

Abstract
Background
Niemann-Pick disease type C (NP-C) is a rare neurovisceral lipid storage disorder characterised by progressive, disabling neurological symptoms and premature death in most patients. During the last decade, national cohort studies have accrued a great deal of data on the symptomatology and natural history of NP-C.
Methods
In an observational cohort study, we present a substantial update based on the clinical presentation and follow-up of all known UK-based patients with a confirmed diagnosis of NP-C who have been tracked on an electronic database at the Department of Genetic Medicine, University of Manchester, UK. Patients were stratified according to accepted age-at-neurological-onset categories. Data on patients’ clinical signs and symptoms, medical history and genetic studies are summarised using descriptive methods.
Results
A total of 146 patients with NP-C were included, representing the full known UK NP-C cohort, as observed from database information between 1999 and the end of 2011: 72 patients (49 %) were alive at the end of the observation period. Among a total of 116 patients (79 %) who possessed at least one identified, disease-causing NP-C gene mutation, 114 (98 %) had NPC1 and two (2 %) had NPC2 mutations. Overall, 53/194 (27 %) identified mutations were novel. Six patients (4 %) had an early, non-neurological neonatal onset form of NP-C. The numbers (%) of patients with accepted age-at-neurological onset forms were: 8 (5 %) early-infantile onset, 51 (35 %) late-infantile onset, 42 (29 %) juvenile onset, and 25 (17 %) adolescent/adult onset. Fourteen patients diagnosed based on visceral symptoms and/or sibling history, confirmed in most cases by genetic analysis, did not have any neurological manifestations at last follow up (11 patients with mean [SD] age at last follow up 2.5 [1.8] years: 3 with mean [SD] age at death 20.8 [15.9] years). A total of 51 patients (35 %) received miglustat therapy. The mean (SD) overall treatment duration up to the end of the observation period was 2.6 (2.3) years.
Conclusions
This UK cohort is the largest national NP-C cohort reported to date, and confirms the wide phenotypic variability of the disease, as reported in other countries. Further analyses are required to assess the impact of miglustat therapy on neurological disease progression.
Electronic supplementary material
The online version of this article (doi:10.1186/s12883-015-0511-1) contains supplementary material, which is available to authorized users.
Keywords: Niemann-Pick disease type C, Natural history, NPC1/NPC2, Miglustat
Background
Niemann-Pick disease type C (NP-C) is a rare neurovisceral lipid storage disorder characterised by progressive, disabling neurological symptoms and premature death in most patients [1–3]. It is caused by autosomal recessive inheritance of mutations in either of two genes (NPC1 and NPC2), and has been estimated to affect one case in every 100,000–120,000 live births [1, 2, 4].
The clinical presentation of NP-C is highly heterogeneous, necessitating a multidisciplinary diagnostic process that takes into account clinical assessments, histological and electron microscopic tests, and biochemical and molecular genetic laboratory studies [1, 5]. Clinical work-up requires detection and recognition of numerous non-specific systemic and neurological signs and symptoms. While ancillary testing helps to narrow the differential diagnosis, final confirmation of NP-C requires demonstration of characteristic intralysosomal accumulation of unesterified cholesterol (based on filipin staining in cultured skin fibroblasts) and/or the identification of one or more disease-causing mutations in either the NPC1 or NPC2 genes [1, 2]. The demonstration of abnormal cholesterol homeostasis with impaired low-density lipoprotein (LDL)-induced cholesterol esterification can also provide supportive data in cases with an uncertain biochemical phenotype, but is rarely performed now [5, 6]. Newer screening and diagnostic tools have recently been developed. The NP-C suspicion index (SI) allows more rapid detection of patients who warrant further testing for NP-C [7–9]. Laboratory measurements of plasma oxysterols (particularly cholestane-3β,5α,6β-triol and 7-ketocholesterol) [10–12], and certain sphingolipids such as lyso-sphingosine [13, 14], have shown promise in allowing more rapid diagnosis in patients with suggestive clinical signs and symptoms. The increasing application of these newer measures is expected to increase the efficiency of diagnosis in NP-C.
Previous studies in cohorts of NP-C patients from France, Spain, Portugal and the USA have accrued data describing the biochemical and clinical phenotypes, genetics and natural history of NP-C [15–17]. NP-C has historically been considered a childhood-onset disease, but patients with late-onset symptoms are increasingly being detected due to the wider application of biochemical and genetic diagnostic techniques. In 2007, a retrospective case note review documented clinical signs and symptoms and subsequent disease course based on 94 NP-C patients diagnosed in the UK between 1999 and 2006 [15]. Data were available from approximately even numbers of patients with neonatal-onset (n = 33), childhood-onset (n = 31) and adolescent/adult-onset disease (n = 30), and a detection rate of 4–5 new cases per year between 1990 and 1999 was reported [15].
Major efforts have been made in the last decade to further consolidate clinical data from all known NP-C patients diagnosed and managed in the UK. This report provides an update of information from the UK NP-C database based on data collected between 1999 and the end of 2011, including a further 52 patients since the previous report in 2006 [15].
Methods
The UK NP-C database
We reviewed retrospective data for all UK-based patients with NP-C whose details were stored in a database maintained by the Niemann-Pick Disease Clinical Nurse Specialist at the Department of Genetic Medicine, University of Manchester, UK. All data were collected during clinical visits forming part of ongoing long-term care. The database contained information from all UK patients with a diagnosis of NP-C that had been confirmed using filipin staining, causal gene mutation analysis and/or esterification studies between 1999 and the end of 2011 (data cut-off). All patients were either referred or self-referred to the Nurse Specialist or Support Group.
Diagnostic information
Laboratory diagnostic data for all patients are included from at least one of three laboratories in the UK and/or France that provide the required specialised testing. Diagnostic Laboratories in the UK and Lyon, France were involved independently in submitting information on cases they diagnosed to the database.
In general diagnostic information included findings from skin biopsy analyses (filipin staining and cholesterol esterification assays) from all UK patients with: 1) clinical symptoms suggestive of NP-C and; 2) raised plasma chitotriosidase (routinely checked alongside white-cell enzymes if laboratory analyses hinted at a possible lysosomal storage disease). NPC1 gene sequencing analyses were conducted in all patients with positive filipin staining and/or cholesterol esterification findings, and some sibling cases. Patients in whom NPC1 mutations were not identified, or on whom complementation studies have not been performed, underwent further investigations and NPC2 gene sequencing.
Clinical manifestations
During the observation period, information on key signs and symptoms of NP-C were recorded up to the last clinical follow up (i.e., last clinic visit and/or update of medical records in the database) in line with international guidelines for the diagnostic assessment and follow-up of the disease [1]. The following data types were included whenever possible: oculomotor signs (e.g. vertical supranuclear gaze palsy [VSGP]), neurological manifestations (e.g., cerebellar signs [ataxia, dysarthria, dysphagia, dyskinesia], seizures/cataplexy); spasticity; childhood developmental status (e.g. psychomotor delay and/or regression, learning disabilities); cognitive loss/problems, psychiatric abnormalities (e.g., psychosis, behavioural abnormalities); and systemic symptoms (e.g., hepatosplenomegaly, lung disease or neonatal cholestatic disease). Information on miglustat therapy was also included, where available, for all treated patients.
Ethical data reporting
All information was accessed in accordance with applicable laws and ethical requirements for the study period concerned, and all study procedures, including informed consent for molecular genetic analyses, were conducted in line with ethical standards of the responsible institutional ethics committees and the Helsinki Declaration (1975) and subsequent revisions. All patients and/or their kin provided written informed consent for publication of individual clinical details, as presented in this report. Data reported previously for patients included in this cohort, based on publications by Lachmann et al. [18], Patterson et al. [19], and Patterson et al. [20], are identified where relevant.
