

Summary
Until now, treatment of primary autoimmune hemolytic anemia of the warm type (wAIHA) is primarily based on immunosuppression. However, many patients do not respond adequately to treatment, and treated patients may develop severe side effects due to uncontrolled, mixed and/or long-lasting immunosuppression. Unfortunately, the newly used therapeutic monoclonal antibodies are unspecific and remain frequently ineffective. Thus, development of a specific therapy for AIHA is necessary. The ideal therapy would be the identification and elimination of the causative origin of autoimmunization and/or the correction or reprogramming of the dysregulated immune components. Blood transfusion is the most rapidly effective measure for patients who develop or may develop hypoxic anemia. Although some effort has been made to guide physicians on how to adequately treat patients with AIHA, a number of individual aspects should be considered prior to treatment. Based on my serological and clinical experience and the analysis of evidence-based studies, we remain far from any optimized therapeutic measures for all AIHA patients. Today, the old standard therapy using controlled steroid administration, with or without azathioprine or cyclophosphamide, is, when complemented with erythropoiesis-stimulating agents, still the most effective therapy in wAIHA. Rituximab or other monoclonal antibodies may be used instead of splenectomy in therapy-refractory patients.
Keywords: Autoimmune hemolysis, AIHA, Azathioprine, Blood transfusion, Corticosteroids, Cyclophosphamide, Cushing's syndrome, Erythropoietin, Intravenous IgG, Mycophenolate, Rituximab, Splenectomy
Introduction
Although autoimmune hemolytic anemia (AIHA) is uncommon, it remains one of the best characterized human autoimmune diseases. It results from the production of specific complement- and/or non-complement-activating autoantibodies (aab) to red blood cells (RBCs). These aab may lead to RBC destruction of variable extent (hemolysis) and anemia. Most importantly, the class and characteristics of the causative aab play a role in the patient's treatment and outcome. However, treatment is usually commenced independent of aab behavior [1,2,3,4,5,6].
In cases where RBC destruction cannot be compensated by an increase of erythropoiesis, the affected patients may develop hypoxemic anemia which invariably requires urgent treatment. However, there is no drug available which can abruptly halt or compensate significant hemolysis. In addition, none of the administered drugs are always effective, and the beneficial effect is not predictable.
Currently, all treatment options are based on small retrospective studies, case reports, recommendations, traditional treatments, or self-empirical experiences [7,8,9,10,11,12]. Moreover, all of the drugs used in the treatment of AIHA are largely or partly directed against the patient's natural immune system, which may, in turn, extensively derail the immune system due to treatment. In addition, many patients are currently well-informed about their disease, treatment options, and risks, including those related to the drugs used for treatment. This may sometimes result in mistrust and impaired compliance of affected patients. Indeed, this is a real dilemma for both the treating physicians and the affected patients in that the success of a treatment cannot be predicted and complications due to treatment cannot be invariably avoided.
In this review, I will focus on the treatment of AIHA of the warm type (wAIHA) and discuss the most relevant data for each treatment. Our results from treated patients during the last two decades will be presented in a separate publication.
Treatment Options
RBC Transfusion
Based on the nature of wAIHA, the affected patients usually have a positive direct antiglobulin test (fixed aab), and about 40% also free (serum) aab which react with the vast majority or the entire available donor RBCs. This scenario of an incompatible blood supply and transfusion is a challenging issue for all participants, including patients, serologists (laboratory side), and physicians (clinical side).
Numerous reports have addressed the risks surrounding blood transfusion in patients with AIHA [13,14,15]. Therefore, considerable efforts are frequently made prior to blood transfusion. These measures may unnecessarily jeopardize severely anemic patients. We have repeatedly demonstrated that the incidence of alloimmunization, as well as the occurrence of adverse hemolytic transfusion reactions, is unnecessarily overestimated in patients with wAIHA and that positive serological cross-matches do not prevent necessary blood transfusion in these patients [16,17,18]. This notion has been supported by other reports encouraging blood transfusion in patients with decompensated AIHA [19,20]. Ultimately, blood transfusion is the most rapidly effective measure in patients with hypoxic anemia, and prior to the introduction of cortisone in the early 1950s, affected patients were treated exclusively with blood transfusion and/or splenectomy. In addition, based on a recent retrospective analysis it has been suggested that whole blood exchange is an effective measure in severely affected patients [21].
The efficacy of blood transfusion in AIHA has been discussed in previous studies and may be explained by the natural limitation of Fc- and C3b-mediated phagocytosis [16,18]. Alternatively, the vast majority, if not all, severely affected patients would be lost due to uncontrolled haemolysis and anaemia. In all cases where the causative aab are incapable of activating the terminal complement components (C5-9), the affected patients may survive for a long period of time without treatment. Indeed, even severely affected patients may initially demonstrate a rapid followed by a rather gradual aggravation of anaemia despite increasing haemolysis. Namely, the capacity of macrophages is limited by its nature and may further diminish due to ongoing hemolysis and RBC transfusions. Thus, a considerable portion of the transfused RBCs may survive, at least, for a short amount of time in the circulation. This may not only somewhat assist in compensating haemolysis and the oxygen supply but may also help in achieving the desired effect of the used therapeutic drugs. Most important, I recommend commencing therapy prior to blood transfusion, i.e., administration of 1-2 mg/kg/day prednisolone. If patients are already under treatment, no additional measures prior to transfusion are required.
Corticosteroids
Although a long-lasting beneficial effect is only achieved in less than 15-20% of patients treated with steroids, these compounds are generally accepted as the first-line therapy in wAIHA [3,4,5,6,7,8,9,10,11,12]. In fact, I am not aware of a single patient who has not, at least initially, been treated with steroids. This may be explained by the long history of steroid use in AIHA, the lack of an alternative, and the abrupt effect, which may achieved within a number of days in rare cases or within 1-3 weeks in the majority of cases [5,8,9,10,11,12]. In addition, there is an obscure phenomenon that some patients may immediately feel well upon administration of steroid therapy (≥100 mg). Although the reason for this subjective feeling remains unclear, this measure remains attractive for the involved physicians at bedside. However, steroids are relatively or entirely contraindicated in many patients with comorbidities, including diabetes and uncontrolled hypertension, obesity, osteoporosis, peptic ulcer, psychosis, cataract, and prematurity (children). In addition, all of these comorbidities may result from steroid treatment.
