

Abstract
SLC39A8 is a membrane transporter responsible for manganese uptake into the cell. Via whole-exome sequencing, we studied a child that presented with cranial asymmetry, severe infantile spasms with hypsarrhythmia, and dysproportionate dwarfism. Analysis of transferrin glycosylation revealed severe dysglycosylation corresponding to a type II congenital disorder of glycosylation (CDG) and the blood manganese levels were below the detection limit. The variants c.112G>C (p.Gly38Arg) and c.1019T>A (p.Ile340Asn) were identified in SLC39A8. A second individual with the variants c.97G>A (p.Val33Met) and c.1004G>C (p.Ser335Thr) on the paternal allele and c.610G>T (p.Gly204Cys) on the maternal allele was identified among a group of unresolved case subjects with CDG. These data demonstrate that variants in SLC39A8 impair the function of manganese-dependent enzymes, most notably β-1,4-galactosyltransferase, a Golgi enzyme essential for biosynthesis of the carbohydrate part of glycoproteins. Impaired galactosylation leads to a severe disorder with deformed skull, severe seizures, short limbs, profound psychomotor retardation, and hearing loss. Oral galactose supplementation is a treatment option and results in complete normalization of glycosylation. SLC39A8 deficiency links a trace element deficiency with inherited glycosylation disorders.
Main Text
The solute carrier (SLC) gene superfamily comprises a group of nearly 400 putative transporter proteins.1, 2 Among them, the SLC39 family consists of 14 zinc- and iron-related proteins (ZIPs) functioning as divalent cation transporters.2
SLC39A8 (MIM: 608732) encodes an electroneutral Mn2+/(HCO3−)2 and Zn2+/(HCO3−)2 influx symporter, most commonly known as ZIP8. The transmembrane protein has 462 amino acids and a wide tissue distribution with highest expression in placenta, lung, and kidney.1, 3 ZIP8 plays a role in manganese reabsorption in the proximal tubule of the kidney4 and in manganese uptake into the brain.5 It localizes mainly to the cell-surface membrane, but also to lysosomal and mitochondrial membranes.3, 6 ZIP8 is able to transport a number of other divalent cations including zinc,7 cadmium,7 iron,8 and cobalt.8 Although zinc uptake is carried out by other members of the ZIP family as well,6 manganese transport might be a principal endogenous function of ZIP8.7
An Slc39a8 hypomorphic mouse model showed intrauterine growth retardation and died either prenatally or within 48 hr after birth.2 Malformed caved-in skulls, hypoplastic hind limbs, underdeveloped eyes, hypoplastic spleen, kidneys, liver, and lung, and anemia were found. Although several other divalent cations showed decreased tissue concentrations, manganese was undetectable.2
Manganese is an essential trace element. In the first 6 months of life, manganese intake is very low because human milk contains only 15 μg/l.9 Recommended daily intake for adults is 2 mg.10 The trace element is taken up from the proximal small gut as Mn2+ via the divalent metal transporter-1 (DMT1; official name SLC11A2).4 In the blood, Mn2+ is oxidized to Mn3+ by coeruloplasmin, binds to transferrin as the major manganese-binding protein,11 and enters the cell via the transferrin receptor into endosomal vesicles where it is reduced to Mn2+ and exported via DMT1 and perhaps other transporters to the cytoplasm.4 Elimination occurs primarily via the bile, and very little Mn2+ is found in urine.9, 12 Manganese overload causes psychiatric symptoms that can progress to a Parkinsonian-like neurological disorder.10 Mutations in SLC30A10 encoding the manganese exporter SLC30A10 (also called ZnT10) lead to severe hypermanganesemia with similar neurological symptoms (MIM: 613280).13, 14
Manganese-containing metalloenzymes include pyruvate carboxylase, which catalyzes an important anaplerotic reaction of the citric acid cycle; in manganese-deficient animals, magnesium can be substituted for manganese without decreasing pyruvate carboxylase activity.12 Superoxide dismutase-2, which combats free-radical formation, is also manganese dependent, as is glutamine synthase, which converts glutamate to glutamine in astrocytes;10 in glutamine synthase, magnesium can substitute for manganese.15, 16 Glycosyltransferases are known to be manganese-containing enzymes; an extensive literature describes the role of manganese in glycosylation.17 Manganese-deficient animals exhibit skeletal abnormalities with shortened limbs caused by diminished production of N-acetylgalactosamine containing chondroitin sulfate.17 The Golgi-localized enzyme β-1,4-galactosyltransferase is manganese dependent.18
Informed consent from the individuals’ parents was obtained. Approval for investigations and therapy was obtained from the local Bioethics Committee.
Individual A is the first-born daughter of unrelated German parents, delivered after 38 weeks of pregnancy, APGAR scores 9/10/10. Her birth weight was 2,670 g (10th percentile), body length was 46 cm (<3rd percentile), and head circumference was 35 cm (68th percentile). Short limbs and cutaneous syndactylies between the second and third toes of both feet were present (Figure 1). Examination of the placenta revealed a single umbilical artery.
Figure 1.

