

Summary
Autoimmune hemolytic anemia (AIHA) is caused by the increased destruction of red blood cells (RBCs) by anti-RBC autoantibodies with or without complement activation. RBC destruction may occur both by a direct lysis through the sequential activation of the final components of the complement cascade (membrane attack complex), or by antibody-dependent cell-mediated cytotoxicity (ADCC). The pathogenic role of autoantibodies depends on their class (the most frequent are IgG and IgM), subclass, thermal amplitude (warm and cold forms),as well as affinity and efficiency in activating complement. Several cytokines and cytotoxic mechanisms (CD8+ T and natural killer cells) are further involved in RBC destruction. Moreover, activated macrophages carrying Fc receptors may recognize and phagocyte erythrocytes opsonized by autoantibodies and complement. Direct complement-mediated lysis takes place mainly in the circulations and liver, whereas ADCC, cytotoxicity, and phagocytosis occur preferentially in the spleen and lymphoid organs. The degree of intravascular hemolysis is 10-fold greater than extravascular one. Finally, the efficacy of the erythroblastic compensatory response can greatly influence the clinical picture of AIHA. The interplay and relative burden of all these pathogenic mechanisms give reason for the great clinical heterogeneity of AIHAs, from fully compensated to rapidly evolving fatal cases.
Keywords: Autoimmune hemolytic anemia, Autoantibodies, Cytokines
Introduction
Autoimmune hemolytic anemia (AIHA) is defined as the increased destruction of red blood cells (RBCs) in the presence of anti-RBC autoantibodies with or without complement activation. Autoantibodies are produced by both tissue and circulating self-reactive B lymphocytes, following cooperation with T helper lymphocytes. A third key player in anti-RBC autoimmunity is the complement system, which is able to induce a direct erythrocyte osmotic lysis through the sequential activation of the membrane attack complex (MAC). Moreover, autoantibodies are able to destroy RBCs by antibody-dependent cell-mediated cytotoxicity (ADCC), which is mediated by cytotoxic CD8+ T cells and natural killer (NK) that carry membrane receptors for the Fc portion of immunoglobulin G (IgG). Finally, activated macrophages carrying Fc receptors may recognize and phagocyte erythrocytes opsonized by autoantibodies and complement. While direct complement-mediated lysis takes place mainly in the circulations and liver, ADCC and phagocytosis occur preferentially in the spleen and lymphoid organs [1].
AIHAs can be distinguished on the basis of the autoantibody Ig class and thermal characteristics, i.e. the optimal temperature of reaction with autologous erythrocytes, in warm (wAIHA), cold (cold agglutinin disease; CAD), and mixed forms, although atypical cases of difficult diagnostic classification are reported with increasing frequency [1,2,3]. Moreover, AIHAs are classified in primary (idiopathic), in which hemolysis dominates the clinical picture in the absence of any other coexisting disorder, and in secondary forms, accompanying and complicating an underlying disease. The majority of warm forms are idiopathic or associated with other autoimmune diseases; acute cold forms are mainly secondary to infections, whereas chronic CADs are frequently associated to lymphoproliferative or neoplastic diseases [1,2,3]. All these factors account for the great clinical heterogeneity of AIHAs, from compensated forms without anemia to fulminating disease. Table 1 shows the main characteristics of AIHAs.
Table 1.
Classification of AIHAs
| 1) Warm autoimmune hemolytic anemia (wAIHA) |
| DAT positive for IgG or IgG+C3d |
| IgG antibody directed mainly against epitopes of the Rh system |
| Idiopathic or secondary to autoimmune, lymphoproliferative, or neoplastic diseases |
| 2) Cold autoimmune hemolytic anemia |
| 2a) Cold agglutinin disease (CAD) |
| DAT positive for C3d |
| IgM directed against the I/i system |
| Idiopathic or secondary to infections (acute transitory) or to lymphoproliferative diseases (chronic) |
| 2b) Paroxysmal cold hemoglobinuria (PCH) |
| Donath-Landsteiner test positive |
| Bithermic IgG directed against the erythrocyte P antigen |
| Idiopathic or secondary to syphilis (chronic) or to other infections (acute transitory) |
| 3) Mixed autoimmune hemolytic anemia |
| DAT positive for IgG and C3d |
| Coexistence of warm IgG autoantibodies and high titer IgM cold agglutinins |
| Idiopathic or secondary to lymphoproliferative or autoimmune diseases |
It is commonly thought that autoimmunity is the result of the interaction of genetic predisposition and environmental factors. While several HLA associations have been reported for various organ and systemic autoimmune diseases (type-1 insulin-dependent diabetes, Hashimoto thyroiditis, Graves disease, multiple sclerosis, myasthenia gravis, pemphigus vulgaris, systemic lupus erythematosus, rheumatoid arthritis and ankylosing spondylitis), no clear HLA linkage has been found for AIHA. Moreover, the mechanisms involved in the breakdown of both central and peripheral tolerance in the development of AIHA are still largely unknown. Central tolerance occurs during the early differentiation of B cells in the bone marrow and T cells in the tymus, where autoreactive cells undergo apoptosis upon binding to self-antigens (negative selection). Peripheral tolerance is an active process, where anergy or suppression against self-antigens is driven by specific CD4+/CD25+ regulatory T cells (Tregs) and CD8+ suppressor T lymphocytes [4].