Data analysis
In recognition of international guidelines for the management of NP-C, patients are assessed based on neonatal presentation of NP-C (characterised mainly by systemic symptoms [splenomegaly, hepatomegaly, neonatal cholestatic disease and liver failure], and hereafter referred to as the ‘neonatal’ form), and as per accepted age subgroups based on onset of neurological manifestations (i.e., early infantile- [<2 years], late infantile- [2 to <6 years], juvenile- [6–15 years] and adolescent/adult-onset [>15 years]). Patients with a confirmed diagnosis but, as yet, no neurological symptoms, were also included in a ‘non neurological’ category.
All data analyses were exploratory in nature, and no statistical analyses of differences between patient subgroups were performed. Data are presented using descriptive statistics (mean, SD, median and range for continuous variables, and n (%) for categorical values). Patients for whom no numerical data values were available were treated as having ‘missing values’.
Results
Overall cohort characteristics
This UK cohort comprised a total of 146 NP-C patients born between 1954 and 2009, among whom 77 (53 %) were female and 69 (47 %) male. Patient demographics and general characteristics per patient subgroup are summarised in Table 1. Among a total of 112 patients (77 %) who possessed at least one identified, disease-causing NP-C gene mutation, 110 (98 %) had NPC1 mutations and two (2 %) had NPC2 mutations. In patients where genetic analyses did not reveal any known or identifiable novel mutations, diagnoses were based on filipin staining and ancillary methods combined with clinical examination findings and medical history.
Table 1.
Patient demographics and general characteristics per age-at-onset subgroup
| Neonatal onset (N = 6) | Early infantile onset (N = 8) | Late-infantile onset (N = 51) | Juvenile onset (N = 42) | Adolescent/adult onset (N = 25) | |
|---|---|---|---|---|---|
| Gender, n (%) female | 3 (50) | 7 (88) | 28 (55) | 25 (60) | 10 (40) |
| Age, years: | |||||
| At neurological onset | |||||
| na | – | 8 | 50 | 40 | 18 |
| Mean (SD) | – | 1.1 (0.7) | 4.1 (1.2) | 9.4 (2.6) | 24.2 (8.8) |
| At diagnosis | |||||
| na | 4c | 7c | 49 | 39 | 24 |
| Mean (SD) | 0.1 (0.1) | 1.3 (1.5) | 4.6 (5.5) | 11.5 (7.4) | 29.3 (9.3) |
| At last follow upb | |||||
| na | – | – | 19 | 21 | 17 |
| Mean (SD) | – | – | 11.6 (8.9) | 20.2 (9.0) | 39.5 (9.2) |
| At death | |||||
| na | 6 | 8 | 30 | 19 | 8 |
| Mean (SD) | 0.19 (0.22) | 5.6 (2.0) | 13.4 (6.7) | 25.9 (8.9) | 33.7 (6.2) |
| Number (%)d with NP-C genetic information | 1 (17) | 8 (100) | 41 (80) | 36 (86) | 16 (64) |
| Number (%)d treated with miglustat | – | 2 (25) | 17 (33) | 20 (48) | 12 (48) |
aNumber of patients with available data; blast follow up (data cut-off at end-2011) in living patients only; cage at diagnosis not relevant for two neonatal patients and one early-infantile patient who were diagnosed post-mortem; dpercentages calculated relative to total number of patients per treatment subgroup
Six patients (4 %) had the visceral neonatal form of NP-C. The numbers (%) of patients per accepted age-at-neurological onset category were: early-infantile onset (n = 8 [5 %]), late-infantile onset (n = 51 [n = 35 %]), juvenile onset (n = 42 [29 %]); and adolescent/adult onset (n = 25 [17 %]). A total of 14 patients (10 %), most of whom were detected due to early visceral symptoms and three of whom had a sibling history of NP-C, had no neurological symptoms and are categorised herein as having non-neurological disease.
The overall mean (SD; range) ages at neurological onset and diagnosis were 8.8 (8.1; 0–40) years and 10.4 (11.5; 0–49.5) years, respectively. In general, age at diagnosis tended to increase in line with age at onset of neurological manifestations (Table 1): NP-C was diagnosed most quickly among patients with infantile onset, with the greatest delays to diagnosis recorded among patients with adolescent/adult onset. A total of 43 patients (29 %) were siblings who were also affected by NP-C. Based on all patients with available data, the mean time period between neurological disease onset and diagnosis was 1.73 (5.80) years, while this period among the sibling subgroup was 1.25 (4.75) years. In many cases, confirmation of a diagnosis of NP-C in one sibling led to a more rapid diagnosis in either younger or older siblings.
Overall, 72/146 (49 %) patients were alive at data cut-off. Among patients with available data, the mean age at last follow up (i.e., the latest clinical assessment before data cut-off at the end of 2011) ranged from 11.6 years in the late-infantile onset subgroup to 39.5 years in the adolescent/adult-onset subgroup. Mean ages at death among the age-at-onset subgroups ranged from 0.19 years among neonatal patients to 33.7 years in the adolescent/adult-onset subgroup. In non-neurological NP-C patients , the mean age at last follow-up among 11 living patients was 2.5 years (range 0.5–6.1). The mean age at death among three non-neurological patients who died was 20.8 (15.9) years (range 4.9–36.7).
Neurological and psychiatric manifestations: overall cohort
The large majority of patients had at least one neurological manifestation commonly associated with NP-C at last follow up. A total of 19 patients (13 %) had no recorded manifestations, five of whom had the neonatal form of the disease.
Figure 1 summarises the prevalence of individual neurological symptoms per age at onset subgroup. Overall, developmental delay (during childhood) and/or cognitive deterioration were recorded in the greatest proportion of patients up to last follow up (in 119/146 patients [82 %]). In order of incidence, other common neurological signs were: ataxia (in 110 patients [76 %]), VSGP (103 patients [71 %]), dysarthria (99 patients [68 %]), dysphagia (93 patients [64 %]) and seizures/cataplexy (72 patients [50 %]).
Fig. 1.
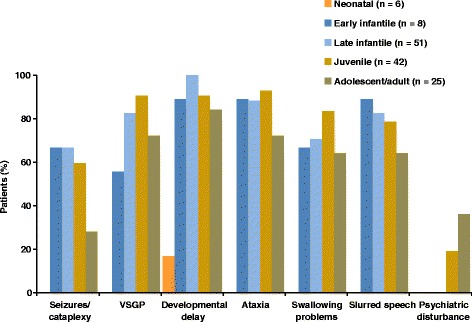
Occurrence of neurological and psychiatric manifestations per patient subgroup Percentages calculated based on numbers of patients with available data
Psychiatric disturbances were recorded in a total of 17 (12 %) patients. As could be expected, all cases in whom psychiatric disturbances were recorded were in the juvenile-onset or adolescent/adult-onset subgroups (incidence per subgroup 8/42 patients [19 %] and 9/25 patients [36 %], respectively).
Visceral symptoms: overall cohort
Visceral symptoms of NP-C were recorded most frequently in patients with the neonatal visceral form of NP-C and in the early infantile-onset and non-neurological subgroups, and least frequently among adolescent/adult-onset patients (Fig. 2). Prolonged neonatal jaundice with or without neonatal liver disease was by far the most common visceral symptom, occurring in all neonatal onset, 6/8 (75 %) early-infantile onset, 29/51 (57 %) late-infantile onset, and 14/42 (33 %) juvenile-onset patients. Only one adolescent/adult-onset patient had a recorded history of neonatal jaundice. Organomegaly (hepatosplenomegaly in most cases) also tended to occur less and less frequently as age at neurological onset increased. All patients in whom no neurological manifestations have yet been recorded had a history of at least one visceral symptom, most commonly prolonged neonatal jaundice (in 5/14 [36 %] patients).
Fig. 2.