If treatment with steroids is tolerable, the standard practice is to commence with a short-term (<2-3 weeks) dose of 1-2 mg/kg/day of prednisolone or prednisone until stabilization. However, a long-lasting stable condition or remission may be achieved in only less than 20% of patients who do not develop significant side effects. The incidence of side effects is significantly higher in patients receiving steroids for a long period of time, and has even been observed in many patients who receive only 5-7.5 mg/day. Thus, Cushing's syndrome is not a rare complication in such cases. To avoid or to minimize these complications, a combination therapy with azathioprine or cyclophosphamide is indicated in almost all cases. After stabilization, corticosteroids should be gradually reduced to a minimum dose or even stopped (table 1).
Table 1.
Current treatment options for primary wAIHA*
| Drug | Dose | Response to treatment (%) |
||
|---|---|---|---|---|
| within | initially, % | long-term**, % | ||
| Prednisolone/prednisone | 1.0–2.0 mg/kg | 1–3 weeks | 70–80 | ≤20 |
| Dexamethasone | 4 × 40 mg/day | few days | 70–80 | ≤20 |
| 1–4 cycles / 2–4 weeks | ||||
| Azathioprine+ | 2–4 mg/kg | 1–3 months | 60–70 | 60–70 |
| Mycophenolate mofetil+ | 1–2 g/day | 4–6 weeks | 60–70 | 60–70 |
| Cyclophosphamide+ | 1–2 mg/kg | 2–4 weeks | 80–90 | 80–90 |
| Rituximab‡ | 4 × 375 mg/m2/week | 1–8 weeks | 60–70 | ≤20 |
| Splenectomy | − | few days | <50 | <50 |
Based on published data and my experience (for comparison see text and cited references).
Significant improvement or remission.
Usually combined with prednisolone or prednisone (after stabilization reduced to low dose ≤ 7.5 mg/day).
Following previous treatment with or in combination with other drugs (see text).
In severely affected patients, treatment with pulsed high-dose dexamethasone at 40 mg/day for 4 days is more rapidly effective than that with prednisone or prednisolone [22]. However, no difference between outcomes has yet been proven.
Azathioprine/Mycophenolate Mofetil
Patients, who do not adequately respond to corticosteroids prior to the development of side effects and/or within 2-3 weeks, and patients, who cannot be treated with steroids, may be treated with azathioprine (3-4 mg/kg/day) alone or in combination with adequate doses of steroids, i.e., elderly patients and/or patients with diabetes. If necessary, treatment may be commenced with high-dose steroid therapy and azathioprine to achieve rapid stabilization. Subsequently, a stepwise reduction of steroid doses is essential in most cases. Replacement of azathioprine with mycophenolate mofetil (1-2 g/day) may be helpful [23,24] in patients who demonstrate signs of azathioprine intolerability, i.e., abdominal pain, significant leukopenia, persistent increase of liver enzymes, or other severe side effects.
Unfortunately, the use of azathioprine in AIHA has rarely been well documented, and valuable information, such as duration of treatment, long-lasting side effects, and most importantly or character of the causative aab, is lacking in almost all of the reported cases. The importance of the latter point is reflected by the difference between Fc- and C3b-mediated phagocytosis. In my experience, azathioprine or mycophenolate mofetil with steroids is effective in most patients (70-80%) with wAIHA (table 1). This is supported by the outcome of a study performed on a large number of dogs treated with azathioprine and prednisolone [25]. This treatment combination resulted in remission of AIHA in the majority of treated dogs.
Cyclophosphamide
Low-dose oral cyclophosphamide (1-2 mg/kg/day) in combination with steroids, as described for azathioprine, is indicated in patients who do not respond well or do not tolerate treatment with azathioprine or mycophenolate mofetil and/or high-dose steroids. In my experience, cyclophosphamide and steroids stabilize the vast majority of patients (80-90%) with AIHA within 1-4 weeks. If the compensation of hemolysis appears to be inadequate or insufficient, as reflected by the number of reticulocytes, a high-pulse treatment with erythropoiesis-stimulating agents is useful in almost all cases (table 1). As soon as hemolysis is stabilized, steroid therapy should be reduced and that of cyclophosphamide when patients develop significant neutropenia and/or reticulocytopenia.
Although this regimen has been used over a number of decades for the treatment of refractory AIHA, available data on the efficacy of this treatment are limited in many aspects, and do not reflect the true results which would be obtained from a consequently applied therapy. The most recognized side effects observed are leukopenia, infections, impaired fertility, teratogenicity, and an increase in the prevalence of neoplasia [4].
The idea to use heroic doses of cyclophosphamide (50 mg/kg/day for 4 days) for the treatment of refractory AIHA [26,27,28] is associated with a number of high risks and unpredictable outcomes. Similarly, the intravenous administration of 0.5-1 g within the span of 1 day is often abortive. In summary, although data concerning treatment with cyclophosphamide remains limited, there is considerable evidence that the drug has been applied much more widely and is more effective than has been reported. Ultimately, AIHA is a rare disease, and the results of reported and most of the treated patients have not been consequently described and remain speculative. During the last decade, treatment with cyclophosphamide has been largely replaced by rituximab, although not by the author.
Hematopoietic Stem Cell Transplantation
Treatment of patients suffering from refractory AIHA [29] with hematopoietic stem cell transplantation is not recommended. In addition, other valuable treatment options, including the use of monoclonal antibodies, are now available.
Rituximab and Other Molecules
Rituximab is a potent anti-CD20 monoclonal antibody that destroys B lymphocytes via complement activation. Subsequently, these cells remain depleted from the circulation for approximately 1 year. The drug is primarily used for the treatment of non-Hodgkin's lymphoma and other hematological malignancies [30]. In AIHA, treatment with rituximab has increasingly been used as a result of uncontrolled studies and, presumably, a battle of publications. Meanwhile, several hundreds of patients have been treated with rituximab, but the results cannot invariably be attributed to monotherapy with this drug. Rituximab is used as a second-line therapy in non-responder patients who have been treated recently with other drugs or who received rituximab during treatment with other drugs [31,32,33,34,35,36,37,38,39,40,41]. Thus, an additive effect cannot always be excluded. Until now, no known studies have addressed the use of rituximab as a first-line monotherapy in AIHA. In addition, there is evidence that rituximab causes, though rarely, severe side effects, including infections, serum sickness, acute respiratory distress, progressive multifocal leukoencephalopathy, and presumably induction of malignant lymphoma [42,43,44,45,46]. Moreover, follow-up studies exceeding 5 years are lacking, and long-term severe side effects cannot be excluded. Most importantly, during the last decade, the drug has also been used extensively in the treatment of autoimmune thrombocytopenia (ITP). A recent multicenter, double-blind, placebo-controlled trial [47] does not support the initial results in ITP patients, which is quite similar to AIHA in roughly most aspects including pathogenesis and treatment [48].
