

Abstract
An emergent hypothesis is that a resistance to the anabolic drive by insulin may contribute to loss of strength and muscle mass in patients with chronic kidney disease (CKD). We tested whether insulin resistance extends to protein metabolism using the forearm perfusion method with arterial insulin infusion in 7 patients with CKD and metabolic acidosis (bicarbonate 19 mmol/l) and 7 control individuals. Forearm glucose balance and protein turnover (2H-phenylalanine kinetics) were measured basally and in response to insulin infused at different rates for 2 h to increase local forearm plasma insulin concentration by approximately 20 and 50 μU/ml. In response to insulin, forearm glucose uptake was significantly increased to a lesser extent (−40%) in patients with CKD than controls. In addition, whereas in the controls net muscle protein balance and protein degradation were decreased by both insulin infusion rates, in patients with CKD net protein balance and protein degradation were sensitive to the high (0.035 mU/kg per min) but not the low (0.01 mU/kg per min) insulin infusion. Besides blunting muscle glucose uptake, CKD and acidosis interfere with the normal suppression of protein degradation in response to a moderate rise in plasma insulin. Thus, alteration of protein metabolism by insulin may lead to changes in body tissue composition which may become clinically evident in conditions characterized by low insulinemia.
Keywords: chronic renal disease, insulin resistance, protein, skeletal muscle
Insulin resistance is a well-known complication of chronic kidney diseases (CKD).1, 2, 3, 4, 5, 6, 7 Although not clinically remarkable, insulin resistance appears to be strongly implicated in the pathogenesis of hypertension, accelerated atherosclerosis, and cardiovascular events.8, 9, 10 Besides its effects on glucose and lipid metabolism, insulin also influences a number of metabolic pathways related to protein metabolism that lead to anabolic or anticatabolic effects.11 Particularly, even small increases in blood insulin levels, well within the physiological range, are associated with pronounced inhibition of protein breakdown.11 An emergent hypothesis is that a resistance to the anabolic drive by insulin may contribute to loss of strength and muscle mass that are observed with the progression of CKD.12, 13 This hypothesis is supported by studies obtained in animal models of CKD. Bailey et al.14 have recently identified a series of abnormal postreceptor signaling changes in the insulin/insulin-like growth factor-1 pathway in the muscle of rats with CKD. These include the occurrence of functional abnormalities in the insulin receptor substrate/phosphatidylinositol 3-kinase (PI3K) cascade that decrease the phosphorylation of the downstream effector Akt. The low phosphorylated Akt activity has been shown to stimulate the expression of specific E3 ubiquitin conjugating enzymes, atrogin-1/MAFbx and MuRF1, to accelerate muscle protein degradation. Furthermore, a decrease in muscle PI3K activity per se activates Bax, leading to the stimulation of caspase-3 activity and increase protein degradation.12, 14, 15 It is interesting that metabolic acidosis, a common complication observed in patients with CKD, also interferes with insulin-induced intracellular signaling by suppressing PI3K activity in muscle, and thus increases protein degradation through an upregulation of the ubiquitin-requiring pathway.16, 17
Despite initial controversial results obtained in human insulin-resistant states (see Tessari et al.11 for review), a recent clinical study has shown that the anticatabolic response to insulin is blunted in obese insulin-resistant women.18 Interestingly, patients with end-stage renal disease due to diabetic nephropathy have an accelerated loss of lean body mass as compared with nondiabetic patients,19 suggesting that insulin resistance accelerates net catabolism. The evidence that insulin resistance also extends to protein metabolism in CKD is suggested by cross-sectional studies in which protein turnover was compared with basal insulin levels or with ‘static' indexes of insulin resistance. Siew et al.20 observed that even lesser degrees of glucose intolerance in the absence of overt diabetes mellitus are associated with both increased whole-body and muscle protein breakdown, suggesting that protein metabolism is scarcely sensitive to basal insulin levels in uremia. In addition, when we studied muscle protein turnover in patients with CKD and metabolic acidosis, we observed that the inverse association between protein degradation and plasma insulin occurring in healthy subjects was lost in CKD patients.21 However, in the few studies22, 23 in which amino acid or protein metabolism was evaluated during the euglycemic hyperinsulinemic clamp (a ‘gold standard' for the detection of insulin resistance), a normal antiproteolytic response to insulin was observed, even in the presence of metabolic acidosis with mean bicarbonate of 17 mmol/l.24 It is interesting that in these studies the effects of insulin were tested in the high (~60–100 μU/ml) insulin physiological range that is needed to test insulin sensitivity regarding glucose. However, protein metabolism appears to be maximally sensitive at only modestly increased (up to 30 μU/ml) insulin concentrations.25 Therefore, the occurrence of defects in muscle insulin's response in uremia could be offset by the high insulin levels attained in previous studies.