Molecular mimicry between self-antigens and foreign antigens is one of several mechanisms by which pathogens might be involved in the initiation and/or acceleration of autoimmunity. This is likely the prevalent mechanism of AIHAs secondary to various bacterial, mycoplasma or virus infections, probably due to the presence of common antigenic epitopes on RBC proteins or carbohydrates. Additional mechanisms involved in the failure to maintain self-tolerance are polyclonal lymphocyte activation, again induced by several viral infections, and the emergence of ‘forbidden clones', as observed in lymphoproliferative diseases. This hypothesis, proposed more than 60 years ago by Burnet and based on the persistence of self-reactive clones of lymphocytes that should have been deleted via immune tolerance, has gained new interest due to the success of B-cell-depleting therapy in several autoimmune diseases [5]. A particular condition is related to the generation of neoantigens by environmental agents or drugs, as observed in drug-induced AIHA, where the autoimmune response persists until they are destroyed and eliminated [6,7]. Moreover, in a large study of 717 patients displaying anti-RBC autoantibodies, about one-third was found positive for alloantibodies, and RBC transfusion was identified as a major cause for autoimmunization [8]. Recently, attention has been focused on the autoimmune/inflammatory syndrome induced by adjuvants, which include several still undefined diseases, such as siliconosis, Gulf War syndrome, Spanish toxic oil syndrome, Ardystil syndrome, and post-vaccination [9]. Above all there is a loss of suppressor mechanisms and the alteration of the regulatory cells and cytokine network involved in the control of self-tolerance [4].
Autoantibodies in the Pathogenesis of AIHA
The most frequent autoantibodies against RBCs are IgG, which mainly determine extravascular hemolysis through the ADCC in the reticuloendothelial system (spleen and to a lesser extent liver). The IgG subclass influences the degree to which these antibodies shorten RBC survival: IgG1 is the most commonly encountered subclass and together with IgG3 shortens the half-life more efficiently than IgG2 and IgG4. Autoantibodies of this class are mostly directed against epitopes of the Rh system. They generally react at 37 °C and, therefore, are responsible for the warm forms of AIHA [1]. IgG are able to activate the complement cascade, with the exception of IgG2 and IgG4 subclasses. IgA autoantibodies are generally associated with IgG and only rarely reported as the sole causative agent of AIHA.
IgM are pentameric autoantibodies able to dramatically fix complement compared with other isotypes, thus prevalently causing intravascular hemolysis, and to a lesser extent C3d-mediated extravascular lysis (mainly in the liver). Autoantibodies of this class are directed against the I/i system. Their optimal temperature of reaction is 4 °C, and thus they are responsible for the cold forms of AIHA [1,2]. However, the thermal amplitude of IgM autoantibodies ranges from 0 to 34 °C, and those with a thermal activity close to physiological temperatures (warm IgM) are the most harmful autoantibodies, able to cause severe forms of AIHA with a reported mortality rate of about 20% [6]. In fact, it is worth reminding that the amount of RBC destruction by intravascular hemolysis has been calculated in 200 ml of RBCs in 1 h, whereas by extravascular hemolysis it is 10-fold lesser (0.25 ml RBCs/kg/h, i.e., for a patient weighing 70 kg = 17.5 ml RBCs/h = 420 ml RBCs / 24 h) [1,2].
Anti-RBC antibodies are usually detected by the direct antiglobulin test (DAT) or Coombs test, generally performed by the traditional tube agglutination technique with polyspecific antisera. Nowadays the use of monospecific anti-IgG, anti-IgM, and anti-C3 antisera is recommended to define the class of the autoantibody and its thermal amplitude. It is worth mentioning a simple test, the spontaneous agglutination of RBCs at 20 °C, a characteristic feature indicating the presence of cold IgM autoantibodies. According to DAT results and to the thermal characteristics of the autoantibody, AIHAs are usually classified in two main groups: wAIHA (∼70% of cases), which displayed IgG only or IgG plus C3d on erythrocytes, and CAD (∼20% of patients), which showed C3d only, along with high serum titer of cold agglutinins of I specificity. There are few cases (7-8% of all AIHAs), which showed a DAT positive for IgG and C3d, with the coexistence of warm autoantibodies and high-titer cold agglutinins (mixed AIHA). However, there is increasing evidence of atypical cases of difficult classification - mainly DAT-negative - which are frequently severe and refractory/relapsed after several therapy lines, and may have a fatal outcome [10,11]. DAT-negative cases may be caused by warm IgM autoantibodies, which can be identified by the dual direct antiglobulin test (DDAT) [12,13]. It is worth mentioning the Donath-Landsteiner autoantibody, a bithermic hemolysin able to fix complement at cold temperatures and to determine RBCs lysis at 37 °C, that is directed against the erythrocyte P antigen and responsible of paroxysmal cold hemoglobinuria, which is rather common in children (30% of cases) but rare in adults (<1% of all AIHA cases) [1,2].
Finally, DAT positivity may also be due to drug-induced antibodies that are associated with a variety of drugs (antimicrobials: 42% of cases, anti-inflammatory drugs: 15%, and anti-neoplastics: 11%) but occur rarely (about 1 in 1 million of the population). Drug-dependent antibodies react in vitro only in the presence of drug bound to RBCs or added to the patient's serum in test systems (typical example penicillin and cefotetan). Drug-independent antibodies can be detected in vitro without adding any drug, thus with characteristics identical to classic autoantibodies. The typical example is methyldopa, which causes the production of RBC autoantibodies in about 15% of the patients receiving the drug, but only about 1% develops AIHA [1,6,7,14,15].
