Figure 3.

Western blot analysis was performed for autophagy- and phospha-tidylinositol 3-kinase (PI3K) pathway-related markers. (A) The cells were treated with XC-302 (8.0–0.5 µM) for 24 h. Cell lysates were prepared, and an immunoblot was performed using antibodies against LC3-I, LC3-II, beclin 1 and p62. (B) The effects of 4 µM XC-302 incubation on the protein expression of LC3-I, LC3-II and p62 in CNE-2 cells at 0, 3, 6, 12, 24 and 36 h. (C) Cells were treated with different concentrations of XC-302 for 24 h. The immunoblot was conducted and probed with anti-AKT, anti-p-AKT, anti-mTOR and anti-p-mTOR. LC, light chain; AKT, protein kinase B; mTOR, mechanistic target of rapamycin.
Subsequently, whether the downstream targets of PI3K could be inhibited by XC-302 was examined in the CNE-2 cell line. As shown in Fig. 3C, following exposure to XC-302 for 24 h, although the levels of AKT and mTOR did not exhibit significant changes, the phosphorylation levels of AKT and mTOR protein were attenuated when treated by XC-302 in a dose-dependent manner.
Autophagy inhibitor 3-MA reduces cell death induced by XC-302 in the CNE-2 cells
3-MA is a known specific inhibitor of autophagy, which is frequently used to detect the role of autophagy under various treatments. Thus, the effects of 3-MA was investigated on the apoptosis, autophagy, proliferation and clone formation of XC-302-treated CNE-2 cells. The apoptosis rate of cells were markedly decreased in cells co-treated with various concentrations of XC-302 and 3-MA, compared with those treated with XC-302 alone (P<0.05) (Fig. 4A and B). To detect the formation of autophagosomes specifically, cells were transfected with the GFP-LC3 plasmid transiently. Following treatment with XC-302, cells showed increased LC3 punctures compared with the control in a dose-dependent manner (Fig. 4C), visualized by fluorescence microscopy. In addition, there was a significant degradation of green fluorescence in XC-302 (8 µM) and 3-MA co-treated cells. These results corroborate the observation that XC-302 induced autophagy, as well as 3-MA decreased the accumulation of autophagosomes, in XC-302-exposed CNE-2 cells. The proliferation of the CNE-2 cells was inhibited by XC-302 and could be reversed by exposure to 3-MA after 24 or 48 h, as revealed by the MTT assay (P<0.05) (Fig. 5A). Subsequently, clone formation assays were used to further ensure that 3-MA reduced the sensitivity of the cells to XC-302. The group combining 5 mM 3-MA with 4 µM XC-302 formed more colonies compared with the individual use of XC-302 (P<0.05) (Fig. 5B and C). These results clearly reveal that autophagy contributes to XC-302-induced cell death in CNE-2 cells.
Figure 4.
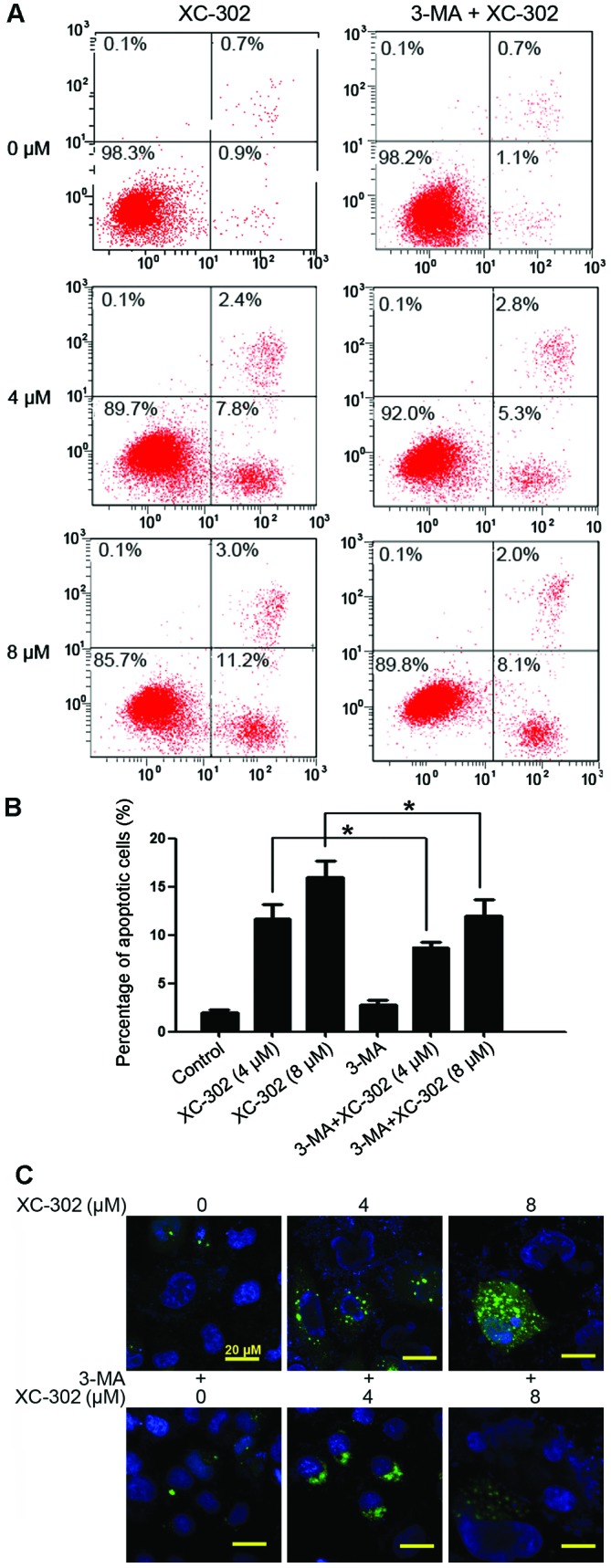