Testing different insulin levels allows to take advantage of the different sensitivity to insulin of protein and glucose metabolism. The present clinical investigation explores the effects of two different insulin levels on two selected end points of its action, such as glucose and protein metabolism, in the muscle of patients with nondiabetic CKD. We hypothesized that the insulin response regarding both protein and glucose metabolism is blunted in the skeletal muscle of CKD patients with metabolic acidosis. We tested this postulate via an interventional design in which insulin levels were raised at two different levels to mimic a local state of low or moderate hyperinsulinemia. Net exchange of glucose and protein turnover across forearm muscle were measured in the basal, postabsorptive state, as well as in the hyperinsulinemic state. Our results show that in patients with CKD the exquisite ability of skeletal muscle protein metabolism to respond to modestly increased insulin levels is blunted, whereas the response to high insulin is preserved.
RESULTS
Serum insulin and forearm blood flow
By the infusion of low-dose insulin (0.01 mU/kg per min), venous insulin was increased on the average from 6 to 29 μU/ml and from 8 to 30 μU/ml in controls and CKD patients, respectively (Table 1). The administration of a higher insulin dose (0.035 mU/kg per min) increased insulin levels by ~50 μU/ml (on the average from 7 to 62 μU/ml and from 10 to 59 μU/ml in controls and patients, respectively). These insulin levels are similar to the postprandial concentrations. Insulin levels measured in the contralateral arm vein were not changed during the study (data not shown).
Table 1. Arterial insulin concentrations (μU/ml) in patients with chronic kidney disease (CKD) and in controls in the baseline and during insulin infusion.
|
Insulin infusion rate (mU/kg per min) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
0.01 |
0.035 |
||||||||
| 0 | 80 | 100 | 120 | 0 | 80 | 100 | 120 min | ||
| Controls (n=5) | 6±1 | 22±4a | 28±3a | 30±1a | Controls (n=7) | 7±1 | 47±4a | 60±4a | 62±3a |
| CKD (n=5) | 8±1 | 26±2a | 26±2a | 29±2a | CKD (n=7) | 10±2 | 44±3a | 56±3a | 59±3a |
P⩽0.05 basal versus insulin-infused period.
Forearm blood flow was not significantly changed by the 0.01 mU/kg per min insulin infusion rates (Table 2). At the highest insulin dose (0.035 mU/kg per min), a 30% increase in forearm blood flow was observed in both controls and CKD subjects.
Table 2. Forearm blood flow (ml/min per 100 ml) in patients with chronic kidney disease (CKD) and in controls in the baseline and during insulin infusion.
|
Insulin infusion rate (mU/kg per min) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
0.01 |
0.035 |
||||||||
| 0 | 80 | 100 | 120 | 0 | 80 | 100 | 120 min | ||
| Controls (n= 5) | 3.5±0.2 | 3.7±0.1 | 3.6±0.2 | 3.7±0.2 | Controls (n=7) | 3.7±0.1 | 4.2±0.3 | 4.6±0.1a | 4.7±0.2a |
| CKD (n= 5) | 4.1±0.2 | 4.0±0.2 | 3.8±0.26 | 4.0±0.3 | CKD (n=7) | 4.2±0.4 | 4.9±0.6 | 4.9±0.4a | 5.1±0.4a |
P⩽0.05 basal versus insulin-infused period.
Effects of local hyperinsulinemia on the forearm uptake of glucose
The 0.01 mU/kg per min insulin infusion did not cause any significant change in muscle glucose uptake in both patients and controls (data not shown). The 0.035 mU/kg per min insulin infusion rate resulted in significant increases of glucose uptake of ~3–4-fold in control subjects (Figure 1). In contrast, the insulin-induced increase in forearm glucose uptake was impaired by ~35% in patients with CKD (Figure 1).
Figure 1.

The effect of insulin on forearm skeletal muscle glucose uptake in patients with chronic kidney disease (CKD) (n=7) and controls (n=7). The 0.035 mU/kg per min insulin infusion rate resulted in significant increases of glucose uptake of ∼3–4-fold in control subjects. The effect of local insulin was reduced by ~40% in patients with CKD. Values are mean±s.e.m. aStatistically significant differences (P⩽0.02) between basal and insulin-infused periods. bStatistically significant differences (P⩽0.03) between patients with CKD and controls.