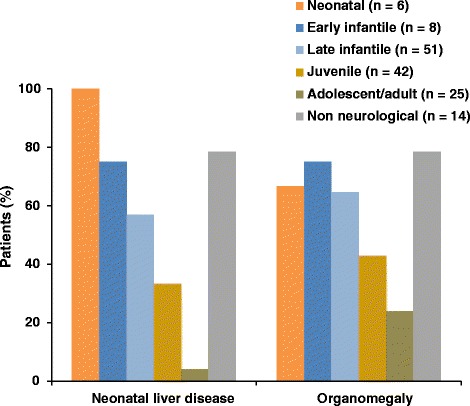
Occurrence of visceral symptoms per patient subgroup Percentages calculated based on numbers of patients with available data
Clinical symptomatology and outcomes in age-at-onset subgroups
Neonatal onset patients
All six patients with the neonatal form of NP-C had liver disease at or soon after birth (mean [SD] age 0.19 [0.22] years), with organomegaly also apparent in four cases and foetal ascites present in two (Table 2). Hepatosplenomegaly was also recorded in four cases. Patients with the severe neonatal-onset form of NP-C have been reported in other national cohorts to have a short lifespan [2, 16], and the same was true in this UK cohort. Two cases were stillborn and the other four died within 1–7 months of birth. Liver disease was the most frequently recorded cause of death (three patients), and one patient died due to failure to thrive and chest infection. One patient (patient 1) had a record of developmental delay from birth.
Table 2.
Patients with neonatal NP-C
| Patient number/Gender | Sibship | Date of birth | Age at diagnosis | Age at last FU/deatha | Neonatal LD | HS/S | Seizures/cataplexy | VSGP | Dev. delay | Ataxia | Swallowing problems | Psychiatric disturbance | Slurred speech | Miglustat? | Age at 1st miglustat start | Miglustat duration | Genetic mutations |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1/M | – | 2000 | 4 m | 4 ma | PJ LD | HS | – | – | From birth | – | – | – | – | No | – | – | – |
| 2/F | a | 2001 | PM | 0a | FA | – | – | – | – | – | – | – | – | No | – | – | – |
| 3/F | a | 2002 | PM | 0a | FA | – | – | – | – | – | – | – | – | No | – | – | – |
| 4/M | b | 1985 | Birth | 1 ma | Yes | Yes | – | – | – | – | – | – | – | No | – | – | c.3501C > G(p.Phe1167Leu)/c.3501C > G(p.Phe1167Leu) |
| 5/M | b | 1985 | Birth | 2 ma | Yes | Yes | – | – | – | – | – | – | – | No | – | – | c.3501C > G(p.Phe1167Leu)/c.3501C > G(p.Phe1167Leu) |
| 6/F | c | 2009 | 2 m | 7 ma | Yes | Yes | – | – | – | – | – | – | – | No | – | – | c.3020C > T(p.Pro1007Leu)/c.3020C > T(p.Pro1007Leu) |
aPatient died; ‘–’, no data/not known; C cataplexy, E epilepsy, FA foetal ascites, HS/S hepatosplenomegaly/splenomegaly, LD liver disease, m months, PJ prolonged jaundice, PM post mortem, y years, w weeks
Early-infantile onset patients
All but one of the early-infantile onset patients were female (Table 3). The mean (SD; range) age at onset of neurological manifestations in this subgroup was 1.1 (0.7; 0–2.0) years. Overall, the mean (SD) time between onset of neurological manifestations and diagnosis was 0.26 (1.49) years, with diagnostic testing commenced based on recognition of visceral symptoms in four patients.
Table 3.
Patients with early-infantile neurological onset
| Patient number/Gender | Sibship | Date of birth | Age at diagnosis | Age at last FU (y, m)/deathb | Neonatal LD | HS/S | Seizures/cataplexy | VSGP | Dev. delay | Ataxia | Swallowing problems | Psychiatric disturbance | Slurred speech | Miglustat? | Age at 1st miglustat start | Miglustat duration | Genetic mutations |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7/F | – | 2000 | 4 m | 3y 4 mb | PJ LD | Yes | C 2y 11 m | – | <18 m | <2y | 13 m | – | <2y | No | – | – | c.3578_3591 + 9del/c.3578_3591 + 9del |
| 8/F | – | 2006 | 19 m | 3y 9 mb | PJ | Yes | No | No | Yes | Yes | 19 m | – | Yes | No | – | – | – |
| 9/F | – | 2005 | 3 m | 4y 1 mb | PJ LD, LTx | Yes | – | – | Yes | Yes | Yes | – | Yes | No | – | – | c.1526A > C(p.Tyr509Ser/? |
| 10/F | – | 2003 | 6 m | 4y 5 mb | PJ LD | Yes | E 3y 5 m | <18 m | Yes | Yes | 3y | – | No speech | No | – | – | c.2801G > A(p.Arg934Gln)/c.2978del(p.Gly993Glu fsX4) |
| 11/M | d | 1990 | PM | 7y 8 mb | No | No | C 2y E 4y | Yes | <2y | <2y | 5y | – | Yes | No | – | – | c.2819C > T(p.Ser940Leu/?a |
| 12/F | e twin | 1998 | 11 m | 8y 5 mb | PJ | Yes | C 5y E 5y 10 m | 3y | Yes | Never mobile | No | – | <3y | Yes | 7y | 1w | c.3107C > T(p.Thr1036Met/c.3557G > A(p.Arg 1186His) |
| 13/F | e twin | 1998 | 11 m | 7y 1 mb | PJ | Yes | C 5y E 5y 10 m | 3y | Yes | Never mobile | No | – | <3y | Yes | 7y | 1w | c.3107C > T(p.Thr1036Met/c.3557G > A(p.Arg 1186His) |
| 14/F | – | 1995 | 4y 5 m | 6y 5 mb | No | No | C 4y E 5y | 4y | 1y 5 m | 4y | 5y | – | Yes | – | – | – | c.3503G > A(p.Cys1168Tyr)/c.3503G > A(p.Cys1168Tyr) |
aSecond mutant allele not found after full genome sequencing; bPatient died; ‘–’, no data/not known; C cataplexy, E epilepsy, FA foetal ascites, HS/S hepatosplenomegaly/splenomegaly, LD liver disease, LTx liver transplant, m months, PJ prolonged jaundice, PM post mortem, y years, w weeks
The mean (SD) age at death was 5.6 (2.0) years. The most common recorded cause of death (7/8 cases [88 %]) was ‘NP-C’, reflecting a gradual and in some cases rapid deterioration, with eventual loss of all skills and bodily functions with no one apparent causative factor. However, the recorded causes of death could reflect the way death certificates are completed in the UK, where the primary disease is often stated as the causative factor.
A total of 6/8 patients (75 %) exhibited both prolonged neonatal jaundice and hepatosplenomegaly (Fig. 2). One patient (patient 9) underwent liver transplantation due to severe cholestatic liver disease.
Developmental delay, ataxia and dysarthria were the most commonly recorded neurological symptoms, each occurring in all eight patients (Fig. 1). Cataplexy/epileptic seizures and swallowing difficulties were each recorded in 6/8 (75 %) patients. Ophthalmic assessments revealed VSGP in 5/8 (63 %) patients.
Late-infantile onset patients
The mean (SD; range) age at neurological onset in this subgroup was 4.1 (1.2; 0.4–8.0) years, and the overall mean (SD) time period between neurological onset and diagnosis among patients with available data was 0.56 (5.25) years. Again, diagnostic testing was commenced in approximately half of this patient subgroup after initial recognition of visceral symptoms.
Overall, 30/51 (59 %) patients died before data cut-off at the end of 2011 (mean [SD] age at death 13.4 [6.7] years). ‘NP-C’ was listed as the cause of death in 26/30 patients (87 %) with available information. Pneumonia was listed specifically as the cause of death in two cases, but any association with dysphagia or previous food aspiration was not recorded.
Neurological manifestations were more common than visceral symptoms in this subgroup (Table 4). Again, developmental delay was recorded most frequently (in all 51 patients), followed by ataxia (in 45/51 [88 %]) and VSGP (in 42/51 [82 %]). Dysarthria, dysphagia and seizures/cataplexy were observed in 42/51 (82 %), 36/51 (71 %) and 34/51 (67 %) patients, respectively (Fig. 1).
Table 4.