Most patients (60-70%) respond well to treatment with rituximab (table 1), but only 20% of patients may achieve a minimum of 5 years in remission [31,36,37]. The effect of treatment with rituximab is unpredictable and is usually not abrupt. Whether the immediate effect of treatment in isolated cases may be related to a transitory saturation of macrophages due to the mass of destroyed B lymphocytes rather than immune modulation remains speculative. Whatever the mechanisms leading to an abrupt or delayed response may be, the data related to AIHA treatment with rituximab have been generated from heterogeneous studies and patients, and no randomized studies have been performed comparing standard therapy with rituximab as monotherapy. In addition, many of the treated patients had chronic lymphocytic leukemia or lymphoma and treatment with rituximab was not only justified but also clearly indicated.
A recent evidence-based focused review showed a partial response rate between 33 and 77% and a complete remission rate between 29 and 55% [11]. Why some authors prefer to use this treatment [10,12] is unclear. Whether or not response differed between patients with non-complement and those with complement-activating aab is currently unknown.
Interestingly, the beneficial effect of rituximab appears to be much better in neonates and children than in adult patients with AIHA [49,50,51,52,53]. The question regarding long-lasting side effects in children remains obscure.
The value of newly developed molecules including ofatumumab and alemtuzumab [54,55], bortezumib [56], anti-FcRn [57], TNT003 (an inhibitor of the serine protease C1S[58], peptide inhibitors of C3 activation [59], IgG-specific endoglycosidase EndoS [60], kinase inhibitors [61], bispecific antibodies [62], or multivalent antibodies [63] has not been well examined in AIHA.
In summary, the actual effect of a rituximab monotherapy is unknown, and there is no possibility to characterize patients who may respond to treatment with rituximab alone as a first- or second-line therapy.
Splenectomy
Although a large number of studies dealing with splenectomy in AIHA have been published during the last century, the results remain somewhat inconclusive and do not allow for a final conclusion to be drawn when splenectomy is clearly indicated. Comparable to data from other therapies, the available data stem from heterogeneous patients who were neither serologically nor clinically well characterized. Theoretically, patients with Fc-mediated phagocytosis may respond better than those with complement-mediated phagocytosis (C3b) or intravascular hemolysis. In addition, the incidence of infections following splenectomy appears to be higher than has yet been suggested [64,65,66]. Most importantly, the outcome is unpredictable, and severe complications including thrombosis, overwhelming infection, and even death may rarely occur. Although laporascopic splenectomy is now increasingly used [67,68], the use of monoclonal antibodies may help to avoid splenectomy in many patients who do not respond to standard therapy [10,12]. Prior to splenectomy, patients should be vaccinated with the quadrivalent meningococcal polysaccharide, polyvalent pneumococcal and Hemophilus influenza type B vaccine [69].
The rate of long-term response to splenectomy is highly variable in published studies and ranges between 25 and 80%, but there are no evidence-based data regarding the true cure rate [4,7,70,71,72,73]. The first series of patients (n = 28) was described by Chertkow and Dacie [70] in 1955. After a 5-year period, only 2 of these patients remained in remission. Allgoad and colleagues [7] reported on a complete response in 17 of 28 patients in 1 year, and 6 were found to have relapsed. Based on the available data, approximately 38-70% of patients with AIHA may respond to splenectomy [5], although data regarding durable remission remain unclear. My own experiences with splenectomy in severely affected and therapy-refractory patients are rather unpromising.
Erythropoiesis-Stimulating Agents
Recombinant erythropoiesis-stimulating agents are used in the treatment of anemia due to diminished erythropoiesis, i.e. renal anemia [74]. Recently, we successfully treated a series of AIHA patients with recombinant erythropoietin and/or erythropoietin biosimilar. The beneficial effect of this treatment may be explained by different mechanisms, including further stimulation of erythropoiesis, decrease in the number of aab per RBC, prolongation of RBC lifespan, and presumably inhibition of eryptosis [75]. Although the mechanism behind this effect remains unknown, the use of these agents is quite encouraging and should be further characterized and optimized.
Other Drugs
An isolated number of patients have been found to benefit from plasmapheresis [76,77], danazol [78], cyclosporine A [79,80], and vincristine-loaded platelets [81]. High-dose intravenous IgG may be effective in children [82,83] rather than in adults [84,85].
Discussion
There is no doubt that advances in the understanding of AIHA have steadily increased. However, our knowledge is, similarly to that of other autoimmune diseases, limited in several aspects. Therefore, we may infrequently make premature decisions that may result in failure. The most optimal therapy is the recognition and elimination of the causative factors of any disease. However, this remains impossible in primary AIHA as all available treatment options are primarily unspecific and directed against our physiological immune components, i.e. macrophages, T and B lymphocytes, or complement. Further attention should be paid to specific therapeutic measures rather than to marketing support and/or the use of unspecific agents. Unfortunately, several reports are abortive and serve either for marketing and/or self-prestige purposes. The motive for data publication must be scientifically and morally sounded, and not of benefit for the author.
As has been descriptively demonstrated, none of the used drugs is specific, and a beneficial effect has not been predictable in any single reported case. Thus, the conclusion from a recent evidence-based focused review is correct that ‘the evidence available for the treatment of AIHA is sparse and of low methodological quality, being predominantly small case series’ [11]. The recommendations made by these authors were of 2 C level (evidence from randomized and observational studies with major methodological flaws or other sources of evidence, e.g. case series).
