

Abstract
Colorectal carcinoma (CRC) is one of the most frequent cancers. Along the surface of the large bowel, several foci of CRC may appear simultaneously or over the time. The development of at least two different tumours has been defined as multiple primary CRC (MPCRC): When more than one tumour is diagnosed at the same time, it is known as synchronous CRC (SCRC), while when a second neoplasm is diagnosed some time after the resection and/or diagnosis of the first lesion, it is called metachronous CRC (MCRC). Multiple issues can promote the development of MPCRC, ranging from different personal factors, such as environmental exposure, to familial predisposition due to hereditary factors. However, most studies do not distinguish this dichotomy. High- and low-pentrance genetic variants are involved in MPCRC. An increased risk for MPCRC has been described in Lynch syndrome, familial adenomatous polyposis, and serrated polyposis. Non-syndromic familial CRCs should also be considered as risk factors for MPCRC. Environmental factors can promote damage to colon mucosae that enable the concurrence of MPCRC. Epigenetics are thought to play a major role in the carcinogenesis of sporadic MPCRC. The methylation state of the DNA depends on multiple environmental factors (e.g., smoking and eating foods cooked at high temperatures), and this can contribute to increasing the MPCRC rate. Certain clinical features may also suggest individual predisposition for MPCRC. Different etiopathogenic factors are suspected to be involved in SCRC and MCRC, and different familial vs individual factors may be implicated. MCRC seems to follow a familial pattern, whereas individual factors are more important in SCRC. Further studies must be carried out to know the molecular basis of risks for MPCRC in order to modify, if necessary, its clinical management, especially from a preventive point of view.
Keywords: Multiple primary colorectal cancer, Synchronous colorectal cancer, Metachronous colorectal cancer, Chromosomal instability, Microsatellite instabillity, CpG island methylator phenotype
Core tip: Multiple primary colorectal cancer (MCRC), both Synchronous and Metachronous tumours, is not deeply studied yet, and also has a great clinical impact. Both genetic and environmental factors may affect in the development of MCRC, collaborating in promoting different foci of dysplasia. In general terms, Metachronous forms are mainly related to family factors whereas Synchronous tumours are linked with individual factors. With the exception of cases of hereditary forms of colorectal carcinoma (CRC), the others appears without a well-known molecular basis, and maybe different from sporadic colorectal cancer. For all these reasons, we present a review focused on the state of the art of these particular forms of CRC.
INTRODUCTION
The colon is one of the localizations where carcinomas most frequently occur. The large bowel mucosa has a great extension. Thus, high- and low-penetrance genetic variants as well as environmental exposure all affect a large field, where several foci of colorectal cancer (CRC) may appear along the surface simultaneously or over time. The development of at least two different tumours has been defined as multiple primary CRC (MPCRC); when more than one tumour is diagnosed at the same time, this is known as synchronous CRC (SCRC), while when a second neoplasm is diagnosed some time after the resection and/or diagnosis of the first lesion, it is called metachronous CRC (MCRC). Initial studies did not distinguished between both concepts, and Moertel et al[1] were the first to describe in 1958 the currently most used criteria. MCRC was defined as a pathologically proven adenocarcinoma, separated from the line of anastomosis, different from recurrence, and diagnosed at a minimal interval of 6 mo after the initial CRC; CRCs diagnosed within 6 mo after the initial diagnosis were considered as SCRC[1].
MPCRCs make up 5%-10% of all CRCs. Estimations of the risk of developing MCRC vary widely in the literature, and range from 1.5% to 9%[2,3], depending on the time interval of the series. Recent series describe a risk of MCRC of 3.4%, 10 years after the first diagnosis[4]. On the other hand, large series of CRC estimate a prevalence of SCRC between 3.1% and 3.9%[5,6].
Multiple primary tumours usually arise on a common etiologic substrate, either genetic or environmental. Recurrence after endoscopic polypectomy is considered a risk factor for the development of multicentric CRC. Different adenoma features such as size, villous component, and number and location of polyps, may predict a high risk of metachronous lesions[7]. Nonetheless, recent findings in molecular colorectal carcinogenetics have provided evidence that chromosomal instability, microsatellite instability, and gene methylation are involved in various predisposing lesions or factors for SCRC and MCRC.
As mentioned before, multiple factors can promote the development of MPCRC ranging from different personal factors such as environmental exposure to familial predisposition due to inheritance. However, most studies do not distinguish this dichotomy. There are different entities that increase the risk of MPCRC. First, there are hereditary CRC syndromes (Lynch syndrome, Familial Adenomatous Polyposis), which present germline mutations and promote the development of several lesions overtime[8]. On the other hand, there are diseases and conditions that affect a large area of the colonic mucosa during specific periods of time and promote the formation of several foci of dysplasia, such as inflammatory bowel disease[9]. However, the origin of most of the cases of MPCRCs is still unclear; nowadays, well-defined factors only explain about 10% of SCRCs[8]. Perhaps the basis of most MPCRCs should be described as a situation in which one of the two main factors (genetic predisposition or environmental influence) predominates, or in which these factors are balanced.
There are two main points in which MPCRC stands out. Firstly, tumour multiplicity provides a good model to examine common molecular alterations and, more specifically, a potential field effect[10]. Secondly, and possibly more importantly, there is the possibility of prevention within this subset of CRC, i.e., the existence of different prophylactic actions such as extensive surgery or chemopreventive treatment[11,12]. As is well known, the extension of surgical resection can be influenced by the presence, or at least the risk, of SCRC or MCRC. Moreover surveillance programs can be tailored if risk factors of MCRC are identified in order to reduce morbidity and even mortality[4,13].
Below we give an overview of the current of knowledge of both hereditary and environmental factors that influence SCRC and MCRC, and the importance of gaining more specific knowledge of these factors is adressed. In Table 1 publications are summarized that show the prevalence of and risk factors for MPCRC. In Table 2 publications are summarized that address the main molecular features analysed for MPCRC.
Table 1.