Clinical Phenotype of SLC39A8 Deficiency
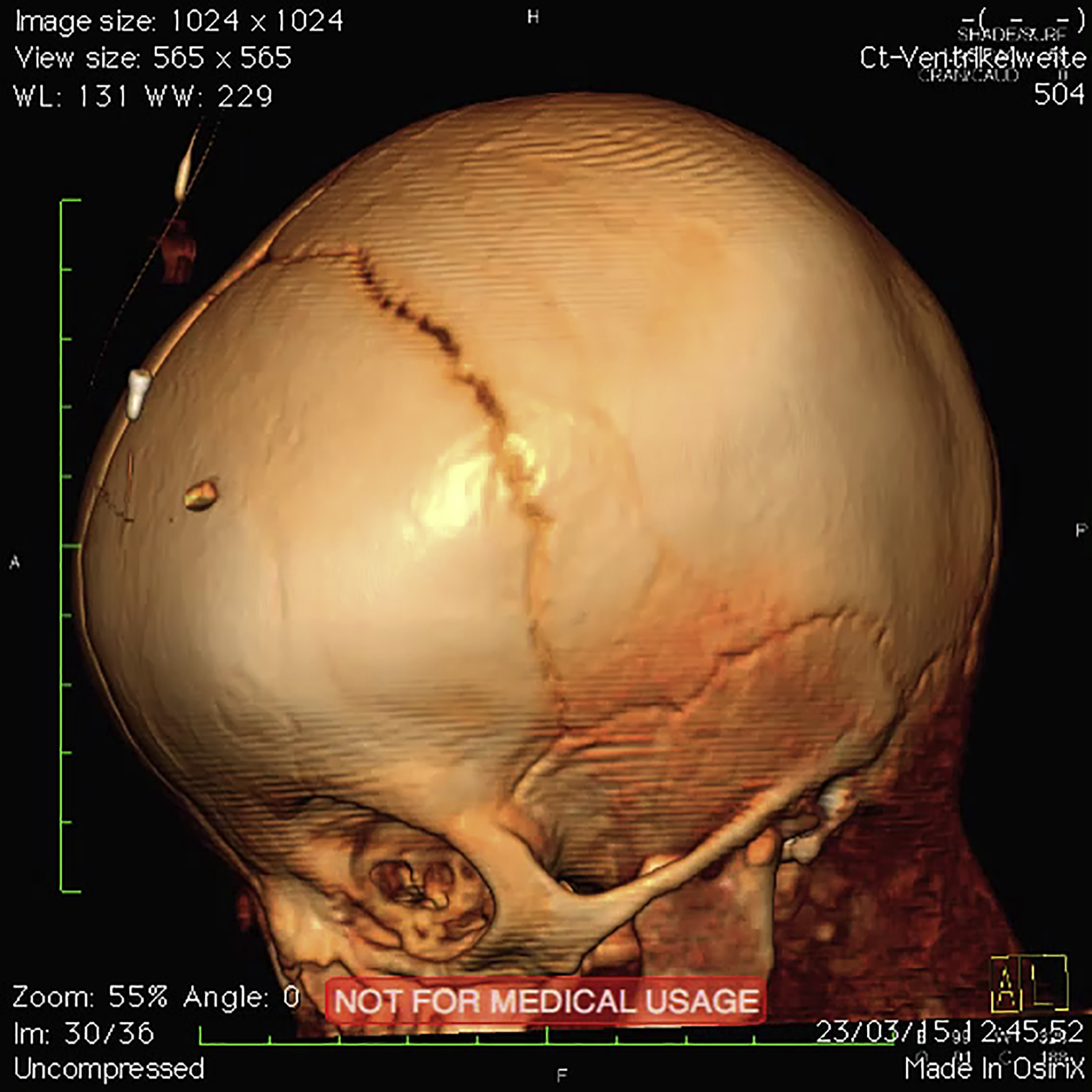
(A) Three-dimensional reconstruction of cranial CT images of individual A at age 4 months demonstrates cranial asymmetry due to premature synostoses of coronary and lambdoid sutures. Note the lacunar skull presenting with regions of apparent thinning on right-hand side of the cranium. Movie S1 shows skull reconstruction.
(B) Cranial MRI at age 6 months revealed asymmetry of the brain with cerebral atrophy of the left hemisphere. The ventricles are enlarged, especially on the left side of the brain. A subdural hygroma is also present.
(C) Photographs of individual A at age 9 months demonstrate several dysmorphic features: divergent strabismus, distinct cranial malformation with asymmetry of the skull, flat face, and low-set ears.
(D) Dysproportionate short stature with short limbs, especially of the lower extremities.
At time of initial referral at 4 months of age, she presented with dysproportionate dwarfism and cranial asymmetry, as well as malformation of the viscerocranium. CT and MRI scans verified craniosynostosis of the coronary and lambdoid sutures with asymmetrical brain atrophy, but normal cerebellum. Intermittent divergent strabismus and absence of visual fixation were noted. Fundoscopy showed no abnormalities. Brain-stem-evoked-response audiometry (BERA) revealed profound hearing impairment of the left ear and moderate impairment of the right ear.
Clusters of infantile spasms with hypsarrythmia on electroencephalography recordings occurred up to five times a day and did not respond to conventional anti-convulsive treatment.
She was started on glucocorticoid therapy,19 but had only temporary improvement. Episodes of apnoea/hypopnoea required administration of oxygen, but declined in frequency and finally resolved in the following months.
At age 7 months, an episode of liver disease occurred with elevated transaminase enzymes (AST: 441 U/l [reference < 80 U/l], ALT: 102 U/l [reference < 55 U/l]) and impaired blood coagulation (INR 1.35 s [reference 0.85–1.15 s], PTT 61 s [reference 28–41 s]). Liver disease resolved over a period of several weeks. Manganese concentrations were always below the detection level in plasma and urine. Serum glutamine (548 μmol/l [reference range 410–760]), glutamate (108 μmol/l [reference range 70–220 μmol/l]), and arginine (38 μmol/l [reference range 40–90]) were unremarkable and did not indicate glutamine synthase deficiency. Lactate (1.61 mmol/l) and pyruvate (0.13 mmol/l) and urinary organic acids were normal without any indication of pyruvate decarboxylase dysfunction. Serum parameters of iron metabolism were unremarkable, as were serum zinc levels (1,137–1,190 μg/l [reference range 750–1,400 μg/l]). Glycosaminoglycan electrophoresis showed normal amounts of chondroitin sulfate, composed of high amounts of N-acetylgalactosamine. Xanthine levels in the urine were elevated.