Apoptosis and autophagy in CNE-2 cells, induced by XC-302, which could be inhibited by exposure to 3-methyladenine (3-MA). (A) Testing of apoptosis was by Annexin V-FITC/propidium iodide assays, and the CNE-2 cells were treated by 4 and 8 µM XC-302 in the absence (left) or presence (right) of 5 mM 3-MA for 24 h. (B) The apoptosis rates are described as the mean of three independent experiments ± standard deviation. P-values were obtained by comparing the apoptosis rates in cells treated with XC-302 alone with those in 3-MA combined with XC-302 (*P<0.05). (C) Images acquired by fluorescence microscopy, green fluorescent protein (GFP)-light chain 3 (LC3)-transfected cells treated with dose increasing XC-302 showed increased GFP-LC3 dots (upper), whereas at an XC-302 concentration of 8 µM, 3-MA significantly decreased the green fluorescence (lower panels).
Figure 5.

3-Methyladenine (3-MA) accelerates the proliferation and clone formation of the CNE-2 cell line by suppressing autophagy. (A) CNE-2 cells were exposed to 4 µM XC-302/5 µM 3-MA, or a co-treatment of XC-302 and 3-MA for 24 and 48 h. The survival rate was compared by the MTT assay. (B and C) Cells were treated with 4 µM XC-302/5 µM 3-MA, or a combination of XC-302 and 3-MA for 6 h, and the clone formation ability was analyzed. Mean of three independent experiments; bars, mean ± standard deviation (*P<0.05).
Knockdown of beclin 1 accelerates proliferation and clone formation of CNE-2 cell line treated by XC-302
Beclin 1 is a critical regulator of autophagy, which can specifically suppress the autophagy pathway by knockdown of it. The beclin 1 gene was further knocked down using siRNA and the efficiency of the knockdown was confirmed by western blotting (Fig. 6A). Using the MTT assay, similar to the effect of 3-MA, the knockdown of beclin 1 resulted in a higher vital rate following exposure to XC-302 (P<0.05) (Fig. 6B). Additionally, clone formation is markedly increased in beclin 1 knockdown cells compared with the scramble control cells following the treatment of XC-302 (Fig. 6C and D). Taken together, these results revealed that the inhibition of autophagy by beclin 1 knockdown promoted proliferation and clone formation of CNE-2 cells that were exposed to XC-302.
Figure 6.

Stimulations of silencing beclin 1 on proliferation and clone formation in the CNE-2 cell line treated by XC-302. (A) CNE-2 cells were transfected with beclin 1 small interfering RNA (siRNA) or a non-target control siRNA for 48 h, and the expression of beclin 1 was verified by western blotting analysis. (B) The MTT assay measured cell viability between four groups (si-control, si-control cells with 4 µM XC-302, si-beclin 1 and si-beclin 1 cells with 4 µM XC-302). (C) Cells were treated with different drugs and siRNA, and the clone formation ability was analyzed. (D) The clone formation abilities are presented as the mean ± standard deviation. The P-value was measured by comparing the clone numbers between XC-302-treated cells silenced by si-control and those by si-beclin (*P<0.05).