Different studies about the prevalence of multiple primary colorectal carcinomaand the main clinical risk factors
| Ref. | Study design | Prevalence of MPCRC No. of cases (% of global) | Risk factors for MPCRC |
| [4] | 10283 CRC patients. Study of MCRC vs solitary CRC | 135 (1.3) | Previous SCRC. OR = 3.4, 95%CI (1.9-5.9) Less frequent in rectum. OR = 0.3, 95%CI (0.1-0.6) Not associated with the development of MCRC: Sex, age, TNM stage, or grade of differentiation of the initial CRC |
| [2] | 1298 CRC patients. Study of MPCRC features | 53 (4) MPCRC 33 (2.5) SCRC 20 (1.5) MCRC | Lynch > sporadic (P < 0.001) MCRC (5.8% vs 1.3%) SCRC (5.8% vs 2.4%) |
| [8] | 1793 CRC patients. Study of SCRC features | 102 (3.6) SCRC | Frequencies of predisposing disease in SCRC patients: 5% FAP (5) 6% SP (6) 2% UC (2) |
| [9] | 1537 CRC patients 69 FAP 780 UC 685 de novo CRC | Prevalence of SCRC in special populations: 21% of CRC in FAP 18% of CRC in UC 2.5% of sporadic CRC | |
| [37] | 382 CRC patients Study of SCRC vs solitary CRC | 28 (7.3%) SCRC 208 (54.5%) synchronous adenomas | Male gender: OR = 1.97, 95%CI (1.13-3.45) Age ≥ 59 yr: OR = 2.57, 95%CI (1.54-4.29) History of adenomas: OR= 3.04, 95%CI: 1.04-8.85 Obstructive tumours: OR = 0.48, 95%CI: 0.27-0.85 |
| [32] | 15562 CRC cases. SCRC vs solitary CRC | 596 (3.8%) SCRC | Male gender: OR = 1.41, 95%CI (1.19 -1.68) Adenomas present: OR = 2.02, 95%CI (1.69-2.41) Aged over 75: OR = 1.31, 95%CI (1.08 -1.59) |
| [12] | 1522 CRC patients. Study of SCRC vs solitary CRC | 27 (1.8%) SCRC | Male gender SCRC (70%); solitary CRC (56%), P = 0.001. Personal history of adenoma SCRC (4%); solitary CRC (1%), P = 0.001 Right sided tumour location SCRC (32%); solitary CRC (25%), P = 0.003 |
| [57] | 382 patients with CRC. Study of MCRC features | 28 (7.3%) | Statistical differences: Older than 59 years OR = 2.57, 95%CI (1.54-4.29) History of adenoma OR = 3.04, 95%CI (1.04-8.85) Obstructive CRC OR = 0.48, 95%CI (0.27-0.85) Alcohol univariate analysis P = 0.006, no significance in multivariate analysis No statistical significance: Personal history of other tumours History of cancer in first-degree family members Revised Bethesda criteria (at least one criterion) BMI Predominant symptom Predominant localitation |
| [13] | 18782 CRC cases MPCRC features | 134 (0.71%) SCRC 300 (1.60%) MCRC | SCRC Men: OR = 1.45; 95%CI: 1.02-2.06 Age older than 65: OR = 1.50, 95%CI (1.02-2.21) Located in proximal colon: OR = 1.70, 95%CI (1.20-2.41) Risk of CRC of first-degree relatives of SCRC patients (OR= 1.86; 95%CI: 1.37-2.53) MCRC: (OR = 2.34; 95%CI: 1.62–3.36) Solitary CRC (OR = 1.75; 95%CI: 1.63-1.88) |
CRC: Colorectal cancer; MCRC: Metachronous CRC; SCRC: Synchronous CRC; SSAs: Sessile serrated adenomas; UC: Ulcerative colitis; SP: Serrated polyposis; BMI: Body mass index; HR: Hazard ratio; OR: Odds ratio.
Table 2.
Summary of studies about the prevalence of multiple primary colorectal carcinoma and the main molecular features of synchronous and metachronous colorectal carcinoma
| Ref. | Study design | Prevalence of MPCRC (% of global) | Risk factors for MPCRC | Carcinogenetic pathways |
| [10] | Solitary (29) MPCRC (12) Study of MPCRC features | No differences: Age Gender Body mass index Tumour location History of CRC of MSI | CIMP-high 17.2% solitary vs 66.7% MPCRC P = 0.004 | |
| [36] | 57 MPCRC Comparison of methylation status of solitary CRC vs MPCRC 57 | Higher methylation for p14 MGMT in MPCRC P < 0.05 Correlations: MINT1 (r = 0.8) p16 (r = 0.8), MLH1 (r = 0.9) MGMT (r = 0.6) at the same site | ||
| [16] | 4760 CRC patients Study of SCRC vs solitary CRC | 58 (1.2%) SCRC: 42 (72%) sporadic 4 (7%) UC 8(14%)Lynch 1 (2%) FAP 3 (5%) SP | Older patients (P = 0.001) Right colon (P = 0.0003) Synchronous polyps (P = 0.0001) Classical adenoma 47% vs 12% SSAs 16% vs 0% | (MSI-H) 36% vs 12%; (P = 0.0005) 92% if SSA precursor |
| [17] | 2884 patients SCRC vs solitary CRC | 77 (2.7%) SCRC | 21 (27%) had a family history of Lynch | 54 (32%) MSI-H (> in women and elderly) congruence (MSS/MSI) Yes: 67 patients (87%) No: 10 patients (13%) |
| [30] | 2884 CRC Study of MPCRC methylation state in SCRC vs MCRC | 33 (1.1%) MCRC 77 (2.6%) SCRC | MSI-H MCRC were younger (64 vs 76 years, P =0.01) | MSI-H tumors in 12 (36%) MCRC 29 (38%) SCRCP Promoter methylation 50% MCRC 83% SCRCP P = 0.03 |
| [35] | 2,068 CRC patients SCRC vs solitary CRC | 47 (2.3%) SCRC | Mean age 68.9 vs 65.5 (P =0.016) No difference: Family history of CRC BMI | MSI-high (P = 0.037). > BRAF (P = 0.0041) > CIMP-high (P = 0.013) Correlation pairs LINE-1 (r = 0.82; P = 0.0072) CpG islands (P < 0.0001) |
CRC: Colorectal cancer; MCRC: Metachronous CRC; SCRC: Synchronous CRC; SSAs: Sessile serrated adenomas; UC: Ulcerative colitis; SP: Serrated polyposis; BMI: Body mass index; HR: Hazard ratio; OR: Odds ratio; MSI: Microsatellite instability; FAP: Familial adenomatous polyposis.
Familial predisposition
There is much evidence that some MPCCRs are linked with a hereditary pattern. Lynch syndrome (LS), also named hereditary nonpolyposis colon cancer (HNPCC), is the most common hereditary CRC syndrome. It has an autosomal dominant hereditary pattern, and it is defined by the presence of a germ-line mutation in one of the four DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6 and PMS2)[14]. MPCRC tends to appear more frequently among patients with LS compared with patients with sporadic CRC.
