

Abstract
Although it is generally accepted that amino acids were present on the prebiotic Earth, the mechanism by which α-amino acids were condensed into polypeptides before the emergence of enzymes remains unsolved. Here, we demonstrate a prebiotically plausible mechanism for peptide (amide) bond formation that is enabled by α-hydroxy acids, which were likely present along with amino acids on the early Earth. Together, α-hydroxy acids and α-amino acids form depsipeptides—oligomers with a combination of ester and amide linkages—in model prebiotic reactions that are driven by wet–cool/dry–hot cycles. Through a combination of ester–amide bond exchange and ester bond hydrolysis, depsipeptides are enriched with amino acids over time. These results support a long-standing hypothesis that peptides might have arisen from ester-based precursors.
Keywords: chemical evolution, day–night cycle, depsipeptides, origins-of-life, proto-peptides
For several decades there has been evidence that amino acids were present on the prebiotic Earth, either by endogenous formation1 or by delivery in meteorites.2 Nevertheless, the process by which amino acids were first converted to peptides is still unclear. A particular challenge is that peptide (amide) bond formation is thermodynamically unfavorable in aqueous solution.3 Several mechanisms for prebiotic peptide formation from amino acids have been proposed, including chemical activation, heating with inorganic catalysts, chemistry at liquid–air interfaces and vapor phase deposition onto catalytic surfaces.4
The difficulty of forming amide bonds in model prebiotic reactions inspired the proposal that polyesters came before polypeptides in an early stage of life.5 Polyesters are readily formed from α-hydroxy acids when subjected to conditions intended to mimic wet–dry (e.g., night–day) environmental cycles on the prebiotic Earth.6 This led us to hypothesize that mixtures of α-hydroxy acids and α-amino acids would produce oligomers that contain both ester and amide linkages (depsipeptides) when subjected to the same environmental cycles. This idea was particularly attractive from an origins-of-life perspective because simple α-hydroxy acids and α-amino acids (e.g., lactic acid, glycine, alanine) have been found together in model prebiotic reaction mixtures1b, 7 and in meteorites.8
A representation of our idea for peptide bond formation is shown in Scheme 1. Briefly, in the dry–hot phase, solvent evaporation promotes ester bond formation and spontaneous ester–amide bond exchange.9 In the cool-wet phase, reactants disperse and ester linkages are expected to preferentially hydrolyze. Therefore, this process is predicted to generate oligomers enriched in amino acids over time, with hydroxy acid residues remaining at the N-terminus (see Supporting Information: Mechanism).
scheme 1.
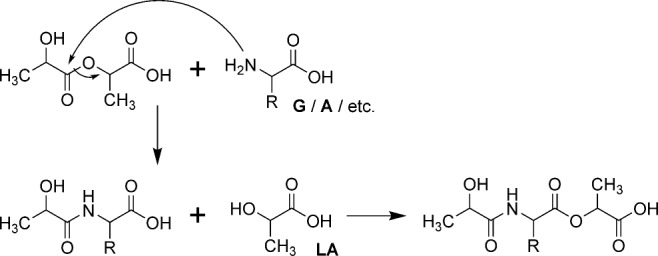
Proposed reaction scheme for lactic acid (LA)-mediated peptide bond formation and depsipeptide elongation by ester–amide exchange (also see SI: Mechanism). Here, a free amino acid (G, A, etc.) with a neutral -NH2 terminus (in equilibrium with -NH3+) reacts with a lactic acid dimer to form amide-linked depsipeptides. Elongation leads to longer oligomers.
When L-lactic acid (LA) is subjected to 4 wet–dry cycles (wet phase 65 °C, 5.5 h; dry phase 85 °C, 18 h) linear polyesters are formed (Figure 1 a), consistent with our previous report.6b When glycine (G) is mixed with LA in a 1:1 mole ratio and subjected to the same wet–dry cycles, essentially all mass spectral signals correspond to depsipeptides (Figure 1 b). G-containing depsipeptides of up to 10 residues in length are observed, containing varying amounts of amide and ester linkages (Figure 1 b). In contrast, when glycine alone is subjected to 4 wet–dry cycles no peptides are observed in the mass spectrum (Figure S1 in the Supporing Information). We note that for samples containing mixtures of LA and G, formation of ester bonds between LA monomers is observed at dry phase temperatures as low as 55 °C, whereas amide bonds are first observed at 65 °C (Figure S2). From this data, we confirm that amide bonds are produced under milder conditions than the protocols of Fox and Harada, which required temperatures above 150 °C,10 and with longer oligomer lengths than produced by salt catalysis,11 observations that make ester-mediated amide bond formation more compatible with some models put forth for the origins of life.