Insulin sensitivity of protein metabolism in patients with CKD
In the basal, postabsorptive state, rates of protein degradation and net protein balance were, as a trend, higher (by ~7 and 9–13%, P<0.08–0.06, respectively) in patients than controls. However, all phenylalanine kinetic parameters were not statistically different between groups. The net protein balances for phenylalanine are shown in Figure 2. In control subjects, the negative net protein balance observed in the postabsorptive state was markedly decreased by insulin infusion at both 0.01 and 0.035 mU/kg per min (Figure 2a and b). Contrariwise, no effect of low-dose (0.01 mU/kg per min) insulin infusion was observed in patients with CKD (Figure 2a). At higher insulin infusion (0.035 mU/kg per min), net protein balance declined in patients similar to control subjects (Figure 2b).
Figure 2.

Effect of insulin at 0.01 and 0.035 mU/kg per min on forearm muscle net protein balance. (a, b) In control subjects, the negative net protein balance observed in the basal, postabsorptive state was markedly decreased by insulin at both 0.01 (n=5) and 0.035 mU/kg per min (n=7). (a) Contrariwise, no effect of low-dose (0.01 mU/kg per min) insulin infusion was observed in patients with chronic kidney disease (CKD) (n=5). (b) At higher insulin infusion rates (0.035 mU/kg per min) net protein balance declined in patients (n=7) similarly to control subjects. Values are mean±s.e.m. aStatistically significant differences (P⩽0.01 or less) between basal and insulin-infused periods. bStatistically significant differences (P⩽0.03 or less) between patients with CKD and controls.
Figure 3 shows the effects of insulin on protein degradation. The 0.01 mU/kg per min insulin infusion produced a significant decrease (~31–34%) of protein degradation in control subjects (Figure 3a). A similar high suppression (~31–37%) of protein degradation was observed in response to the higher insulin infusion rates (Figure 3b). Protein degradation was not affected by low-dose insulin (Figure 3a), whereas a normal suppression was observed in response to the higher insulin infusion rates (Figure 3b) in CKD patients.
Figure 3.

Effect of insulin at 0.01 and 0.035 mU/kg/min on forearm muscle protein degradation. (a) In control subjects the 0.01 mU/kg per min insulin infusion produced a significant ~31–34% decrease of protein degradation while it was only modestly influenced in patients with chronic kidney disease (CKD) (n=5); (b) A similar suppression of protein degradation in response to the higher insulin infusion rates was observed in control subjects (n=7) and patients with CKD (n=7). Values are mean±s.e.m. aStatistically significant differences (P⩽0.01) between basal and insulin-infused periods.
Similar to what was previously observed,25 no change in muscle protein synthesis was observed at both insulin infusion rates in controls and CKD patients (Figure 4a and b).
Figure 4.

Effect of insulin at 0.01 and 0.035 mU/kg per min on forearm muscle protein synthesis. (a, b) No change in muscle protein synthesis was observed at both insulin infusion rates in controls and patients with chronic kidney disease (CKD). NS, nonsignificant.
Figure 5 depicts the dose–response relationships between net protein balance across the forearm and insulin levels. To determine the dose–response characteristics for the effect of insulin on muscle protein metabolism, the net balance of phenylalanine across the forearm was plotted versus the corresponding steady-state plasma insulin concentrations (only the 100 and 120 min infusion data point considered) during insulin infusion. The fitted curve is a nonlinear least squares best fit to a logarithmic function. In control subjects net protein balance decreased steeply from basal, postabsorptive insulin concentrations of ~8 μU/ml until insulin concentrations of ~30 μU/ml, declining more modestly thereafter. Thus, the maximal effect of insulin occurred at concentrations of ~30 μU/ml. A similar trend was observed for CKD patients, but for each insulin level between basal and 30 μU/ml the insulin response was shifted to the right. The curve of insulin sensitivity in patients with CKD tended to overlap with the curve obtained in controls in the high insulin physiological range.
Figure 5.

Dose–response relationships between net protein balance across the forearm and insulin levels. To determine the dose–response characteristics for the effect of insulin on muscle protein metabolism, the net balance of phenylalanine across the forearm was plotted versus the corresponding steady-state plasma insulin concentrations (only the 100 and 120 min infusion data point considered) during insulin infusion. The fitted curve is a nonlinear least squares best fit to a logarithmic function. In control subjects, net protein balance decreased steeply from −17 nmol/min per 100 ml in the basal, postabsorptive state to −8 nmol/min per 100 ml when insulin concentrations were raised to 30 μU/ml. With further increases in insulin concentrations to ~100 μU/ml, net protein balance decreased only to −5 nmol/min per 100 ml. Thus, the maximal effect of insulin occurred at insulin concentrations between 15 and 30 μU/ml. In CKD patients the insulin response was shifted downward with higher net protein balance at moderate hyperinsulinemia. The curve of insulin sensitivity of protein balance in patients with chronic kidney disease (CKD) overlapped with the same curve obtained in controls in the high insulin physiological range.