Patients with late-infantile neurological onset
| Patient number/Gender | Sibship | Date of birth | Age at diagnosis | Age at last FU (y, m)/deathb | Neonatal LD | HS/S | Seizures/cataplexy | VSGP | Dev. delay | Ataxia | Swallowing problems | Psychiatric disturbance | Slurred speech | Miglustat? | Age at 1st miglustat start | Miglustat duration | Genetic mutations |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 15/M | f | 2000 | 2y | 5 y 4 mb | – | <1y 8 m | E <4y 11 m | – | <4y 6 m | <4y 6 m | <4y 6 m | – | <3y | No | – | – | c.3020C > T(p.Pro1007Leu)/c.3020C > T(p.Pro1007Leu) |
| 16/F | – | 2004 | 11 m | 7y 2 m | PJ | S | No | 5y | 5y | 4y | No | – | Mild <4y | Yes | 6y | 1y | – |
| 17a/M | – | 2005 | 1y 2 m | 6y 7 m | HS | – | No | <6y | <5y | No | No | – | <6y | Yes | 5y | 1y | c.1526A > C(p.Tyr509Ser)/c.688_693del(p.Ser230_Val231del |
| 18/F | – | 2002 | 6 m | 6y 4 mb | PJ | S | C 3y | Yes | 3y | Yes | 3y | – | 3y | No | – | – | c.2008_2011del(p.Cys670ProfsX17/?d |
| 19a/M | – | 1990 | 15y | 21y 2 m | – | – | E 15y | Yes | 5y | Yes | – | – | – | Yes | 16y | 5y | c.2292G > A(p.Gly764Ala)/? |
| 20/F | – | 1987 | 6y | 13y 6 mb | No | 4y | – | 7y | 5y 5 m | 7y | 4y | – | Yes | No | – | – | c.2324A > C(p.Gln775Pro)/c.2956G > A(p.Gly986Ser) |
| 21/M | g | 2003 | 2y 11 m | 7yb | PJ | HS | C 4y | 2y 11 m | 3y | <4y | Yes | – | <4y | No | – | – | c.2464_2465insT(p.Lys822IlefsX48)/c.2201G > T(p.ser734Ile) |
| 22a/F | g | 2005 | 4y | 6y 10 m | Slight jaundice | No | No | <6y | <5y | <6y | No | – | <6y | Yes | 3y | 3y | c.2464_2465insT(p.Lys822IlefsX48)/c.2201G > T(p.ser734Ile) |
| 23/F | d | 1997 | 3y 9 m | 12y 9 mb | No | No | C 6y | 6y | 6y 5 m | 4y 7 m | 7y | – | Yes | Yes | – | – | c.2819C > T(P.Ser940Leu)/? |
| 24a/F | – | 2001 | 7y | 10y 10 m | No | No | E 9y C 8y | 9y | <5y | 9y | 6y | – | <6y | Yes | 8y | 2y | c.3019C > g(p.Pro1007Ala)/? |
| 25a/F | c/twin | 2006 | At birth | 5y 6 m | PJ LD | HS | No | 4y | <4y | <4y | No | – | 4y | Yes | 4y | 1y | c.3020C > T(p.Pro1007Leu)/c.3020C > T(p.Pro1007Leu) |
| 26a/F | c/twin | 2006 | At birth | 5y 6 m | PJ LD | HS | No | 4y | <4y | <4y | No | – | 4y | Yes | 4y | 1y | c.3020C > T(p.Pro1007Leu)/c.3020C > T(p.Pro1007Leu) |
| 27/F | f | 2008 | At birth | 3y | Yes | Yes | No | No | <3y | Mild | No | – | No | Yes | 0.5y | 2.5y | c.3020C > T(p.Pro1007Leu)/c.3020C > T(p.Pro1007Leu) |
| 28/M | – | 1994 | 9 m | 9y 3 mb | PJ LD | HS | C 4y E 7y | 4y | 1y | <5y | 5y | – | 5y | No | – | – | c.3182 T > C(p.Ile1061Thr)/?d |
| 29/F | – | 1990 | 9 m | 9y 5 mb | PJ LD | No | Yes | <6y | <6y | 6y 5 m | – | – | – | No | – | – | c.3182 T > C(p.Ile1061Thr)/? |
| 30/F | – | 1975 | 8y | 28yb | PJ LD | HS | E 17y | 8y | 13y | 5y | Yes | – | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/? |
| 31/F | – | 2003 | 3 m | 8y 6 m | PJ LD | At birth | E 8y | 4y | <4y | <4y | <7y | – | 5y | Yes | – | – | c.3182 T > C(p.Ile1061Thr)/?d |
| 32//M | h | 1989 | 2y | 11y 2 mb | PJ LD | No | E 6y 5 m | Yes | 4y | Yes | Yes | – | – | No | – | – | c.3182 T > C(p.Ile1061Thr)/? |
| 33/M | h | 1988 | 1y | 8y 5 mb | PJ LD | No | C 6y E 6y | No | <5y | – | 8y | – | – | No | – | – | c.3182 T > C(p.Ile1061Thr)/? |
| 34/F | – | 1990 | 4y 9 m | 14yb | No | 4y | C 7y E 8y | 7y | 6y | 4y | 8y | – | 7y | No | – | – | c.3182 T > C(p.Ile1061Thr)/? |
| 35/F | – | 1989 | 7y | 16yb | No | At birth | C 8y E 10y | 5y | 4y | 4y 5 m | 9y | – | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/? |
| 36/M | – | 1987 | 8 m | 12y 1 mb | PJ LD | At birth | C 5y 5 m E 7y 8 m | 5y | 5y | 5y | 9y | – | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/? |
| 37/M | – | 1991 | 4y 3 m | 9yb | PJ LD | HS 3y | C 5y | 4y | 4y 6 m | 2y | 5y | – | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/? |
| 38/M | – | – | 16y | 22y 11 mb | PJ | No | No | 16y | 8y | 4y | 17y | – | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/? |
| 39/M | – | 2001 | 8 m | 10y 6 m | PJ | HS | No | Yes | 4y 6 m | Yes | No | – | No | Yes | 7y | 3y | c.3182 T > C(p.Ile1061Thr)/del10bp962 |
| 40/M | – | 1995 | 10 m | 10yb | PJ LD | S | C 4y 9 m | 4y | >4y | Yes | Yes | – | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/c.1142G > A(p.Trp381X) |
| 41/F | – | 1996 | 12 m | 11y 9 mb | No | HS 9 m | C 5y 6 m | 5y | 5y | 5y 6 m | No | – | 11y | Yes | 7y | 4y | c.3182 T > C(p.Ile1061Thr)/c.2656G > C(p.Gly886Arg) |
| 42/F | – | 1983 | 9y 5 m | 25y 7 mb | No | S 9y 6 m | E 5y | Yes | Yes | 12y | Yes | – | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/c.3019C > G(p.Pro1007Ala) |
| 43/F | – | 1993 | 6y | 9y 6 mb | PJ | 5y | C 5y E6y | 5y | 5y | 5y | 6y | – | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/c.3107C > T(p.Thr1036Met) |
| 44a/M | – | 2007 | 4 m | 4y 1 m | NC | Yes | No | No | <3y | <3y | No | – | <3y | Yes | 2y | 2y | c.3182 T > C(p.Ile1061Thr)/c.3107C > T(p.Thr1036Met) |
| 45a/M | – | 1998 | 4 m | 13y 9 m | PJ | Yes | C 8y | 5y | 9y | No | No | – | No | Yes | 5y | 8y | c.3182 T > C(p.Ile1061Thr)/c.3175C > T(p.Arg1059X) |
| 46/F | – | 1996 | 7y 8 m | 15y 6 mb | No | No | C 8y | 7y 8 m | 7y 8 m | 5y | 8y 5 m | – | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/c.3182 T > C(p.Ile1061Thr) |
| 47a/M | – | 2006 | At birth | 5y | PJ LD | Yes | C <4y | No | 3y | Yes | <5y | – | <4y | Yes | 4y | 1y | c.3182 T > C(p.Ile1061Thr)/c.3422 T > G(p.Val1141Gly) |
| 48/F | – | 1982 | 3y | 29y 4 m | PJ | HS at birth | No | 3y | 3y | 5y | 18y | – | Yes | Yes | – | – | c.3182 T > C(p.Ile1061Thr)/c.3467A > G(p.Asn1156Ser) |
| 49a/F | – | 2001 | 12 m | 10y 7 mb | No | S | C 9y | 4y | 4y | 4y | 7y | – | 6y | No | – | – | c.3182 T > C(p.Ile1061Thr)/c.3591 + 4delA |
| 50a/M | – | 2004 | 1y 11 m | 7y 5 mb | No | 6 m | C 4y | 4y | 3y 5 m | 3y | 4y 5 m | – | Yes | Yes | 5y | 2y | c.3259 T > C(p.Phe1087Leu)/c.2516 T > G(p.Ile839Arg) |