Prior to the era of rituximab, treatment of AIHA was largely based on corticosteroids with or without the use of azathioprine or cyclophosphamide. Since 1980, I have treated many patients with AIHA and have also been involved in the management of several hundreds of AIHA patients. Although hemolysis was initially quite difficult to control in several patients, the vast majority, if not all, survived or had the opportunity to survive hemolysis per se. If the patient deceased, the reason was found to be related to failures in patient management rather than to uncontrolled hemolysis, misdiagnosis [86], inadequate treatment, treatment complications, or refusal of blood transfusion due to in vitro serological incompatibilities [18]. Replacement of the aforementioned standard therapy by rituximab, in my opinion, is currently unlikely. First, an abrupt therapeutic effect required for severely affected patients is unpredictable and may only occur in isolated cases. Second, the beneficial effect of rituximab without any recent or concomitant treatment with steroids and/or other drugs remains unknown. In addition, rituximab is not yet licensed for the treatment of AIHA, and may cause immediate or sometimes severe side effects, which may overweight AIHA, such as multifocal leukoencephalopathy. Similarly, an escalation of therapy by using rituximab plus dexamethasone or other immunosuppressive drugs [87,88] is somewhat difficult to justify in patients with benign diseases.
I would like to emphasize that rituximab may be helpful only in some instances. In a previous study, we reported on the first use of rituximab in patients with therapy-refractory AIHA [89,90], but the overwhelming success observed in those patients may be attributed to an additive effect to previous therapies or to an exceptional observation.
Recommendations to Treatment of Primary wAIHA
Current treatment options are predominantly based on expert opinions. However, none of the published recommendations is able to cover all aspects which should be obligatory to consider prior to commencement of any treatment. We must take into account that primary wAIHA is a benign rather than a malignant disease, and the outcome of any treatment should be acceptable or at least objectively justified. Thus, treatment of wAIHA remains to be individualized on the basis of severity of hemolysis and anemia, comorbidity, age, sex, lifestyle, and patient's compliance. Future studies should focus on the role of aab and their capability to activate in vivo complement.
Based on my experiences in this field, patients with significant hemolysis are, if tolerable, initially treated with prednisolone or prednisone (1-2 mg/kg) (fig. 1). If necessary, a better effect may be achieved with pulsed high dexamethasone administration (40 mg/day for 4 days). Once the patient has been stabilized, the corticosteroid prednisolone or prednisone dose should be reduced to 15 mg/day and following complete remission to less than 5-7.5 mg/day. Patients who may not tolerate treatment with corticosteroids or who do not respond to high doses of corticosteroids within 2 weeks should be treated with azathioprine (2-4 mg/kg) as soon as possible, particularly elderly patients. An alternative to azathioprine is mycophenolate mofetil (1-2 g/day). However, the drug of choice in refractory cases is cyclophosphamide (60 mg/m2/day). In all cases, a combination with low-dose corticosteroids (after stabilization, ≤7.5 mg/day) and erythropoietin is often helpful (fig. 1, 2). Rituximab is indicated in patients refractory to the aforementioned therapeutic combinations. Only few or isolated patients benefit from splenectomy, plasmapheresis, danazol, or cyclosporine A. High-dose intravenous IgG has been effective in children with AIHA secondary to infections. Finally blood transfusion remains the most effective life-saving measure in patients with hypoxic anemia.
Fig. 1.

How I treat patients with AIHA of warm type. Prior to treatment, all conditions including age, sex, comorbidity, and severity of hemolysis/anemia have to be considered.
Fig. 2.
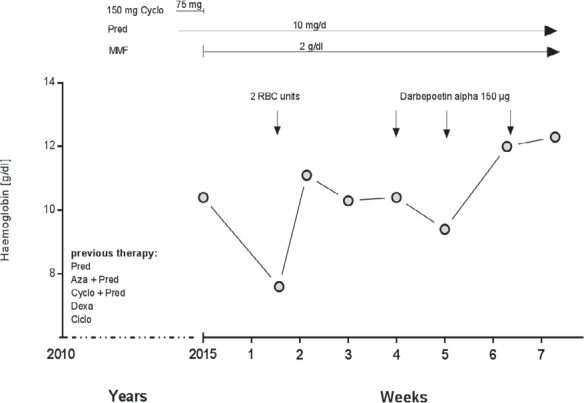
An example of a typical case with refractory AIHA of warm type. Prior to treatment with darbepoetin patient's hemoglobin has never exceed 11.0 g/dl during oberservation. Cyclo = Cyclophosphamide, Pred = prednisolone, MMF = mycophenolate mofetil, Aza = azathioprine, Dexa = dexamethasone, ciclo = ciclosporin.
Disclosure Statement
The author does not have any conflicts of interest regarding this article.
Acknowledgement
I thank Dr. Hans-Gert Heuft for his constructive comments and suggestions which have been considered in this article.
References
- 1.Jandl JH, Jones AR, Castle WB. The destruction of red cells by antibodies in man. I. Observations of the sequestration and lysis of red cells altered by immune mechanisms. J Clin Invest. 1957;36:1428–1459. doi: 10.1172/JCI103542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atkinson JP, Frank MM. Studies on the in vivo effects of antibody. Interaction of IgM antibody and complement in the immune clearance and destruction of erythrocytes in man. J Clin Invest. 1974;54:339–344. doi: 10.1172/JCI107769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salama A, Ahrens N, Kiesewetter H. Serological and clinical aspects of autoimmune hemoytic anemias. Infus Ther Transfus Med. 2002;29:206–217. [Google Scholar]
- 4.Petz LD, Garraty G. Immune Hemolytic Anemias. New York, Churchill Livingstone 2004.
