

Abstract
A new catalytic asymmetric desymmetrization reaction for the synthesis of enantioenriched derivatives of 2-azabicyclo[3.3.1]nonane, a key motif common to many alkaloids, has been developed. Employing a cyclohexanediamine-derived primary amine organocatalyst, a range of prochiral cyclohexanone derivatives possessing an α,β-unsaturated ester moiety linked to the 4-position afforded the bicyclic products, which possess three stereogenic centers, as single diastereoisomers in high enantioselectivity (83–99 % ee) and in good yields (60–90 %). Calculations revealed that stepwise C–C bond formation and proton transfer via a chair-shaped transition state dictate the exclusive endo selectivity and enabled the development of a highly enantioselective primary amine catalyst.
Keywords: desymmetrization, enamine catalysis, Michael addition, organocatalysis, quantum-chemical calculations
The morphan structural motif (2-azabicyclo[3.3.1]nonane) is common to many biologically relevant alkaloid natural products. This core subunit is found within over 300 natural products, including the strychnos, daphniphyllum, and madangamine families.1 Furthermore, it is present in many other biologically relevant molecules, such as the immunosuppressant FR901483, the cytotoxic agent aspernomine, and the analgesic morphine.1a
As part of our research program directed towards the synthesis of various alkaloid natural products, including daphniphyllum and manzamine2 targets, we sought to develop a new catalytic asymmetric method to access the morphan motif with high efficiency and selectivity. Retrosynthetic analysis revealed that a direct approach could exploit a desymmetrization3 of prochiral ketone I by an intramolecular Michael addition reaction to an α,β-unsaturated ester under enamine catalysis.4 Although aldol variants of this type are known,5 such a catalytic asymmetric Michael reaction has not been reported to date, despite its potential to directly provide morphan skeleton II, which possesses three stereocenters and a two carbon appendage useful for subsequent synthetic manipulation (Scheme 1).6 Accordingly, we viewed this proposed desymmetrization reaction as a good opportunity to unveil new reactivity in organocatalysis whilst accessing key bicyclic building blocks that are useful for the synthesis of morphan-like natural product libraries.
scheme 1.
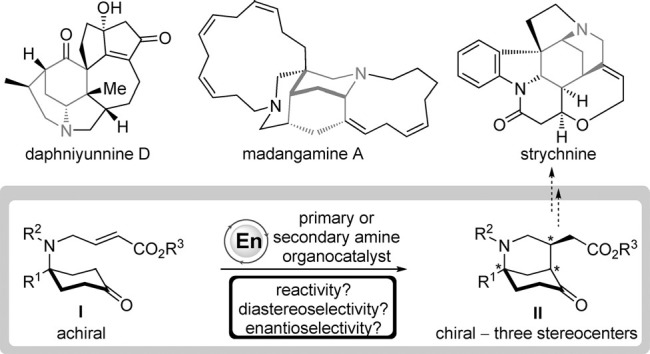
Desymmetrization strategy for the generation of morphans.
Initially, substrate 2 a was chosen as a model system to test our concept; its precursor 1 was accessible on scale,7 the spirocyclic pyrrolidinone backbone would place reactive functionality in close proximity,8 and its synthesis by cross metathesis would provide a point of diversity if the desymmetrization proved successful.
Proof of concept was established quickly and unexpectedly; the attempted purification of 2 a from ruthenium residues that had remained from the cross metathesis reaction with QuadraSil AP, a propylamine-functionalized silica gel scavenger, facilitated the formation of cyclized product (±)-3 a in quantitative yield and excellent diastereoselectivity (Scheme 2). Control experiments using propylamine in CH2Cl2 at room temperature, with and without additional benzoic acid as a co-catalyst, afforded the same product in high yields as a single diastereomer, and the primary amine functionality was thus identified as a catalytically competent species.
scheme 2.