| 51/F | i | 1984 | 8y 6 m | 27y 5 m | No | 2y | C 5y | 8y | 13y | 5y | 13y | – | 11y | No | – | – | c.3467A > G(p.Asn1156Ser)/?d |
| 52/F | i | 1978 | 14y 5 m | 33y 5 mb | No | No | E 13y | 14y | 5y | 13y | 13y | – | 12y | No | – | – | c.3467A > G(p.Asn1156Ser)/?d |
| 53/F | j | 1995 | 8y 4 m | 16y 11 m | No | No | E 7y 5 m C 8y | 6y 9 m | 3y 5 m | 4y | 8y 6 m | – | Yes | No | – | – | c.3591 + 4delA/?d |
| 54a/M | – | 2006 | At birth | 5y 5 m | PJ LD | Yes | No | No | Yes | No | Yes | – | Yes | No | – | – | c.58G > T(p.Glu20X)/c.58G > T(p.Glu20X) [NPC2] |
| 55/M | k | 2000 | – | 10y 5 mb | No | No | C 6y | 5y | 5y | 5y | 5y | – | Yes | No | – | – | – |
| 56/M | – | 2005 | 3y 10 m | – | Yes | – | – | – | 4y | – | – | – | 4y | No | – | – | – |
| 57/F | l | – | 2y | 6yb | No | HS 6 m | No | 3y | 2y 5 m | 2y 5 m | 5y | – | No | No | – | – | – |
| 58/M | – | – | 9y 5 m | 15yb | No | No | C + E 5y | 6y | 6y | 4y | 10y | – | Yes | No | – | – | – |
| 59/F | – | 1987 | 8y | 17y 6 mb | No | 4y | C 5y E 10y | 7y | 5y | 6y | 11y | – | Yes | No | – | – | – |
| 60/M | b | 1983 | 8y | 16yb | PJ | No | E 16y | Yes | 8y | 5y | 12y | – | Yes | No | – | – | c.3501C > G(p.Phe1167Leu)/c.3501C > G(p.Phe1167Leu) |
| 61/M | l | 1990 | 7y 1 m | 17y 5 mb | No | S 2y 6 m | C 10y | 7y 1 m | 4y 5 m | 5y 5 m | 10y | – | Yes | No | – | – | – |
| 62/F | – | 1993 | 5y | 9y 5 mb | PJ | HS 5y 5 m | C 5y | No | 18 m | 4y | No | – | Yes | No | – | – | – |
| 63/M | k | 1998 | – | – | – | – | – | Yes | Yes | Yes | Yes | – | Yes | No | – | – | – |
| 64/F | – | 1982 | 29y | 29y 6 m | No | No | No | 29y | Yes | Yes | 20y | – | 24y | No | – | – | c.3022A > C(p.Asn108His)/c.182 T > C(p.Ile 1061Thr) |
| 65/F | – | 2008 | 2y | 3y 9 m | Yes | HS | No | No | Yes | No | No | – | No | No | – | – | [NPC2]c |
aPatients included in previously reported NPC Registry baseline characteristics study [19]; bpatient died; cmutation found but information not accessible during observation period; dsecond mutant allele not found after full genome sequencing. ‘–’, no data/not known; C cataplexy, E epilepsy, FA foetal ascites, HM hepatomegaly, HS/S hepatosplenomegaly/splenomegaly, LD liver disease, LTx liver transplant, m months, NC neonatal cholestasis, PJ prolonged jaundice, PM post mortem, y years, w weeks
The only two patients in the UK cohort who had NPC2 mutations were in this age-at-onset subgroup (patients 54 and 65), both of whom displayed neonatal jaundice and hepatosplenomegaly, as was common among other late-infantile onset patients. However, both of these patients displayed relatively few typical neurological manifestations: patient 54 had developmental delay, dysarthria and dysphagia, and only developmental delay was recorded in patient 65.
Overall, hepatosplenomegaly was the most common visceral symptom in this patient subgroup, recorded in 32/51 (63 %) patients. A history of neonatal jaundice was recorded in 29/51 (57 %) (Fig. 2).
Juvenile-onset patients
Among juvenile-onset patients, neurological manifestations were first noted at a mean (SD; range) age of 9.4 (2.6; 5.0–15.0) years. The mean (SD) time between neurological onset and diagnosis was 1.64 (6.09) years. In this subgroup, diagnostic testing was commenced based on the appearance of neurological signs in the majority (approximately three-quarters) of cases.
In total, 19/42 (45 %) patients had died by data cut-off (mean [SD] age at death, 25.9 [8.9] years) (Table 5). As for late-infantile onset patients, most deaths (11/18 evaluable patients [61 %]) in this subgroup were recorded as being due to ‘NP-C’. Four of the 18 (22 %) patients with available information died due to respiratory-related complications but again, no associations with dysphagia or previous aspiration were noted.
Table 5.
Patients with juvenile neurological onset
| Patient number/Gender | Sibship | Date of birth | Age at diagnosis | Age at last FU (y, m)/deathc | Neonatal LD | HS/S | Seizures/cataplexy | VSGP | Dev. delay/cognitive problems | Ataxia | Swallowing problems | Psychiatric disturbance | Slurred speech | Miglustat? | Age at 1st miglustat start | Miglustat duration | Genetic mutations |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 66/F | – | 1970 | 24y 5 m | 41y 2 m | No | Yes | C 24y | 12y | 11y | 24y | 24y | 32y | 24y | Yes | 32y | – | c.1211G > A(p.Arg404Gln)/?d |
| 67/F | – | 1995 | 10y 1 m | 16y 3 m | No | No | E 10y 3 m | 10y 1 m | 8y | 10y | 13y | No | Yes | Yes | 12y | 4y | c.1552C > T(p.Arg518Trp)/c.283 T > C(p.Ser95Pro) |
| 68/F | – | 1996 | 10y 6 m | 15y 1 m | – | – | <10y | Yes | <10y | <10y | <10y | No | <10y | No | – | – | c.2848G > A(p.Val950Met)/? |
| 69/F | – | 1994 | 14y | 17y 10 m | No | No | No | Yes | 10y | Yes | Yes | – | 6y | No | – | – | c.2974G > T(p.Gly992Trp)/? |
| 70/F | – | 1983 | 14y | 21y 11 mc | No | No | E 12y | Yes | 8y | 11y | 17y | No | Yes | No | – | – | c.3019C > G(p.Pro1007Ala)/? |
| 71a/M | – | 1991 | 17y | 20y 4 m | – | – | E 12y | Yes | At birth | Yes | Yes | Yes | Yes | Yes | 18y | 2y | c.3019C > G(p.Pro1007Ala)/c.1553G > A(p.Arg518Gln |
| 72/F | – | 1986 | 2 m | 25y 10 m | FA LD | At birth | E 14y | 11y | 11y | 14y | 15y | No | <14y | No | – | – | c.3176G > A(p.Arg1059Gln)/? |
| 73/M | – | 1996 | 14y | 15y 5 m | No | No | No | 12y | 7y | 7y | 13y | No | 12y | Yes | 14y | 1y | c.3182 T > C(p.Ile1061Thr)/? |
| 74/M | – | – | 7y 5 m | 20yc | PJ | HS | No | 7y | 6y 5 m | 7y | 8y | No | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/? |
| 75/F | – | 1986 | 10y | 19y 2 mc | No | No | C 11y | 10y | 7y | 7y | 11y | No | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/? |
| 76/M | – | 2000 | 7y | 11y 1 mc | – | – | E 7y | Yes | Yes | Yes | Yes | No | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/? |