DISCUSSION
Three major observations are made from this study. First, in accordance with early studies4, 5, 6, 26 the muscle response of glucose metabolism to insulin is blunted in CKD patients. Second, as a new finding, insulin sensitivity of muscle protein metabolism is overall preserved at insulin levels in the high (~8–9-fold higher than basal) physiological range. This observation is in keeping with studies on whole-body protein metabolism during euglycemic hyperinsulinemic clamp.24, 25, 26 Third, the muscle response of protein metabolism to insulin in the low (~3-fold higher than basal) physiological range is markedly impaired in patients with CKD. Therefore, the results of this study indicate that in patients with CKD skeletal muscle protein metabolism is truly insulin resistant at low hormone levels, whereas the sensitivity of muscle protein metabolism to high insulin levels is preserved.
Early studies have suggested that insulin resistance of glucose metabolism in uremia is not because of impaired insulin binding to its receptor but rather due to defects in intracellular signaling processes.27, 28 Insulin enhances glucose uptake in skeletal muscle through the activation of the PI3K–Akt signaling pathway, a pathway that is inhibited in skeletal muscle in uremia.12, 29 In our study the response to insulin of glucose metabolism in the muscle of CKD patients was impaired at insulin levels of ~60 μU/ml, levels that are physiologically observed after a mixed meal. Therefore, our findings are in accordance with what was shown several years ago by Alvestrand et al.5 who observed a marked impairement of glucose uptake by peripheral tissues at systemic high insulin levels in chronically uremic subjects. Our results are also in keeping with what was shown recently by Miyamoto et al.30 who observed that, despite a markedly elevated insulin response (~50 μU/ml), hemodialysis patients are unable to maintain normal postprandial blood glucose levels after a carbohydrate-rich meal. Meal consumption-induced hyperglycemia has been associated with an inflammatory response predicting cardiovascular disease and mortality in patients with diabetes mellitus and is putatively an important factor in uremic oxidative stress.30
In vivo, insulin has both anabolic (in the presence of amino acid availability) and anticatabolic (during fasting) effects.11 Using tracers, several laboratories reported that in the basal, postabsorptive state, physiological doses of insulin inhibit whole-body protein and muscle degradation.31, 32, 33 However, the molecular mechanisms by which insulin reduces muscle protein degradation are only in part known. In myotubes, insulin decreases the activity of the ubiquitin–proteasome pathway through changes in the phosphorylation status of members of the Akt-dependent signaling pathway.34 In rats there is evidence that insulin-stimulated Akt activity induces phosphorylation of the FOXO family of transcription factors.35, 36 FOXO-1 and -3 regulate the transcription of components of the ubiquitin–proteasome system, including the ubiquitin ligases muscle atrophy F-box (MAFbx) and muscle-specific RING finger-1 (MuRF-1), that are central to the control of muscle protein breakdown.37 In humans, when muscle protein kinetics in association with intracellular signaling were studied in response to insulin, decreases in the amounts of MAFbx protein and the C2 proteasome unit (but not of MuRF-1) were observed. Certainly, these findings provide a mechanistic explanation for the fall in insulin-induced muscle proteolysis.38 It is interesting that metabolic acidosis stimulates the activity of different proteases including caspase-3 and the ubiquitin–proteasome system that increase muscle protein degradation.12 Therefore, insulin and acidosis appear to target in an opposite way the same signaling pathway in muscle, as also shown in a cultured muscle cell model.39
In our work we set out to study insulin sensitivity of protein metabolism in CKD patients with untreated acidosis because, in previous studies, acidosis has been shown to increase protein degradation,16, 17 an effect that is reversed by bicarbonate supplementation.40 In addition, acidosis decreases insulin sensitivity of glucose metabolism.24 Besides acidosis, different complications of CKD such as inflammation15 and elevated angiotensin II15 interfere with insulin- and insulin-like growth factor-1-induced intracellular signaling to promote muscle catabolism.
The first effect of insulin is a fast, nitric oxide-dependent microvascular recruitment that precedes increases in blood flow.41 Both muscle structure and blood flow are altered in CKD patients because of dissociation between capillary supply and muscle cell requirements, presumably secondary to decreased capillary density.42 Although the measure of capillary recruitment was not feasible in this study, it is possible that the different effects of the two insulin doses in CKD patients were caused by differential effects on muscle perfusion, with the higher dose being the only one able to stimulate microvascular recruitment.
