

Abstract
At the water–trihexyl(tetradecyl)phosphonium tris(pentafluoroethyl)trifluorophosphate ([P14,6,6,6][FAP]) ionic liquid interface, the unusual electrochemical transfer behavior of protons (H+) and deuterium ions (D+) was identified. Alkali metal cations (such as Li+, Na+, K+) did not undergo this transfer. H+/D+ transfers were assisted by the hydrophobic counter anion of the ionic liquid, [FAP]−, resulting in the formation of a mixed capacitive layer from the filling of the latent voids within the anisotropic ionic liquid structure. This phenomenon could impact areas such as proton-coupled electron transfers, fuel cells, and hydrogen storage where ionic liquids are used as aprotic solvents.
Keywords: capacitance, liquid–liquid interfaces, proton transfer, room temperature ionic liquids, voltammetry
Proton transfer reactions occur widely in nature and technology. Important energy conversions in nature require coupled reactions involving transfer of electrons and protons (for example, cell respiration and photosynthesis),1 which have stimulated the mimicking of these systems in order to achieve highly efficient energy convertors2 or energy storage3 cells. Recently, room temperature ionic liquids (RTILs) have received intense scrutiny owing to their unique physicochemical properties,4 which make them highly attractive for various studies including electrochemistry,5 with available potential windows as wide as 7 V at solid electrodes.6 Since the pioneering study by Quinn et al.,7 electrochemistry at water/RTIL (H2O/RTIL) interfaces has become a new platform to study interfacial charge transfer processes, including simple8 and facilitated ion transfers.9 While proton transfers have been of interest at polarized water/oil interfaces,10 including proton transfer catalyzed reactions,11 proton transfers into ionic liquids have not yet been addressed.
Herein, we explore the behavior of protons at the interface between water and trihexyl(tetradecyl)phosphonium tris(pentafluorethyl)trifluorphosphate (H2O/[P14,6,6,6][FAP]), building on previous work on the formation of a H2O/RTIL microinterface array for organic cation transfer12 and protein behavior.13 Surprisingly, a remarkable ion transfer process was observed when alkali cation solutions were replaced with an acidic (hydrogen or deuterium cation) aqueous solution. The proton–RTIL interactions are believed to be the result of interfacial transfers into voids in the RTIL phase. These results will be of particular interest in areas where aprotic RTILs are utilized in protonated environments (for example, electrocatalytic reactions, proton-coupled electron transfers, fuel cells, and hydrogen storage).
At an array of microscale H2O/RTIL interfaces, [P14,6,6,6][FAP] showed the largest potential window compared to the RTILs trihexyl(tetradecyl)phosphonium bis(trifluoromethylsulfonyl)imide [P14,6,6,6][NTf2], 1-hexyl-3-methylimidazolium tris(pentafluoro-ethyl)trifluorophosphate [C6mim][FAP], and 1-butyl-1-methy-l-pyrrolidinium tris(pentafluoroethyl)trifluorophosphate [C4mpyrr][FAP] (Supporting Information, Figure S1). When LiCl was replaced with HCl as the aqueous electrolyte at the H2O/[P14,6,6,6][FAP] interface (Figure 1), an increase in current at −0.1 V in the forward sweep of the cyclic voltammograms (CVs) was observed. This response to aqueous phase hydrogen cations is quite different than the behavior of acidic aqueous solutions at H2O/organic solvent interfaces.14 Furthermore, this charge transfer process is absent when the aqueous phase consists of LiCl, NaCl, or KCl (Figure 1, inset), but is evident when the aqueous phase contains deuterium ions (D+); the latter behave similarly to protons. The CVs in Figure 1 are quite different from those expected for a diffusion-controlled ion transfer reaction at microinterface arrays.12, 15
Figure 1.
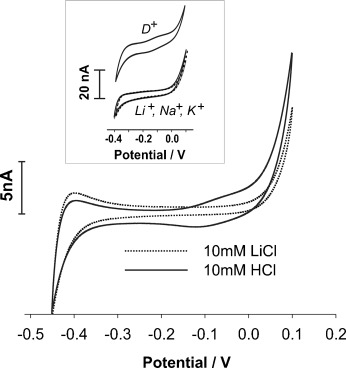
Cyclic voltammograms of the H2O/[P14,6,6,6][FAP] microinterface array, where the aqueous phase is 0.01 m LiCl (••••) or HCl (—). Inset: 0.1 m LiCl, NaCl, KCl in H2O or DCl in D2O. Scan rate: 10 mV s−1.