Discussion
The PI3K/AKT/mTOR signaling pathway has a crucial role in the tumorigenesis of human cancers, including NPC (17,18), which is extremely attractive for targeted, molecular drug therapies. The NPC cell lines and tissues also overexpressed phosphorylated AKT (19-21). The two well-studied PI3K inhibitors are wortmannin and LY294002 (22). However, due to toxic responses, and poor pharmacological properties, their application has been primarily restricted. As a novel multiple target point inhibitor, including PI3K, which was developed independently, XC-302 exhibited promising inhibition of NPC cell proliferation, and not only induced apoptotic cell death but also autophagy in the present study.
As well as apoptosis, autophagy is a type of programmed cell death that is mediated by activation of the autophagy-related gene proteins. When autophagy is induced, the characteristic structure change is the formation of acidic vesicular organelles with double-membrane structures engulfing bulk cytoplasmic organelles. Numerous antitumor agents have been reported to induce autophagy (23). The results of the present study showed the autophagosome accumulations in the CNE-2 cells and MDC-positive stain following exposure to XC-302. LC3-II, beclin 1 and p62 are the reliable biomarkers of autophagy (24). As shown by western blotting analysis, XC-302 augmented LC3-II and beclin 1 levels, and downregulated p62 in the CNE-2 cells. Currently, increasing studies have suggested that the PI3K/AKT pathway has a vital role in the regulation of autophagy (25–27). The consequences of protein expression are the hypothesis that the inhibition of the PI3K/AKT/mTOR pathway is involved in XC-302 induced autophagy. In the present study, XC-302 caused autophagy in the CNE-2 cell line, which was activated when PI3K/AKT/mTOR was inhibited. In particular, 3-MA suppressed the XC-302-induced accumulation of autophagosomes. As opposed to triggering the type I PI3K by XC-302, 3-MA inhibits autophagy in mammalian cells through inhibiting class III PI3K (28,29), which may explain the diverse effects of XC-302 and 3-MA in autophagy. XC-302 treatment also triggered apoptosis, as evidenced by Annexin V-PI double staining.
However, whether autophagy represents a survival mechanism or contributes to cell death remains controversial (30). The present study has defined the role of XC-302-mediated autophagy in CNE-2 cell functions. Inhibition of autophagy by 3-MA could markedly suppress XC-302-induced cell death. Consistent with this, the present findings were further strengthened by transduction of specific target siRNAs to block autophagy, and knockdown of beclin 1 in combination with XC-302 markedly accelerated proliferation and clone formation of the CNE-2 cell line compared to using XC-302 alone. Taken together, these results propose that autophagy provided a survival disadvantage for these NPC cells.
Notably, the promoted effect of apoptosis due to XC-302 treatment was reversed when autophagy was suppressed by 3-MA in the CNE-2 cells. In NPC, the associations of apoptosis with autophagy in response to PI3Ks inhibition remain unknown. The present results demonstrate that autophagy, as well as apoptosis, participate commonly in the death of CNE-2 cells induced by XC-302, and furthermore, autophagy may serve as a significant role in the process of apoptotic cell death. The mechanisms connecting autophagy with apoptosis are not entirely clear, and studies have generated inconsistent results. However, B-cell lymphoma-2 (Bcl-2) has been defined as a central regulator of autophagic and apoptotic signaling pathways (31–34). Beclin 1 was discovered by a yeast two-hybrid system as a binding partner of Bcl-2 (Bcl-2-beclin 1 complex) (31). Recent discoveries have suggested that certain apoptotic proteins modulate autophagy (35). In the present study, the phenomenon of mitochondrial swelling was observed when cells were exposed to XC-302. Tolkovsky et al (36) considered that the cytochrome c released by injured mitochondria could activate autophagy and apoptosis when the permeability of mitochondria was altered, which prompted the membrane potential. Therefore, it is possible that the mitochondria switches the cellular program from one to another.
In conclusion, to the best of our knowledge, this is the first study to report that XC-302 may induce autophagy, which promotes the program of CNE-2 cell death probably by inhibition of the PI3K/AKT/mTOR signaling pathway. Thus, XC-302 may be a potential novel candidate for treating human NPC, and enhancing autophagy is a new way to strengthen the effects of antitumor therapies. Furthermore, XC-302 also promoted apoptosis in CNE-2 cells, which could be reversed when autophagy was suppressed. While autophagy has been served as a possible therapeutic target in NPC treated with XC-302, the exact effects and association between autophagy and apoptosis remain unclear. More studies are required in the future to elucidate the associations.