The Role of Complement in the Pathogenesis of AIHA
The complement system plays a role in both warm AIHA and CAD. In the former cases IgG bound to erythrocytes are able to fix complement protein complex C1 and thus activate the classical complement pathway; IgG3 subclass is the more efficient, followed by IgG1, whereas IgG2 and IgG4 are less or not active. Warm IgM autoantibodies are potent complement activators but they are usually not found on RBCs. In fact, they temporary bind, activate complement, and then detach from the membrane, where only complement factors may be demonstrated. Following complement activation, phagocytosis of C3b-opsonized erythrocytes occurs mainly in the liver, although complete intravascular hemolysis mediated by the terminal lytic complex may occur as well [12,16,17,18]. DAT positivity for C3 fragments is a marker of complement involvement in wAIHA.
The role of the complement system in CAD is also significant, provided that the thermal amplitude of the IgM autoantibody is close to values found on the surface of the body areas exposed to external cold temperatures. IgM-cold autoantibody binds C1 and thereby initiates the classical complement pathway [19]: C1 esterase activates C4 and C2, generating C3 convertase, which results in the cleavage of C3 to C3a and C3b. When RBCs return to the central parts of the body with a temperature of 37 °C, the IgM autoantibody detaches from the cell surface, while C3b remains bound. A part of the C3b-coated RBCs is sequestered in the liver, whereas the other erythrocytes are lysed through the activation of C5 by C3b. The final formation of the MAC determines intravascular hemolysis [20,21,22]. This occurs in severe acute exacerbations and in some severe hemolytic patients, as evidenced by the finding of hemoglobinuria, a typical finding of paroxysmal nocturnal hemoglobinuria (PNH). This pathogenic mechanism accounts for the beneficial effect of C5 inhibition with the monoclonal antibody eculizumab in some cases [23,24]. Further evidence for the potential use of complement inhibitors in the treatment of AIHA is the finding that TNT003, a mouse monoclonal antibody targeting complement protein C1s, prevents induction of in vitro hemolysis by cold agglutinins [25]. Moreover, a C1-esterase inhibitor concentrate was found to protect erythrocytes from complement-mediated destruction in AIHA [26]. Further complement inhibitors, such as compstatin Cp40, a low-molecular-peptide that prevents cleavage of C3, and TT30, which prevents C3 convertase activation, have shown promising effects on complement-mediated lysis of erythrocytes in PNH [27,28], and may be potential treatments suitable for acute hemolysis in AIHA.
It is worth reminding that cold IgM antibodies are frequently monoclonal and, in more than 90% of the patients, show light chain restriction [29]. Moreover, CAD patients have frequently an associated clonal B-cell lymphoproliferative disorder (lymphoplasmacytic lymphoma, marginal zone lymphoma), or at least unclassified clonal lymphoproliferation or reactive lymphocytosis [20,21]. These findings account for the rationale of treating refractory and severe CAD cases with rituximab [30], alone or in combination with fludarabine [31] or bendamustine, particularly when associated with chronic lymphocytic leukemia [32], or with bortezomib [33]. Since most RBC destruction does not occur in the spleen, it is not surprising that splenectomy is usually ineffective in CAD [20,21,22].
T Lymphocytes and Immunoregulatory Cytokines in the Pathogenesis of AIHA
The T-cell mediated regulation of the humoral immune system has been shown to play a critical role in loss of self-tolerance in AIHA. In the past years increased in vitro proliferative response of T cells and increased cytotoxicity of natural killer cells against autologous RBCs have been described in AIHA [34,35]. Until a few years ago, CD4+ T helper cells were subdivided into two major T-cell subsets, termed the Th1 and Th2 subsets. More recently, Th17 cells, which are characterized by secretion of IL-17, have been identified as key effectors in the development of many autoimmune diseases, including AIHA [40]. Elevated frequency of Th17 cells and increased IL-17 secretion were found closely correlated with the disease activity in AIHA patients. Consistently, in the rat model of AIHA, the adoptive transfer of Th17 cells heightened anti-RBC antibody responses and increased the onset of AIHA, whereas disease onset was abrogated by in vivo neutralization of IL-17 [36].
Moreover, CD4+ CD25+ Tregs are now recognized as key players in immunologic tolerance and, thereby, in preventing AIHA and other polyclonal autoimmune disorders. Ward et al. [37] firstly described autoantigen-specific, IL-10-secreting Treg clones recovered ex vivo from a patient with AIHA. These clones recognized a peptide, 72H-86L, derived from the Rh RBC autoantigen and expressed different T regulatory markers depending on activation state: high levels of CD25 and LAG-3 when expanding nonspecifically, but FoxP3 after activation by the autoantigen they recognize [38]. More recently, circulating CD4+ Tregs were found reduced in patients with AIHA, along with high levels of IL-10 and IL-12 [39]. It is known that Tregs produce IL-10 and transforming growth factor β (TGF-β), and the IL-10/IL-12 imbalance may thus play an essential role in the onset and/or maintenance of AIHA. Several other abnormalities of immunoregulatory cytokines have been reported in AIHA: IL-1α, IL-2/IL-2R, IL-6, and IL-21 serum levels were found increased; in culture experiments, the Th1 cytokines IL-2 and IL-12 were reported elevated, whereas IFN-γ was found reduced. Moreover, the Th2 cytokines IL-4 and IL-13 were reported increased, together with elevated production of IL-6, IL-10, and IL-17 [36,40,41]. Patients with active hemolysis showed further reduction of IFN-γ and increased secretion of TGF-β. Moreover, addition of IL-4, IL-6, IL-10, IL-13, and TGF-β increased autoantibody secretion in culture [40]. All these data are in favor of a reduced Th1- and a predominant Th2-like profile in the immunopathogenesis of AIHA, in contrast to the prevalent role of CD8+ IFN-γ-secreting cytotoxic T cells in organ-specific autoimmunity (i.e multiple sclerosis, experimental autoimmune encephalomyelitis, type 1 diabetes, and autoimmune thyroiditis). Moreover, elevated levels of TGF-β favor the differentiation of a Th17 subset, which amplifies the pro-inflammatory and autoimmune response. On the contrary, decreased levels of both IFN-γ and Tregs resulted in decreased inhibition of Th2 response, i.e. an amplification of the autoantibody-mediated autoimmune disease (fig. 1). Notwithstanding these cytokine abnormalities, no difference was reported in the phenotypic distributions of polymorphisms of the TNF-α, IL-10, IL-12, and CTLA-4 between AIHA patients and controls. As regards therapy, immunoregulatory cytokines were evaluated after low-dose rituximab (100 mg fixed dose for 4 weekly infusions) [42]. The clinical response was correlated with an increase of IFN-γ, and a reduction of IL-4 and IL-17, suggesting that the drug exerts an immunomodulating activity together with the well-known B-depleting effect. Finally, recent unpublished observations report increased serum values of BAFF and APRIL in AIHA, opening future therapeutic options with monoclonal antibodies directed against this factors, e.g. belimumab. Table 2 summarizes the main immunological abnormalities involved in AIHA pathogenesis.