The influence of HCl concentration (in the range 1–500 mm) was investigated (Supporting Information, Figure S2). Although not detectable at 1 mm, at 5 mm a shoulder was evident at the positive end of the CV. Higher concentrations of HCl were easily detected (Figure 2). These voltammograms exhibit some notable features: i) the current on the forward sweep reaches a steady-state, consistent with radial diffusion to microinterfaces, but it increases with the scan rate; ii) the reverse scans exhibit a peak-shaped voltammogram, consistent with linear diffusion or an adsorption/desorption process. The diffusion-limited steady-state current (Ilim) at a micro-interface array is described by Equation (1):
| 1 |
Figure 2.
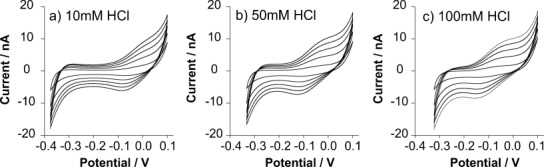
Cyclic voltammetry at the micro-interface array between water and the RTIL [P14,6,6,6][FAP]. The aqueous phase is a) 10 mm HCl, b) 50 mm HCl, or c) 100 mm HCl. Scan rates: 5, 10, 25, 50, 75, and 100 mV s−1 (inner to outer).
where Np is the number of microinterfaces, z, D, and C are the charge, diffusion coefficient, and bulk concentration of the transferring species, and r is the radius of one interface.
For the diffusion-controlled transfer of protons at a H2O/RTIL interface array consisting of 30 microinterfaces, taking into account the bulk aqueous diffusion coefficient of protons, DH+∼10−4 cm2 s−1,16 the limiting current is expected to be 13, 65, and 130 μA at each of the concentrations shown in Figure 2 (10–100 mm); the experimental currents are, in fact, several orders of magnitude lower than these predictions. Furthermore, the limiting currents unexpectedly change with scan rate [see Eq. (1)]. Scan rate data for the five cations studied are provided in the Supporting Information (Figure S3). Also, it is notable that the proton transfer potential is dependent on the proton concentration in the aqueous phase (Supporting Information, Figure S4). The half-wave potential for proton transfer varied linearly with the logarithm of proton concentration, with a slope of −64 mV dec−1 (Supporting Information, Figure S4), close to the expected −59 mV dec−1 for a single-positive charged species. This shift in the potential can be attributed to a facilitated proton transfer mechanism driven by H+-[FAP]− ion-pairing. Mirkin and co-workers proposed a facilitated transfer, from water to low permittivity solvents, of hydrophilic ions by hydrophobic counter ions.17 This process involves a transient ion-pairing that facilitates the transfer across the interface, which agrees with the phenomenon observed here, although it fails to explain the exclusion of alkali metal cations.
Electrochemical impedance spectroscopy (EIS) was employed to probe the capacitive and resistive properties of the H2O/RTIL system. Figure 3 a shows Nyquist plots of the H2O/RTIL system, where the aqueous phase is 1 mm LiCl in the absence (solid line) or presence of 50 mm HCl (dotted line) polarized at −0.2 V (no charge transfer). Figure 3 b shows the Nyquist plots where the H2O/RTIL interface with acidic aqueous phase is polarized at −0.2 V (dotted line) or 0.0 V (red solid line): interfacial proton transfer occurs only at the latter potential. The corresponding equivalent circuits, which show the combinations of series and parallel resistances from solution, membrane and diffusion (Rs, Rm, and Rd) and capacitances (non-ideal CPEm, CPEd, and ideal Cnew) that best fit the experimental data, are shown beneath each plot. In these circuits, Rs is the solution resistance, while Rm and CPEm are attributed to the silicon membrane that supports the H2O/RTIL microinterfaces and the Ag/AgCl electrodes, since these are constant and independent of the applied potential. Rd and CPEd correspond to a diffusion process at lower frequencies (right hand side of the Nyquist plot).
Figure 3.