Win et al[15] investigated the MCRC risk in a retrospective cohort of 79 patients with previous rectal adenocarcinoma and with germline MMR gene mutations. Twenty-seven per cent of MMR mutation carriers were diagnosed with MCRC. Cumulative risk of MCRC was 19% (95%CI: 9%-31%) at 10 years, 47% (95%CI: 31%-68%) at 20 years, and 69% (95%CI: 45%-89%) at 30 years after surgical resection. In spite of colonoscopy surveillance, 22% of MCRC cases were diagnosed at stage II, and 6% at stage III. Fante et al[2] investigated a subset of 1298 registered patients with CRC, and 53 patients (4.1%) were identified with MPCRC. The frequency in LS patients rose to 11.5% (5.8% SCRC, 5.8% MCRC), whereas the frequency in sporadic CRC was 3.6%, the most commonly reported frequency in most studies[4]. Differences were greater when only metachronous lesions were compared; these were four times more frequent in LS cases (5.8% vs 1.3%; P < 0.001)[2]. Hu et al[16] reported similar findings: in a study that included 54 SCRC cases, more than 14% were in the context of LS. Moreover, 27.6% of patients with SCRC had first-degree relatives with different LS-related cancers[17].
Being affected by multiple primary neoplasms is a criterion for screening for LS according to the revised Bethesda guidelines[18]. MPCRC is a very important parameter in PREMM126, a computer model to estimate the overall cumulative probability of having a mismatch repair gene mutation. Males under 78 years old with MPCRC have at least a 5% cumulative risk of an MMR gene mutation, calculated by the PREMM126 model, independent of information regarding their relatives. Five per cent is the cut-off recommended for referral for genetic evaluation and/or for considering molecular testing of tumour samples for microsatellite instability (MSI) or immunohistochemistry[19]. As is well known, identifying LS is essential in order to intensify screening colonoscopy.
Familial adenomatous polyposis (FAP) is an autosomal dominant disorder showing mutations in the adenomatous polyposis coli (APC) gene. It is characterized by the presence of hundreds to thousands of adenomas that can become malignant. Hu et al[16] reported that 2% of patients with SCRC suffer from FAP. However, the frequency of SCRC identified in that series seems to be greater than the current findings in global series of CRC, where FAP represents less than 1% of all CRCs. The common surgical management of FAP includes prophylactic resection of the entire colon. When a more conservative approach is taken (for example when the rectum is preserved, or in attenuated forms of FAP), strict surveillance should be done in order to avoid the development of MCRC[20].
Different studies have suggested an association of MPCRC with the serrated pathway, as a consequence of a field defect arising in the mucosa of patients with large sessile serrated adenoma (SSA), resulting in increased risk for SCRC[16]. Serrated polyposis syndrome refers to a condition characterized by multiple, large and proximal hyperplastic polyps[21]. Although there are still many doubts about the significance and the pathogenesis of serrated polyposis, the main consensus criteria for the diagnosis of serrated polyposis include the possibility of a familial pattern. According to the World Health Organization[22], serrated polyposis is diagnosed if any of the following criteria is met: (1) at least five serrated polyps proximal to the sigmoid colon, two or more of which have a diameter of greater than 10 mm; (2) any number of serrated polyps proximal to the sigmoid colon in an individual who has a first-degree relative with serrated polyposis; and (3) more than 20 serrated polyps of any size but distributed throughout the colon.
A study of 58 patients with SCRC identified 13 patients whose tumours were derived from SSA (SSA-associated SCRC): Three of them (23%) (SSA-associated SCRC) met criteria for serrated polyposis[16]. Moreover, a family history of CRC in patients with serrated polyposis syndrome has been reported in different studies, ranging from 33% to 59% of patients[21,23,24]. Nevertheless, no germline mutation associated with serrated polyposis syndrome has yet been identified. The main carcinogenetic pathway related with serrated adenocarcinoma is the DNA methylation pathway CpG Island Methylation Phenotype (CIMP). CIMP is often related with environmental exposure, and therefore some authors hypothesize about the possibility of an inherited abnormality of epigenetic regulation[25]. Such an abnormality would lead into the accumulation of somatic methylation events in tumour suppressor genes and would synergize with somatic oncogenic activation of BRAF, resulting in the development of premalignant serrated lesions. Other investigations found a weak association with mutations in MUTYH or MBD4 genes, especially when adenomas and serrated polyps are simultaneously present[23]. The presence of conventional adenomas in serrated polyposis is also associated with an increased risk of CRC[26].
Excluding the high-penetrance inherited CRC syndromes, around 10% of CRC patients have a family history of the disease. These non-syndromic familial CRCs have been defined as “apparently sporadic forms of the disease that occur in families more often than expected by chance”[27,28]. Samadder et al[13] found an increased risk of CRC in first-degree relatives of patients with MPCRC, whereas a relative of a sporadic CRC patient has an increased risk of 1.75 (OR = 1.75, 95%CI: 1.63-1.88) compared with no affected relatives: first-degree relatives of SCRC patients had an about 1.9-fold increased risk of CRC (OR = 1.86, 95%CI: 1.37-2.53), and first-degree relatives of MCRC patients had a 2.5-fold increased risk (OR = 2.34, 95%CI: 1.62-3.36). The differences in risk between solitary CRC and MCRC were statistically significant (OR = 1.41, 95%CI: 1.05-1.91)[13].
When talking about familial predisposition in MPCRC, differences between SCRC and MCRC should be taken into account. Whereas MCRC can be facilitated by some inherited predisposition, with a continuous possibility during the entire lifespan of a person to develop a carcinoma, SCRC appears more likely to be the result of damage through some environmental factor during a specific period of time that enables the concurrence of two tumours at the same time. SCRC tends to be diagnosed at an advanced age compared with sporadic CRC, whereas MCRC tends to appear at an earlier age.This also supports the predominantly familial pattern of MCRC[13]. We have explained above how the risk of having an MCRC in LS is higher than that of having an SCRC[2]. The pathological features of MCRC also suggest a hereditary pattern. It is known that mucinous adenocarcinoma is a typical histologic feature of LS-associated CRC: In a large series of 102 patients with SCRC and 56 patients with MCRC, no differences in the incidence of mucinous carcinomas between SCRC and solitary CRC were found; nevertheless they are more common in metachronous forms[29].
Another factor supporting the hypothesis of the weaker relationship of SCRC with hereditary forms is the higher relation with sporadic MSI forms. Compared with solitary CRC, where about 10%-20% of patients show high MSI, the two types of MPCRC show more than 30% of MSI tumours. In some series, MSI was present in 36% of MCRC cases and in 38% of SCRC cases[30]. Up to 81% per cent of those SCRC cases lose MLH1 protein expression because of promoter hypermethylation; thus, the frequency in SCRC is twice as high as in MCRC (81% vs 41%). Both multiple forms associated with MLH1 promoter hypermethylation were more likely to be diagnosed at an older age and showed less frequently CRC in a first-degree relatives[30]. On the other hand, the high prevalence of MSI tumours also explains the most frequent distribution of MPCRC: both SCRC and MCRC used to be located in the proximal colon[31].