Fig. 1.
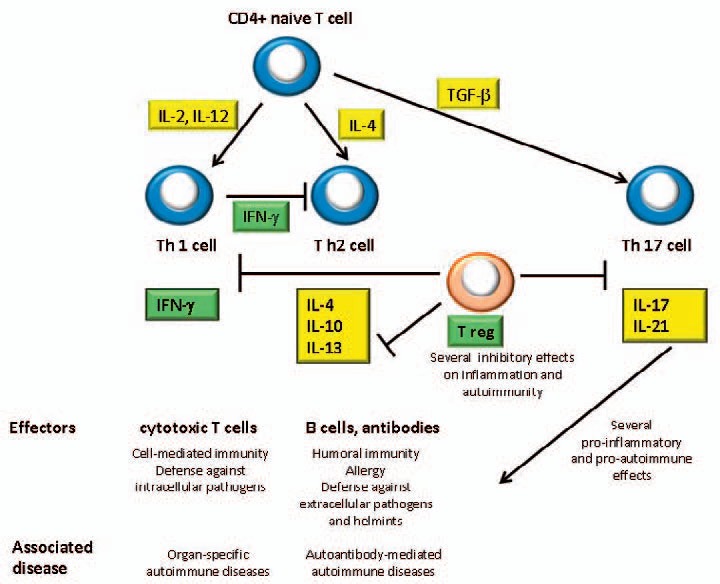
T-lymphocyte subsets and cytokine interactions in AIHA. Cytokines that were found elevated are highlighted in yellow: IL-2 and IL-12, that induce the differentiation of CD4+ naïve T cells into Th1 subset, and IL-4, that promotes Th2 switch. Elevated levels of TGF-β favor the differentiation of Th17 subset, which amplifies the pro-inflammatory and autoimmune response. On the contrary, decreased levels of both IFN-γ and Tregs were found (highlighted in green). The former resulted in decreased inhibition of Th2 response, i.e. an amplification of the autoantibody-mediated autoimmune disease, and the latter may cause a lack of down-regulation of inflammatory and autoimmune pathways.
Table 2.
Main immunological abnormalities involved in AIHA pathogenesis
| Pathogenic mechanism | |
|---|---|
| Autoantibodies | |
| IgG (all subclasses) | antibody-dependent cellular cytotoxicity (ADCC) |
| IgG1 and IgG3 | complement activation |
| IgM | strong complement activation |
| Complement system | membrane attack complex (MAC) |
| phagocytosis of C3b-opsonized erythrocytes | |
| Cellular immunity | increased cytotoxicity of natural killer cells |
| activated autoreactive CD4+ T helper 1 | |
| increased number of Th17 cells | |
| reduced peripheral CD4+ T-reg | |
| Immunoregulatory cytokines | high serum levels of IL-1α, IL-2/IL-2R, IL-6, IL-21 |
| increased production in culture of T helper 1 cytokine IL-2 and IL-12 | |
| reduced production of IFN-γ in culture | |
| high levels of T helper 2 cytokines IL-4 and IL-13 in culture | |
| increased production in culture of IL-6 and IL-10 | |
| elevated levels of IL-17 in culture | |
| increased production in culture of TGF-β in active patients | |
Role of the Spleen in the Pathogenesis of AIHA
The spleen is a major organ for antibody synthesis and immune destruction of IgG-coated erythrocytes. This is the reason why it is considered the most effective conventional second-line treatment of warm AIHA [43]. However, splenectomy is ineffective in about 30% of cases, presumably because there is still an elevated synthesis of autoantibodies in other lymphoid organs, and the liver replaces the spleen's function of RBC destruction. Unfortunately, the extent of splenic sequestration assessed by radiolabeled erythrocytes as well as the disease duration and the response to steroids are not reliable predictors of its effectiveness [44]. The rate of splenectomy is about 10-15% of all AIHA cases but is nowadays decreasing given the availability of equally effective second-line medical treatments, primarily rituximab. It is worth reminding that splenectomy is usually ineffective in CAD since most RBC destruction occurs in the liver [20,21]. An interesting recently reported finding is the emergence of long-lived autoreactive plasma cells in the spleen of primary wAIHA patients treated with rituximab [45]. The spleen was obtained after surgery for relapse or failure of rituximab and analyzed by microscopy and flow cytometry, showing the presence of Bcl6+ germinal-center B cells. The residual population of CD19+ B cells includes non-proliferative memory B cells and plasma cells, which secreted anti-RBC IgG in vitro. Furthermore, the cytokine BAFF was increased in the supernatants of spleen cell cultures, suggesting that it may play a role in the differentiation of short-lived into long-lived plasma cells. The authors conclude that the presence of splenic long-lived autoreactive plasma cells may explain why some wAIHA patients cannot achieve a response after rituximab. The B-cell depletion induced by rituximab itself promotes a suitable environment for the maturation of autoimmune long-lived plasma cells in the spleen. Targeting specifically some factors such as BAFF right after rituximab injection might be an interesting therapeutic option in the future.