Figure 1.

Demonstration of depsipeptide formation from lactic acid (LA) and glycine (G) through ester-amide exchange. a) Negative electrospray mass spectrum of LA after 4 wet–dry cycles (wet phase 65 °C, 5.5 h; dry phase 85 °C, 18 h). Polyesters are observed with n≥10. b) Mass spectrum of a 1:1 mol LA/G mixture after 4 cycles. The most intense signals correspond to mixed depsipeptides. LA-G depsipeptides are observed up to n=10 (inset).
MS analysis revealed depsipeptides undergo a chemical transition over the course of multiple environmental cycles, enriching for peptide linkages at the expense of ester linkages. This phenomenon is illustrated by the products of a 1:1 mixture of LA and L-alanine (A) (Figure 2 a). LA and A neutral residue masses differ by approximately 1 Da. Thus, for a LA-A depsipeptide of a given residue length, a shift toward lower molecular weight corresponds to A enrichment. Copolymerization of LA and A could produce a mixture of depsipeptides with all possible combinations. For example, the pentamer series (n=5) could contain the 5LA polyester, along with the 4LA+1A, 3LA+2A, 2LA+3A, and 1LA+4A depsipeptides, each separated by 1 Da. After 1 cycle we observe pentamers with each of these possible LA/A combinations, except for the 1LA+4A depsipeptide (Figure 2 a, top). As the number of cycles increases, the distribution of these species shifts to lower mass-to-charge values, corresponding to increasing incorporation of A (Figure 2 a, subsequent spectra). After 12 environmental cycles, the pentamer series contains 1LA+4A, 2LA+3A, and 3LA+2A depsipeptides. The 4LA+1A depsipeptide and 5LA polyester are no longer present. Following 20 cycles, the average composition of the depsipeptides is approximately 65 % A and 35 % LA. In Figure 2 b a theoretical limit of A abundance for each oligomer is provided for comparison, based on the assumption that one LA residue remains attached to the N-terminus of the peptide. We expect amino acid abundance would continue to approach this theoretical limit with additional cycling, as long as catalytic hydroxy acids continue to be present.
Figure 2.

Amino acid/peptide bond enrichment in depsipeptides following environmental cycling. a) Mass spectral region showing LA-A pentamers after 1, 4, 8, and 12 cycles. LA residue mass=72.0211 Da; A residue mass=71.0371 Da. As the number of cycles increases, the pentamer series is enriched in A residues, as evidenced by a mass decrease of 0.9840 Da per exchanged residue. Also see Figure S5 and S6. b) Isotopically-corrected MS data for n=2 through n=8 oligomers shows the relative amounts of A incorporated as a function of environmental cycles. The theoretical limit of A in each oligomer, shown in black, corresponds to n−1 residues as the N-terminal lactamide is not thought to exchange. c) Emergence of amide bonds as demonstrated by IR spectroscopy. Spectra are of (orange) LA + glycine (G) monomers before cycling, (yellow) LA-G mixture after 1 cycle, (light green) LA-G mixture after 8 cycles, and (green) LA-G mixture after 16 cycles, where LA was re-added after the 8th cycle. The band in top spectrum at 1710 cm−1 corresponds to free carboxylic acids of monomers. Upon cycling, reduction of the carboxylic acid band is observed, and the formation of ester bonds is observed (shoulder at 1730 cm−1). Eventually, amide bond bands are observed (Amide I, 1640 cm−1; Amide II, 1530 cm−1), similar to those of a polyglycine spectrum (black, Ref. 13). Full IR spectra are provided in Figure S7–S9.
A number of studies on prebiotic amide bond formation have noted the generation of diketopiperazines (DKPs), or cyclic amino acid dimers,1c, 4a,f, 11, 12 which appear less reactive than free amino acids (Figure S3). We tentatively identify cyclic LA (cyclic 2LA) and cyclic mixed LA-A (cyclic 1LA+1A) species in our MS data, but have not observed the 2A DKP to date (Figure S4). We hypothesize the 2LA and 1LA+1A cyclic species can reversibly interchange with their respective linear forms, whereas we expect the rate of interchange of the 2A DKP with its linear form to be slower. Whether or not DKPs are involved in depsipeptide formation remains unclear at this time; however, it is clear DKP formation is not an obstacle to forming linear depsipeptides and, eventually, peptide sequences.