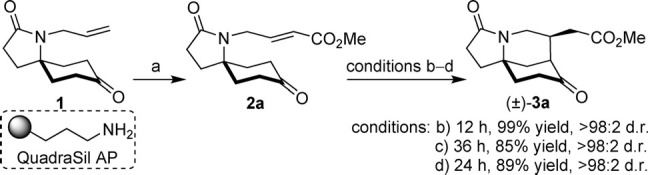
Synthesis of a model substrate and proof-of-concept transformations. Reagents and conditions: a) Hoveyda–Grubbs II catalyst, methyl acrylate, CH2Cl2, 45 °C, 48 h; b) QuadraSil AP, 0.5 mg per mg of substrate, CH2Cl2, RT; c) propylamine (20 mol %), CH2Cl2, RT; d) propylamine (20 mol %), PhCO2H (20 mol %), CH2Cl2, RT.
Consequently, a range of commonly used chiral single-enantiomer primary9 and secondary10 amine organocatalysts 4 a–4 e were screened at 20 mol % loading against the model system in the presence of benzoic acid as a co-catalyst11 (Table 1). In terms of reactivity and enantioselectivity, (1R,2R)-cyclohexanediamine (4 e) was the most promising lead, and accordingly, derivatives were sought with the aim to boost enantioselectivity. Commercially available (1R,2R)-trans-N-Boc-1,2-cyclohexanediamine (4 j) gave similar results to 4 e. However, Jacobsen’s thiourea catalyst 4 k,4d with its increased hydrogen-bond-donor ability arising from the thiourea moiety,12 resulted in a significant increase in enantioselectivity whilst maintaining a short reaction time and high diastereoselectivity; the major diastereomeric product 3 a was obtained in 90 % ee and >98:2 d.r. The loading of the primary amine catalyst could be reduced to 5 mol % with a nominal increase in enantioselectivity albeit with a longer reaction time, which was subsequently overcome by elevating the reaction temperature to 45 °C.
Table 1.
Reaction development and optimization.
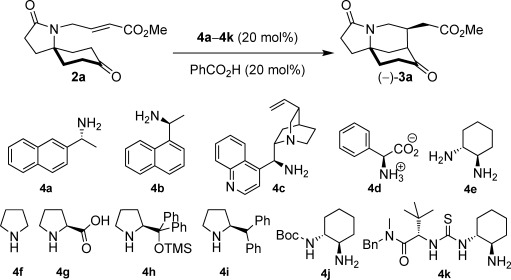
| Entry | Cat. | t | Yield[a] [%] | d.r.[b] | ee[c] [%] |
|---|---|---|---|---|---|
| 1 | 4 a | 5 days | 72 | >98:2 | 63 |
| 2 | 4 b | 8 days | 74 | >98:2 | 69[d] |
| 3 | 4 c | 7 days | 69 | >98:2 | 31 |
| 4 | 4 d | NR | – | – | – |
| 5 | 4 e | 24 h | 82 | >98:2 | 63 |
| 6 | 4 f–4 i | NR | – | – | – |
| 7 | 4 j | 22 h | 87 | >98:2 | 64 |
| 8 | 4 k | 26 h | 86 | >98:2 | 90 |
| 9[e] | 4 k | 25 h | 78 | >98:2 | 90 |
| 10[f] | 4 k | 96 h | 80 | >98:2 | 92 |
| 11[g] | 4 k | 48 h | 88 | >98:2 | 93 |
Yield of isolated product after flash column chromatography.
Diastereomeric ratios (d.r.) were determined by 1H NMR spectroscopy.
The ee values were determined by HPLC analysis on a chiral stationary phase.
(+)-3 a was obtained.
CHCl3 as the solvent.
4 k (5 mol %), benzoic acid (1.25 mol %), RT.
4 k (5 mol %), benzoic acid (1.25 mol %), 45 °C. See the Supporting Information for details. Bn=benzyl, Boc=tert-butyloxycarbonyl, NR=no reaction. TMS=trimethylsilyl.