- 5.Baros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210. doi: 10.1016/j.tmrv.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Berentsen S, Sundic T. Red blood cell destruction in autoimmune hemolytic anemia: role of complement and potential new targets for therapy. Biomed Res Int. 2015;2015:363278. doi: 10.1155/2015/363278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allgood JW, Chaplin H., Jr Idiopathic acquired autoimmune hemolytic anemia. A review of forty-seven cases treated from 1955 through 1965. Am J Med. 1967;43:254–273. doi: 10.1016/0002-9343(67)90168-4. [DOI] [PubMed] [Google Scholar]
- 8.Murphy S, LoBuglio AF. Drug therapy of autoimmune hemolytic anemia. Semin Hematol. 1976;13:323–334. [PubMed] [Google Scholar]
- 9.Packman CH. Hemolytic anemia due to warm autoantibodies. Blood Rev. 2008;22:17–31. doi: 10.1016/j.blre.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 10.Lechner K, Jäger U. How I treat autoimmune hemolytic anemias in adults. Blood. 2010;116:1831–1838. doi: 10.1182/blood-2010-03-259325. [DOI] [PubMed] [Google Scholar]
- 11.Crowther M, Chan YL, Garbett IK, Lim W, Vickers MA, Crowther MA. Evidence-based focused review of the treatment of idiopathic warm immune hemolytic anemia in adults. Blood. 2011;118:4036–4040. doi: 10.1182/blood-2011-05-347708. [DOI] [PubMed] [Google Scholar]
- 12.Zanella A, Barcellini W. Treatment of autoimmune hemolytic anemias. Haematologica. 2014;99:1547–1554. doi: 10.3324/haematol.2014.114561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buetens OW, Ness PM. Red blood cell transfusion in autoimmune hemolytic anemia. Curr Opin Hematol. 2003;10:429–433. doi: 10.1097/00062752-200311000-00006. [DOI] [PubMed] [Google Scholar]
- 14.Petz LD. ‘Least incompatible’ units for transfusion in autoimmune hemolytic anemia: should we eliminate this meaningless term? A commentary for clinicians and transfusion medicine professionals. Transfusion. 2003;43:1503–1507. doi: 10.1046/j.1537-2995.2003.00583.x. [DOI] [PubMed] [Google Scholar]
- 15.King KE, Ness PM. Treatment of autoimmune hemolytic anemia. Semin Hematol. 2005;42:131–136. doi: 10.1053/j.seminhematol.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Salama A, Berghöfer H, Mueller-Eckhardt C. Red blood cell transfusion in warm-type autoimmune haemolytic anaemia. Lancet. 1992;340:1515–1517. doi: 10.1016/0140-6736(92)92766-9. [DOI] [PubMed] [Google Scholar]
- 17.Ahrens N, Pruss A, Kähne A, Kiesewetter H, Salama A. Coexistence of autoantibodies and alloantibodies to red blood cells due to blood transfusion. Transfusion. 2007;47:813–816. doi: 10.1111/j.1537-2995.2007.01194.x. [DOI] [PubMed] [Google Scholar]
- 18.Yürek S, Almahallawi M, Mayer B, Pruss A, Salama A. Precautions surrounding blood transfusion in autoimmune haemolytic anemias are overestimated. Blood Transfus 2015; doi: 10.2450/2015.0326-14. [DOI] [PMC free article] [PubMed]
- 19.Petz LD. A physician's guide to transfusion in autoimmune haemolytic anaemia. Br J Haematol. 2004;124:712–716. doi: 10.1111/j.1365-2141.2004.04841.x. [DOI] [PubMed] [Google Scholar]
- 20.Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859–1862. doi: 10.1111/j.1537-2995.2006.00990.x. [DOI] [PubMed] [Google Scholar]
- 21.Li BJ, Yuan X, Jiang YJ, Ning Li, Shu XW, Liu KL. Retrospective analysis of 30 severe autoimmune hemolytic anemia patients treated by whole blood exchange transfusion. Transfusion 2015; doi: 10.1111/trf.13122. [DOI] [PubMed]
- 22.Meyer O, Stahl D, Beckhove P, Huhn D, Salama A. Pulsed high-dose dexamethasone in chronic autoimmune haemolytic anaemia of warm type. Br J Haematol. 1997;98:860–862. doi: 10.1046/j.1365-2141.1997.3203137.x. [DOI] [PubMed] [Google Scholar]
- 23.Howard J, Hoffbrand AV, Prentice HG, Mehta A. Mycophenolate mofetil for the treatment of refractory auto-immune haemolytic anaemia and auto-immune thrombocytopenia purpura. Br J Haematol. 2002;117:712–715. doi: 10.1046/j.1365-2141.2002.03430.x. [DOI] [PubMed] [Google Scholar]
- 24.Kotb R, Pinganaud C, Trichet C, Lambotte O, Dreyfus M, Delfraissy JF, Tchernia G, Goujard C. Efficacy of mycophenolate mofetil in adult refractory auto-immune cytopenias: a single center preliminary study. Eur J Haematol. 2005;75:60–64. doi: 10.1111/j.1600-0609.2005.00437.x. [DOI] [PubMed] [Google Scholar]
- 25.Piek CJ, Junius G, Dekker A, Schrauwen E, Slappendel RJ, Teske E. Idiopathic immune-mediated hemolytic anemia: treatment outcome and prognostic factors in 149 dogs. J Vet Intern Med. 2008;22:366–373. doi: 10.1111/j.1939-1676.2008.0060.x. [DOI] [PubMed] [Google Scholar]
- 26.Brodsky RA, Petri M, Smith BD, Seifter EJ, Spivak JL, Styler M, Dang CV, Brodsky I, Jones RJ. Immunoablative high-dose cyclophosphamide without stem-cell rescue for refractory, severe autoimmune disease. Ann Intern Med. 1998;129:1031–1035. doi: 10.7326/0003-4819-129-12-199812150-00007. [DOI] [PubMed] [Google Scholar]
- 27.Moyo VM, Smith D, Brodsky I, Crilley P, Jones RJ, Brodsky RA. High-dose cyclophosphamide for refractory autoimmune hemolytic anemia. Blood. 2002;100:704–706. doi: 10.1182/blood-2002-01-0087. [DOI] [PubMed] [Google Scholar]