IR spectroscopy confirms the emergence of peptide bonds (Figure 2 c). The summed IR spectrum of G and LA monomers contains a strong band at 1710 cm−1, corresponding to carboxylic acids. After subjecting a 1:1 mixture of LA and G to 1 wet–dry cycle, the intensity of the carboxylic acid band is reduced and a shoulder appears at 1730 cm−1, indicative of ester bonds. With continued cycling, we observe growth of Amide I and Amide II bands (1640 cm−1 and 1530 cm−1, respectively), and the spectrum of the sample subjected to 16 cycles begins to resemble that of polyglycine.13 These observations are fully supportive of our proposed mechanism of peptide bond enrichment through repeated ester–amide exchange.
2D NMR spectroscopy further confirmed the formation of amide bonds between G residues and between G and LA residues (Figure S10–S15). Based on 1H NMR analysis, the combined yield of LA-G and G-G amides is shown in Figure 3 a. After 8 cycles total amide bond yield is ca. 10 %. Notably, LA monomer appears to deplete with repeated cycling; we hypothesize this is due to incorporation into depsipeptides and by-products, and also evaporation in our open reaction system. With a single replenishment of LA at the 8th cycle, the total amide bond yield achieved after 16 cycles was 40 %.
Figure 3.

a) Amide bond yield as a function of wet–dry cycles, determined by 1H NMR spectroscopy. After 8 cycles, approximately 10 % of G monomer has been incorporated into depsipeptides. Amide bond yields increase significantly when LA is re-added after cycle 8, resulting in approximately 40 % of G being incorporated into depsipeptides. b–d) Sequencing of depsipeptides by tandem MS reveals internal amino acid incorporation. b) Sequencing of 2LA+4G, theoretical precursor ion [M−H]−=389.13 Da, formed after 8 cycles. CID collision energy=20 eV. The primary sequence is LA-G-G-G-G-LA. c) Sequencing of 2LA+3A, theoretical precursor ion [M−H]−=374.16 Da, formed after 12 cycles. CID collision energy=15 eV. The primary sequence is LA-A-A-A-LA. d) Sequencing of 2 LA+2G+1A, theoretical precursor ion [M−H]−=346.13 Da, formed after 4 cycles. CID collision energy=20 eV. LA-A-G-G-LA, LA-G-A-G-LA, and LA-G-G-A-LA sequences are observed.
Depsipeptide residue sequences were determined using tandem MS. Consistent with the proposed reaction mechanism, LA residues are progressively displaced from the core of depsipeptides, leading to continuous peptide sequences predominantly in the middle of the oligomers sequence. Examples of internal peptide sequences are shown in Figure 3 b–d. Specifically, the G-G-G-G internal peptide sequence is observed after 8 cycles in the LA/G reaction system (Figure 3 b); the three-amino acid residue A-A-A sequence is observed after 12 cycles in the LA/A reaction system (Figure 3 c); and the three possible 2Gly+1Ala internal sequences are detected after only 4 cycles of the glycine–LA/G/A reaction system (Figure 3 d). Based on data such as that shown in Figure 3 d, the sequences of the peptide core in depsipeptides appear to be random.
In the experiments described above, reaction mixtures were initially around pH 3 due to unbuffered carboxylic acid moieties. While acidic pH can facilitate ester bond formation, protonation of the amine also reduces its ability to act as a nucleophile in the ester-amide exchange reaction. Moreover, from an origin of life perspective, prebiotic environments of pH 3 would have likely been rare.14 To test the impact of pH on amide bond formation, sodium hydroxide, ammonium hydroxide, and triethylamine were used to adjust the pH to 7 for a 1:1 mixture of LA and A. Depsipeptides were formed with all three bases; however, depsipeptide formation was more efficient with ammonium hydroxide or triethylamine than sodium hydroxide (Figure S16–S18). In addition to the ability of the ammonium ion and protonated triethylamine to act as general acids, the volatility of ammonia and triethylamine during evaporative cycles is also expected to facilitate ester bond formation. Upon rehydration, the samples neutralized with ammonia or triethylamine exhibited a decrease in pH from 7 to approximately 3.5. To account for this effect, base was re-added in each rehydration step to return the solution to pH 7. After 1 triethylamine-adjusted cycle, both LA oligoesters and mixed depsipeptides are observed (Figure 4 a). Following 8 cycles, enrichment of A-containing depsipeptides is observed (Figure 4 b), although at a slower rate than in pH-unadjusted samples. An internal di-alanine sequence (A-A) is observed in the 2LA+2A tetramer, formed after 8 triethylamine cycles (Figure 4 b).