With optimized conditions established, the scope of the reaction with respect to the scaffold and the ester group was assessed (Table 2). Initially, changes to the ester group on the spirocyclic pyrrolidinone-containing construct were investigated (2 a—2 d), and pleasingly, the results for various esters were consistently good; enantioselectivities ranged from 90 to 93 % ee, and yields above 80 % were achieved (entries 1–4). Variations to the prochiral scaffold were next investigated. The spirocyclic pyrrolidine-containing α,β-unsaturated esters 2 e–2 h were excellent substrates, although it was necessary to increase the benzoic acid co-catalyst loading to 2.5 mol % to maintain good reaction rates (entries 5–8). Pleasingly a non-spirocyclic substrate possessing N-ethyl and 4-methyl substituents underwent cyclization with equally high diastereo- and enantiocontrol (96 % ee) but at a marginally diminished reaction rate (entry 9). Related secondary amine substrates 2 j and 2 k possessing larger C4 substituents also reacted to afford the N-unprotected morphan products 3 j and 3 k as single diastereomers with 84 and 83 % ee, respectively (entries 10 and 11). Substrates with a hydrogen atom at the C4 position would be the most relevant to natural product synthesis (Scheme 1), and accordingly, this scaffold type was assessed in the desymmetrization reaction. A series of ester substrates, 2 l–2 q, each possessing an N-benzyl protecting group, were then examined. Pleasingly, these were found to give the highest enantioselectivities (96-99 % ee) of all of the scaffolds tested, albeit with diminished reaction rates (entries 12–17). Furthermore, a range of differentially N-protected substrates 2 r–2 v gave the desired cyclized products 3 r–3 v with excellent enantioselectivities (95–98 % ee; entries 18–22). In total, 22 unactivated α,β-unsaturated esters substrates with three points of diversity successfully cyclized under the action of catalyst 4 k to give the bicyclic products with the morphan skeleton in high diastereo- and enantioselectivity. The relative stereochemical configuration of 3 p and the absolute stereochemical configuration of a sulfonamide derivative of 3 r were established by single-crystal X-ray analysis (see the Supporting Information).
Table 2.
Scope of the intramolecular desymmetrization.[a]
| Entry | Product | R | t [h] | Yield[b] [%] | ee[c] [%] | |
|---|---|---|---|---|---|---|
| 1 |  |
3 a | Me | 40 | 88 | 93 |
| 2 | 3 b[d] | Et | 40 | 89 | 90 | |
| 3 | 3 c | tBu | 45 | 87 | 92 | |
| 4 | 3 d | Bn | 40 | 81 | 92 | |
| 5 | 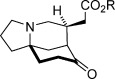 |
3 e | Me | 36 | 83 | 94 |
| 6 | 3 f | Et | 36 | 89 | 94 | |
| 7 | 3 g | Bn | 36 | 85 | 95 | |
| 8 | 3 h | Ph | 36 | 83 | 94 | |
| 9 | 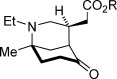 |
3 i | Et | 96 | 83 | 96 |
| 10 | 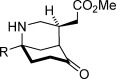 |
3 j | Ph(CH2)2 | 96 | 92 | 84 |
| 11 | 3 k | CH3(CH2)16 | 96 | 86 | 83 | |
| 12 | 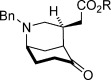 |
3 l | Et | 84 | 84 | 96 |
| 13 | 3 m | Bn | 84 | 89 | 97 | |
| 14 | 3 n | Cy | 120 | 85 | 96 | |
| 15 | 3 o | iPr | 120 | 76 | 99 | |
| 16 | 3 p | Me | 96 | 90 | 97 | |
| 17 | 3 q | Ph | 96 | 72 | 97 | |
| 18 | 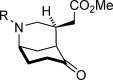 |
3 r | Boc | 96 | 85 | 98 |
| 19 | 3 s | allyl | 96 | 87 | 97 | |
| 20 | 3 t | DPP | 96 | 79 | 95 | |
| 21 | 3 u | CH2(CH)2CO2Me | 96 | 90 | 97 | |
| 22 | 3 v | Me | 96 | 84 | 96 |
Catalyst 4 k (5 mol %), PhCO2H (1.25 mol % for 3 a–d, 2.5 mol % for 3 e–v), CH2Cl2 (0.2 m), 45 °C (3 a–i) or 50 °C (3 j–v).
Yield of isolated product after flash column chromatography (d.r. >98:2 for all products as determined by 1H NMR spectroscopy).
The ee values were determined by HPLC analysis on a chiral stationary phase.
The Z isomer of 2 b gave (±)-3 b with >98:2 d.r. and in 82 % yield. Cy=cyclohexyl, DPP=diphenylphosphinoyl.