Due to the complex clinical phenotype, selective metabolic screening included investigation of glycosylation of serum transferrin, the common biomarker used to screen for congenital disorders of glycosylation (CDG).20, 21, 22 Glycosylation was investigated by isoelectric focusing (IEF), immunoprecipitation followed by SDS-PAGE, and high-performance liquid chromatography (HPLC), as described previously.23 Serum transferrin has two N-linked sugar side chains, each bearing two terminal, negatively charged sialic acid residues (tetrasialo-transferrin) (Figure 2A). Loss of a side chain leads to the loss of two sialic acid residues (type I pattern); truncation of the side chain results in uneven loss of sialic acid residues (type II pattern). Because loss of sialic acids results in an overall alteration in the charge of the transferrin molecule, N-glycosylation abnormalities can be visualized by IEF. In contrast to control, transferrin in individual A revealed the loss of one to four sialic acid residues (CDG type II pattern) (Figure 2B). HPLC analysis was performed to quantify transferrin isoforms and showed that only 10.2% of transferrin was present as the correctly glycosylated tetrasialo-transferrin (Figure 3A, day 0). HPLC reference values for transferrin isoforms are as follows: asialo-transferrin 0%, monosialo-transferrin 0%, disialo-transferrin 0.38%–1.82%, trisialo-transferrin 1.16%–6.36%, tetrasialo-transferrin 85.7%–94.0%, and pentasialo-transferrin 2.6%–10.2%.
Figure 2.

Schematic Presentation of Transferrin Glycosylation and Glycosylation Profile of Individual A
(A) Transferrin is glycosylated at two asparagine (Asn) residues. The attached glycans consist of N-acetylglucosamine (blue square), mannose (green circle), galactose (yellow circle), and sialic acid (purple rhomboid). Terminal sialic acid residues are negatively charged, so that truncation or loss of a glycan side chain results in an overall change in charge of the transferrin molecule. The main transferrin species in healthy individuals is tetrasialo-transferrin, having four terminal sialic acids.
(B) The glycosylation profile of individual A is a type II CDG pattern with increased amounts of trisialo-, disialo-, monosialo-, and asialo-transferrin.
Figure 3.

Changes in N-Glycosylation with Galactose Therapy
(A) Transferrin isoforms were quantitated by HPLC from serum samples and expressed as percentage of total; during galactose supplementation, normalization of glycosylation is seen. Galactose was given on day 1–29, interrupted from day 30 to 43, and then reinstated. Note the steep decline in tetrasialo-transferrin in the short interval between the two therapy intervals. Tetrasialo-transferrin reached stable values within the normal range approximately 2 months after initiation of the second therapy interval.
(B) ESI-TOF mass spectra, before and after 120 days of therapy, identifies the partially defective N-glycans attached to serum transferrin. Whereas the mass spectrum before therapy identified mostly transferrin isoforms with defective galactosylation, 120 days of therapy resulted in a normal glycosylation profile with correct galactosylation. The different N-glycan species are indicated below; only one (of sometimes several) possible structures is depicted.
Detailed N-glycan analysis of serum transferrin by electrospray ionization time-of-flight mass spectrometry (ESI-TOF MS) was carried out, as described elsewhere.24 ESI-TOF mass spectrometry of transferrin confirmed the loss of terminal sialic acids (Figure 3B, day 0). However, in addition to sialic acid, the majority of abnormal transferrin isoforms lacked one or more galactose residues, indicating a primary problem in galactosylation rather than sialylation (Figure 3B, day 0: G > F, E > D, C > B).
DNA was isolated with the PAXGene Blood DNA system (PreAnalytiX GmbH) and DNA concentration was measured with the Qubit 2.0 fluorometer (Thermo Fisher Scientific). Whole-exome sequencing (Illumina 2 × 100-bp paired-end reads) was performed on individual A, as described previously.25 Mapping to hg19 (UCSC Genome Browser) delivered 305,077 entries for SNPs and 51,022 for indels, in some cases containing multiple entries for single loci due to multiple transcripts/names. The average coverage was 275× and the total number of reads was 271,309,084. There were no variants in the panel of known CDG subtypes, thus excluding the recognized disorders of galactosylation, B4GALT1-CDG (MIM: 607091) and SLC35A2-CDG (MIM: 300896).26, 27, 28, 29 Due to clinical similarity to SLC35A2-CDG, SLC35A2 transcripts were analyzed in cDNA from individual A’s fibroblasts but exhibited normal size and sequence, thus excluding any intronic splice variants. After several steps of data reduction (2% cut-off regarding minor allele frequency, minor allele versus major allele coverage ratio ≥ 15%, minimal coverage of at least four reads, focusing on variants with a likely functional effect [including potential splice variants], and evaluating them with MutationTaster30), we obtained a list of 141 genes with at least 2 different mutations, 43 genes with mutations that were homozygous or X-chromosomal, and 39 genes with a single mutation that were on imprinted genes or had the potential of dominant effects. Using GeneDestiller 2014 to obtain in-depth information for all genes and reducing them to genes with a potential effect on galactosylation, SLC39A8 was identified as the most likely candidate. SLC39A8 variants (GenBank: NM_022154.5) were c.112G>C (p.Gly38Arg) in exon 1 with a coverage of 257× and c.1019T>A (p.Ile340Asn) in exon 6 with a coverage of 147× in the whole exome sequencing data. Both altered amino-acid residues are highly conserved in the protein (Figure 4C). The p.Gly38Arg missense variant was identified in 2 out of 15,930 alleles from non-Finnish European individuals in the ExAC database. The resulting frequency of 0.0001255 makes this variant very rare among the European population. The p.Ile340Asn variant was not identified previously. Sanger sequencing confirmed the variants in SLC39A8. The mother was heterozygous for the c.1019T>A variant, and the father was heterozygous for the c.112G>C variant. Based on the deleterious-appearing nature of the variants, segregation data, and biochemical evidence of Mn deficiency and the pathological link to glycosylation abnormalities in the individual, SLC39A8 is a strong candidate gene for the disorder in individual A.