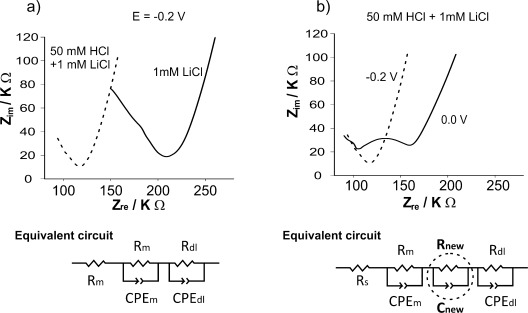
EIS of the H2O/RTIL system: a) The aqueous phase is 50 mm HCl in 1 mm LiCl (– – –) or 1 mm LiCl (—), at −0.2 V (no interfacial transfer); b) 50 mm HCl in 1 mm LiCl at −0.2 V (– – –) or 0.0 V (—). Below the graphs are the corresponding equivalent circuits which fit the experiments with w of 50 mm HCl in 1 mm LiCl at either a) −0.2 V or b) 0.0 V. Frequency range shown: 500 kHz to 80 Hz.
EIS reveals a new semi-circle feature between 100 and 160 kΩ (Figure 3 b) when implemented at a potential (0.0 V) where the transfer process occurs. This new feature (Figure 3 b, Rnew and Cnew), reaches an impedance maximum of circa 30 kΩ and occurs at high frequencies. This is an indication of a process two or more orders of magnitude faster than the diffusion process observed at lower frequencies (Rd and CPEd). Experiments for different HCl concentrations produced similar Nyquist plots (Supporting Information, Figure S5). The magnitude of the new semi-circle feature is independent of the proton concentration above 10 mm HCl.
This new feature fits to a circuit with a third capacitor, which suggests a change at the H2O/RTIL interface owing to the transfer of protons and their interaction with [P14,6,6,6][FAP]. Relative dielectric permittivity (εr) values reported for a wide range of ionic liquids vary between 8 and 16.18 Assuming that the relative dielectric permittivity of [P14,6,6,6][FAP] is within this range and the geometric area (A) of the microinterface array is 1.18×10−4 cm2, then the thickness of this new capacitor can be estimated by [Eq. (2)].
| 2 |
where C is the capacitance, εr is the relative dielectric permittivity, ε0 is the electric constant (8.85×10−12 F m−1), d is the thickness, and A is the microinterface array area. The new capacitor value (C) is circa 3.7×10−10 F, as obtained from the equivalent circuit fitting (Cnew, Figure 3 b), and corresponds to a new layer of thickness 2.3–4.5 nm at the H2O/RTIL microinterface array.
Alternating current (AC) voltammetry of the H2O/RTIL system with either LiCl or HCl as aqueous electrolytes was undertaken at different phase angles (0° and 90°). This analysis presented a negative shift of the potential of zero charge (Epzc) in the presence of the acidic aqueous phase,19 which indicates an interaction of positively-charged species with the H2O/[P14,6,6,6][FAP] microinterface (Supporting Information, Figure S6). Furthermore, similar CV behavior was observed when the RTIL phase was replaced with a solution of RTIL dissolved in an organic solvent (Supporting Information, Figure S7), although the magnitude of the effect was diminished owing to the dilution factor. Conventional electrolytes in organic solvents do not exhibit the effect seen here with [P14,6,6,6][FAP]. Furthermore, ionic effects were investigated in both phases, although the presence of [NTf2]−, [C6mim]+, and [C4mpyrr]+ in the RTIL limited the available potential window (≤0.25 V). Nevertheless, when replacing Cl− with SO42− in the aqueous phase, the same phenomenon was observed in the presence of H+ (Supporting Information, Figure S8).