Interestingly, when Z-configured Michael acceptor (Z)-2 b was subjected to the optimized reaction conditions, it afforded the same morphan product (±)-3 b as the racemate indicating that geometrically pure trans-configured starting materials were crucial to achieving the high enantioselectivities observed. Taken together, these results clearly demonstrate that diastereoselectivity is a result of inherent substrate control and not a consequence of the chiral catalyst employed, which governs enantioselectivity. To understand the mechanism and origins of the high stereocontrol, quantum-chemical calculations were performed for the racemic and enantioselective series of our reaction.13 Stationary points were optimized at the M06-2X/6-311+G(d,p) level of theory;14 implicit solvation15 by CH2Cl2 was included using a conductor-like polarizable continuum model (CPCM). These results were corroborated by other computational methods (see the Supporting Information for details).16 In the interest of tractability, calculations were performed on N-methyl substrate 2 v with a methylamine catalyst as a model for propylamine. The s-cis enamine conformation is more stable than the s-trans conformation by 2.9 kcal mol−1; however, we found that only the latter is able to undergo Michael addition as the enamine N–H must be oriented towards the ester: Proton transfer to the oxygen atom occurs along the (intrinsic) reaction coordinate, which is not possible for the s-cis conformer. A concerted ene reaction can be dismissed, as this step has an unfeasibly high activation barrier of 33.4 kcal mol−1. From the s-trans enamine, formation of the Michael endo diastereomer was computed to occur via chair-shaped TS-A, with a staggered conformation about the incipient C–C bond (Figure 1). In this transition state, proton transfer occurs asynchronously with C–C bond formation, giving enol adduct E. The keto tautomer results from a 1,3-prototropic shift in TS-C, assisted by the imine N atom to form endo adduct F. Formation of the alternative exo diastereomer is possible via TS-B. In this transition state, the forming six-membered ring adopts a boat conformation with greater eclipsing interactions about the incipient C–C bond than in endo-TS-A. The exo pathway is kinetically disfavored by 1.7 kcal mol−1, but more importantly, intramolecular proton transfer to the ester α-carbon atom is geometrically impossible for this diastereomer. We thus predict that exo enol G will revert back to the more stable starting enamine and not proceed to the keto tautomer. The exclusive endo selectivity results from an irreversible, kinetically favored pathway. Transition states corresponding to the formation of cyclobutane17 and cyclic enol ether intermediates18 were also located. Only cyclobutane I was computed to be more stable than the starting enamine, however, its formation is disfavored (TS-D) with respect to proton transfer to TS-C and is thus unlikely to constitute a significant resting state in the catalytic cycle.
Figure 1.
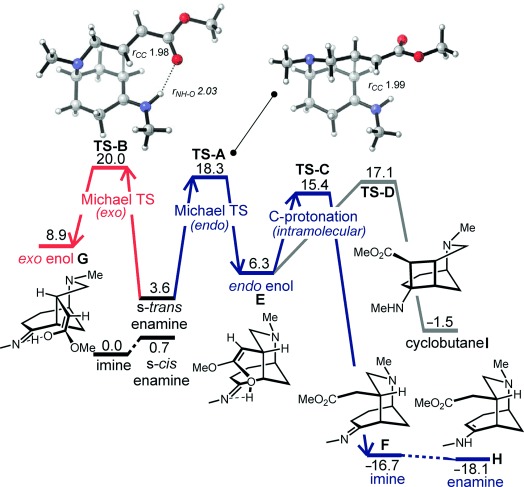
Free-energy profile for the cyclization of 2 v catalyzed by methylamine at the CPCM-M06-2X/6-311+G(d,p) level of theory (Grel values in kcal mol−1 at 45 °C, 1 mol L−1).