Additional Factors That Influence the Clinical Characteristics of AIHA
The major determinants of the clinical severity of AIHA are the autoantibody pathogenicity (related to their class, subclass, thermal amplitude as well as affinity and efficiency in activating complement) and the activity of the reticuloendothelial system. However, there are other factors that influence the clinical picture of AIHA, in particular the efficacy of erythroblastic compensatory response, represented by the absolute number of reticulocytes in the peripheral blood. Reticulocytopenia is not rare in AIHA, particularly in children and in severe cases [46,47,48], and may be due to autoimmune reaction against bone marrow erythroid precursors. There is emerging evidence that the administration of erythropoietin may be useful to overcome the temporary bone marrow failure in compensating hemolysis [49], which is in line with the efficacy of thrombopoietin agonists in primary immune thrombocytopenia [50]. Moreover, the use of erythropoietin may reduce transfusion need and avoid hemolysis related to overtransfusion.
The relative weight of all factors involved in the pathogenesis of AIHA could account for the great heterogeneity of the disease. A recent large multicenter study of 308 AIHA cases showed that 60% were wAIHA, 27% CAD, 8% mixed, and 5% atypical (mostly DAT-negative). A severe presentation (Hb levels < 6 g/dl) was observed in about one-third of cases, mainly warm IgG+C, mixed and atypical forms, with frequently associated reticulocytopenia [10]. As regards therapy, the majority of wAIHA patients received first-line steroid therapy only, whereas patients with mixed and atypical forms had an increased risk of relapse and were more frequently treated with 2 or more therapy lines. Thrombotic events were associated with a severe onset, intravascular hemolysis and previous splenectomy, and predictors of a fatal outcome were severe infections, particularly in splenectomized cases, acute renal failure, Evans syndrome, and multi-treatment (4 or more lines) [10].
Moreover, we recently described 13 acute and very severe cases of AIHA, displaying multi-drug refractoriness and related complications, of whom 7 were fatal. These challenging AIHAs showed a highly unpredictable clinical courses, suggesting that rapidly evolving cases deserve a scrupulous attention by experienced clinicians to avoid a delay in potentially life-saving strategies. In particular, the presence of intravascular hemolysis, hemoglobinuria, DAT positivity for IgG+C, reticulocytopenia, and/or Evans syndrome should alarm clinicians to generously give steroid boluses, to early utilize rituximab, and to consider plasma exchange in critical conditions. We also suggest performing splenectomy soon after rituximab failure, because of the poor prognosis of surgery after multiple immunosuppressive therapies. Moreover, we suggest the use of erythropoietin to reduce overtransfusion and an anti-thrombotic prophylaxis, particularly in splenectomized patients [11]. Currently we use low-molecular-weight heparins at prophylactic doses, i.e. 30-50% of the anticoagulant dose, as long as the hemolytic process is active [51].
An additional factor that influence the clinical picture of AIHA is the association with lymphoproliferative diseases. It is known that about 10% of patients with chronic lymphocytic leukemia and 2-3% of those with non-Hodgkin's and Hodgkin's lymphoma develop AIHA. Moreover, in a population-based study of 86 patients with CAD, a clonal bone marrow lymphoproliferation was demonstrated in 75% and 90% of the patients by bone marrow biopsy and flow cytometry, respectively [20]. Moreover, there is increasing evidence that severe and fatal AIHA may be a complication of hematopoietic stem cell transplantation. A recent large study at King's College Hospital [52] described an overall incidence of 3.6% and an association with hematopoietic stem cell transplantation from unrelated donors. About two-thirds of cases required multiple treatment, but only half achieved complete resolution of AIHA. Ten cases were fatal, and death was directly attributable to AIHA in 4 cases, indicating that post-transplantation AIHA had a higher overall mortality (HR 2.48) than primary AIHA.
Finally, it is worth reminding the passenger lymphocyte syndrome, which is due to the production of antibodies directed against recipient RBCs by passively transferred lymphocytes transfused with the stem cell, most often observed when the donor is group O and the patient group A. A transient immune hemolysis has also been associated with solid organ transplantation, with a risk and degree of hemolysis in proportion to the mass of lymphocytes transplanted being lowest in kidney (antibody in 17%, hemolysis in 9%), next higher in liver (antibody in 40%, hemolysis in 29%), and highest in heart-lung transplants (antibody in 70%, hemolysis in 70%) [53,54].
Conclusions
AIHA is an heterogeneous condition including warm, cold, and mixed forms, along with atypical cases of difficult diagnostic classification (DAT-negative, warm IgM), which are reported with increasing frequency. It can be primary or secondary to lymphoproliferative syndromes, infections, immunodeficiency, and tumors. Moreover, AIHA is described with increasing frequency following hematopoietic stem cell transplantation, and in recipients of solid organ transplants. The clinical presentation of primary AIHA may vary from mild to very severe forms, with a reported mortality of 11% in older series, and of 4% in a more recent studies; secondary forms are usually more severe, with an increased mortality, particularly in post-transplant forms. Several immunologic mechanisms are involved in the pathogenesis of AIHA: autoantibodies, ADCC, phagocytes, B and T lymphocytes, Tregs, cytokines, and the complement system. New insights in this complex scenario may give hints for future therapeutic options specifically aimed at correcting the main pathogenic mechanism involved in the different forms of AIHA.