Figure 4.

Overview of the Identified Variants and Their Effect on Glycosylation
(A) High-performance liquid chromatography (HPLC) spectra of serum transferrin show an increase in trisialo-transferrin for all affected individuals, when compared to those of a healthy control. The most severe hypoglycosylation is found in the person carrying the p.[Gly38Arg];[p.Ile340Asn] double variant.
(B) Isolectric focusing (IEF) of serum transferrin from three of the individuals described by Boycott et al. in this issue (B2, D4, D5) carrying only the homozygous p.Gly38Arg variant,31 and the two individuals described herein with variants p.[Val33Met; p.Ser335Thr];[p.Gly204Cys] and p.[Gly38Arg];[p.Ile340Asn], respectively. IEF shows increased trisialo-transferrin in all affected persons.
(C) Alignment of the five identified variants in SLC39A8 with amino acid exchanges highlighted in yellow. Indicated in red are amino acids that are conserved throughout all analyzed species. Blue denotes amino acids with inter-species variation. The number indicates the position of the exchanged amino acid. All identified variants result in an amino-acid exchange in highly conserved regions of the ZIP8 protein encoded by SLC39A8.
To identify further evidence that mutations in SLC39A8 cause a disorder of manganese transport and glycosylation, we screened our group of unsolved cases with impaired glycosylation to identify additional affected individuals. We identified a second individual (individual B) with variants in SLC39A8: c.97G>A (p.Val33Met) and c.1004G>C (p.Ser335Thr) on the paternal allele and c.610G>T (p.Gly204Cys) on the maternal allele. Affected amino acids have been conserved during evolution (Figure 4C). Both mutations on the paternal allele are possibly causative although the p.Ser335Thr lies within the transmembrane domain V of ZIP8, which is part of the ion channel. Tetrasialo-transferrin was lowered to 77.74%, trisialo-transferrin was increased to 19.48%, and monosialo-transferrin was increased to 1.27% (Figures 4A and 4B). Individual 2, a girl, is the first child of a non-consanguineous couple born at term after an uneventful pregnancy with a birth weight of 2,720 g (3rd percentile), a length of 46 cm (<3rd percentile), and a head circumference of 34 cm (<97th percentile). Global psychomotor retardation became evident in the first year of life. In early childhood, epilepsy occurred and she was treated with anticonvulsants. MRI revealed cerebellar atrophy. Currently, at 19 years of age, the young woman is hypotonic without spasticity, shows severe scoliosis, and is confined to a wheelchair without being able to sit or walk without support. She no longer has seizures and can form two-word sentences. Head circumference is 54.5 cm (50th percentile), height is 122 cm (>3rd percentile), and weight is 22 kg (>3rd percentile). Increased laxity of the wrists and mild contractures of the elbow and knee are present. Previously inverted nipples have everted. Ophthalmological signs include hyperopia, astigmatism, and strabismus. Nystagmus occurs sporadically after voluntary eye movements. Assessment of manganese in whole blood and urine revealed manganese concentrations below the detection level.
At this time we became aware of a second group of individuals with variants in SLC39A8 and an autosomal-recessive syndrome characterized by intellectual disability and cerebellar atrophy (described by Boycott et al. in an accompanying paper31). Glycosylation studies were then carried out in persons B2, D4, and D5 from this study. All of them had abnormal transferrin glycosylation patterns with decreased tetrasialo-transferrin (81.94%–83.67% [reference range 85.7%–94.0%]), increased trisialo-transferrin (11.88%–12.85% [reference range 1.16%–6.36%]), and increased monosialo-transferrin (0.31%–0.46% [reference range 0%]) (Figures 4A and 4B).
Given the severe clinical presentation of individual A, we attempted to improve the impaired galactosylation during N-glycan formation by increasing her dietary galactose intake. Daily galactose intake was raised from 1 g to 2 g/kg body weight within 2 weeks, given in 5 daily doses. After 4 weeks, galactose was discontinued for 2 weeks. Galactose therapy was reinitiated and increased to a daily dose of 3.75 g/kg body weight given over 22 hr via gastrointestinal pump feeding. Uridine (150 mg/kg bodyweight) was added to ensure that sufficient uridine was available for UDP-galactose formation.32 Stool sampling for reducing substances was negative––indicating complete galactose uptake. Galactose and uridine were supplied by Vitaflo Pharma.
Galactose supplementation resulted in dramatic improvements in glycosylation. The amount of normally glycosylated tetrasialo-transferrin before treatment was severely decreased to 10.16% (HPLC reference range 85.7%–94.0%) with strong elevations of hypogalactosylated transferrin isoforms. After galactose therapy, glycosylation became completely normalized with a tetrasialo-transferrin of 87.74% (Figure 3A). Only trisialo-transferrin was still slightly increased after 4 months of treatment (7.7% [HPLC reference range 1.16%–6.36%]). Improvement of galactosylation was instantaneous, apparently dependent only on transferrin biosynthesis, which has a biological half life of 7–10 days.33 ESI-TOF mass spectrometry of transferrin confirmed the complete normalization of previously severely hypoglycosylated transferrin (Figure 3B, day 120).