Individual predisposition
The screening guidelines for LS list the presence of SCRC as a risk factor. However, LS-associated SCRC accounts for a minority of all SCRC cases. Hu et al[16] described LS only in the 38% of SCRC cases. Thus, apart from the already known hereditary forms of CRC, there is an important proportion of MPCRC without a clear basis of inheritance. Some clinical features may suggest individual predisposition for MPCRC. The risk of sporadic CRC increases with age, and therefore an increased prevalence of multiple tumours would be expected in the elderly population. As mentioned before, SCRCs are identified in older patients compared with solitary CRC (median age 70 years vs 60 years; P = 0.001)[16], and other studies support these findings[5,13]. This association between SCRC and age can be explained by cumulative environmental damage, because hereditary patterns usually lead to an early onset of the disease. Nevertheless, other studies did not find any differences regarding to the age at presentation[8,32].
As we described above, MSI is associated with SCRC. Although it is frequent in LS, it is not exclusive of it, and MSI is identified in about 10%-15% of sporadic CCR cases[33]. Several small tumour-based studies have found that MSI and abnormal methylation (CIMP-High and BRAF mutations) were more frequent in sporadic SCRC and MCRC[13]. Overall chromosomal instability, MSI, and CIMP are implicated in developing various predisposing lesions for MPCRC. In a molecular study of SCRC, Lam et al[8] found a 60% positive status for chromosomal instability; 10% was MSI and CIMP-0, whereas 30% was MSI and CIMP positive. Hu et al[16] also reported similar findings: They described a signinficantly increased rate of MSI, up to 36% (21/58) vs 12% (13/109) in solitary tumours (P = 0.0005), and they also found a difference of 20% in the prevalence of precursor SSA: 22% (13/58) in SCRCs vs 2% in solitary CRCs (2/109)) (P = 0.0001). Along these same lines, some authors point out that the pathway by which MPCRC shows MSI differs from the classical mutations of LS[34]. Epigenetics are thought to play a major role in the carcinogenesis of sporadic MPCRC. Between 31% and 62% of SCRC tumours have lost of MLH1 protein expression because of the hypermethylation of the promotor[16,30]. SSAs are the most common lesions of the methylation pathway, as Hu et al[16] showed that most (13/21, 62%) of the MSI SCRCs were associated with precursor SSA lesions and were apparently sporadic, with concurrent loss of MLH1 and PMS2 expression, and positive for the BRAF V600E mutation. They also found that 22% of SCRCs developed from an SSA, as opposed to only the 2% of solitary CRCs (P = 0.0001). In agreement with these findings, Gonzalo et al[10] found a close association between MPCRC and CIMP, identifying 102 CpG sites that showed significant hypermethylation in multiple tumours compared with solitary CRCs: 66.6% of MPCRCs were CIMP-high, whereas 5 of 29 (17.2%) solitary tumours showed CIMP (P = 0.004). Nosho et al[35] also described differences in methylation-related features in MPCRC: They found alterations in six of the eight CIMP methylation panel markers, with higher methylation levels of the long interspersed nuclear element 1 (LINE-1) and a higher frequency of BRAF mutations. Konishi et al[36] analysed the methylation status of a limited number of makers in 57 MPCRCs and 69 solitary CRCs, and found that the methylation status of p14 and MGMT was significantly higher in multiple tumours; in addition, they described concordant methylation within tumour pairs at the same colonic site. Nosho et al[35] also found CIMP correlation between tumours at the same colonic site, and no correlation was found for pairs of tumours located in discordant locations. In summary, as the methylation state depends on multiple environmental factors (smoking, eating foods cooked at high temperatures), it may contribute to increasing the rate of MPCRC.
There are several studies that correlate environmental exposure with increased risk of MPCRC. Borda et al[37] studied possible risk factors for developing this entity, and they proposed alcohol intake as a risk factor for MCRC and SCRC. The relation between CRC and alcohol intake has been described before, even with moderate intake (about 20-40 g/d)[38]. Moreover, a cumulative alcohol intake of more than 9800 g, calculated as weekly average alcohol intake multiplied by years of drinking, has been described as a risk factor for SCRC[39]. Another study showed that risk of MCRC was not associated with gender, age at diagnosis, country of recruitment, cigarette smoking status, maximum dimension of primary tumour, and histological grade of the rectal cancer[15]. This study associated the risk of MCRC exclusively with the higher stage at diagnosis of the tumour (HR = 6.14, 95%CI: 1.21–13.14; P = 0.03) and a prior diagnosis of SCRC (HR = 11.54, 95%CI: 10.6-12.5; P = 0.04). Tobacco smoke contains a variety of genotoxic substances, including polycyclic aromatic hydrocarbons, nitrosamines, and heterocyclic and aromatic amines. Compounds in cigarette smoke activate the aromatic hydrocarbon receptor which can lead to DNA methylation[40]. Samowitz et al[41] described how heavy smoking could be related with CIMP and BRAF mutation, but no differences were found in MSI rate, although other authors did find an association[42].
SCRC is more frequently associated with adenomas than solitary CRC. Nowadays, precursor entities such as multiple serrated sessile polyposis are more commonly related with SCRC[43]. Tobacco can lead to an MPCRC because an increased number of polyps, either adenomas or serrated polyps, has been found in patients with direct exposure to tobacco[44]. However, a special pathogenic role has been described for serrated polyposis[21]. The tendency for these lesions to be multiple, when there is an association with smoking, and the frequency of BRAF mutation and CIMP, point to a defect that may result from interactions between environment with a low penetrance genetic predisposition[25].
As we have seen, tobacco and alcohol consumption have been adressed in several studies, and both are considered risk factors for CRC. When Borda et al[37] investigated both habits in relation with SCRC they found that both SCRC and tobacco were associated with male gender, but they did not find a statistically significant difference using multivariate analysis[37]. A study by Piñol et al[12] supports the observation that SCRC is independently associated with gender (P < 0.001). Another risk factor studied by Borda et al[37] was body mass index (BMI), and they found that a BMI of less than 21 was a protective factor for MPCRC.