Disclosure Statement
The author has no conflict of interest to declare.
Acknowledgement
This work was supported by research funding from Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, grant number RC 2014 and by Ministry of Health, grant number RF 2010 convention N. 141/RF-2010-2303934.
References
- 1.Petz LD, Garratty G. Immune Hemolytic Anemias, 2nd ed. Philadelphia, Churchill Livingstone, 2004.
- 2.Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309–317. doi: 10.1111/j.1365-2141.2011.08643.x. [DOI] [PubMed] [Google Scholar]
- 3.Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258–271. doi: 10.1002/ajh.10062. [DOI] [PubMed] [Google Scholar]
- 4.Abbas AK, Lichtman AH, Pillai S. Cellular and Molecular Immunology, Philadelphia, Saunders, 2012.
- 5.McQueen F. A B cell explanation for autoimmune disease: the forbidden clone returns. Postgrad Med J. 2012;88:226–233. doi: 10.1136/postgradmedj-2011-130364. [DOI] [PubMed] [Google Scholar]
- 6.Garratty G, Arndt PA. Drugs that have been shown to cause drug-induced immune hemolytic anemia or positive direct antiglobulin tests: some interesting findings since 2007. Immunohematology. 2014;30:66–79. [PubMed] [Google Scholar]
- 7.Salama A. Drug-induced immune hemolytic anemia. Expert Opin Drug Saf. 2009;8:73–79. doi: 10.1517/14740330802577351. [DOI] [PubMed] [Google Scholar]
- 8.Ahrens N, Pruss A, Kähne A, Kiesewetter H, Salama A. Coexistence of autoantibodies and alloantibodies to red blood cells due to blood transfusion. Transfusion. 2007;47:813–816. doi: 10.1111/j.1537-2995.2007.01194.x. [DOI] [PubMed] [Google Scholar]
- 9.Alijotas-Reig J. Human adjuvant-related syndrome or autoimmune/inflammatory syndrome induced by adjuvants. Where have we come from? Where are we going? A proposal for new diagnostic criteria. Lupus 2015;2015, doi: 10.1177/0961203315579092. [DOI] [PubMed]
- 10.Barcellini W, Fattizzo B, Zaninoni A, Radice T, Nichele I, Di Bona E, Lunghi M, Tassinari C, Alfinito F, Ferrari A, Leporace AP, Niscola P, Carpenedo M, Boschetti C, Revelli N, Villa MA, Consonni D, Scaramucci L, De Fabritiis P, Tagariello G, Gaidano G, Rodeghiero F, Cortelezzi A, Zanella A. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: a GIMEMA study of 308 patients. Blood. 2014;124:2930–2936. doi: 10.1182/blood-2014-06-583021. [DOI] [PubMed] [Google Scholar]
- 11.Fattizzo B, Zaninoni A, Nesa F, Sciumbata VM, Zanella A, Cortelezzi A, Barcellini W. Lessons from very severe, refractory and fatal primary autoimmune hemolytic anemias. Am J Hematol. 2015;90:E149–151. doi: 10.1002/ajh.24047. [DOI] [PubMed] [Google Scholar]
- 12.Arndt PA, Leger RM, Garratty G. Serologic findings in autoimmune hemolytic anemia associated with immunoglobulin M warm autoantibodies. Transfusion. 2009;49:235–242. doi: 10.1111/j.1537-2995.2008.01957.x. [DOI] [PubMed] [Google Scholar]
- 13.Bartolmas T, Salama A. A dual antiglobulin test for the detection of weak or nonagglutinating immunoglobulin M warm autoantibodies. Transfusion. 2010;50:1131–1134. doi: 10.1111/j.1537-2995.2009.02533.x. [DOI] [PubMed] [Google Scholar]
- 14.Bass GF, Tuscano ET, Tuscano JM. Diagnosis and classification of autoimmune hemolytic anemia. Autoimmun Rev. 2014;13:560–564. doi: 10.1016/j.autrev.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 15.Garratty G. Immune hemolytic anemia caused by drugs. Expert Opin Drug Saf. 2012;11:635–642. doi: 10.1517/14740338.2012.678832. [DOI] [PubMed] [Google Scholar]
- 16.Packman CH. Hemolytic anemia due to warm autoantibodies. Blood Rev. 2008;22:17–31. doi: 10.1016/j.blre.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 17.Dacie J. Auto-immune haemolytic anaemia [AIHA]: pathogenesis; in Dacie J (ed): The Haemolytic Anaemias. Vol. 3. London, Churchill Livingstone, 1992, pp. 392-451.