What is the full meaning of our work? In humans, insulin plays a pivotal role in maintaining muscle protein mass during fasting, during which proteins are lost, and in the fed state, during which protein gains take place. We show here that acidemic CKD patients need higher than normal levels of insulin to inhibit proteolysis. The degree of insulinemia achieved when a low dose of insulin was administered in this study (~28–30 μU/ml) is comparable with that expected after a low glycemic index meal, such as breakfast, and thus it turns from these results that the normal suppression of proteolysis by insulin at this concentration is impaired in CKD. In contrast, the sensitivity to insulin at higher insulin concentrations, as may occur with meals containing refined carbohydrates, appears to be preserved. Defects in the regulation by insulin of protein metabolism in uremia on one hand may lead to changes in body tissue composition, metabolic rates, and individual amino acid metabolic steps that may become clinically evident in conditions characterized by low insulinemia, such as during fasting and/or low energy intakes. On the other hand, defects in the regulation in protein metabolism described here could overlap with alterations in insulin sensitivity occurring in normal aging43 and also interact with other factors such as dietary and lifestyle habits (physical activity) to boost protein catabolism. Therefore, preventing or treating muscle catabolism favored by insulin resistance may potentially prove to be a new target for the nutritional treatment of patients with CKD.
MATERIALS AND METHODS
Study participants
Seven patients with CKD and seven control subjects were studied (Table 3). The causes of renal disease were: chronic glomerulonephritis (three patients), interstitial nephritis (two patients), and hypertensive nephrosclerosis (two patients). None of the patients had any history of diabetes or major organ disease with the exception of CKD. There were no differences between groups as for age, gender, body mass index, fat-free and fat mass, normalized protein nitrogen appearance, estimated calorie intake, Subjective Global Assessment score, C-reactive protein, and serum albumin. Estimated glomerular filtration rate44 and hemoglobin levels were lower in patients. The average forearm volume was 927±55 and 901±48 ml, respectively, in patients and controls (P=nonsignificant).
Table 3. Clinical characteristics of controls and chronic kidney disease (CKD) subjects.
| Controls | CKD subjects | |
|---|---|---|
| Number of subjects | 7 | 7 |
| Age (years) | 50±3 | 55±2 |
| Gender (M/F) | 4/3 | 4/3 |
| BMI (kg/m2) | 24±2 | 23±1 |
| FFM (kg) | 51±2 | 49±3 |
| Fat mass (kg) | 24±1 | 23±2 |
| nPNA (g/kg) | 1.1±0.1 | 1.2±0.1 |
| Energy intake (kcal/kg) | 31±1 | 32±1 |
| SGA score | 7 (6–7) | 7 (6–7) |
| CRP (mg/l) | 3 (2–3) | 3 (2–5) |
| Estimated GFR (ml/min per 1.73 m2) | 109±6 | 17±1b |
| Blood glucose (mmol/l) | 4.0±0.1 | 4.3±0.1 |
| (HCO3) (mmol/l) | 24±1 | 19±1 |
| Albumin (g/dl) | 4.2±0.1 | 4.1±0.1 |
| Hemoglobin (g/dl) | 13.5±1 | 11.5±0.5a |
| BUN (mg/dl) | 20±2 | 88±4b |
Abbreviations: BMI, body mass index; BUN, blood urea nitrogen; CRP, C-reactive protein; F, female; FFM, fat-free mass; GFR, glomerular filtration rate; M, male; nPNA, normalized protein nitrogen appearance; SGA, Subjective Global Assessment.
Data are mean±s.e.m. or median (range). Significance of difference versus control subjects:
P<0.05;
P<0.01.
The protocol described here was approved by the Ethics Committee of the Department of Internal Medicine of the University of Genoa. All subjects were informed about the nature, purposes, procedures, and possible risks of the study before their informed consents were obtained. The procedures were in accordance with the Declaration of Helsinki Principles regarding ethics of human research.