| 77a/F | – | 1996 | 4y 5 m | 15y 4 m | PJ | HS 4y 5 m | E 12y | 7y | 7y | 6y | No | No | No | Yes | 7y | 8y | c.3182 T > C(p.Ile1061Thr)/c.2801G > A(p.Arg934Gln) |
| 78a/F | – | 2002 | 2 m | 9y 10 m | PJ | HS | C 6y | Yes | 6y | No | No | No | No | Yes | 7y | 2y | c.3182 T > C(p.Ile1061Thr)/c.2819C > T(p.Ser940Leu) |
| 79/F | – | 1985 | 4y 1 m | 26y 4 m | PJ LD | HS 11y | C 7y E 17y | 11y | 13y | 13y | 16y | No | No | Yes | – | – | c.3182 T > C(p.Ile1061Thr)/c.2974G > T(p.Gly992Trp) |
| 80/M | m | 1978 | 12y | 18y 8 mc | No | S 2–10y | E 11y | 10y | 8y | 10y | 18y | No | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/c.3019C > G(p.Pro1007Ala |
| 81/M | – | 2003 | 6 m | 8y 8 m | PJ LD | HS | – | – | No | – | – | – | – | Yes | 7y | 1y | c.3182 T > C(p.Ile1061Thr)/ c.3182 T > C(p.Ile1061Thr) |
| 82/M | – | 1970 | At birth | 32yc | No | No | E 22y | Yes | 12y | Yes | 27y | No | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/ c.3182 T > C(p.Ile1061Thr) |
| 83/F | – | 1991 | 13y | 18y 2 mc | No | No | No | Yes | 10y | 14y | 14y 5 m | No | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/ c.3182 T > C(p.Ile1061Thr) |
| 84b/M | – | 1988 | 12y | 23y 7 m | No | S 7 y | No | 11y 9 m | 17y | 17y | No | No | No | Yes | 21y | 2y | c.3182 T > C(p.Ile1061Thr)/ c.3182 T > C(p.Ile1061Thr) |
| 85b/F | – | 1991 | 6y 5 m | 20y 1 m | No | S 5y 5 m | No | 8y 3 m | 11y 1 m | 9y | 19y | No | No | Yes | 12y | 8y | c.3182 T > C(p.Ile1061Thr)/c.3493G > A(p.Val1165Met) |
| 86/F | – | 2005 | 8w | 6y 6 m | PJ LD | HS | No | Mild 6y | No | No | No | No | No | Yes | 4y | 2y | c.3182 T > C(p.Ile1061Thr)/c.350-351delAG |
| 87/F | – | 1986 | 17y | 25y 5 m | No | No | E 16y | Yes | 5y (FAS) | <16y | <16y | – | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/c.3566A > G(p.Glu1189Gly) |
| 88/M | – | 1985 | 18y | 26y 7 m | No | S 18 y | No | 15y | 13y | 13y | 18y | 18y 6 m | 18y | Yes | – | – | c.3182 T > C(p.Ile1061Thr)/c.3566A > G(p.Glu1189Gly) |
| 89a/F | – | 1994 | 10y 4 m | 17y 9 m | No | No | C <4y E <17y | <17y | <10y 4 m | <10y 4 m | <17y | No | <17y | Yes | 14y | 3y | c.3182 T > C(p.Ile1061Thr)/c.410C > T(p.Thr137Met) |
| 90/F | – | 1983 | 15y | 22y 8 mc | PJ | Birth | E 14y | 11y | 8y | 11y | 16y | No | Yes | No | – | – | c.3263A > G(p.Tyr1088Cys)/c.1201C > A(p.Pro401Thr) |
| 91/M | – | 1989 | 17y | 23y | No | No | No | Yes | 8y | 9y | 18y | 17y | 17y | Yes | 18y | 3 m | c.3493A > G(p.Val1165Met)/c.3493A > G(p.Val1165Met) |
| 92/F | b | 1990 | 3 m | 17y 2 mc | PJ | No | E 15y | 11y | 10y | 10y 5 m | 15y | No | Yes | Yes | – | – | c.3501C > G(p.Phe1167Leu)/c.3501C > G(p.Phe1167Leu) |
| 93/F | j | 1998 | 10y | 13y 6 m | No | No | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | 10y | 3y | c.3591 + 4delAla/? |
| 94/F | – | 1997 | 10y | 14y 8 mc | No | No | C + E 10y | No | <10y | <10y | Yes Gx | No | Yes | No | – | – | c.3019C > G(p.Pro1007Ala)/c.2464-2465insT |
| 95/M | – | 1998 | 12y | 13y 6 m | No | No | C 11y | 11y | 9y | Tremor 11y | No | No | <5y | Yes | 13y | 6 m | – |
| 96/F | – | 1996 | 9y | 15y 8 mc | Mild jaundice | S | – | Yes | Yes | Yes | Yes Gx | No | 9y | No | – | – | – |
| 97/F | – | 1991 | 16y | 17y 6 mc | – | – | S, C | Yes | <15y | <15y | <17y | No | Yes | No | – | – | – |
| 98/M | – | 1967 | PM | 25yc | PJ | S 2 y | No | Yes | 11y | 2y | 14y | 25y | Yes | No | – | – | – |
| 99/F | – | 1972 | 16y | 39y 11 m | PJ | No | No | 16y | 11y | 28y | 30y | 30y | Yes | Yes | – | – | c.1211G > A(p.Arg404Gln)/c.1133 T > C(P.Val378Ala) |
| 100/M | – | 1971 | 14y | 29y 2 mc | No | 13y 5 m | 13y | 13y 5 m | 13y | 13y 5 m | 13y | No | Yes | No | – | – | – |
| 101/F | – | 1963 | 18y | 40y 3 mc | PJ | No | E 31y | 16y | 12y | 16y | 23y | 16y | Yes | No | – | – | c.1843C > T(p.Arg615Cys)/c.2972-2973del(p.Gln991ArgfsX15) |
| 102/F | n | – | Teens | 23yc | No | No | – | No | Teens | Teens | Teens | 23y | – | No | – | – | c.3182 T > C(p.Ile1061Thr)/? |
| 103/M | – | 1987 | 20y | 24y 5 m | No | No | No | <19y | No | 13y | 22y | No | 13y | Yes | 22y | 2y | c.2861C > T(p.Ser954Leu)/c.3107C > T(p.Thr1036Met) |
| 104/M | – | 1972 | 6y | 39y 8 m | No | HS 5y | No | No | No | 6y | No | No | No | No | – | – | c.1844G > T(p.Arg615Leu)/c.1844G > T(p.Arg615Leu) |
| 105/M | – | 1969 | Early 20s | 37y 8 mc | PJ | S 4y | E 30y | Yes | 11y | 20s | Yes | No | Yes | No | – | – | – |
| 106a/F | o | 1976 | 27y | 35y 10 m | No | No | No | 27y | 13y | 26y | 26y | No | 27y | Yes | – | – | c.1552C > T(p.Arg518Trp)/c.1552C > T(p.Arg518Trp) |
| 107/M | o | 1972 | 30y | 37y 2 mc | No | No | Teens | 26y | 26y | 26y | 27y | 25y | 26y | No | – | – | c.1552C > T(p.Arg518Trp)/c.1552C > T(p.Arg518Trp) |
aPatients included in previously reported NPC Registry baseline characteristics study [19]; bPatients included in previously reported NPC Registry baseline and longitudinal data study [19, 20]; cpatient died; dsecond mutant allele not found after full genome sequencing. ‘–’, no data/not known; C cataplexy, E epilepsy, FA foetal ascites, Gx gastrostomy, HM hepatomegaly, HS/S hepatosplenomegaly/splenomegaly, LD liver disease, LTx liver transplant, m months, NC neonatal cholestasis, PJ prolonged jaundice, PM post mortem, y years, w weeks
Neurological manifestations were substantially more common than visceral symptoms in this subgroup. Ataxia was the most common neurological manifestation (in 39/42 [93 %] patients), followed by VSGP and childhood developmental delay or cognitive problems (both in 38/42 [91 %]), dysphagia (35/42 [83 %]), dysarthria (33/42 [79 %]) and seizures/cataplexy (25/42 [60 %]) (Fig. 1). Unlike younger patient subgroups, psychiatric disturbances were recorded in juvenile-onset patients (8/42 [19 %] cases). The age at onset of psychiatric disturbances ranged from 16 to 32 years.