We then considered the asymmetric induction arising from thiourea catalyst 4 k. Our computations considered an enamine derived from substrate 2 v with a simplified, truncated thiourea catalyst 4 l. Low-energy conformations for each stationary point along the potential energy surface were located with Monte Carlo conformational searches19 employing semi-empirical PM6-DH2 calculations20 and subsequent refinement with M06-2X/6-311+G(d) optimizations (Figure 2). The reactive enamine geometry differs between the two pathways, with the s-cis enamine yielding the major enantiomer and the s-trans enamine yielding the minor enantiomer, as these conformations enable both pathways to benefit from stabilizing hydrogen bonding interactions between ester and thiourea. Energetic discrimination between Michael transition states TS-J and TS-K results from differing cyclohexylthiourea conformations: Rotation about the C(cyclohexyl)–N(thiourea) bond reveals a 4 kcal mol−1 conformational preference for the thiourea C–N bond to be syn-coplanar with the cyclohexyl C–H bond (ϕCNCH=30°) over the antiperiplanar conformation (ϕCNCH=180°). In the favored transition state TS-J, the catalyst adopts this preferred conformation (ϕCNCH=31°) whereas in disfavored TS-K, the less stable form is adopted (ϕCNCH=178°). The thiourea catalyst stabilizes ester enolate formation such that C–C bond formation and proton transfer now occur in two separate steps (see the Supporting Information for a full energy profile). The calculated enantioselectivity imparted by catalyst 4 l amounts to 96 % ee based on a ΔΔG≠ value of 2.4 kcal mol−1 between the selectivity-determining transition states along the two pathways, which agrees with the absolute sense and magnitude (96 % ee) obtained with 4 k. Our computational studies predicted that thiourea catalyst 4 l would thus be as competent as 4 k despite being greatly simplified. The computed transition state TS-J suggests a lack of any significant contribution from the tert-leucine fragment in catalyst 4 k, as it would be oriented away from the substrate into space. Accordingly, 4 l was then synthesized and tested in the cyclization of 2 v to validate the computational prediction of enantioselectivity (Scheme 3). Pleasingly, product 3 v was obtained in 83 % yield and 97 % ee as a single diastereomer, showing excellent agreement between experiment and theory.
Figure 2.
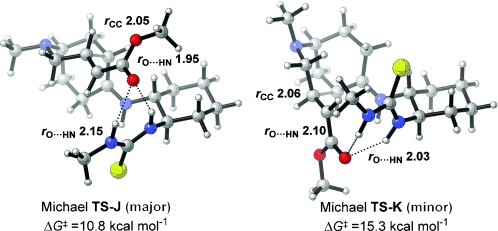
Transition states of the aminothiourea-catalyzed Michael reaction forming enantiomeric adducts of the endo diastereomer computed at the CPCM-M06-2X/6-311+G(d,p) level of theory.
scheme 3.
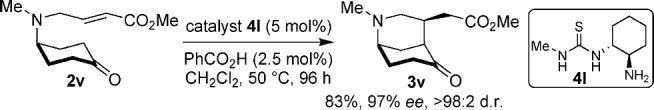
Computer-aided catalyst design of 4 l.
In summary, we have developed a highly enantioselective primary amine catalyzed Michael addition of a ketone to unactivated α,β-unsaturated esters. The reaction benefits from three points of diversity—the C4 substituent, the nitrogen group, and the ester moiety—and provides access to the morphan scaffold in high enantio- and diastereoselectivity (up to 99 % ee and >98:2 d.r.). Computational studies to probe the origins of the high enantiocontrol have been performed, and the results of the calculations identified a new low-molecular-weight catalyst that can impart the same level of enantioselectivity. The application of this new enantioselective desymmetrization method to complex natural product synthesis is ongoing in our group, and the findings will be reported in due course.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.For a review on natural products that contain a morphan core and strategies addressing their synthesis, see:
- 1a.Bonjoch J, Diaba F, Bradshaw B. Synthesis. 2011:993. for a recent synthesis of daphenylline, see: [Google Scholar]
- 1b.Lu Z, Li Y, Deng J, Li A. Nat. Chem. 2013;5:679. doi: 10.1038/nchem.1694. for a recent synthesis of (+)-madangamine D, see: [DOI] [PubMed] [Google Scholar]
- 1c.Ballette R, Pérez M, Proto S, Amat M, Bosch J. Angew. Chem. Int. Ed. 2014;53:6202. doi: 10.1002/anie.201402263. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014;126 for a recent review on strychnine, see: [Google Scholar]
- 1d.Cannon JS, Overman LE. Angew. Chem. Int. Ed. 2012;51:4288. doi: 10.1002/anie.201107385. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]
- 2.Kang B, Jakubec P, Dixon DJ. Nat. Prod. Rep. 2014;31:550. doi: 10.1039/c3np70115h. [DOI] [PubMed] [Google Scholar]
- 3.For a review on desymmetrization reactions, see:
- 3a.Willis M. J. Chem. Soc. Perkin Trans. 1. 1999:1765. for selected examples of organocatalytic desymmetrization reactions, see: [Google Scholar]
- 3b.Hayashi Y, Gotoh Y, Tamura T, Yamaguchi H, Masui R, Shoji M. J. Am. Chem. Soc. J. Am. Chem. Soc. 2005;127:16028. doi: 10.1021/ja055740s. [DOI] [PubMed] [Google Scholar]
- 3c.Vo N, Pace R, O’Hara F, Gaunt M. J. Am. Chem. Soc. 2008;130:404. doi: 10.1021/ja077457u. [DOI] [PubMed] [Google Scholar]
- 3d.Liu Q, Rovis T. J. Am. Chem. Soc. 2006;128:2552. doi: 10.1021/ja058337u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e.Gu Q, Rong Z, Zheng C, You S. J. Am. Chem. Soc. 2010;132:4056. doi: 10.1021/ja100207s. [DOI] [PubMed] [Google Scholar]
- 3f.Wu W, Li X, Huang H, Yuan X, Lu J, Zhu K, Ye J. Angew. Chem. Int. Ed. 2013;52:1743. doi: 10.1002/anie.201206977. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013;125 [Google Scholar]
- 3g.Takizawa S, Nguyen TM-N, Grossmann A, Enders D, Sasai H. Angew. Chem. Int. Ed. 2012;51:5423. doi: 10.1002/anie.201201542. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]
- 3h.Miyamae N, Watanabe N, Moritaka M, Nakano K, Ichikawa Y, Kotsuki H. Org. Biomol. Chem. 2014;12:5847. doi: 10.1039/c4ob00733f. for desymmetrization reactions of prochiral cyclohexanones, see: by Michael additions with nitroolefins: [DOI] [PubMed] [Google Scholar]
- 3i.Xu Y, Zou W, Sundén H, Ibrahem I, Córdova A. Adv. Synth. Catal. 2006;348:418. [Google Scholar]
- 3j.Luo S-Z, Zhang L, Mi X-L, Qiao Y-P, Cheng J-P. J. Org. Chem. 2007;72:9350. doi: 10.1021/jo7020357. by Michael additions with vinyl sulfones: [DOI] [PubMed] [Google Scholar]
- 3k.Chen YM, Lee P-H, Lin J, Chen K. Eur. J. Org. Chem. 2013:2699. by aldol reactions: [Google Scholar]
- 3l.Jiang J, He L, Luo S-W, Cun L-F, Gong L-Z. Chem. Commun. 2007:736. doi: 10.1039/b615043h. [DOI] [PubMed] [Google Scholar]
- 3m.Companyó X, Valero G, Crovetto L, Moyano A, Rios R. Chem. Eur. J. 2009;15:6564. doi: 10.1002/chem.200900488. by the α-alkylation of ketones and aldehydes using chiral ionic liquids: [DOI] [PubMed] [Google Scholar]
- 3n.Zhang L, Cui L, Li X, Li J, Luo S, Cheng J-P. Eur. J. Org. Chem. 2010:4876. by deprotonation to give silyl enol ethers: [Google Scholar]