Recent models to explain the electrical double layer (EDL) formed at electrode/RTIL interfaces have taken into account the finite size of the ions,20 the concentrated nature of the electrolyte,21 as well as steric effects,22 which were not addressed in the Gouy–Chapman theory. Fedorov and Kornyshev used the formation of “voids” in the ionic liquid structure to explain the camel-shaped capacitance of the EDL at metal/RTIL interfaces.23 It was suggested that ionic liquids may contain voids, owing to the typically anisotropic nature of the bulky ions present, which can accommodate small charged molecules. This effect could provide a cavity for hosting of ions transferred from an adjoining immiscible phase, leading to the formation of a new capacitive layer and a change in capacitive current, as observed here. Such a change explains why the CV currents (Figures 1, 2) vary with sweep rate despite having a steady-state form. Further transfer of these ions is hindered by the large non-polar alkyl groups of the RTIL cation ([P14,6,6,6]+), which reduces the mobility of the proton within the ionic liquid. Furthermore, this process is possible only for small cations, either solvated or unsolvated, because H+ and D+ (radii ∼0.037 nm and ∼0.036 nm, respectively;24 hydronium radius ∼0.14 nm) can physically occupy the cavities in the RTIL phase within the EDL because of their dimensions. The RTIL void radii were estimated to be r∼0.17–0.19 nm (see the Supporting Information), which can accommodate hydronium ions, but not hydrated alkali cations (>0.3 nm). This resembles LaF3 solid-state membranes where only fluoride can occupy vacancies in the crystal lattice.25 It is also known that diffusion of small gas molecules, such as H2, in RTIL occurs by hopping between RTIL interstices.26 In the present studies, no chemical reactions between transferred cation and components of the RTIL were observed, which is in agreement with the reversible solvation of H+ by [FAP]− reported by Silvester et al. in studies of H2(g) oxidation.27
In conclusion, voltammetry and electrochemical impedance spectroscopy suggests that solvated H+ and D+ transfer cross the H2O/[P14,6,6,6][FAP] microinterface, forming a capacitive thin film layer, but larger cations (Li+, Na+, and K+) do not transfer. This new capacitor-like layer is formed as H+/D+ fill the latent voids within the RTIL, but further transfer into the bulk RTIL is limited by the steric effect of the RTIL cations. This is in agreement with recent theoretical models of the EDL at electrode/RTIL interfaces, where crowding effects, finite size of the ions, and steric effects have been incorporated to expand the Gouy–Chapman model. The ability to transport ions into cavities within a liquid phase may have implications for proton transfer reactions of importance in proton-coupled electron transfers, fuel cells, hydrogen storage, and protein extraction and stabilization.
Acknowledgments
The authors thank Curtin University for a scholarship to EAdE. The micropore array membranes were a gift from Tyndall National Institute, Cork, Ireland.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Hammes-Schiffer S. Acc. Chem. Res. 2009;42:1881–1889. doi: 10.1021/ar9001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Vries S, Dörner K, Strampraad MJF, Friedrich T. Angew. Chem. Int. Ed. 2015;54:2844–2848. doi: 10.1002/anie.201410967. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015;127 [Google Scholar]
- 3.Andrews J, Mohammadi SS. Int. J. Hydrogen Energy. 2014;39:1740–1751. [Google Scholar]
- 4.Barrosse-Antle LE, Bond AM, Compton RG, O’Mahony AM, Rogers EI, Silvester DS. Chem. Asian J. 2010;5:202–230. doi: 10.1002/asia.200900191. [DOI] [PubMed] [Google Scholar]