Inflammatory bowel disease (IBD) can increase the risk of CRC. The prevalence of CRC in ulcerative colitis (UC) is 3.7%, increasing to 5.4% in those cases with pancolitis[45]. Continuous inflammation induces molecular damage and causes a hypermethylation state of the colon mucosa that can promote carcinogenesis. Colonoscopy surveillance may be carried out in order to diagnose the precancerous state of dysplasia. Chromosomal instability and MSI pathways also have a major role in UC-associated CRC. However, there are differences in the instance and frequency of these alterations. APC loss of function is less frequent and usually occurs later in the UC-associated dysplasia-carcinoma sequence than in the classical adenoma-carcinoma sequence. By contrast in patients with UC, p53 mutations occur earlier and are often detected even in mucosa without dysplasia or still indefinite for it. Issa et al studied the CpG methylation status of four genes (ER, MYOD, p16 exon 1, and CSPG2) in patients with UC[46]. The methylation status was higher in high-grade dysplasia patients both in apparent normal mucosa and in high-grade dysplasia areas, suggesting that methylation precedes dysplasia; increased methylation is also widespread in the mucosa of high-risk patients. Several polymorphisms of genes related with the inflammatory response may increase this CpG methylation state[47]. SCRCs have been frequently found in the colons of patients who had total colectomy for low-grade dysplasia[9]. Therefore it is not coincidental that MPCRC often appears in the context of IBD: SCRCs are present in 18% of UC-related CRCs[9]. In a retrospective study of 64 patients with Crohn´s disease and CRC, Maser et al[48] found MCRC in 39% of patients 7 years after segmental of subtotal resection, which increased and at a rate of 0.5% per year after 8 to 10 years. Duration and extension of colitis are the two major features associated with increased risk of CRC[49]. From a review of the literature, Eaden et al[45] derived incidence rates of CRC in UC patients which corresponded to cumulative probabilities of 2% by 10 years, 8% by 20 years, and 18% by 30 years. These data suggest an accumulated methylation over time, provoking a field defect that reflects acquired predisposition to CRC and that might promote MPCRC as well[46].
Simultaneous individual and familial predisposition
An important proportion of MPCRC should be described as a whole, in which hereditary component cannot be distinguished from acquired alterations. Although SCRC and MCRC are different entities, they are linked. Patients with MCRC received a diagnosis of SCRC at the time of the initial CRC more often than patients with a solitary CRC (11.1% vs 3.1%; P = 0.001)[4]. Other studies described the increased risk of developing a MCRC after a SCRC with a hazard ratio 11.54 (HR = 11.54, 95 %CI: 10.6-12.5; P = 0.04)[15]. Synchronous adenomas represent a multifocal disease affecting the colonic mucosa. In spite of resection of all the synchronous adenomas, another lesion may appear from a damaged mucosa and develop into a MCRC[50]. The susceptibility to develop two primary neoplasms, simultaneously or consecutively, in the large bowel can be explained in different but not exclusive ways. Firstly, inheritance can mediate cumulative molecular defects that lead to dysplasia at different sites of the colon. On the other hand, one defined enviromental exposure may be implicated in the genesis of synchronous tumours, and continued exposure could result in another consecutive neoplasm. Nowadays, apart from hereditary forms such as LS or FAP, in most cases we do not know what the genetic inherited predisposition for MPCRC is. Whereas MCRC could be mediated by a constitutional factor that generates different polyps over time with a continuous possibility of developing a carcinoma, the carcinogenetic process begins at the same time in two different places of the mucosa for SCRC. This situation arises because of the exposure of a large area of mucosa to the same ethiology factors for a time sufficient to initiate carcinogenesis. Regardless of individual or hereditary causes, once the carcinogenic process has begun it can continue generating new lesions in the remaining colon. The risk for MCRC after diagnosis of the first CRC is higher than the prevalence of single CRC in a sex- and age-matched population, and the risk is higher in the first years following diagnosis, up to 61% in the first three years[4]. The risk is even higher when SCRC was previously identified, so that a close follow up must be carried out.
As we mentioned above, different etiopathogenic factors are suspected to be involved for synchronous and metachronous CRC, and different familial vs individual factors may be implicated (Figure 1). MCRC seems to follow a familial pattern, whereas individual factors are more important in SCRC[43].
Figure 1.
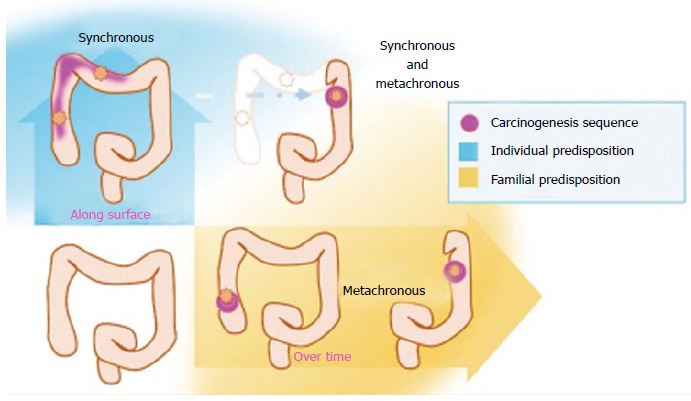
Both familial and individual factors may influence the genesis of multiple primary colorectal cancer. Genetic predisposition can promote the carcinogenesis sequence over time, whereas enviromental factors promote formation of different tumours along the colon surface at the same time.
Since SCRC and MCRC can appear in the same individual, in some occasions both familial and individual conditions concur. A study performed by Greenstein et al[9] reported that SCRC accounted for 2.5% of “de novo” CRC, and SCRC occurred in 18% of UC patients, and in 21% of patients with FAP[9]. Familial predisposition seems to be important in this development. The methylation state of LINE-1 appears concordant between SCRCs in the same individual, indistinctly of congruent or incongruent localization of the tumours[35]. Moreover, the methylation state of LINE-1 was also high in normal mucosa of these cases[35]. These findings suggest a similar substrate of the mucosa in relation with environmental exposure, where probably heritable factors modify the phenotype, by mutation of BRAF, hypermethylation of promoters of MMR genes (MLH1), or chromosome instability. Both genetic and environmental factors may influence serrated neoplasia as well to develop into CRC, which in these cases acquires MSI status. A polymorphism in the MLH1 promoter (-93 G>A) gives an increased risk for loss of MLH1 function in serrated polyps. This polymorphism does not increased the number of serrated polyps, but appears to promote their malignancy[40]. Moreover, smoking promotes the development of MSI in serrated polyps[25].
HETEROGENEITY IN CRC
Finally, according to this feature of CRC, SCRC and MCRC may be defined as clinical entities and sometimes may not reflect the condition of the tumor at the molecular level. Genetic heterogeneity plays a fundamental role in this case. Sometimes MPCRCs show similar clinicopathological features, as shown by Huan et al[51], who explored a large cohort with 5346 patients with synchronous, metachronous and solitary CRC, finding similar clinicopathological features between SCRC and MCRC. Moreover, analysing primary CRCs and their metastatic sites[52], checking for mutations KRAS, NRAS, BRAF, PIK3CA, and TP53 genes in 615 patients found for most cases the same mutations between both link.