- 28.Thabet AF, Faisal M. Pulse cyclophosphamide therapy in refractory warm autoimmune hemolytic anemia: a new perspective. Indian J Hematol Blood Transfus. 2014;30:313–318. doi: 10.1007/s12288-013-0290-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Passweg JR, Rabusin M, Musso M, Beguin Y, Cesaro S, Ehninger G, Espigado I, Iriondo A, Jost L, Koza V, Lenhoff S, Lisukov I, Locatelli F, Marmont A, Philippe P, Pilatrino C, Quartier P, Stary J, Veys P, Vormoor J, Wahlin A, Zintl F, Bocelli-Tyndall C, Tyndall A, Gratwohl A, Autoimmune Disease Working Party of the EBMT Haematopoetic stem cell transplantation for refractory autoimmune cytopenia. Br J Haematol. 2004;125:749–755. doi: 10.1111/j.1365-2141.2004.04978.x. [DOI] [PubMed] [Google Scholar]
- 30.Cvetković RS, Perry CM. Rituximab: a review of its use in non-Hodgkin's lymphoma and chronic lymphocytic leukaemia. Drugs. 2006;66:791–820. doi: 10.2165/00003495-200666060-00005. [DOI] [PubMed] [Google Scholar]
- 31.Garvey B. Rituximab in the treatment of autoimmune haematological disorders. Br J Haematol. 2008;141:149–169. doi: 10.1111/j.1365-2141.2008.07054.x. [DOI] [PubMed] [Google Scholar]
- 32.Dierickx D, Verhoef G, Van Hoof A, Mineur P, Roest A, Triffet A, Kentos A, Pierre P, Boulet D, Bries G, Lê PQ, Janssens A, Delannoy A. Rituximab in auto-immune haemolytic anaemia and immune thrombocytopenic purpura: a Belgian retrospective multicentric study. J Intern Med. 2009;266:484–491. doi: 10.1111/j.1365-2796.2009.02126.x. [DOI] [PubMed] [Google Scholar]
- 33.Gómez-Almaguer D, Solano-Genesta M, Tarín-Arzaga L, Herrera-Garza JL, Cantú-Rodríguez OG, Gutiérrez-Aguirre CH, Jaime-Pérez JC. Low-dose rituximab and alemtuzumab combination therapy for patients with steroid-refractory autoimmune cytopenias. Blood. 2010;116:4783–4785. doi: 10.1182/blood-2010-06-291831. [DOI] [PubMed] [Google Scholar]
- 34.Peñalver FJ, Alvarez-Larrán A, Díez-Martin JL, Gallur L, Jarque I, Caballero D, Díaz-Mediavilla J, Bustelos R, Fernández-Aceñero MJ, Cabrera JR. Rituximab is an effective and safe therapeutic alternative in adults with refractory and severe autoimmune hemolytic anemia. Ann Hematol. 2010;89:1073–1080. doi: 10.1007/s00277-010-0997-y. [DOI] [PubMed] [Google Scholar]
- 35.Barcellini W, Zaja F, Zaninoni A, Imperiali FG, Battista ML, Di Bona E, Fattizzo B, Consonni D, Cortelezzi A, Fanin R, Zanella A. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biologic studies. Blood. 2012;119:3691–3697. doi: 10.1182/blood-2011-06-363556. [DOI] [PubMed] [Google Scholar]
- 36.Maung SW, Leahy M, O'Leary HM, Khan I, Cahill MR, Gilligan O, Murphy P, McPherson S, Jackson F, Ryan M, Hennessy B, McHugh J, Goodyer M, Bacon L, O'Gorman P, Nee A, O'Dwyer M, Enright H, Saunders J, O'Keeffe D. A multi-centre retrospective study of rituximab use in the treatment of relapsed or resistant warm autoimmune haemolytic anaemia. Br J Haematol. 2013;163:118–122. doi: 10.1111/bjh.12486. [DOI] [PubMed] [Google Scholar]
- 37.Roumier M, Loustau V, Guillaud C, Languille L, Mahevas M, Khellaf M, Limal N, Noizat-Pirenne F, Godeau B, Michel M. Characteristics and outcome of warm autoimmune hemolytic anemia in adults: new insights based on a single-center experience with 60 patients. Am J. Hematol. 2014;89:E150–155. doi: 10.1002/ajh.23767. [DOI] [PubMed] [Google Scholar]
- 38.Barcellini W, Fattizzo B, Zaninoni A, Radice T, Nichele I, Di Bona E, Lunghi M, Tassinari C, Alfinito F, Ferrari A, Leporace AP, Niscola P, Carpenedo M, Boschetti C, Revelli N, Villa MA, Consonni D, Scaramucci L, De Fabritiis P, Tagariello G, Gaidano G, Rodeghiero F, Cortelezzi A, Zanella A. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: a GIMEMA study of 308 patients. Blood. 2014;124:2930–2936. doi: 10.1182/blood-2014-06-583021. [DOI] [PubMed] [Google Scholar]
- 39.Reynaud Q, Durieu I, Dutertre M, Ledochowski S, Durupt S, Michallet AS, Vital-Durand D, Lega JC. Efficacy and safety of rituximab in auto-immune hemolytic anemia: a meta-analysis of 21 studies. Autoimmun Rev. 2015;14:304–313. doi: 10.1016/j.autrev.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 40.Dierickx D, De Rycke A, Vanderschueren S, Delannoy A. New treatment options for immune-mediated hematological disorders. Eur J Intern Med. 2008;19:579–586. doi: 10.1016/j.ejim.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 41.Gürcan HM, Keskin DB, Stern JN, Nitzberg MA, Shekhani H, Ahmed AR. A review of the current use of rituximab in autoimmune diseases. Int Immunopharmacol. 2009;9:10–25. doi: 10.1016/j.intimp.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 42.Dunleavy K, Tay K, Wilson WH. Rituximab-associated neutropenia. Semin Hematol. 2010;47:180–186. doi: 10.1053/j.seminhematol.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gea-Banacloche JC. Rituximab-associated infections. Semin Hematol. 2010;47:187–198. doi: 10.1053/j.seminhematol.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 44.Kelesidis T, Daikos G, Boumpas D, Tsiodras S. Does rituximab increase the incidence of infectious complications? A narrative review. Int J Infect Dis. 2011;15:e2–e16. doi: 10.1016/j.ijid.2010.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shetty S, Ahmed AR. Preliminary analysis of mortality associated with rituximab use in autoimmune diseases. Autoimmunity. 2013;46:487–496. doi: 10.3109/08916934.2013.838563. [DOI] [PubMed] [Google Scholar]
- 46.Peuvrel L, Chiffoleau A, Quéreux G, Brocard A, Saint-Jean M, Batz A, Jolliet P, Dréno B. Melanoma and rituximab: an incidental association? Dermatology. 2013;226:274–278. doi: 10.1159/000350681. [DOI] [PubMed] [Google Scholar]