Neonatal jaundice and/or cholestatic liver disease were recorded in 14/42 (33 %) patients, and organomegaly was seen in 18/42 (43 %) patients (Fig. 2). While no specific neurological signs were recorded in one patient (patient 81), evidence of neurological involvement was documented at the local treatment centre. However, it is not possible to report this patient’s presenting neurological manifestation as he was lost to follow up.
Adolescent/adult-onset patients
Overall, patients with neurological onset during adolescence or adulthood were characterized by insidious onset and slow disease progression (Table 6). Neurological onset in this older age subgroup occurred at a mean (SD; range) age of 24.2 (8.8; 15.0–40.0) years. The mean (SD) period between neurological onset and diagnosis was 6.0 (6.26) years, and all evaluable patients in this subgroup were diagnosed after the appearance of neurological symptoms. By data cut-off, a total of 8/24 (32 %) patients had died (mean [SD] age at death, 33.7 [6.2] years). Among patients with available records, seven were recorded as being due to NP-C.
Table 6.
Patients with adolescent-adult neurological onset
| Patient number/Gender | Sibship | Date of birth | Age at diagnosis | Age at last FU (y, m)/deathc | Neonatal LD | HS/S | Seizures/cataplexy | VSGP | Dev. delay/cognitive problems | Ataxia | Swallowing problems | Psychiatric disturbance | Slurred speech | Miglustat? | Age at 1st miglustat start | Miglustat duration | Genetic mutations |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 108b/M | m | 1976 | 23y | 39y | PJ | No | No | 17y | 17y | 25y | 25y | No | Yes | Yes | 32y | 3y | c.3182 T > C(p.Ile1061Thr)/c.3019C > G(p.Pro1007Ala) |
| 109/F | – | 1975 | 18y | 30yc | No | No | E 18y 6 m | 18y | 15y | 15y | 24y | 18y | Yes | No | – | – | c.3182 T > C(p.Ile1061Thr)/c.3019C > G(p.Pro1007Ala) |
| 11/M | – | 1978 | 20y | 27y 3 mc | No | No | No | <24y | <20y | No | 23y | Yes | – | No | – | – | – |
| 111/F | – | 1975 | 25y 6 m | 30yc | No | No | C 25y | 25y | 16y | 24y | 23y | No | 23y | No | – | – | – |
| 112/M | n | 1971 | 24y | 40y 2 m | No | No | No | 16y | 24y | <24y | No | 29y | 32y | No | – | – | c.3182 T > C(p.Ile1061Thr)/? |
| 113/M | p | 1968 | 19y | 43y 2 mc | No | S 18y | E 17y | 25y | 18y | 18y | 25y | 17y | 18y | No | – | – | c.1843C > T(p.Arg615Cys)/c.3289-3291del(p.Asp1097del) |
| 114a/M | – | 1987 | 22y | 24y 11 m | No | No | No | 22y | 15y | 15y | 23y | No | 18y | Yes | 24y | 0.5y | – |
| 115/M | p | 1969 | 18y | 28y 8 mc | No | S 1y 4 m | No | Yes | Yes | Yes | 25y | 28y | Yes | No | – | – | c.1843C > T(p.Arg615Cys)/c.3289-3291del(p.Asp1097del) |
| 116/F | – | 1963 | 38y | 48y 6 m | No | No | No | <34y | 37y | 34y | 39y | No | 34y | Yes | 39ye | – | c.1133 T > C(p.Val378Ala)/c.422_423dup(p.Lys142X) |
| 117a/F | o | 1978 | 25y | 33y 11 m | No | No | No | No | 25y | 25y | 32y | No | 32y | Yes | 29y | 4y | c.1552C > T(p.Arg518Trp)/c.1552C > T(p.Arg518Trp) |
| 118/M | – | – | 27y | 35y 4 m | – | Yes | Teens | No | Yes | Yes | Yes | 20s | Yes | No | – | – | – |
| 119d/F | – | 1966 | 32y | 42y | No | No | No | Yes | <18y | 29y | 38y | No | 38y | Yes | 37y | 8 | c.3182 T > C(p.Ile1061Thr)/? |
| 120/M | – | 1972 | 29y | 39y 10 mc | – | – | – | – | Yes | – | – | – | – | No | – | – | – |
| 121a/M | – | 1982 | 20y | 29y 2 m | No | No | No | – | Yes | – | – | – | Yes | No | – | – | – |
| 122/M | – | 1964 | 31y | 48y 6 m | No | Yes | No | Yes | 46y | No | Yes | No | No | Yes | 42y | 5y | – |
| 123a/M | q | 1967 | 40y | 44y 5 m | – | No | Tremor | <40y | <40y | <40y | No | No | No | Yes | 43y | 1y | c.1408G > C(p .Ala 470 Pro/c.1816G > C (p.Glu608Gln) |
| 124/F | – | 1961 | 49y | 50y 11 m | No | No | No | 46y | 42y | 42y | 49y | 40y | 42y | Yes | 49y | 1y | c.2000C > G(p.Ser667Trp)/? |
| 125/F | r | 1981 | 29y | 30y 3 mc | No | 20y | No | 20s | 20s | 20s | 20s | Teens | 20s | – | – | – | c.2764C > T(p.Gln922X)/c.1133 T > C(p.Val378Ala) |
| 126/M | r | 1985 | 25y | 26y 8 m | No | No | No | 25y | Mild 25y | No | No | No | No | Yes | 25y | 1y | c.2764C > T(p.Gln922X)/c.1133 T > C(p.Val378Ala) |
| 127/M | – | 1969 | 42y | 42y 6 m | – | No | Tremor (20s) | <34y | <34y | Yes | 40y | No | Yes | No | 43y | – | c.2903A > G(p.Asn968Ser)/c.3182 T > C(p.Ile1061Thr) |
| 128/F | – | 1954 | 49y 6 m | 57y 6 m | No | No | No | Yes | Yes | 20s | No | No | 20s | No | – | – | c.3182 T > C(p.Ile1061Thr)/? |
| 129/F | – | – | PM | 40yc | – | – | – | – | – | – | – | – | – | – | – | – | c.3182 T > C(p.Ile1061Thr)/c.2861C > T(p.Ser954Leu) |
| 130/M | q | – | 40y | 41y 9 m | – | 40y | Tremor (35y) | Yes | No | Yes | 40y | No | No | Yes | 39y | – | c.1408G > C(p .Ala 470 Pro/c.1816G > C (p.Glu608Gln) |
| 131/F | – | – | 27y | 27y 8 m | No | No | No | No | No | 25y | No | 27y | No | No | – | – | c.3022A > C p (Asn 1008 His)/c.3182 T > C p.(Ile 1061Thr) |
| 132a/M | s | 1974 | Early 30s | – | – | – | – | – | 20y | – | – | – | – | Yes | – | – | c.2336del (p.Phe779SerfsX2)/c.2621A > T(p.Asp874Val) |
aPatients included in previously reported NPC Registry baseline characteristics study [11]; bPatients included in previously reported NPC Registry baseline and longitudinal data study [11, 12]; cpatient died; dpatient 119 F previously subject of case report by Lachmann et al. [10]; etreatment interrupted (first treatment start at 39 y, second treatment start at 46 y); ‘–’, no data/not known; C cataplexy, E epilepsy, FA foetal ascites, HM hepatomegaly, HS/S hepatosplenomegaly/splenomegaly, LD liver disease, LTx liver transplant, m months, NC neonatal cholestasis, PJ prolonged jaundice, PM post mortem, y years, w weeks
A history of developmental delay and/or cognitive deterioration was the most frequently recorded neurological manifestation (in 21/25 [84 %] patients), followed by VSGP and ataxia (both in 18/25 [72 %] patients). Dysarthria, dysphagia and seizures/cataplexy were present in 28–64 % of patients. Among those with available information on time of onset of specific manifestations, the majority of neurological signs seemed to appear during adulthood. In particular, patient 124 had no observable neurological symptoms up until her 4th decade of life, during which a full spectrum of characteristic neurological signs as well as psychiatric problems occurred. Patient 129 had a long history of neurological problems but no data on age at onset or recorded details on specific neurological manifestations. Patient 132 displayed cognitive deterioration since his twenties, but again, no information is available regarding specific neurological manifestations.