Figure 4.

Depsipeptide formation and enrichment within an alanine-lactic acid sample that had an initial pH of 7. a,b) Mass spectra of n=4 alanine-lactic acid depsipeptides formed after 1 cycle and after 8 cycles. Before the first cycle, the solution was titrated to pH 7 by the addition of triethylamine. Because the sample became more acidic as solvent and base were evaporated (i.e., pH 3.5), triethylamine was added before each subsequent cycle to bring the sample back to pH 7. After 8 cycles, we observe enrichment of the 2LA+2A depsipeptide and reduction of the 4LA oligoester. c) Tandem MS sequencing of 2LA+2A. Theoretical precursor ion [M−H]−=303.12 Da, formed after 8 cycles. CID collision energy=12 eV. The primary sequence is LA-A-A-LA.
Thus far, we have primarily focused on systems where additional hydroxy acids or amino acids are not added in the rehydration phases. With this approach, the longest oligomers we have observed are n=14, and contain up to seven (likely non-consecutive) amide bonds after 8 cycles (Figure S19–S20). However, ester–amide exchange appears to be most efficient when LA is in slight excess, and therefore peptide formation can be enhanced by the addition of hydroxy acids during the hydration phase of a cycle (Figure 3 a). These results suggest that open systems, in which hydroxy acids are re-introduced during each cycle, are attractive for exploration. Such systems could represent a model of weather/tidal cycles on the early Earth by which volatile forms of reactants (e.g., α-hydroxy acids) were regularly delivered as part of the water cycle.
Having demonstrated that a mixture of hydroxy acids and amino acids can give rise to depsipeptides with homo-peptide regions in a plausible prebiotic reaction, it is now of interest to determine the scope and limitations of this reaction. For example, we have verified the production of depsipeptides with various starting concentrations and ratios of hydroxy acids and amino acids (Figure S21–S22), with amino acids of mixed stereochemistry, at lower dry-state temperatures, and with a different α-hydroxy acid. These preliminary results have revealed that leucine, a more hydrophobic amino acid, is incorporated into depsipeptides with LA, and on apparently an equal basis with A and G in reactions that contain LA and all three amino acids (Figure S23–S26). Serine, a more polar amino acid, is also incorporated into depsipeptides, but reaction products are more complex, possibly due to the functional side chain of serine (Figure S27–S28). Depsipeptide formation appears to be even more efficient when glycolic acid is used as the catalyst, in place of lactic acid (Figure S29–S30). Finally, the reaction does not appear to show a stereochemical preference, at least for pentamers and shorter (Figure S31). Clearly, a comprehensive mapping of the chemical space of α-hydroxy acid-catalyzed peptide formation will require a systematic investigation and substantial effort, and doing so may provide valuable clues regarding the chemical pressures that shaped the sequences and structures of peptides formed on the prebiotic Earth.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1a.Miller SL. Science. 1953;117:528–529. doi: 10.1126/science.117.3046.528. [DOI] [PubMed] [Google Scholar]
- 1b.Miller SL, Urey HC. Science. 1959;130:245–251. doi: 10.1126/science.130.3370.245. [DOI] [PubMed] [Google Scholar]
- 1c.Parker ET, Zhou M, Burton AS, Glavin DP, Dworkin JP, Krishnamurthy R, Fernández FM, Bada JL. Angew. Chem. Int. Ed. 2014;53:8132–8136. doi: 10.1002/anie.201403683. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014;126 [Google Scholar]