But in most cases, we have to take on account the proposed role of genetic heterogeneity in individual predisposition to CRC described by Galvan et al[53]. Either a single genetic defects or the polygenic conditions produce a cancer-prone condition in the human normal tissue; individual risk of cancer might be further modulated by environmental factors, leading to somatic mutations and ultimately to cancer. This is very striking when talking about the demethylation grade from normal mucosa of healthy people, single cancer patient and multiple CCR, emerging an increased rate of demethylation among normal mucosa of older patients, and in normal mucosa of patients with multiple CRC risk from younger patients, suggesting an inherited predisposition for the apparent field cancerization effect of somatic demethylation[54]. This influence of environmental conditions among hereditary predisposition is clearly shown by Rosty et al[55]. They found that the majority of CRCs arising in individuals with serrated polyposis (SP) do not harbor molecular hallmarks of serrated pathway CRCs but show a diverse range of molecular profiles. This suggests that CRC in SP patients may develop from non-serrated polyps through either a derivative of the traditional adenoma pathway. SP could therefore, potentially, be considered a disorder associated with a hypermature or inappropriately aged colonic mucosa, possibly secondary to an alteration in DNA methylation, with a resultant propensity to the development of early onset multiple serrated polyps and conventional adenomas.
Finally, another aspect thar supports the heterogeous condition of CRC is described by Zauber et al[56] who evaluated by a set of 6 different markers (LOH for APC, DCC, and mutations of KRAS, BRAF, MSI and methylation of MMR genes) 50 patients with SCRC and 5 MCRCs. They found that genetic changes may vary considerably, particularly when the tumors are found in different colon segments. Frequent differences in the molecular findings are also seen between SCRCs sharing the identical microenvironment of the same colon segment. These findings support the hypothesis that MPCRC may follow different pathways of carcinogenesis in the same patient.
CONCLUSION
MPCRC is a rare event but its prevalence is not negligible, and it has a great clinical impact. Both genetic and environmental factors may affect in the development of MCRC, collaborating in promoting different foci of dysplasia. If lesions progress at the same time, SCRCs arise, whereas MCRCs appear if they develop at different time. In general terms, MCRCs are mainly related to family factors whereas SCRC are linked with individual factors. Nevertheless, in some cases both entities arise in the same individual. Due to the heterogeneity of current studies, conclusive information on the molecular basis of MPCRC is scant, with the exception of cases of hereditary forms of CRC. For this reason it is difficult to draw definitive conclusions at this time. New studies focusing on the carcinogenic mechanism must be done in order to better understand the molecular basis of MPCRC.
ACKNOWLEDGMENTS
We thank Ron Hartong for linguistic and editorial assistance.
Footnotes
Supported by Mutua Madrileña Foundation, No. 2012-0036.
Conflict-of-interest statement: Authors declare no conflict of interests for this article.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 1, 2015
First decision: September 14, 2015
Article in press: October 27, 2015
P- Reviewer: Morandi L S- Editor: Qi Y L- Editor: A E- Editor: Wu HL
References
- 1.Moertel CG, Bargen JA, Dockerty MB. Multiple carcinomas of the large intestine: a review of the literature and a study of 261 cases. Gastroenterology. 1958;34:85–98. [PubMed] [Google Scholar]
- 2.Fante R, Roncucci L, Di GregorioC MG, Losi L, Benatti P, Pedroni M, Percesepe A, De Pietri S, Ponz de Leon M. Frequency and clinical features of multiple tumors of the large bowel in the general population and in patients with hereditary colorectal carcinoma. Cancer. 1996;77:2013–2021. doi: 10.1002/(SICI)1097-0142(19960515)77:10<2013::AID-CNCR8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 3.Fajobi O, Yiu CY, Sen-Gupta SB, Boulos PB. Metachronous colorectal cancers. Br J Surg. 1998;85:897–901. doi: 10.1046/j.1365-2168.1998.00800.x. [DOI] [PubMed] [Google Scholar]
- 4.Mulder SA, Kranse R, Damhuis RA, Ouwendijk RJ, Kuipers EJ, van Leerdam ME. The incidence and risk factors of metachronous colorectal cancer: an indication for follow-up. Dis Colon Rectum. 2012;55:522–531. doi: 10.1097/DCR.0b013e318249db00. [DOI] [PubMed] [Google Scholar]
- 5.Mulder SA, Kranse R, Damhuis RA, de Wilt JH, Ouwendijk RJ, Kuipers EJ, van Leerdam ME. Prevalence and prognosis of synchronous colorectal cancer: a Dutch population-based study. Cancer Epidemiol. 2011;35:442–447. doi: 10.1016/j.canep.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Kaibara N, Koga S, Jinnai D. Synchronous and metachronous malignancies of the colon and rectum in Japan with special reference to a coexisting early cancer. Cancer. 1984;54:1870–1874. doi: 10.1002/1097-0142(19841101)54:9<1870::aid-cncr2820540917>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 7.Hart AR, Kudo S, Mackay EH, Mayberry JF, Atkin WS. Flat adenomas exist in asymptomatic people: important implications for colorectal cancer screening programmes. Gut. 1998;43:229–231. doi: 10.1136/gut.43.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lam AK, Carmichael R, Gertraud Buettner P, Gopalan V, Ho YH, Siu S. Clinicopathological significance of synchronous carcinoma in colorectal cancer. Am J Surg. 2011;202:39–44. doi: 10.1016/j.amjsurg.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 9.Greenstein AJ, Slater G, Heimann TM, Sachar DB, Aufses AH. A comparison of multiple synchronous colorectal cancer in ulcerative colitis, familial polyposis coli, and de novo cancer. Ann Surg. 1986;203:123–128. doi: 10.1097/00000658-198602000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalo V, Lozano JJ, Alonso-Espinaco V, Moreira L, Muñoz J, Pellisé M, Castellví-Bel S, Bessa X, Andreu M, Xicola RM, et al. Multiple sporadic colorectal cancers display a unique methylation phenotype. PLoS One. 2014;9:e91033. doi: 10.1371/journal.pone.0091033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parry S, Win AK, Parry B, Macrae FA, Gurrin LC, Church JM, Baron JA, Giles GG, Leggett BA, Winship I, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut. 2011;60:950–957. doi: 10.1136/gut.2010.228056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piñol V, Andreu M, Castells A, Payá A, Bessa X, Jover R. Synchronous colorectal neoplasms in patients with colorectal cancer: predisposing individual and familial factors. Dis Colon Rectum. 2004;47:1192–1200. doi: 10.1007/s10350-004-0562-7. [DOI] [PubMed] [Google Scholar]