References
- 1.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 2.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: Rationale and promise. Cancer Cell. 2003;4:257–262. doi: 10.1016/S1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- 3.Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28:1075–1083. doi: 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang ET, Adami HO. The enigmatic epidemiology of nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prev. 2006;15:1765–1777. doi: 10.1158/1055-9965.EPI-06-0353. [DOI] [PubMed] [Google Scholar]
- 5.Spratt DE, Lee N. Current and emerging treatment options for nasopharyngeal carcinoma. Onco Targets Ther. 2012;5:297–308. doi: 10.2147/OTT.S28032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin S, Pan J, Han L, Guo Q, Hu C, Zong J, Zhang X, Lu JJ. Update report of nasopharyngeal carcinoma treated with reduced-volume intensity-modulated radiation therapy and hypothesis of the optimal margin. Radiother Oncol. 2014;110:385–389. doi: 10.1016/j.radonc.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 7.Xiao WW, Huang SM, Han F, Wu SX, Lu LX, Lin CG, Deng XW, Lu TX, Cui NJ, Zhao C. Local control, survival, and late toxicities of locally advanced nasopharyngeal carcinoma treated by simultaneous modulated accelerated radiotherapy combined with cisplatin concurrent chemotherapy: Long-term results of a phase 2 study. Cancer. 2011;117:1874–1883. doi: 10.1002/cncr.25754. [DOI] [PubMed] [Google Scholar]
- 8.Gupta AK1, McKenna WG, Weber CN, Feldman MD, Goldsmith JD, Mick R, Machtay M, Rosenthal DI, Bakanauskas VJ, Cerniglia GJ, et al. Local recurrence in head and neck cancer: relationship to radiation resistance and signal transduction. Clin Cancer Res. 2002;8:885–892. [PubMed] [Google Scholar]
- 9.Liu T, Sun Q, Li Q, Yang H, Zhang Y, Wang R, Lin X, Xiao D, Yuan Y, Chen L, et al. Dual PI3K/mTOR inhibitors, GSK2126458 and PKI-587, suppress tumor progression and increase radio-sensitivity in nasopharyngeal carcinoma. Mol Cancer Ther. 2014;14:429–439. doi: 10.1158/1535-7163.MCT-14-0548. [DOI] [PubMed] [Google Scholar]
- 10.Yang F, Qian XJ, Qin W, Deng R, Wu XQ, Qin J, Feng GK, Zhu XF. Dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 has a therapeutic potential and sensitizes cisplatin in nasopharyngeal carcinoma. PLoS One. 2013;8:e59879. doi: 10.1371/journal.pone.0059879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Chen LH, Yuan YW, Li QS, Sun AM, Guan J. Activation of AKT is associated with metastasis of nasopharyngeal carcinoma. Tumour Biol. 2012;33:241–245. doi: 10.1007/s13277-011-0272-4. [DOI] [PubMed] [Google Scholar]
- 12.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng Q, Yu X, Xiao L, Hu Z, Luo X, Tao Y, Yang L, Liu X, Chen H, Ding Z, et al. Neoalbaconol induces energy depletion and multiple cell death in cancer cells by targeting PDK1-PI3-K/Akt signaling pathway. Cell Death Dis. 2013;4:e804. doi: 10.1038/cddis.2013.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao YY, Tian Y, Zhang J, Xu F, Yang YP, Huang Y, Zhao HY, Zhang JW, Xue C, Lam MH, et al. Effects of an oral allosteric AKT inhibitor (MK-2206) on human nasopharyngeal cancer in vitro and in vivo. Drug Des Devel Ther. 2014;8:1827–1837. doi: 10.2147/DDDT.S67961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munafó DB, Colombo MI. A novel assay to study autophagy: Regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114:3619–3629. doi: 10.1242/jcs.114.20.3619. [DOI] [PubMed] [Google Scholar]
- 17.Ma BB, Lui VW, Hui EP, Lau CP, Ho K, Ng MH, Cheng SH, Tsao SW, Chan AT. The activity of mTOR inhibitor RAD001 (everolimus) in nasopharyngeal carcinoma and cisplatin-resistant cell lines. Invest New Drugs. 2010;28:413–420. doi: 10.1007/s10637-009-9269-x. [DOI] [PubMed] [Google Scholar]