- 5.Armand M, Endres F, MacFarlane DR, Ohno H, Scrosati B. Nat. Mater. 2009;8:621–629. doi: 10.1038/nmat2448. [DOI] [PubMed] [Google Scholar]
- 6a.Ignat’ev NV, Welz-Biermann U, Kucheryna A, Bissky G, Willner H. J. Fluorine Chem. 2005;126:1150–1159. [Google Scholar]
- 6b.Nishi N, Imakura S, Kakiuchi T. Anal. Chem. 2006;78:2726–2731. doi: 10.1021/ac052152o. [DOI] [PubMed] [Google Scholar]
- 7.Quinn BM, Ding ZF, Moulton R, Bard AJ. Langmuir. 2002;18:1734–1742. [Google Scholar]
- 8.Wang YX, Kakiuchi T, Yasui Y, Mirkin MV. J. Am. Chem. Soc. 2010;132:16945–16952. doi: 10.1021/ja1066948. [DOI] [PubMed] [Google Scholar]
- 9.Langmaier J, Samec Z. Anal. Chem. 2009;81:6382–6389. doi: 10.1021/ac9008258. [DOI] [PubMed] [Google Scholar]
- 10.Sabela A, Marecek V, Samec Z, Fuoco R. Electrochim. Acta. 1992;37:231–235. [Google Scholar]
- 11a.Peljo P, Qiao L, Murtomaki L, Johans C, Girault HH, Kontturi K. ChemPhysChem. 2013;14:311–314. doi: 10.1002/cphc.201200953. [DOI] [PubMed] [Google Scholar]
- 11b.Partovi-Nia R, Su B, Li F, Gros CP, Barbe JM, Samec Z, Girault HH. Chem. Eur. J. 2009;15:2335–2340. doi: 10.1002/chem.200801807. [DOI] [PubMed] [Google Scholar]
- 12.Silvester DS, Arrigan DWM. Electrochem. Commun. 2011;13:477–479. [Google Scholar]
- 13.de Eulate EA, Silvester DS, Arrigan DWM. Chem. Asian J. 2012;7:2559–2561. doi: 10.1002/asia.201200390. [DOI] [PubMed] [Google Scholar]
- 14.Scanlon MD, Strutwolf J, Arrigan DWM. Phys. Chem. Chem. Phys. 2010;12:10040–10047. doi: 10.1039/c003323e. [DOI] [PubMed] [Google Scholar]
- 15a.Strutwolf J, Scanlon MD, Arrigan DWM. Analyst. 2009;134:148–158. doi: 10.1039/b815256j. [DOI] [PubMed] [Google Scholar]
- 15b.Shao Y, Girault HH. J. Electroanal. Chem. 1991;282:59–72. [Google Scholar]
- 16.Medvedev ES, Stuchebrukhov AA. J. Phys. Condens. Matter. 2011;23:234103. doi: 10.1088/0953-8984/23/23/234103. [DOI] [PubMed] [Google Scholar]
- 17a.Sun P, Laforge FO, Mirkin MV. J. Am. Chem. Soc. 2007;129:12410. doi: 10.1021/ja075774v. [DOI] [PubMed] [Google Scholar]
- 17b.Laforge FO, Sun P, Mirkin MV. J. Am. Chem. Soc. 2006;128:15019–15025. doi: 10.1021/ja0656090. [DOI] [PubMed] [Google Scholar]
- 18a.Wakai C, Oleinikova A, Ott M, Weingartner H. J. Phys. Chem. B. 2005;109:17028–17030. doi: 10.1021/jp053946+. [DOI] [PubMed] [Google Scholar]
- 18b.Daguenet C, Dyson PJ, Krossing I, Oleinikova A, Slattery J, Wakai C, Weingärtner H. J. Phys. Chem. B. 2006;110:12682–12688. doi: 10.1021/jp0604903. [DOI] [PubMed] [Google Scholar]
- 19.Jensen H, Fermin DJ, Moser JE, Girault HH. J. Phys. Chem. B. 2002;106:10908–10914. [Google Scholar]
- 20.Kornyshev AA. J. Phys. Chem. B. 2007;111:5545–5557. doi: 10.1021/jp067857o. [DOI] [PubMed] [Google Scholar]
- 21.Kilic MS, Bazant MZ, Ajdari A. Phys. Rev. E. 2007;75:021502. doi: 10.1103/PhysRevE.75.021502. [DOI] [PubMed] [Google Scholar]
- 22.Yining H, Shanghui H, Tianying Y. J. Phys. Condens. Matter. 2014;26:284103. [Google Scholar]
- 23a.Fedorov MV, Georgi N, Kornyshev AA. Electrochem. Commun. 2010;12:296–299. [Google Scholar]
- 23b.Georgi N, Kornyshev AA, Fedorov MV. J. Electroanal. Chem. 2010;649:261–267. [Google Scholar]
- 24a.Choi P, Jalani NH, Datta R. J. Electrochem. Soc. 2005;152:E123–E130. [Google Scholar]
- 24b.Marcus Y. J. Chem. Soc. Faraday Trans. 1991;87:2995–2999. [Google Scholar]
- 25.Frant MS, Ross JW. Science. 1966;154:1553–1555. doi: 10.1126/science.154.3756.1553. [DOI] [PubMed] [Google Scholar]
- 26.Meng Y, Aldous L, Compton RG. J. Phys. Chem. C. 2011;115:14334–14340. [Google Scholar]
- 27.Silvester DS, Ward K, Aldous L, Hardacre C, Compton RG. J. Electroanal. Chem. 2008;618:53–60. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information