- 3o.Xlaraz A, Oudeyer S, Levacher V. Tetrahedron: Asymmetry. 2013;24:764. by a Friedlander synthesis using 4-substituted cyclohexanones: [Google Scholar]
- 3p.Li L, Seidel D. Org. Lett. 2010;12:2010. doi: 10.1021/ol1023932. by deprotonation with chiral bases: [DOI] [PubMed] [Google Scholar]
- 3q.McGrath N, Binner JR, Markopoulos G, Brichacek M, Njardarson JT. Chem. Commun. 2011;47:209. doi: 10.1039/c0cc01419b. by a Baeyer–Villiger oxidation: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3r.Zhou L, Liu X, Ji J, Zhang Y, Hu X, Lin L, Feng X. J. Am. Chem. Soc. 2012;134:17023. doi: 10.1021/ja309262f. by a desymmetrization–fragmentation process of cyclic ketones: [DOI] [PubMed] [Google Scholar]
- 3s.Dickmeiss G, De Sio V, Udmark J, Poulsen TB, Marcos V, Jørgensen KA. Angew. Chem. Int. Ed. 2009;48:6650. doi: 10.1002/anie.200903253. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009;121 [Google Scholar]
- 4.For a review, see:
- 4a.Mukherjee S, Yang JW, Hoffmann S, List B. Chem. Rev. 2007;107:5471. doi: 10.1021/cr0684016. for reviews on enamine-catalyzed Michael additions, see: [DOI] [PubMed] [Google Scholar]
- 4b.Tsogoeva SB. Eur. J. Org. Chem. 2007:1701. [Google Scholar]
- 4c.Zhang Y, Wang W. Catal. Sci. Technol. 2012;2:42. for reports on the addition of ketones to nitroolefins, see: [Google Scholar]
- 4d.Huang H, Jacobsen EN. J. Am. Chem. Soc. 2006;128:7170. doi: 10.1021/ja0620890. [DOI] [PubMed] [Google Scholar]
- 4e.Tsogoeva SB, Wei S. Chem. Commun. 2006:1451. doi: 10.1039/b517937h. [DOI] [PubMed] [Google Scholar]
- 4f.Yalalov DA, Tsogoeva SB, Schmatz S. Adv. Synth. Catal. 2006;348:826. [Google Scholar]
- 5.For a desymmetrization process featuring an aldol reaction, see:
- 5a.Diaba F, Bonjoch J. Org. Biomol. Chem. 2009;7:2517. doi: 10.1039/b906835j. for a related all-carbon-scaffold aldol reaction, see: [DOI] [PubMed] [Google Scholar]
- 5b.Itagaki N, Sugahara T, Iwabuchi Y. Org. Lett. 2005;7:4185. doi: 10.1021/ol051569d. for an alternative enantioselective approach, see: [DOI] [PubMed] [Google Scholar]
- 5c.Bradshaw B, Parra C, Bonjoch J. Org. Lett. 2013;15:2458. doi: 10.1021/ol400926p. [DOI] [PubMed] [Google Scholar]
- 6.For superstoichiometric enamine additions to unactivated esters, see:
- 6a.Hirai Y, Terada T, Yamazaki T. J. Am. Chem. Soc. 1988;110:958. [Google Scholar]
- 6b.Hirai Y, Terada T, Yamazaki T, Momose T. J. Chem. Soc. Perkin Trans. 1. 1992:509. for an intramolecular nitro-Michael addition to α,β-unsaturated esters, see: [Google Scholar]
- 6c.Nodes WJ, Nutt DR, Chippindale AM, Cobb AJA. J. Am. Chem. Soc. 2009;131:16016. doi: 10.1021/ja9070915. [DOI] [PubMed] [Google Scholar]
- 7.Kan T, Fujimoto T, Ieda S, Asoh Y, Kitaoka H, Fukuyama T. Org. Lett. 2004;6:2729. doi: 10.1021/ol049074w. [DOI] [PubMed] [Google Scholar]
- 8.Jung ME, Piizzi G. Chem. Rev. 2005;105:1735. doi: 10.1021/cr940337h. [DOI] [PubMed] [Google Scholar]
- 9.For reviews on primary amine catalysis, see:
- 9a.Peng F, Shao Z. J. Mol. Catal. A. 2008;285:1. [Google Scholar]
- 9b.Xu L-W, Luo J, Lu Y. Chem. Commun. 2009:1807. doi: 10.1039/b821070e. for a specific example of primary amine catalysis, see: [DOI] [PubMed] [Google Scholar]
- 9c.Xu Y, Córdova A. Chem. Commun. 2006:460. doi: 10.1039/b514783m. [DOI] [PubMed] [Google Scholar]
- 10.For selected examples, see:
- 10a.Sakthivel K, Notz W, Bui T, Barbas CF., III J. Am. Chem. Soc. 2001;123:5260. doi: 10.1021/ja010037z. , ; [DOI] [PubMed] [Google Scholar]