The underlying pathomechanism of SLC39A8 deficiency links a trace element deficiency to an inherited glycosylation disorder. Glycosylation is an essential co- and post-translational modification that occurs in about half the proteins synthesized in the cell. N-glycosylation takes place on asparagine residues that acquire a preassembled oligosaccharide side chain in the endoplasmic reticulum, which is subsequently modified extensively in the Golgi apparatus. Ultimately, the proteins obtain branched oligosaccharide side chains with terminal galactose and sialic-acid residues.
Complete inhibition of N-glycosylation is incompatible with life. Leaky mutations in genes coding for any of the glycosyltransferases or sugar transporters involved in the process are responsible for a group of severe inherited metabolic disorders called congenital disorders of glycosylation (CDG). Disruption of SLC35A2, which encodes the UDP-galactose transporter that ensures Golgi import of UDP-galactose, leads to a particularly severe disorder presenting with severe seizures beginning in infancy, profound statomotor retardation, brain atrophy, impaired vision and hearing, and severe scoliosis.29 SLC35A2 is located on the X chromosome; only boys with mosaicism and girls have been described with this disease. In boys without mosaicism, the disease is probably lethal during intrauterine development. Transferrin, a serum protein often used for selective CDG screening, is hypogalactosylated in SLC35A2 deficiency.29
The fact that complete inhibition of N-glycosylation is incompatible with life might reflect the finding that the mouse and human SLC39A8 is expressed as early as the gastrula stage,34 and in visceral endoderm at GD7.5,35 and in fact is used as a potential indicator of cell differentiation (self-renewal-related signaling) in embryonic stem cells.36 That the loss of this gene is incompatible with life is consistent with mouse embryos having total ablation of Slc39a8: embryonic lethality of Slc39a8–/– knockout pups takes place no more than 2–3 days after implantation.37
SLC39A8 deficiency in its severe form has a striking similarity to UDP-galactose transporter deficiency. In fact, clinical presentation and transferrin hypogalactosylation led to the initial suspicion of SLC35A2-CDG in individual A described herein.
Galactose transfer to a vast number of acceptor proteins is carried out by the so-called galactosyltransferases (GalT), a group of enzymes located in the Golgi apparatus.38 Galactosylation is carried out mainly by the enzymes UDP-Gal:N-acetylglucosamine β-1,4-galactosyltransferase I (B4GALT1; EC 2.4.1.22) and UDP-Gal:N-acetylglucosamine β-1,4-galactosyltransferase II (B4GALT2; EC 2.4.1.22).39
Several experimental studies have investigated the importance of divalent cations for galactosyltransferases. GalT activation is absolutely metal-ion dependent, with divalent manganese being the most potent and natural activator.40, 41 Manganese attaches to two different binding sites. The high-affinity site I binds manganese with a Km value of 0.4 μM in intact Golgi vesicles; complete absence of manganese leads to no residual enzyme activity.41 The low-affinity activator site II binds manganese with a Km value of 440 μM.41 Site I will accept zinc instead of manganese, resulting in enzyme activity that is decreased by more than 60%; it does not bind magnesium or calcium.40, 41 In the absence of manganese, calcium can substitute at site II, resulting in 25-fold poorer affinity for UDP-galactose.40
There were no detectable serum or urinary manganese levels in individual A. Due to the manganese dependence of β-1,4-galactosyltransferase involved in N-glycosylation, severe transferrin hypogalactosylation occurred. The enzyme activity curve at low manganese concentrations has a hyperbolic shape, so that small changes in manganese concentration will have strong effects on the galactosylation activity.41 Accordingly, the case subjects described by Boycott et al. with diminished (but measurable) manganese concentrations had impaired, but less severe, hypoglycosylation defects. Whereas the p.Gly38Arg substitution was found in homozygous form in more mildly affected individuals,31 the severe phenotype in the subject described here, and complete absence of manganese in body fluids, must therefore reflect the p.Ile340Asn substitution, which affects the highly conserved transmembrane domain V of ZIP8; this domain is part of the ion channel.3
Dietary supplementation of galactose alone, or galactose in combination with uridine, improves galactosylation by increasing the intracellular UDP-galactose pool.42 In SLC39A8 deficiency, low manganese concentrations probably will decrease the affinity of galactosyltransferase for UDP-galactose. Increasing the UDP-galactose pool by galactose and uridine supplementation completely restores galactosylation. The similarities of SLC39A8 and SLC35A2 deficiencies suggest that the secondary glycosylation abnormality has a major role in pathogenesis of this disease, which might become normalized by simple dietary changes.
Dietary manganese deficiency leads to bone and connective tissue disease in animals.43 Although skeletal abnormalities were present in individual A, N-acetylgalactosamine containing chondroitin sulfate was found in normalconcentrations in the urine. There were no biochemical indications of impairment of other manganese-dependent metalloenzymes, e.g., arginase,44 glutamine synthase,15, 16 or pyruvate carboxylase.45 However, elevated xanthine excretion in the urine indicated decreased activity of manganese-dependent xanthine oxidase.46
Although the pleiotropic phenotype varies, the known affected individuals share common features, which might facilitate the diagnosis of SLC39A8 deficiency in further individuals. The known affected individuals share delayed milestones, severe psychomotor retardation, seizures, strabismus, and impaired growth in the majority of affected individuals. Whereas most of the individuals presented by Boycott et al., as well as individual B of this study, exhibit severe cerebellar atrophy, this was not detected in individual A who had generalized brain atrophy with severe hypsarrhythmia. Skeletal abnormalities—although not detected in every affected individual—might facilitate recognition of additional cases at an early stage. The particularly severe phenotype observed in individual A possibly reflects the impact of the amino acid substitutions in highly conserved regions of ZIP8, especially within the transmembrane domain V affecting the ion channel.