Experimental protocol
The methods for the measurement of muscle protein turnover are reported extensively elsewhere.21, 45 Briefly, muscle protein metabolism was estimated by the forearm perfusion technique associated with 2H-phenylalanine kinetics.21, 25 The study was performed in the postabsorptive, overnight fasted state (Figure 6). At 0700 h, a forearm vein was cannulated with a 18-gauge catheter to receive a continuous infusion of L-(ring-2H5)-phenylalanine (D5-Phe) (0.035 μmol/kg per min). Catheters were inserted into a brachial artery and in a retrograde manner into the ipsilateral, deep forearm vein. After a 150-min tracer equilibration period, insulin (Humulin R, Eli Lilly, Indianapolis, IN) (diluted in a solution of normal saline and albumin) was infused for 120 min into the brachial artery at 0.010 and, on a separate day, at 0.035 mU/kg per min. Five patients and five control subjects performed both the 0.010 and the 0.035 mU/kg per min insulin infusion studies, whereas all the subjects performed the 0.035 mU/kg per min insulin infusion study. Arterial and deep venous blood samples were obtained at 20-min intervals during the last hour of both the basal and the insulin infusion periods. Blood flow across the forearm was determined after each arteriovenous sampling.21 Body composition was estimated by anthropometry46 and energy intake by dietetic diaries.
Figure 6.

The design of the study.
Assays
Blood samples were deproteinized with perchloric acid (20% wt/vol). Phenylalanine concentrations were measured by ion-exchange chromatography.21 The D5-Phenylalanine mole percentage enrichments in the supernatant of deproteinized blood were determined as previously described.45 Isotope concentrations were calculated by multiplying E times substrate concentrations. All other serum chemical measurements were determined by routine clinical chemistry laboratory procedures.
Calculations
Arterial concentrations and enrichments (Figure 7) of phenylalanine remained stable between the basal and insulin-infused periods, indicating that the amino acid tracers were at steady state throughout the study.
Figure 7.

Plasma mass percent enrichment (MPE) of phenylalanine at study time points. CKD, chronic kidney disease.
Muscle phenylalanine rate of appearance (protein breakdown) and disposal (protein synthesis) were calculated as previously described21, 45 using the A-V model.21 The net forearm balance (NB) for phenylalanine was calculated as follows:
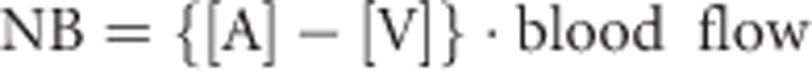 |
where [A] and [V] are the phenylalanine arterial and venous concentrations, respectively. The forearm protein breakdown represented by phenylalanine rate of appearance (Raphe) was calculated as it follows:
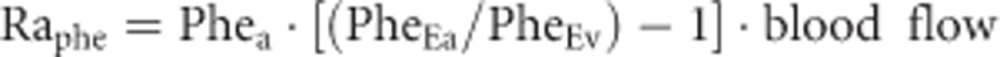 |
in which PheEa and PheEv represent phenylalanine isotopic enrichment in arterial and venous blood, respectively. The local rate of disappearance (Rdphe) of phenylalanine, which represents the muscle protein synthesis rate, was calculated as:
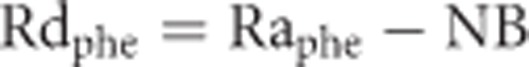 |
Statistical analyses
All data are presented as the mean±s.e. of the mean or median (range). To establish the differences in substrate concentrations between patients and controls, the unpaired t-test and analysis of variance were used. To compare arterial with venous data, statistical analysis was performed using the two-tailed t-test. The effect of insulin on the response variables was analyzed using analysis of variance for repeated measures, the main effects being group (low vs. high insulin) and time (basal, insulin infusion). Linear and nonlinear regression and correlation were used to evaluate the relationship between two variables. Statistical analysis was performed with the Statview Statistical Package (Abacus, Berkeley, CA).
Acknowledgments
This study was supported by grants from the Ministero dell'Università e della Ricerca Scientifica e Tecnologica, and by the University of Genoa (Cofinanziamento di Ateneo).
All the authors declared no competing interests.