- 13.Samadder NJ, Curtin K, Wong J, Tuohy TM, Mineau GP, Smith KR, Pimentel R, Pappas L, Boucher K, Garrido-Laguna I, et al. Epidemiology and familial risk of synchronous and metachronous colorectal cancer: a population-based study in Utah. Clin Gastroenterol Hepatol. 2014;12:2078–84.e1-2. doi: 10.1016/j.cgh.2014.04.017. [DOI] [PubMed] [Google Scholar]
- 14.Peltomäki P. Lynch syndrome genes. Fam Cancer. 2005;4:227–232. doi: 10.1007/s10689-004-7993-0. [DOI] [PubMed] [Google Scholar]
- 15.Win AK, Parry S, Parry B, Kalady MF, Macrae FA, Ahnen DJ, Young GP, Lipton L, Winship I, Boussioutas A, et al. Risk of metachronous colon cancer following surgery for rectal cancer in mismatch repair gene mutation carriers. Ann Surg Oncol. 2013;20:1829–1836. doi: 10.1245/s10434-012-2858-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu H, Chang DT, Nikiforova MN, Kuan SF, Pai RK. Clinicopathologic features of synchronous colorectal carcinoma: A distinct subset arising from multiple sessile serrated adenomas and associated with high levels of microsatellite instability and favorable prognosis. Am J Surg Pathol. 2013;37:1660–1670. doi: 10.1097/PAS.0b013e31829623b8. [DOI] [PubMed] [Google Scholar]
- 17.Dykes SL, Qui H, Rothenberger DA, García-Aguilar J. Evidence of a preferred molecular pathway in patients with synchronous colorectal cancer. Cancer. 2003;98:48–54. doi: 10.1002/cncr.11445. [DOI] [PubMed] [Google Scholar]
- 18.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 19.Kastrinos F, Steyerberg EW, Mercado R, Balmaña J, Holter S, Gallinger S, Siegmund KD, Church JM, Jenkins MA, Lindor NM, et al. The PREMM(1,2,6) model predicts risk of MLH1, MSH2, and MSH6 germline mutations based on cancer history. Gastroenterology. 2011;140:73–81. doi: 10.1053/j.gastro.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balmaña J, Castells A, Cervantes A. Familial colorectal cancer risk: ESMO Clinical Practice Guidelines. Ann Oncol. 2010;21 Suppl 5:v78–v81. doi: 10.1093/annonc/mdq169. [DOI] [PubMed] [Google Scholar]
- 21.Buchanan DD, Sweet K, Drini M, Jenkins MA, Win AK, Gattas M, Walsh MD, Clendenning M, McKeone D, Walters R, et al. Phenotypic diversity in patients with multiple serrated polyps: a genetics clinic study. Int J Colorectal Dis. 2010;25:703–712. doi: 10.1007/s00384-010-0907-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Snover DC, Jass JR, Fenoglio-Preiser C, Batts KP. Serrated polyps of the large intestine: a morphologic and molecular review of an evolving concept. Am J Clin Pathol. 2005;124:380–391. doi: 10.1309/V2EP-TPLJ-RB3F-GHJL. [DOI] [PubMed] [Google Scholar]
- 23.Chow E, Lipton L, Lynch E, D’Souza R, Aragona C, Hodgkin L, Brown G, Winship I, Barker M, Buchanan D, et al. Hyperplastic polyposis syndrome: phenotypic presentations and the role of MBD4 and MYH. Gastroenterology. 2006;131:30–39. doi: 10.1053/j.gastro.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 24.Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010;138:2088–2100. doi: 10.1053/j.gastro.2009.12.066. [DOI] [PubMed] [Google Scholar]
- 25.Young J, Jass JR. The case for a genetic predisposition to serrated neoplasia in the colorectum: hypothesis and review of the literature. Cancer Epidemiol Biomarkers Prev. 2006;15:1778–1784. doi: 10.1158/1055-9965.EPI-06-0164. [DOI] [PubMed] [Google Scholar]
- 26.Rosty C, Hewett DG, Brown IS, Leggett BA, Whitehall VL. Serrated polyps of the large intestine: current understanding of diagnosis, pathogenesis, and clinical management. J Gastroenterol. 2013;48:287–302. doi: 10.1007/s00535-012-0720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niittymäki I, Kaasinen E, Tuupanen S, Karhu A, Järvinen H, Mecklin JP, Tomlinson IP, Di Bernardo MC, Houlston RS, Aaltonen LA. Low-penetrance susceptibility variants in familial colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2010;19:1478–1483. doi: 10.1158/1055-9965.EPI-09-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aaltonen L, Johns L, Järvinen H, Mecklin JP, Houlston R. Explaining the familial colorectal cancer risk associated with mismatch repair (MMR)-deficient and MMR-stable tumors. Clin Cancer Res. 2007;13:356–361. doi: 10.1158/1078-0432.CCR-06-1256. [DOI] [PubMed] [Google Scholar]
- 29.King-Yin Lam A, Ong K, Ho YH. Colorectal mucinous adenocarcinoma: the clinicopathologic features and significance of p16 and p53 expression. Dis Colon Rectum. 2006;49:1275–1283. doi: 10.1007/s10350-006-0650-y. [DOI] [PubMed] [Google Scholar]
- 30.Velayos FS, Lee SH, Qiu H, Dykes S, Yiu R, Terdiman JP, Garcia-Aguilar J. The mechanism of microsatellite instability is different in synchronous and metachronous colorectal cancer. J Gastrointest Surg. 2005;9:329–335. doi: 10.1016/j.gassur.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 31.Yeoman A, Young J, Arnold J, Jass J, Parry S. Hyperplastic polyposis in the New Zealand population: a condition associated with increased colorectal cancer risk and European ancestry. N Z Med J. 2007;120:U2827. [PubMed] [Google Scholar]
- 32.Latournerie M, Jooste V, Cottet V, Lepage C, Faivre J, Bouvier AM. Epidemiology and prognosis of synchronous colorectal cancers. Br J Surg. 2008;95:1528–1533. doi: 10.1002/bjs.6382. [DOI] [PubMed] [Google Scholar]
- 33.Shibata D, Peinado MA, Ionov Y, Malkhosyan S, Perucho M. Genomic instability in repeated sequences is an early somatic event in colorectal tumorigenesis that persists after transformation. Nat Genet. 1994;6:273–281. doi: 10.1038/ng0394-273. [DOI] [PubMed] [Google Scholar]