Defective glycosylation of serum transferrin is a shared laboratory parameter and should be tested for in any case of developmental delay and dysmorphic features of unknown origin.
In summary, our data show that disruption at highly conserved sites of the ZIP8 symporter alters function of the protein, resulting in low serum manganese levels, probably through insufficient renal reabsorption, intestinal manganese resorption, or both. ZIP8 appears to be more important for manganese than zinc homeostasis. Decreased serum manganese concentrations impair the function of galactosyltransferases, linking for the first time a trace element deficiency with inherited glycosylation disorders. Correcting hypogalactosylation by dietary galactose supplementation might well be the single most important therapeutic step for individuals with SCL39A8 deficiency. Because transferrin is the major manganese-binding protein in vascular circulation, correction of transferrin glycosylation, prior to attempting manganese supplementation, might also be reasonable,11 especially because manganese uptake across the blood-brain-barrier is known to be transferrin dependent.4 Additional benefits might occur with manganese supplementation. Manganese sulfate is the most soluble salt and the one normally used in nutritional supplements.47 Given the long-term consequences of the severe presentations of SLC39A8 deficiency such as seizures on the developing brain, early galactose and manganese supplementation should be considered. The promising effects observed from dietary galactose supplementation on glycosylation by serum transferrin analysis will require further evaluation, particularly with regard to the clinical course.
Acknowledgments
We are grateful to the families for their support during the conception of this study. This work was supported, in part, by NIH grants R01 ES010416 and P30 ES06096 (to D.W.N.).
Published: December 3, 2015
Footnotes
Supplemental Data include one movie and can be found with this article online at http://dx.doi.org/10.1016/j.ajhg.2015.11.003.
Accession Numbers
The accession numbers for the data reported in this paper are ClinVar: SCV000256232, SCV000256231, and SCV000256230.
Web Resources
The URLs for data presented herein are as follows:
ExAC Browser, http://exac.broadinstitute.org/
GeneDistiller 2014, http://www.genedistiller.org
MutationTaster, http://www.mutationtaster.org/
NCBI Nucleotide, http://www.ncbi.nlm.nih.gov/nuccore/
OMIM, http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
Supplemental Data
Note the synostoses of the coronary and lambdoid suture.
{kind=link}
References
- 1.He L., Vasiliou K., Nebert D.W. Analysis and update of the human solute carrier (SLC) gene superfamily. Hum. Genomics. 2009;3:195–206. doi: 10.1186/1479-7364-3-2-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gálvez-Peralta M., He L., Jorge-Nebert L.F., Wang B., Miller M.L., Eppert B.L., Afton S., Nebert D.W. ZIP8 zinc transporter: indispensable role for both multiple-organ organogenesis and hematopoiesis in utero. PLoS ONE. 2012;7:e36055. doi: 10.1371/journal.pone.0036055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jenkitkasemwong S., Wang C.Y., Mackenzie B., Knutson M.D. Physiologic implications of metal-ion transport by ZIP14 and ZIP8. Biometals. 2012;25:643–655. doi: 10.1007/s10534-012-9526-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tuschl K., Mills P.B., Clayton P.T. Manganese and the brain. Int. Rev. Neurobiol. 2013;110:277–312. doi: 10.1016/B978-0-12-410502-7.00013-2. [DOI] [PubMed] [Google Scholar]
- 5.Rivera-Mancía S., Ríos C., Montes S. Manganese accumulation in the CNS and associated pathologies. Biometals. 2011;24:811–825. doi: 10.1007/s10534-011-9454-1. [DOI] [PubMed] [Google Scholar]
- 6.Eide D.J. Zinc transporters and the cellular trafficking of zinc. Biochim. Biophys. Acta. 2006;1763:711–722. doi: 10.1016/j.bbamcr.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 7.He L., Girijashanker K., Dalton T.P., Reed J., Li H., Soleimani M., Nebert D.W. ZIP8, member of the solute-carrier-39 (SLC39) metal-transporter family: characterization of transporter properties. Mol. Pharmacol. 2006;70:171–180. doi: 10.1124/mol.106.024521. [DOI] [PubMed] [Google Scholar]
- 8.Wang C.Y., Jenkitkasemwong S., Duarte S., Sparkman B.K., Shawki A., Mackenzie B., Knutson M.D. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J. Biol. Chem. 2012;287:34032–34043. doi: 10.1074/jbc.M112.367284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anonymous. Manganese deficiency in humans: fact or fiction? Nutr. Rev. 1988;46:348–352. doi: 10.1111/j.1753-4887.1988.tb05360.x. [DOI] [PubMed] [Google Scholar]