References
- 1Rigalleau V, Gin H. Carbohydrate metabolism in uraemia. Curr Opin Clin Nutr Metab Care 2005; 8: 463–469. [DOI] [PubMed] [Google Scholar]
- 2Fliser D, Pacini G, Engelleiter R. et al. Insulin resistance and hyperinsulinemia are already present in patients with incipient renal disease. Kidney Int 1998; 53: 1343–1347. [DOI] [PubMed] [Google Scholar]
- 3Kobayashi S, Maesato K, Moriya H et al. Insulin resistance in patients with chronic kidney disease. Am J Kidney Dis 2005; 45: 275–280. [DOI] [PubMed] [Google Scholar]
- 4DeFronzo RA, Smith D, Alvestrand A. Insulin action in uremia. Kidney Int 1983; 24: S102–S114. [PubMed] [Google Scholar]
- 5Alvestrand A, Mujagic M, Wajngot A et al. Glucose intolerance in uremic patients: the relative contributions of impaired beta-cell function and insulin resistance. Clin Nephrol 1989; 31: 175–183. [PubMed] [Google Scholar]
- 6DeFronzo RA, Alvestrand A, Smith D et al. Insulin resistance in uremia. J Clin Invest 1981; 67: 563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7Deferrari G, Garibotto G, Robaudo C et al. Glucose interorgan exchange in chronic renal failure. Kidney Int 1983: S115–S120. [PubMed]
- 8Chen J, Muntner P, Lee Ham L et al. Insulin resistance and the risk of chronic kidney disease in nondiabetic US adults. J Am Soc Nephrol 2003; 14: 469–477. [DOI] [PubMed] [Google Scholar]
- 9Shinohara K, Shoji T, Emoto M et al. Insulin resistance as an independent predictor of cardiovascular mortality in patients with end-stage renal disease. J Am Soc Nephrol 2002; 13: 1894–1900. [DOI] [PubMed] [Google Scholar]
- 10Teta D. Insulin resistance as a therapeutic target for chronic kidney disease. J Ren Nutr 2015; 25: 226–229. [DOI] [PubMed] [Google Scholar]
- 11Tessari P, Cecchet D, Cosma A et al. Insulin resistance of amino acid and protein metabolism in type 2 diabetes. Clin Nutr 2011; 30: 267–272. [DOI] [PubMed] [Google Scholar]
- 12Wang XH, Mitch WE. Mechanisms of muscle wasting in chronic kidney disease. Nat Rev Nephrol 2014; 10: 504–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13Deger SM, Sundell MB, Siew ED et al. Insulin resistance and protein metabolism in chronic hemodialysis patients. J Ren Nutr 2013; 23: e59–e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14Bailey JL, Zheng B, Hu Z et al. Chronic kidney disease causes defects in signalling through the insulin receptor substrate/phosphatidylinositol 3-kinase/Akt pathway: implications for muscle atrophy. J Am Soc Nephrol 2006; 17: 1388–1394. [DOI] [PubMed] [Google Scholar]
- 15Thomas SS, Dong Y, Zhang L et al. Signal regulatory protein-α interacts with the insulin receptor contributing to muscle wasting in chronic kidney disease. Kidney Int 2013; 84: 308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16Rajan V, Mitch WE. Ubiquitin, proteasomes and proteolytic mechanisms activated by kidney disease. Biochim Biophys Acta 2008; 1782: 795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17Reaich D, Channon SM, Scrimgeour CM et al. Correction of acidosis in humans with CRF decreases protein degradation and amino acid oxidation. Am J Physiol 1993; 265: E230–E235. [DOI] [PubMed] [Google Scholar]
- 18Chevalier S, Marliss EB, Morais JA et al. Whole-body protein anabolic response is resistant to the action of insulin in obese women. Am J Clin Nutr 2005; 82: 355–365. [DOI] [PubMed] [Google Scholar]
- 19Pupim LB, Flakoll PJ, Majchrzak KM et al. Increased muscle protein breakdown in chronic hemodialysis patients with type 2 diabetes mellitus. Kidney Int 2005; 68: 1857–1865. [DOI] [PubMed] [Google Scholar]
- 20Siew ED, Pupim LB, Majchrzak KM et al. Insulin resistance is associated with skeletal muscle protein breakdown in non-diabetic chronic hemodialysis patients. Kidney Int 2007; 71: 146–152. [DOI] [PubMed] [Google Scholar]
- 21Garibotto G, Russo R, Sofia A et al. Skeletal muscle protein synthesis and degradation in patients with chronic renal failure. Kidney Int 1994; 45: 1432–1439. [DOI] [PubMed] [Google Scholar]
- 22Alvestrand A, Defronzo RA, Smith D et al. Influence of hyperinsulinaemia on intracellular amino acid levels and amino acid exchange across splanchnic and leg tissues in uraemia. Clin Sci 1988; 74: 155–163. [DOI] [PubMed] [Google Scholar]
- 23Castellino P, Luzi L, Giordano M et al. Effects of insulin and amino acids on glucose and leucine metabolism in CAPD patients. J Am Soc Nephrol 1999; 10: 1050–1058. [DOI] [PubMed] [Google Scholar]
- 24Reaich KA, Graham KA, Channon SM et al. Insulin-mediated changes in PD and glucose uptake after correction of acidosis in humans with CRF. Am J Physiol Endocrinol Metab 1995; 268: E121–E126. [DOI] [PubMed] [Google Scholar]