- 47.Ghanima W, Khelif A, Waage A, Michel M, Tjønnfjord GE, Romdhan NB, Kahrs J, Darne B, Holme PA. Rituximab as second-line treatment for adult immune thrombocytopenia (the RITP trial): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:1653–1661. doi: 10.1016/S0140-6736(14)61495-1. [DOI] [PubMed] [Google Scholar]
- 48.Salama A. Current treatment options for primary immune thrombocytopenia. Expert Rev Hematol. 2011;4:107–118. doi: 10.1586/ehm.10.76. [DOI] [PubMed] [Google Scholar]
- 49.Quartier P, Brethon B, Philippet P, Landman-Parker J, Le Deist F, Fischer A. Treatment of childhood autoimmune haemolytic anaemia with rituximab. Lancet. 2001;358:1511–1513. doi: 10.1016/s0140-6736(01)06573-4. [DOI] [PubMed] [Google Scholar]
- 50.Ng PC, Lee KK, Lo AF, Li CK, Fok TF. Anti B cell targeted immunotherapy for treatment of refractory autoimmune haemolytic anaemia in a young infant. Arch Dis Child. 2003;88:337–339. doi: 10.1136/adc.88.4.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zecca M, Nobili B, Ramenghi U, Perrotta S, Amendola G, Rosito P, Jankovic M, Pierani P, De Stefano P, Bonora MR, Locatelli F. Rituximab for the treatment of refractory autoimmune hemolytic anemia in children. Blood. 2003;101:3857–3861. doi: 10.1182/blood-2002-11-3547. [DOI] [PubMed] [Google Scholar]
- 52.Svahn J, Fioredda F, Calvillo M, Molinari AC, Micalizzi C, Banov L, Schmidt M, Caprino D, Marinelli D, Gallisai D, Dufour C. Rituximab-based immunosuppression for autoimmune haemolytic anaemia in infants. Br J Haematol. 2009;145:96–100. doi: 10.1111/j.1365-2141.2009.07594.x. [DOI] [PubMed] [Google Scholar]
- 53.Ansari S, Tashvighi M, Arbani BD, Salimi AB, Golpaygani M. Rituximab for child with chronic relapsing autoimmune hemolytic anemia. Pediatr Hematol Oncol. 2011;28:164–166. doi: 10.3109/08880018.2010.518339. [DOI] [PubMed] [Google Scholar]
- 54.Cheung WW, Hwang GY, Tse E, Kwong YL. Alemtuzumab induced complete remission of autoimmune hemolytic anemia refractory to corticosteroids, splenectomy and rituximab. Haematologica. 2006;91(5 suppl):ECR13. [PubMed] [Google Scholar]
- 55.Liu B, Gu W. Immunotherapy treatments of warm autoimmune hemolytic anemia. Clin Dev Immunol. 2013;2013:561852. doi: 10.1155/2013/561852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dasanu CA. Bortezomib friend or foe of hemolytic anemia? J Oncol Pharm Pract. 2011;17:233–235. doi: 10.1177/1078155210374240. [DOI] [PubMed] [Google Scholar]
- 57.Wang Y, Tian Z, Thirumalai D, Zhang X. Neonatal Fc receptor (FcRn): a novel target for therapeutic antibodies and antibody engineering. J Drug Target. 2014;22:269–278. doi: 10.3109/1061186X.2013.875030. [DOI] [PubMed] [Google Scholar]
- 58.Shi J, Rose EL, Singh A, Hussain S, Stagliano NE, Parry GC, Panicker S. TNT003, an inhibitor of the serine protease C1s, prevents complement activation induced by cold agglutinins. Blood. 2014;123:4015–4022. doi: 10.1182/blood-2014-02-556027. [DOI] [PubMed] [Google Scholar]
- 59.Risitano AM, Ricklin D, Huang Y, Reis ES, Chen H, Ricci P, Lin Z, Pascariello C, Raia M, Sica M, Del Vecchio L, Pane F, Lupu F, Notaro R, Resuello RR, DeAngelis RA, Lambris JD. Peptide inhibitors of C3 activation as a novel strategy of complement inhibition for the treatment of paroxysmal nocturnal hemoglobinuria. Blood. 2014;123:2094–2101. doi: 10.1182/blood-2013-11-536573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Allhorn M, Briceño JG, Baudino L, Lood C, Olsson ML, Izui S, Collin M. The IgG-specific endoglycosidase EndoS inhibits both cellular and complement-mediated autoimmune hemolysis. Blood. 2010;115:5080–5088. doi: 10.1182/blood-2009-08-239020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tan SL, Liao C, Lucas MC, Stevenson C, DeMartino JA. Targeting the SYK-BTK axis for the treatment of immunological and hematological disorders: recent progress and therapeutic perspectives. Pharmacol Ther. 2013;138:294–309. doi: 10.1016/j.pharmthera.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 62.Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol 2015; doi: 10.1016/j.molimm.2015.01.003. [DOI] [PubMed]
- 63.Nuñez-Prado N, Compte M, Harwood S, Álvarez-Méndez A, Lykkemark S, Sanz L, Álvarez-Vallina L. The coming of age of engineered multivalent antibodies. Drug Discov Today. 2015;20:588–594. doi: 10.1016/j.drudis.2015.02.013. [DOI] [PubMed] [Google Scholar]
- 64.Edgren G, Almqvist R, Hartman M, Utter GH. Splenectomy and the risk of sepsis: a population-based cohort study. Ann Surg. 2014;260:1081–1087. doi: 10.1097/SLA.0000000000000439. [DOI] [PubMed] [Google Scholar]
- 65.Barmparas G, Lamb AW, Lee D, Nguyen B, Eng J, Bloom MB, Ley EJ. Postoperative infection risk after splenectomy: a prospective cohort study. Int J Surg. 2015;17:10–14. doi: 10.1016/j.ijsu.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 66.Moulis G, Sailler L, Sommet A, Lapeyre-Mestre M, Derumeaux H, Adoue D. Rituximab versus splenectomy in persistent or chronic adult primary immune thrombocytopenia: an adjusted comparison of mortality and morbidity. Am J Hematol. 2014;89:41–46. doi: 10.1002/ajh.23580. [DOI] [PubMed] [Google Scholar]
- 67.Bresler L, Guerci A, Brunaud L, Ayav A, Sebbag H, Tortuyaux JM, Lederlin P, Boissel P. Laparoscopic splenectomy for idiopathic thrombocytopenia purpura: outcome and long-term results. World J Surg. 2002;26:111–114. doi: 10.1007/s00268-001-0190-5. [DOI] [PubMed] [Google Scholar]
- 68.Keider A, Feldman M, Szold A. Analysis of outcome of laparoscopic splenectomy for idiopathic thrombocytopenia purpura by platelet count. Am J Hematol. 2005;80:95–100. doi: 10.1002/ajh.20433. [DOI] [PubMed] [Google Scholar]