- 10.Avila D.S., Puntel R.L., Aschner M. Manganese in health and disease. Met. Ions Life Sci. 2013;13:199–227. doi: 10.1007/978-94-007-7500-8_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herrera C., Pettiglio M.A., Bartnikas T.B. Investigating the role of transferrin in the distribution of iron, manganese, copper, and zinc. J. Biol. Inorg. Chem. 2014;19:869–877. doi: 10.1007/s00775-014-1118-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leach R.M., Lilburn M.S. Manganese metabolism and its function. World Rev. Nutr. Diet. 1978;32:123–134. doi: 10.1159/000401764. [DOI] [PubMed] [Google Scholar]
- 13.Tuschl K., Clayton P.T., Gospe S.M., Jr., Gulab S., Ibrahim S., Singhi P., Aulakh R., Ribeiro R.T., Barsottini O.G., Zaki M.S. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am. J. Hum. Genet. 2012;90:457–466. doi: 10.1016/j.ajhg.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quadri M., Federico A., Zhao T., Breedveld G.J., Battisti C., Delnooz C., Severijnen L.A., Di Toro Mammarella L., Mignarri A., Monti L. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am. J. Hum. Genet. 2012;90:467–477. doi: 10.1016/j.ajhg.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maurizi M.R., Pinkofsky H.B., Ginsburg A. ADP, chloride ion, and metal ion binding to bovine brain glutamine synthetase. Biochemistry. 1987;26:5023–5031. doi: 10.1021/bi00390a021. [DOI] [PubMed] [Google Scholar]
- 16.Krajewski W.W., Collins R., Holmberg-Schiavone L., Jones T.A., Karlberg T., Mowbray S.L. Crystal structures of mammalian glutamine synthetases illustrate substrate-induced conformational changes and provide opportunities for drug and herbicide design. J. Mol. Biol. 2008;375:217–228. doi: 10.1016/j.jmb.2007.10.029. [DOI] [PubMed] [Google Scholar]
- 17.Leach R.M., Jr. Role of manganese in mucopolysaccharide metabolism. Fed. Proc. 1971;30:991–994. [PubMed] [Google Scholar]
- 18.Ramakrishnan B., Ramasamy V., Qasba P.K. Structural snapshots of beta-1,4-galactosyltransferase-I along the kinetic pathway. J. Mol. Biol. 2006;357:1619–1633. doi: 10.1016/j.jmb.2006.01.088. [DOI] [PubMed] [Google Scholar]
- 19.Lux A.L., Edwards S.W., Hancock E., Johnson A.L., Kennedy C.R., Newton R.W., O’Callaghan F.J., Verity C.M., Osborne J.P. The United Kingdom Infantile Spasms Study comparing vigabatrin with prednisolone or tetracosactide at 14 days: a multicentre, randomised controlled trial. Lancet. 2004;364:1773–1778. doi: 10.1016/S0140-6736(04)17400-X. [DOI] [PubMed] [Google Scholar]
- 20.Jaeken J. Congenital disorders of glycosylation (CDG): it’s (nearly) all in it! J. Inherit. Metab. Dis. 2011;34:853–858. doi: 10.1007/s10545-011-9299-3. [DOI] [PubMed] [Google Scholar]
- 21.Freeze H.H. Understanding human glycosylation disorders: biochemistry leads the charge. J. Biol. Chem. 2013;288:6936–6945. doi: 10.1074/jbc.R112.429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marquardt T., Denecke J. Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur. J. Pediatr. 2003;162:359–379. doi: 10.1007/s00431-002-1136-0. [DOI] [PubMed] [Google Scholar]
- 23.Zühlsdorf A., Park J.H., Wada Y., Rust S., Reunert J., DuChesne I., Grüneberg M., Marquardt T. Transferrin variants: pitfalls in the diagnostics of congenital disorders of glycosylation. Clin. Biochem. 2015;48:11–13. doi: 10.1016/j.clinbiochem.2014.09.022. [DOI] [PubMed] [Google Scholar]
- 24.Park J.H., Zühlsdorf A., Wada Y., Roll C., Rust S., Du Chesne I., Grüneberg M., Reunert J., Marquardt T. The novel transferrin E592A variant impairs the diagnostics of congenital disorders of glycosylation. Clin. Chim. Acta. 2014;436:135–139. doi: 10.1016/j.cca.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 25.Biskup S. Hochdurchsatz-Sequenzierung in der Humangenetischen Diagnostik. Next-generation sequencing in genetic diagnostics. J. Lab. Med. 2010;34:305–309. [Google Scholar]
- 26.Guillard M., Morava E., van Delft F.L., Hague R., Körner C., Adamowicz M., Wevers R.A., Lefeber D.J. Plasma N-glycan profiling by mass spectrometry for congenital disorders of glycosylation type II. Clin. Chem. 2011;57:593–602. doi: 10.1373/clinchem.2010.153635. [DOI] [PubMed] [Google Scholar]
- 27.Hansske B., Thiel C., Lübke T., Hasilik M., Höning S., Peters V., Heidemann P.H., Hoffmann G.F., Berger E.G., von Figura K., Körner C. Deficiency of UDP-galactose:N-acetylglucosamine beta-1,4-galactosyltransferase I causes the congenital disorder of glycosylation type IId. J. Clin. Invest. 2002;109:725–733. doi: 10.1172/JCI14010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng B.G., Buckingham K.J., Raymond K., Kircher M., Turner E.H., He M., Smith J.D., Eroshkin A., Szybowska M., Losfeld M.E., University of Washington Center for Mendelian Genomics Mosaicism of the UDP-galactose transporter SLC35A2 causes a congenital disorder of glycosylation. Am. J. Hum. Genet. 2013;92:632–636. doi: 10.1016/j.ajhg.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dörre K., Olczak M., Wada Y., Sosicka P., Grüneberg M., Reunert J., Kurlemann G., Fiedler B., Biskup S., Hörtnagel K. A new case of UDP-galactose transporter deficiency (SLC35A2-CDG): molecular basis, clinical phenotype, and therapeutic approach. J. Inherit. Metab. Dis. 2015;38:931–940. doi: 10.1007/s10545-015-9828-6. [DOI] [PubMed] [Google Scholar]