- 25Louard RJ, Fryburg DA, Gelfand RA et al. Insulin sensitivity of protein and glucose metabolism in human forearm skeletal muscle. J Clin Invest 1992; 90: 2348–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26Westervelt FB Jr. Abnormal carbohydrate metabolism in uremia. Am J Clin Nutr 1968; 21: 423–524. [DOI] [PubMed] [Google Scholar]
- 27Smith D, DeFronzo RA. Insulin resistance in uremia mediated by postbinding defects. Kidney Int 1982; 22: 54–62. [DOI] [PubMed] [Google Scholar]
- 28DeFronzo RA, Tobin JD, Rowe JW et al. Glucose intolerance in uremia: quantification of pancreatic beta cell sensitivity to glucose and tissue sensitivity to insulin. J Clin Invest 1978; 62: 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29Verzola D, Procopio V, Sofia A et al. Apoptosis and myostatin mRNA are upregulated in the skeletal muscle of patients with chronic kidney disease. Kidney Int 2011; 79: 773–782. [DOI] [PubMed] [Google Scholar]
- 30Miyamoto T, Rashid Qureshi A, Yamamoto T et al. Postprandial metabolic response to a fat- and carbohydrate-rich meal in patients with chronic kidney disease. Nephrol Dial Transplant 2011; 26: 2231–2237. [DOI] [PubMed] [Google Scholar]
- 31Fukagawa NK, Minaker KL, Rowe JW et al. Insulin-mediated reduction of whole body protein breakdown: dose-response effects on leucine metabolism in postabsorptive man. J Clin Invest 1985; 76: 2306–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32Tessari P, Trevisan R, Inchiostro S. et al. Dose-response curves of effects of insulin on leucine kinetics in humans. Am J Physiol Endocrinol Metab 1986; 251: E334–E342. [DOI] [PubMed] [Google Scholar]
- 33Liu Z, Long W, Hillier T et al. Insulin regulation of protein metabolism in vivo. Diabetes Nutr Metab 1999; 12: 421–428. [PubMed] [Google Scholar]
- 34Sandri M, Sandri C, Gilbert A et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004; 117: 399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35Kim DH, Kim JY, Yu BP et al. The activation of NF-kappaB through Akt-induced FOXO1 phosphorylation during aging and its modulation by calorie restriction. Biogerontology 2008; 9: 33–47. [DOI] [PubMed] [Google Scholar]
- 36Tang ED, Nunez G, Barr FG et al. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem 1999; 274: 16741–16746. [DOI] [PubMed] [Google Scholar]
- 37Bodine SC, Latres E, Baumhueter S et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001; 294: 1704–1708. [DOI] [PubMed] [Google Scholar]
- 38Greenhaff PL, Karagounis LG, Peirce N et al. Disassociation between the effects of amino acids and insulin on signaling, ubiquitin ligases, and protein turnover in human muscle. Am J Physiol Endocrinol Metab 2008; 295: E595–E604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39Franch HA, Raissi S, Wang X et al. Acidosis impairs insulin receptor substrate-1-associated phosphoinositide 3-kinase signaling in muscle cells: consequences on proteolysis. Am J Physiol Renal Physiol 2004; 287: F700–F706. [DOI] [PubMed] [Google Scholar]
- 40Löfberg E, Gutierrez A, Anderstam B et al. Effect of bicarbonate on muscle protein in patients receiving hemodialysis. Am J Kidney Dis 2006; 48: 419–429. [DOI] [PubMed] [Google Scholar]
- 41Vincent MA, Dawson D, Clark AD et al. Skeletal muscle microvascular recruitment by physiological hyperinsulinemia precedes increases in total blood flow. Diabetes 2002; 5: 42–48. [DOI] [PubMed] [Google Scholar]
- 42Bradley JR, Anderson JR, Evans DB et al. Impaired nutritive skeletal muscle blood flow in patients with chronic renal failure. Clin Sci 1990; 79: 239–247. [DOI] [PubMed] [Google Scholar]
- 43Rasmussen BB, Fujita S, Wolfe R et al. Insulin resistance of muscle protein metabolism in aging. Diabetologia 2009; 52: 1889–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44Levey AS, Stevens LA. Estimating GFR using the CKD epidemiology collaboration (CKD-EPI) creatinine equation: more accurate GFR estimates, lower CKD prevalence estimates, and better risk predictions. Am J Kidney Dis 2010; 55: 622–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45Tessari P, Deferrari G, Robaudo C et al. Phenylalanine hydroxylation across the kidney in humans. Kidney Int 1999; 56: 2168–2172. [DOI] [PubMed] [Google Scholar]
- 46Heymsfield SB, Mc Manus C, Smith J et al. Anthropometric measurement of muscle mass: revised equations for calculating bone-free arm muscle area. Am J Clin Nutr 1982; 36: 680–690. [DOI] [PubMed] [Google Scholar]