- 69.American Academy of Pediatrics. Committee on Infectious Diseases Policy statements: recommendations for the prevention of pneumococcal infections, including the use of pneumococcal conjugate vaccine (Prevnar), pneumococcal polysaccharide vaccine, and antibiotic prophylaxis. Pediatrics. 2000;106:362–366. doi: 10.1542/peds.106.2.362. [DOI] [PubMed] [Google Scholar]
- 70.Chertkow G, Dacie JV. Results of splenectomy in auto-immune haemolytic anaemia. Br J Haematol. 1956;2:237–249. [PubMed] [Google Scholar]
- 71.Bowdler AJ. The role of the spleen and splenectomy in autoimmune hemolytic disease. Semin Hematol. 1976;13:335–348. [PubMed] [Google Scholar]
- 72.Coon WW. Splenectomy in the treatment of hemolytic anemia. Arch Surg. 1985;120:625–628. doi: 10.1001/archsurg.1985.01390290099017. [DOI] [PubMed] [Google Scholar]
- 73.Akpek G, McAneny D, Weintraub L. Comparative response to splenectomy in Coombs-positive autoimmune hemolytic anemia with or without associated disease. Am J Hematol. 1999;61:98–102. doi: 10.1002/(sici)1096-8652(199906)61:2<98::aid-ajh4>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 74.Sinclair AM. Erythropoiesis stimulating agents: approaches to modulate activity. Biologics. 2013;7:161–174. doi: 10.2147/BTT.S45971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Salama A, Hartnack D, Lindemann HW, Lange HJ, Rummel M, Loew A. The effect of erythropoiesis-stimulating agents in patients with therapy-refractory autoimmune hemolytic anemia. Transfus Med Hemother. 2014;41:462–468. doi: 10.1159/000366244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ruivard M, Tournilhac O, Montel S, Fouilhoux AC, Quainon F, Lénat A, Travade P, Philippe P. Plasma exchanges do not increase red blood cell transfusion efficiency in severe autoimmune hemolytic anemia: a retrospective case-control study. J Clin Apher. 2006;21:202–206. doi: 10.1002/jca.20096. [DOI] [PubMed] [Google Scholar]
- 77.McLeod BC. Evidence based therapeutic apheresis in autoimmune and other hemolytic anemias. Curr Opin Hematol. 2007;14:647–654. doi: 10.1097/MOH.0b013e3282c8ca66. [DOI] [PubMed] [Google Scholar]
- 78.Pignon JM, Poirson E, Rochant H. Danazol in autoimmune haemolytic anaemia. Br J Haematol. 1993;83:343–345. doi: 10.1111/j.1365-2141.1993.tb08293.x. [DOI] [PubMed] [Google Scholar]
- 79.Fiernkranz E, Lasch HG, Salama A, Müller-Eckhardt C. Therapieresistente Autoimmunhämolyse mit Retikoluzytopenie bei systemischem Lupus erythematodes (SLE) Med Welt. 1988;39:174–176. [Google Scholar]
- 80.Emilia G, Messora C, Longo G, Bertesi M. Long-term salvage treatment by cyclosporin in refractory autoimmune haematological disorders. Br J Haematol. 1996;93:341–344. doi: 10.1046/j.1365-2141.1996.4871026.x. [DOI] [PubMed] [Google Scholar]
- 81.Shvidel L, Sigler E, Shtalrid M, Berrebi A. Vincristine-loaded platelet infusion for treatment of refractory autoimmune hemolytic anemia and chronic immune thrombocytopenia: rethinking old cures. Am J Hematol. 2006;81:423–425. doi: 10.1002/ajh.20632. [DOI] [PubMed] [Google Scholar]
- 82.Bussel JB, Cunningham-Rundles C, Abraham C. Intravenous treatment of autoimmune hemolytic anemia with very high dose gammaglobulin. Vox Sang. 1986;51:264–269. doi: 10.1111/j.1423-0410.1986.tb01967.x. [DOI] [PubMed] [Google Scholar]
- 83.Hoppe B, Gaedicke G, Kiesewetter H, Salama AR. Response to intravenous immunoglobulin G in an infant with immunoglobulin A-associated autoimmune haemolytic anaemia. Vox Sang. 2004;86:151–153. doi: 10.1111/j.0042-9007.2004.00392.x. [DOI] [PubMed] [Google Scholar]
- 84.Salama A, Mahn I, Neuzner J, Graubner M, Mueller-Eckhardt C. IgG therapy in autoimmune haemolytic anaemia of warm type. Blut. 1984;48:391–392. doi: 10.1007/BF00319969. [DOI] [PubMed] [Google Scholar]
- 85.Mueller-Eckhardt C, Salama A, Mahn I, Kiefel V, Neuzner J, Graubner M. Lack of efficacy of high-dose intravenous immunoglobulin in autoimmune haemolytic anaemia: a clue to its mechanism. Scand J Haematol. 1985;34:394–400. doi: 10.1111/j.1600-0609.1985.tb00767.x. [DOI] [PubMed] [Google Scholar]
- 86.Salama A. Clinically and/or serological misleading findings surrounding immune haemolytic anaemias. Transfus Med Hemother 2015; DOI: 10.1159/000438960. [DOI] [PMC free article] [PubMed]
- 87.Birgens H, Frederiksen H, Hasselbalch HC, Rasmussen IH, Nielsen OJ, Kjeldsen L, Larsen H, Mourits-Andersen T, Plesner T, Rønnov-Jessen D, Vestergaard H, Klausen TW, Schöllkopf C. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013;163:393–399. doi: 10.1111/bjh.12541. [DOI] [PubMed] [Google Scholar]
- 88.Rossignol J, Michallet AS, Oberic L, Picard M, Garon A, Willekens C, Dulery R, Leleu X, Cazin B, Ysebaert L. Rituximab-cyclophosphamide-dexamethasone combination in the management of autoimmune cytopenias associated with chronic lymphocytic leukemia. Leukemia. 2011;25:473–478. doi: 10.1038/leu.2010.278. [DOI] [PubMed] [Google Scholar]
- 89.Ahrens N, Kingreen D, Seltsam A, Salama A. Treatment of refractory autoimmune haemolytic anaemia with anti-CD20 (rituximab) Br J Haematol. 2001;114:244–245. doi: 10.1046/j.1365-2141.2001.02873-4.x. [DOI] [PubMed] [Google Scholar]
- 90.Ahrens N, Heymann G, Meyer O, Kiesewetter H, Salama A. Results of treatment with rituximab (anti-CD20) in three patients with autoimmune haemolytic anemia and/or immune thrombocytopenia and a concise review of reported cases. Infus Ther Transfus Med. 2002;29:277–281. [Google Scholar]