- 18.Kurlander RJ, Rosse WF, Logue GL. Quantitative influence of antibody and complement coating of red cells on monocyte-mediated cell lysis. J Clin Invest. 1978;61:1309–1319. doi: 10.1172/JCI109048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berentsen S, Beiske K, Tjønnfjord GE. Primary chronic cold agglutinin disease: an update on pathogenesis, clinical features and therapy. Hematology. 2007;12:361–370. doi: 10.1080/10245330701445392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berentsen S, Ulvestad E, Langholm R, Beiske K, Hjorth-Hansen H, Ghanima W, Sørbø JH, Tjønnfjord GE. Primary chronic cold agglutinin disease: a population based clinical study of 86 patients. Haematologica. 2006;91:460–466. [PubMed] [Google Scholar]
- 21.Swiecicki PL, Hegerova LT, Gertz MA. Cold agglutinin disease. Blood. 2013;122:1114–1121. doi: 10.1182/blood-2013-02-474437. [DOI] [PubMed] [Google Scholar]
- 22.Ulvestad E, Berentsen S, Mollnes TE. Acute phase haemolysis in chronic cold agglutinin disease. Scand J Immunol. 2001;54:239–242. doi: 10.1046/j.1365-3083.2001.00960.x. [DOI] [PubMed] [Google Scholar]
- 23.Roth A, Huttmann A, Rother RP, Duhrsen U, Philipp T. Long-term efficacy of the complement inhibitor eculizumab in cold agglutinin disease. Blood. 2009;113:3885–3886. doi: 10.1182/blood-2009-01-196329. [DOI] [PubMed] [Google Scholar]
- 24.Hill A, Rother RP, Arnold L, Kelly R, Cullen MJ, Richards SJ, Hillmen P. Eculizumab prevents intravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria and unmasks low-level extravascular hemolysis occurring through C3 opsonization. Haematologica. 2010;95:567–573. doi: 10.3324/haematol.2009.007229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi J, Rose EL, Singh A, Hussain S, Stagliano NE, Parry GC, Panicker S. TNT003, an inhibitor of the serine protease C1s, prevents complement activation induced by cold agglutinins. Blood. 2014;123:4015–4022. doi: 10.1182/blood-2014-02-556027. [DOI] [PubMed] [Google Scholar]
- 26.Wouters D, Stephan F, Strengers P, de Haas M, Brouwer C, Hagenbeek A, van Oers MH, Zeerleder S. C1-esterase inhibitor concentrate rescues erythrocytes from complement-mediated destruction in autoimmune hemolytic anemia. Blood. 2013;121:1242–1244. doi: 10.1182/blood-2012-11-467209. [DOI] [PubMed] [Google Scholar]
- 27.Risitano AM, Notaro R, Pascariello C, Sica M, del Vecchio L, Horvath CJ, Fridkis-Hareli M, Selleri C, Lindorfer MA, Taylor RP, Luzzatto L, Holers VM. The complement receptor 2/factor H fusion protein TT30 protects paroxysmal nocturnal hemoglobinuria erythrocytes from complement-mediated hemolysis and C3 fragment. Blood. 2012;119:6307–6316. doi: 10.1182/blood-2011-12-398792. [DOI] [PubMed] [Google Scholar]
- 28.Risitano AM, Ricklin D, Huang Y, Reis ES, Chen H, Ricci P, Lin Z, Pascariello C, Raia M, Sica M, Del Vecchio L, Pane F, Lupu F, Notaro R, Resuello RR, DeAngelis RA, Lambris JD. Peptide inhibitors of C3 activation as a novel strategy of complement inhibition for the treatment of paroxysmal nocturnal hemoglobinuria. Blood. 2014;123:2094–2101. doi: 10.1182/blood-2013-11-536573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harboe M, van Furth R, Schubothe H, Lind K, Evans RS. Exclusive occurrence of K chains in isolated cold haemagglutinins. Scand J Haematol 1965:2:259-266. [DOI] [PubMed]
- 30.Berentsen S, Tjønnfjord GE. Diagnosis and treatment of cold agglutinin mediated autoimmune hemolytic anemia. Blood Rev. 2012;26:107–115. doi: 10.1016/j.blre.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 31.Berentsen S, Randen U, Vågan AM, Hjorth-Hansen H, Vik A, Dalgaard J, Jacobsen EM, Thoresen AS, Beiske K, Tjønnfjord GE. High response rate and durable remissions following fludarabine and rituximab combination therapy for chronic cold agglutinin disease. Blood. 2010;116:3180–3184. doi: 10.1182/blood-2010-06-288647. [DOI] [PubMed] [Google Scholar]
- 32.Quinquenel A, Willekens C, Dupuis J, Royer B, Ysebaert L, De Guibert S, Michallet AS, Feugier P, Guieze R, Levy V, Delmer A. Bendamustine and rituximab combination in the management of chronic lymphocytic leukemia-associated autoimmune hemolytic anemia: a multicentric retrospective study of the French CLL intergroup (GCFLLC/MW and GOELAMS) Am J Hematol. 2015;90:204–207. doi: 10.1002/ajh.23909. [DOI] [PubMed] [Google Scholar]
- 33.Carson KR, Beckwith LG, Mehta J. Successful treatment of IgM-mediated autoimmune hemolytic anemia with bortezomib. Blood. 2010;115:915. doi: 10.1182/blood-2009-09-242917. [DOI] [PubMed] [Google Scholar]
- 34.Fagiolo E. Immunological tolerance loss vs. erythrocyte self antigens and cytokine network disregulation in autoimmune hemolytic anaemia. Autoimmun Rev. 2004;3:53–59. doi: 10.1016/S1568-9972(03)00085-5. [DOI] [PubMed] [Google Scholar]
- 35.Gilsanz F, de La Serna J, Moltó L, Alvarez-Mon M. Hemolytic anemia in chronic large granular lymphocytic leukemia of natural killer cells: cytotoxicity of natural killer cells against autologous red cells is associated with hemolysis. Transfusion. 1996;36:463–466. doi: 10.1046/j.1537-2995.1996.36596338025.x. [DOI] [PubMed] [Google Scholar]