- 2a.Kvenvolden K, Lawless J, Pering K, Peterson E, Flores J, Ponnampe C, Kaplan IR, Moore C. Nature. 1970;228:923–926. doi: 10.1038/228923a0. [DOI] [PubMed] [Google Scholar]
- 2b.Cronin JR, Pizzarello S. Science. 1997;275:951–955. doi: 10.1126/science.275.5302.951. [DOI] [PubMed] [Google Scholar]
- 2c.Bernstein MP, Dworkin JP, Sandford SA, Cooper GW, Allamandola LJ. Nature. 2002;416:401–403. doi: 10.1038/416401a. [DOI] [PubMed] [Google Scholar]
- 3a.Martin RB. Biopolymers. 1998;45:351–353. [Google Scholar]
- 3b.Deamer D, Weber AL. Cold Spring Harbor Perspect. Biol. 2010;2:004929. doi: 10.1101/cshperspect.a004929. , a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4a.Lahav N, White D, Chang S. Science. 1978;201:67–69. doi: 10.1126/science.663639. [DOI] [PubMed] [Google Scholar]
- 4b.Huber C, Wachtershauser G. Science. 1998;281:670–672. doi: 10.1126/science.281.5377.670. [DOI] [PubMed] [Google Scholar]
- 4c.Rode BM. Peptides. 1999;20:773–786. doi: 10.1016/s0196-9781(99)00062-5. [DOI] [PubMed] [Google Scholar]
- 4d.Leman L, Orgel L, Ghadiri MR. Science. 2004;306:283–286. doi: 10.1126/science.1102722. [DOI] [PubMed] [Google Scholar]
- 4e.Brack A. Chem. Biodiver. 2007;4:665–679. doi: 10.1002/cbdv.200790057. [DOI] [PubMed] [Google Scholar]
- 4f.Shanker U, Bhushan B, Bhattacharjee G, Kamaluddin Origins Life Evol. B. 2012;42:31–45. doi: 10.1007/s11084-012-9266-5. [DOI] [PubMed] [Google Scholar]
- 4g.Griffith EC, Vaida V. Proc. Natl. Acad. Sci. USA. 2012;109:15697–15701. doi: 10.1073/pnas.1210029109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4h.Danger G, Plasson R, Pascal R. Chem. Soc. Rev. 2012;41:5416–5429. doi: 10.1039/c2cs35064e. [DOI] [PubMed] [Google Scholar]
- 4i.Martra G, Deiana C, Sakhno Y, Barberis I, Fabbiani M, Pazzi M, Vincenti M. Angew. Chem. Int. Ed. 2014;53:4671–4674. doi: 10.1002/anie.201311089. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014;126 [Google Scholar]
- 5.Rich A. In: Chemical Evolution and the Origin of Life. Buvet R, Ponnamperuma C, editors. Amsterdam: North-Holland; 1971. pp. 180–196. [Google Scholar]
- 6a.Weber AL. Origins Life Evol. Biospheres. 1989;19:7–19. doi: 10.1007/BF01808284. [DOI] [PubMed] [Google Scholar]
- 6b.Mamajanov I, MacDonald PJ, Ying J, Duncanson DM, Dowdy GR, Walker CA, Engelhart AE, Fernández FM, Grover MA, Hud NV, Schork FJ. Macromolecules. 2014;47:1334–1343. [Google Scholar]
- 7.Huber C, Wächtershäuser G. Science. 2006;314:630–632. doi: 10.1126/science.1130895. [DOI] [PubMed] [Google Scholar]
- 8.Peltzer ET, Bada JL. Nature. 1978;272:443–444. [Google Scholar]
- 9a.Goodman M, Stueben KC. J. Am. Chem. Soc. 1959;81:3980–3983. [Google Scholar]
- 9b.Pillon LZ, Utracki LA. Polym. Eng. Sci. 1984;24:1300–1305. [Google Scholar]
- 9c.Hoegberg T, Strom P, Ebner M, Ramsby S. J. Org. Chem. 1987;52:2033–2036. [Google Scholar]
- 10.Fox SW, Harada K. Science. 1958;128:1214–1214. doi: 10.1126/science.128.3333.1214. [DOI] [PubMed] [Google Scholar]
- 11.Rode BM, Schwendinger MG. Origins Life Evol. Biospheres. 1990;20:401–410. [Google Scholar]
- 12a.Orgel LE. J. Mol. Evol. 1989;29:465–474. doi: 10.1007/BF02602917. [DOI] [PubMed] [Google Scholar]
- 12b.Nagayama M, Takaoka O, Inomata K, Yamagata Y. Origins Life Evol. Biospheres. 1990;20:249–257. doi: 10.1007/BF01808107. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki S, Iwashita Y, Shimanou T, Tsuboi M. Biopolymers. 1966;4:337–350. [Google Scholar]
- 14.Kua J, Bada JL. Origins Life Evol. Biospheres. 2011;41:553–558. doi: 10.1007/s11084-011-9250-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information