- 30.Schwarz J.M., Cooper D.N., Schuelke M., Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods. 2014;11:361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 31.Boycott K.M., Beaulieu C.L., Kernohan K.D., Gebril O.H., Mhanni A., Chudley A.E., Redl D., Qin W., Hampson S., Küry S., Care4Rare Canada Consortium Autosomal-recessive intellectual disability with cerebellar atrophy syndrome caused by mutation of the manganese and zinc transporter gene SLC39A8. Am. J. Hum. Genet. 2015;97:886–893. doi: 10.1016/j.ajhg.2015.11.002. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manis F.R., Cohn L.B., McBride-Chang C., Wolff J.A., Kaufman F.R. A longitudinal study of cognitive functioning in patients with classical galactosaemia, including a cohort treated with oral uridine. J. Inherit. Metab. Dis. 1997;20:549–555. doi: 10.1023/a:1005357622551. [DOI] [PubMed] [Google Scholar]
- 33.Kim B.J., Zhou J., Martin B., Carlson O.D., Maudsley S., Greig N.H., Mattson M.P., Ladenheim E.E., Wustner J., Turner A. Transferrin fusion technology: a novel approach to prolonging biological half-life of insulinotropic peptides. J. Pharmacol. Exp. Ther. 2010;334:682–692. doi: 10.1124/jpet.110.166470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harrison S.M., Dunwoodie S.L., Arkell R.M., Lehrach H., Beddington R.S. Isolation of novel tissue-specific genes from cDNA libraries representing the individual tissue constituents of the gastrulating mouse embryo. Development. 1995;121:2479–2489. doi: 10.1242/dev.121.8.2479. [DOI] [PubMed] [Google Scholar]
- 35.Moore-Scott B.A., Opoka R., Lin S.C., Kordich J.J., Wells J.M. Identification of molecular markers that are expressed in discrete anterior-posterior domains of the endoderm from the gastrula stage to mid-gestation. Dev. Dyn. 2007;236:1997–2003. doi: 10.1002/dvdy.21204. [DOI] [PubMed] [Google Scholar]
- 36.Zhu H., Yang H., Owen M.R. Combined microarray analysis uncovers self-renewal related signaling in mouse embryonic stem cells. Syst. Synth. Biol. 2007;1:171–181. doi: 10.1007/s11693-008-9015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang B., He L., Dong H., Dalton T.P., Nebert D.W. Generation of a Slc39a8 hypomorph mouse: markedly decreased ZIP8 Zn2+/(HCO3–)2 transporter expression. Biochem. Biophys. Res. Commun. 2011;410:289–294. doi: 10.1016/j.bbrc.2011.05.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hennet T. The galactosyltransferase family. Cell. Mol. Life Sci. 2002;59:1081–1095. doi: 10.1007/s00018-002-8489-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo S., Sato T., Shirane K., Furukawa K. Galactosylation of N-linked oligosaccharides by human beta-1,4-galactosyltransferases I, II, III, IV, V, and VI expressed in Sf-9 cells. Glycobiology. 2001;11:813–820. doi: 10.1093/glycob/11.10.813. [DOI] [PubMed] [Google Scholar]
- 40.Powell J.T., Brew K. Metal ion activation of galactosyltransferase. J. Biol. Chem. 1976;251:3645–3652. [PubMed] [Google Scholar]
- 41.Kuhn N.J., Ward S., Leong W.S. Submicromolar manganese dependence of Golgi vesicular galactosyltransferase (lactose synthetase) Eur. J. Biochem. 1991;195:243–250. doi: 10.1111/j.1432-1033.1991.tb15700.x. [DOI] [PubMed] [Google Scholar]
- 42.Tegtmeyer L.C., Rust S., van Scherpenzeel M., Ng B.G., Losfeld M.E., Timal S., Raymond K., He P., Ichikawa M., Veltman J. Multiple phenotypes in phosphoglucomutase 1 deficiency. N. Engl. J. Med. 2014;370:533–542. doi: 10.1056/NEJMoa1206605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hidiroglou M., Ivan M., Bryan M.K., Ribble C.S., Janzen E.D., Proulx J.G., Elliot J.I. Assessment of the role of manganese in congenital joint laxity and dwarfism in calves. Ann. Rech. Vet. 1990;21:281–284. [PubMed] [Google Scholar]
- 44.Kuhn N.J., Ward S., Piponski M., Young T.W. Purification of human hepatic arginase and its manganese (II)-dependent and pH-dependent interconversion between active and inactive forms: a possible pH-sensing function of the enzyme on the ornithine cycle. Arch. Biochem. Biophys. 1995;320:24–34. doi: 10.1006/abbi.1995.1338. [DOI] [PubMed] [Google Scholar]
- 45.Keen C.L., Ensunsa J.L., Watson M.H., Baly D.L., Donovan S.M., Monaco M.H., Clegg M.S. Nutritional aspects of manganese from experimental studies. Neurotoxicology. 1999;20:213–223. [PubMed] [Google Scholar]
- 46.Schroeder H.A., Balassa J.J., Tipton I.H. Essential trace metals in man: manganese. A study in homeostasis. J. Chronic Dis. 1966;19:545–571. doi: 10.1016/0021-9681(66)90094-4. [DOI] [PubMed] [Google Scholar]
- 47.Wong-Valle J., Henry P.R., Ammerman C.B., Rao P.V. Estimation of the relative bioavailability of manganese sources for sheep. J. Anim. Sci. 1989;67:2409–2414. doi: 10.2527/jas1989.6792409x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note the synostoses of the coronary and lambdoid suture.