- 36.Xu L, Zhang T, Liu Z, Li Q, Xu Z, Ren T. Critical role of Th17 cells in development of autoimmune hemolytic anemia. Exp Hematol. 2012;40:994–1004. doi: 10.1016/j.exphem.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 37.Ward FJ, Hall AM, Cairns LS, Leggat AS, Urbaniak SJ, Vickers MA, Barker RN. Clonal regulatory T cells specific for a red blood cell autoantigen in human autoimmune hemolytic anemia. Blood. 2008;111:680–687. doi: 10.1182/blood-2007-07-101345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 39.Ahmad E, Elgohary T, Ibrahim H. Naturally occurring regulatory T cells and interleukins 10 and 12 in the pathogenesis of idiopathic warm autoimmune hemolytic anemia. J Investig Allergol Clin Immunol. 2011;21:297–304. [PubMed] [Google Scholar]
- 40.Barcellini W, Clerici G, Montesano R, Taioli E, Morelati F, Rebulla P, Zanella A. In vitro quantification of anti-red blood cell antibody production in idiopathic autoimmune haemolytic anaemia: effect of mitogen and cytokine stimulation. Br J Haematol. 2000;111:452–460. doi: 10.1046/j.1365-2141.2000.02380.x. [DOI] [PubMed] [Google Scholar]
- 41.Toriani-Terenzi C, Fagiolo E. IL-10 and the cytokine network in the pathogenesis of human autoimmune hemolytic anemia. Ann N Y Acad Sci. 2005;1051:29–44. doi: 10.1196/annals.1361.044. [DOI] [PubMed] [Google Scholar]
- 42.Barcellini W, Zaja F, Zaninoni A, Imperiali FG, Battista ML, Di Bona E, Fattizzo B, Consonni D, Cortelezzi A, Fanin R, Zanella A. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biologic studies. Blood. 2012;119:3691–3697. doi: 10.1182/blood-2011-06-363556. [DOI] [PubMed] [Google Scholar]
- 43.Lechner K, Jager U. How I treat autoimmune hemolytic anemias in adults. Blood. 2010;16:1831–1838. doi: 10.1182/blood-2010-03-259325. [DOI] [PubMed] [Google Scholar]
- 44.Crowther M, Chan YL, Garbett IK, Lim W, Vickers MA, Crowther MA. Evidence-based focused review of the treatment of idiopathic warm immune hemolytic anemia in adults. Blood. 2011;118:4036–4040. doi: 10.1182/blood-2011-05-347708. [DOI] [PubMed] [Google Scholar]
- 45.Mahévas M, Michel M, Vingert B, Moroch J, Boutboul D, Audia S, Cagnard N, Ripa J, Menard C, Tarte K, Mégret J, Le Gallou S, Patin P, Thai L, Galicier L, Bonnotte B, Godeau B, Noizat-Pirenne F, Weill JC, Reynaud CA. Emergence of long-lived autoreactive plasma cells in the spleen of primary warm auto-immune hemolytic anemia patients treated with rituximab. J Autoimmun 2015; doi: 10.1016/j.jaut.2015.05.006. [DOI] [PubMed]
- 46.Conley CL, Lippman SM, Ness P. Autoimmune hemolytic anemia with reticulocytopenia. A medical emergency. JAMA. 1980;244:1688–1690. [PubMed] [Google Scholar]
- 47.Liesveld JL, Rowe JM, Lichtman MA. Variability of the erythrocyte response in autoimmune hemolytic anemias: analysis of 109 cases. Blood. 1987;69:820–826. [PubMed] [Google Scholar]
- 48.Aladjidi N, Leverger G, Leblanc T, Picat MQ, Michel G, Bertrand Y, Bader-Meunier B, Robert A, Nelken B, Gandemer V, Savel H, Stephan JL, Fouyssac F, Jeanpetit J, Thomas C, Rohrlich P, Baruchel A, Fischer A, Chêne G, Perel Y, Centre de Référence National des Cytopénies Auto-immunes de l'Enfant (CEREVANCE) New insights into childhood autoimmune hemolytic anemia: a French national observational study of 265 children. Haematologica. 2011;96:655–663. doi: 10.3324/haematol.2010.036053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arbach O, Funck R, Seibt F, Salama A. Erythropoietin may improve anemia in patients with autoimmune hemolytic anemia associated with reticulocytopenia. Transfus Med Hemother. 2012;39:221–223. doi: 10.1159/000339260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuter DJ. The biology of thrombopoietin and thrombopoietin receptor agonists. Int J Hematol. 2013;98:10–23. doi: 10.1007/s12185-013-1382-0. [DOI] [PubMed] [Google Scholar]
- 51.Akl EA, Ramly EP, Kahale LA, Yosuico VE, Barba M, Sperati F, Cook D, Schünemann H. Anticoagulation for people with cancer and central venous catheters. Cochrane Database Syst Rev. 2014;10:CD006468. doi: 10.1002/14651858.CD006468.pub5. [DOI] [PubMed] [Google Scholar]
- 52.Wang M, Wang W, Abeywardane A, Adikarama M, McLornan D, Raj K, de Lavallade H, Devereux S, Mufti GJ, Pagliuca A, Potter VT, Mijovic A. Autoimmune hemolytic anemia after allogeneic hematopoietic stem cell transplantation: analysis of 533 adult patients who underwent transplantation at King's College Hospital. Biol Blood Marrow Transplant. 2015;21:60–66. doi: 10.1016/j.bbmt.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 53.Hoffman PC. Immune hemolytic anemia - selected topics. Hematology Am Soc Hematol Educ Program 2009:80-86. [DOI] [PubMed]
- 54.ElAnsary M, Hanna MO, Saadi G, ElShazly M, Fadel FI, Ahmed HA, Aziz AM, ElSharnouby A, Kandeel MM. Passenger lymphocyte syndrome in ABO and Rhesus D minor mismatched liver and kidney transplantation: A prospective analysis. Hum Immunol. 2015;76:447–452. doi: 10.1016/j.humimm.2015.03.006. [DOI] [PubMed] [Google Scholar]