- 10b.Hayashi Y, Gotoh H, Hayashi T, Shoji M. Angew. Chem. Int. Ed. 2005;44:4212. doi: 10.1002/anie.200500599. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005;117 [Google Scholar]
- 10c.Franzén J, Marigo M, Fielenbach D, Wabnitz TC, Kjærsgaard A, Jorgensen KA. J. Am. Chem. Soc. 2005;127:18296. doi: 10.1021/ja056120u. [DOI] [PubMed] [Google Scholar]
- 11.Patil MP, Sunoj RB. J. Org. Chem. 2007;72:8202. doi: 10.1021/jo071004q. [DOI] [PubMed] [Google Scholar]
- 12.For reviews, see:
- 12a.Connon SJ. Chem. Eur. J. 2006;12:5418. doi: 10.1002/chem.200501076. [DOI] [PubMed] [Google Scholar]
- 12b.Takemoto Y. Org. Biomol. Chem. 2005;3:4299. doi: 10.1039/b511216h. for a review on primary amine thiourea organocatalysis, see: [DOI] [PubMed] [Google Scholar]
- 12c.Serdyuk OV, Heckel CM, Tsogoeva SB. Org. Biomol. Chem. 2013;11:7051. doi: 10.1039/c3ob41403e. [DOI] [PubMed] [Google Scholar]
- 13.Gaussian 09 rev. D.01, Frisch M, Gaussian, Inc., Wallingford CT, 2009. Images prepared with CYLview, 1.0b; Legault C.Y, Université de Sherbrooke, 2009. For a full list of computational citations, see the Supporting Information
- 14.Zhao Y, Truhlar DG. Acc. Chem. Res. 2008;41:157. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 15.Barone V, Cossi M. J. Phys. Chem. A. 1998;102:1995. [Google Scholar]
- 16.For computations of primary enamine catalysis, see:
- 16a.Lam YH, Houk KN, Scheffler U, Mahrwald R. J. Am. Chem. Soc. 2012;134:6286. doi: 10.1021/ja2118392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b.Lam YH, Houk KN. J. Am. Chem. Soc. 2014;136:9556. doi: 10.1021/ja504714m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16c.Cheong PH, Legault CY, Um JM, Çelebi-Ölçüm N, Houk KN. Chem. Rev. 2011;111:5042. doi: 10.1021/cr100212h. , and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17a.Patora-Komisarska K, Benohoud M, Ishikawa H, Seebach D, Hayashi Y. Helv. Chim. Acta. 2011;94:719. [Google Scholar]
- 17b.Burés J, Armstrong A, Blackmond DG. J. Am. Chem. Soc. 2011;133:8822. doi: 10.1021/ja203660r. [DOI] [PubMed] [Google Scholar]
- 17c.Burés J, Armstrong A, Blackmond DG. J. Am. Chem. Soc. 2012;134:6741. doi: 10.1021/ja300415t. [DOI] [PubMed] [Google Scholar]
- 17d.Seebach D, Sun X, Ebert M, Schweizer WB, Purkayastha N, Beck AK, Duschmale J. Helv. Chim. Acta. 2013;96:795. [Google Scholar]
- 18.Sahoo G, Rahaman H, Madarász A, Pápai I, Melarto M, Valkonen A, Pihko PM. Angew. Chem. Int. Ed. 2012;51:13144. doi: 10.1002/anie.201204833. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]
- 19.Rotatable torsions in each diastereomeric transition state were sampled using a locally modified version of a uniform-usage directed MCMM conformational search (1000 steps) interfaced to a version of Mopac, during which the forming bond was constrained. Conformers within 3 kcal mol−1 of the global minimum were reoptimized to genuine saddle points at the M062X/6-311+G(d,p) level of theory; see: MOPAC2012, J. J. P. Stewart, Stewart Computational Chemistry, Colorado Springs, CO, USA, http://OpenMOPAC.net (2012)
- 20a.Korth M, Pitonák M, Rezác J, Hobza P. J. Chem. Theory Comput. 2010;6:344. doi: 10.1021/ct900541n. [DOI] [PubMed] [Google Scholar]
- 20b.Rezác J, Fanfrlik J, Salahub D, Hobza P. J. Chem. Theory Comput. 2009;5:1749. doi: 10.1021/ct9000922. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information