- 34.Lawes DA, Pearson T, Sengupta S, Boulos PB. The role of MLH1, MSH2 and MSH6 in the development of multiple colorectal cancers. Br J Cancer. 2005;93:472–477. doi: 10.1038/sj.bjc.6602708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nosho K, Kure S, Irahara N, Shima K, Baba Y, Spiegelman D, Meyerhardt JA, Giovannucci EL, Fuchs CS, Ogino S. A prospective cohort study shows unique epigenetic, genetic, and prognostic features of synchronous colorectal cancers. Gastroenterology. 2009;137:1609–1620.e1-3. doi: 10.1053/j.gastro.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Konishi K, Shen L, Jelinek J, Watanabe Y, Ahmed S, Kaneko K, Kogo M, Takano T, Imawari M, Hamilton SR, et al. Concordant DNA methylation in synchronous colorectal carcinomas. Cancer Prev Res (Phila) 2009;2:814–822. doi: 10.1158/1940-6207.CAPR-09-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borda A, Martínez-Peñuela JM, Muñoz-Navas M, Prieto C, Betés M, Borda F. [Synchronous neoplastic lesions in colorectal cancer. An analysis of possible risk factors favouring presentation] Rev Esp Enferm Dig. 2008;100:139–145. doi: 10.4321/s1130-01082008000300003. [DOI] [PubMed] [Google Scholar]
- 38.Otani T, Iwasaki M, Yamamoto S, Sobue T, Hanaoka T, Inoue M, Tsugane S. Alcohol consumption, smoking, and subsequent risk of colorectal cancer in middle-aged and elderly Japanese men and women: Japan Public Health Center-based prospective study. Cancer Epidemiol Biomarkers Prev. 2003;12:1492–1500. [PubMed] [Google Scholar]
- 39.Maekawa SJ, Aoyama N, Shirasaka D, Kuroda K, Tamura T, Kuroda Y, Kasuga M. Excessive alcohol intake enhances the development of synchronous cancerous lesion in colorectal cancer patients. Int J Colorectal Dis. 2004;19:171–175. doi: 10.1007/s00384-003-0516-x. [DOI] [PubMed] [Google Scholar]
- 40.Samowitz WS, Curtin K, Wolff RK, Albertsen H, Sweeney C, Caan BJ, Ulrich CM, Potter JD, Slattery ML. The MLH1 -93 G& gt; A promoter polymorphism and genetic and epigenetic alterations in colon cancer. Genes Chromosomes Cancer. 2008;47:835–844. doi: 10.1002/gcc.20584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Samowitz WS. The CpG island methylator phenotype in colorectal cancer. J Mol Diagn. 2007;9:281–283. doi: 10.2353/jmoldx.2007.070031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Limsui D, Vierkant RA, Tillmans LS, Wang AH, Weisenberger DJ, Laird PW, Lynch CF, Anderson KE, French AJ, Haile RW, et al. Cigarette smoking and colorectal cancer risk by molecularly defined subtypes. J Natl Cancer Inst. 2010;102:1012–1022. doi: 10.1093/jnci/djq201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lam AK, Chan SS, Leung M. Synchronous colorectal cancer: clinical, pathological and molecular implications. World J Gastroenterol. 2014;20:6815–6820. doi: 10.3748/wjg.v20.i22.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Botteri E, Iodice S, Raimondi S, Maisonneuve P, Lowenfels AB. Cigarette smoking and adenomatous polyps: a meta-analysis. Gastroenterology. 2008;134:388–395. doi: 10.1053/j.gastro.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 45.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526–535. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001;61:3573–3577. [PubMed] [Google Scholar]
- 47.Tahara T, Shibata T, Nakamura M, Okubo M, Yamashita H, Yoshioka D, Yonemura J, Kmiya Y, Ishizuka T, Fujita H, et al. Host genetic factors, related to inflammatory response, influence the CpG island methylation status in colonic mucosa in ulcerative colitis. Anticancer Res. 2011;31:933–938. [PubMed] [Google Scholar]
- 48.Maser EA, Sachar DB, Kruse D, Harpaz N, Ullman T, Bauer JJ. High rates of metachronous colon cancer or dysplasia after segmental resection or subtotal colectomy in Crohn’s colitis. Inflamm Bowel Dis. 2013;19:1827–1832. doi: 10.1097/MIB.0b013e318289c166. [DOI] [PubMed] [Google Scholar]
- 49.Greenstein AJ. Cancer in inflammatory bowel disease. Mt Sinai J Med. 2000;67:227–240. [PubMed] [Google Scholar]
- 50.Borda A, Martínez-Peñuela JM, Borda F, Muñoz-Navas M, Jiménez FJ, Carretero C. Drawing up an individual risk index for development of metachronous neoplastic lesions in resected colorectal cancer. Rev Española Enfermedades Dig. 2012;104:291–7. doi: 10.4321/s1130-01082012000600002. [DOI] [PubMed] [Google Scholar]
- 51.Huang CS, Yang SH, Lin CC, Lan YT, Chang SC, Wang HS, Chen WS, Lin TC, Lin JK, Jiang JK. Synchronous and metachronous colorectal cancers: distinct disease entities or different disease courses? Hepatogastroenterology. 2015;62:286–290. [PubMed] [Google Scholar]
- 52.Vakiani E, Janakiraman M, Shen R, Sinha R, Zeng Z, Shia J, Cercek A, Kemeny N, D’Angelica M, Viale A, et al. Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J Clin Oncol. 2012;30:2956–2962. doi: 10.1200/JCO.2011.38.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Galvan A, Ioannidis JP, Dragani TA. Beyond genome-wide association studies: genetic heterogeneity and individual predisposition to cancer. Trends Genet. 2010;26:132–141. doi: 10.1016/j.tig.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamiyama H, Suzuki K, Maeda T, Koizumi K, Miyaki Y, Okada S, Kawamura YJ, Samuelsson JK, Alonso S, Konishi F, et al. DNA demethylation in normal colon tissue predicts predisposition to multiple cancers. Oncogene. 2012;31:5029–5037. doi: 10.1038/onc.2011.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosty C, Walsh MD, Walters RJ, Clendenning M, Pearson SA, Jenkins MA, Win AK, Hopper JL, Sweet K, Frankel WL, et al. Multiplicity and molecular heterogeneity of colorectal carcinomas in individuals with serrated polyposis. Am J Surg Pathol. 2013;37:434–442. doi: 10.1097/PAS.0b013e318270f748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zauber P, Huang J, Sabbath-Solitare M, Marotta S. Similarities of molecular genetic changes in synchronous and metachronous colorectal cancers are limited and related to the cancers’ proximities to each other. J Mol Diagn. 2013;15:652–660. doi: 10.1016/j.jmoldx.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 57.Borda A, Jimenez-Perez J, Munoz-Navas MA, Martinez-Penuela JM, Carretero C, Borda F. Analysis of diagnostic timing of metachronous adenomas in colorectal cancer. Gastroenterology. 2009;136:A769. [Google Scholar]