- 18.Ma BB, Lui VW, Hui CW, Lau CP, Wong CH, Hui EP, Ng MH, Tsao SW, Li Y, Chan AT. Preclinical evaluation of the AKT inhibitor MK-2206 in nasopharyngeal carcinoma cell lines. Invest New Drugs. 2013;31:567–575. doi: 10.1007/s10637-012-9896-5. [DOI] [PubMed] [Google Scholar]
- 19.Morrison JA, Gulley ML, Pathmanathan R, Raab-Traub N. Differential signaling pathways are activated in the Epstein-Barr virus-associated malignancies nasopharyngeal carcinoma and Hodgkin lymphoma. Cancer Res. 2004;64:5251–5260. doi: 10.1158/0008-5472.CAN-04-0538. [DOI] [PubMed] [Google Scholar]
- 20.Huang XM, Dai CB, Mou ZL, Wang LJ, Wen WP, Lin SG, Xu G, Li HB. Overproduction of cyclin D1 is dependent on activated mTORC1 signal in nasopharyngeal carcinoma: Implication for therapy. Cancer Lett. 2009;279:47–56. doi: 10.1016/j.canlet.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 21.Chen J, Hu CF, Hou JH, Shao Q, Yan LX, Zhu XF, Zeng YX, Shao JY. Epstein-Barr virus encoded latent membrane protein 1 regulates mTOR signaling pathway genes which predict poor prognosis of nasopharyngeal carcinoma. J Transl Med. 2010;8(30) doi: 10.1186/1479-5876-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tekirdag KA, Korkmaz G, Ozturk DG, Agami R, Gozuacik D. MIR181A regulates starvation- and rapamycin-induced autophagy through targeting of ATG5. Autophagy. 2013;9:374–385. doi: 10.4161/auto.23117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. 2004;36:2491–2502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 25.Fujiwara K, Iwado E, Mills GB, Sawaya R, Kondo S, Kondo Y. Akt inhibitor shows anticancer and radiosensitizing effects in malignant glioma cells by inducing autophagy. Int J Oncol. 2007;31:753–760. [PubMed] [Google Scholar]
- 26.Hung JY, Hsu YL, Li CT, Ko YC, Ni WC, Huang MS, Kuo PL. 6-Shogaol, an active constituent of dietary ginger, induces autophagy by inhibiting the AKT/mTOR pathway in human non-small cell lung cancer A549 cells. J Agric Food Chem. 2009;57:9809–9816. doi: 10.1021/jf902315e. [DOI] [PubMed] [Google Scholar]
- 27.Shin SY, Lee KS, Choi YK, Lim HJ, Lee HG, Lim Y, Lee YH. The antipsychotic agent chlorpromazine induces autophagic cell death by inhibiting the Akt/mTOR pathway in human U-87MG glioma cells. Carcinogenesis. 2013;34:2080–2089. doi: 10.1093/carcin/bgt169. [DOI] [PubMed] [Google Scholar]
- 28.Seglen PO, Gordon PB. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA. 1982;79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 30.Eisenberg-Lerner A, Kimchi A. The paradox of autophagy and its implication in cancer etiology and therapy. Apoptosis. 2009;14:376–391. doi: 10.1007/s10495-008-0307-5. [DOI] [PubMed] [Google Scholar]
- 31.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 32.Levine B, Sinha S, Kroemer G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang NC, Nguyen M, Germain M, Shore GC. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J. 2010;29:606–618. doi: 10.1038/emboj.2009.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marquez RT, Xu L. Bcl-2:Beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am J Cancer Res. 2012;2:214–221. [PMC free article] [PubMed] [Google Scholar]
- 35.Mukhopadhyay S, Panda PK, Sinha N, Das DN, Bhutia SK. Autophagy and apoptosis: where do they meet? Apoptosis. 2014;19:555–566. doi: 10.1007/s10495-014-0967-2. [DOI] [PubMed] [Google Scholar]
- 36.Tolkovsky AM, Xue L, Fletcher GC, Borutaite V. Mitochondrial disappearance from cells: A clue to the role of autophagy in programmed cell death and disease? Biochimie. 2002;84:233–240. doi: 10.1016/S0300-9084(02)01371-8. [DOI] [PubMed] [Google Scholar]