As in the juvenile-onset subgroup, psychiatric disturbances and/or cognitive deterioration were prominent in the adolescent/adult-onset subgroups, recorded in a total of nine patients (36 %). The age at onset of psychiatric disturbances ranged between 17 and 40 years.
There was only one historical record of neonatal jaundice in this subgroup, and organomegaly was recorded in a total of 6/25 (24 %) patients. Splenomegaly or hepatosplenomegaly were observed in three patients during adulthood (at age 18–40 years; patients 113, 125 and 130), and splenomegaly was recorded during the neonatal period in one patient (patient 115 at age 16 months).
Patients with no neurological manifestations
There were a total of 14 patients who had not displayed any neurological manifestations by data cut-off (Table 7). Genetic analyses have revealed NPC1 mutations in the majority (n = 10; 71 %) of these patients, confirming NP-C. Diagnoses among those without identified gene mutations were confirmed based on filipin staining. All of these patients had at least one early visceral symptom of NP-C: neonatal liver disease (mostly prolonged jaundice) and organomegaly both occurred in 11/14 (79 %) patients in this subgroup. Two patients (141 and 143) died in their 2nd and 3rd decades of life due to non NP-C related causes, and patient 146 died aged 4 years due to liver cancer. The remainder of non-neurological patients were alive at data cut-off. The mean (SD) age at last follow up prior to data cut off was 2.5 (1.8) years (range 0.5–6.1 years; n = 11). Among the three patients who died, mean age at death was 20.8 (15.9) years (range 4.9–36.7).
Table 7.
Patients with no neurological symptoms
| Patient number/Gender | Sibship | Date of birth | Age at diagnosis | Age at last FU (y, m)/deatha | Neonatal LD | HS/S | Seizures/cataplexy | VSGP | Dev. delay/cognitive problems | Ataxia | Swallowing problems | Psychiatric disturbance | Slurred speech | Genetic mutations |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 133/M | – | 2010 | 4 m | 6 m | Yes | No | No | No | No | No | No | No | – | – |
| 134/M | – | 2010 | 8 m | 8 m | Yes | – | – | – | – | – | – | – | – | c.3182 T > C(p.Ile1061Thr)/? |
| 135/M | – | 2010 | 1y | 1y | Yes (LTx) | Yes | No | No | No | No | No | No | No | – |
| 136/M | – | 2006 | 6 m | 2y 5 m | PJ LD | <4y | No | No | No | No | No | No | No | c.3182 T > C (p.Ile1061Thr)/c.3182 T > C (p.Ile1061Thr) |
| 137/M | – | 2003 | 2y6m | 6y 1 m | No | Yes | No | No | No | No | No | No | No | c.3182 T > C (p.Ile1061Thr)/c.3467A > G (p.Asn1156Ser) |
| 138/M | – | 2009 | 4 m | 2y 1 m | PJ LD | Yes | No | No | No | No | No | No | No | c.3182 T > C (p.Ile1061Thr)/? |
| 139/M | t | 2010 | <1y | 1y | Yes | Yes | No | No | No | No | No | No | No | c.3182 T > C (p.Ile1061Thr)/c.3289G > A (p.Asp1097Asn) |
| 140/M | t | 2006 | – | 5y | No | No | No | No | No | No | No | No | No | c.3182 T > C (p.Ile1061Thr)/c.3289G > A (p.Asp1097Asn) |
| 141/F | s | 1971 | 1y5m | 36y 8 ma | PJ LD | Birth | No | No | Yes | No | No | No | No | c.2336del (p.Phe779SerfsX2)/c.2621A > T (p.Asp874Val) |
| 142/F | – | 2008 | 2y5m | 3y | Yes | Yes | No | No | No | No | No | No | No | c.3182 T > C (p.Ile1061Thr)/c.3259 T > C (p.Phe1087Leu) |
| 143/M | – | 1987 | 16y | 20y 10 ma | PJ | Birth | No | No | No | No | No | No | No | c.2621A > T (p.Asp874Val)/c.3591 + 4 delA |
| 144/M | – | 2007 | 1y9m | 4y | No | Yes | No | No | No | No | No | No | No | – |
| 145/M | – | 2009 | 4 m | 2y | Yes | Yes | No | No | No | No | No | No | No | – |
| 146/F | – | 2004 | 7 m | 4y 11 ma | PJ | Yes | No | No | No | No | No | – | No | c.3083-3084delP.Gly1028AlafsX22)/c.2201G > T(p.Ser734Ile) |
aPatient died; ‘–’, no data/not known; HS/S hepatosplenomegaly/splenomegaly, LD liver disease, LTx liver transplant, m months, PJ prolonged jaundice, y years, w weeks. Note that none of the non-neurological patients had received miglustat by data cut-off end-2011
Genetics
Overall, 116/146 (79 %) patients had at least one identified NP-C gene mutation (Tables 2–6). Two mutations were recorded in 78/146 (53 %) patients and one mutation was recorded in 38/146 (26 %) patients. Most patients (98 %) with recorded mutant alleles had NPC1 mutations, and two patients (2 %) had NPC2 mutations (one with homozygous c.58G > T(p.Glu20X) mutant alleles and one with a single identified mutation for which information was not available). Thirty patients (21 %) had no recorded information regarding identified NP-C gene mutations. This number may be reduced in future with the increasing availability of next-generation sequencing methods.
Amongst a total of 194 identified mutant NP-C gene alleles, 53 have been classified as novel mutations. The common I1061Thr mutation was recorded in a total of 55 (38 %) patients in the whole cohort (heterozygous in 89 % and homozygous in 11 % of cases). Among the age-at-neurological onset subgroups, I1061Thr mutant alleles were mainly observed in patients with the late infantile- (23/51 [45 %]) and juvenile-onset forms (18/42 [43 %]). A lower proportion of patients in the adolescent-adult onset group had this mutation (7/25 [28 %]). However, the prevalence of I1061Thr mutations was particularly high among non-neurological patients (n = 7 [50 %]; six heterozygotes and one homozygote). One patient with a I1061Thr allele (patient 73), also had a c.882-40 T > A mutant allele, and family studies detected this genotype in the child’s father. However, it is not currently known if the c.882-40 T > A allele is a pathogenic mutation.
Eight patients with available genetic information had the p.Pro1007Ala mutation. Cases were fairly evenly distributed between the late-infantile, juvenile and adolescent/adult-onset subgroups.
Full genome sequencing performed in a number of patients with one identified mutation failed to detect any second mutation. These patients (and their listed genotypes) were: 11 (c.2819C > T(p.Ser940Leu/?), 18 (c.2008_2011del(p.Cys670ProfsX17/?), 28 (c.3182 T > C(p.Ile1061 Thr)/?), 31 (c.3182 T > C(p.Ile1061Thr)/?), 51 (c.3467A > G(p.Asn1156Ser)/?), 52 (c.3467A > G(p.Asn1156Ser)/?), 53 (c.3591 + 4delA/?) and 66 (c.1211G > A(p.Arg404Gln)/?).
Miglustat use
The information regarding miglustat use should be regarded in recognition of the year during which miglustat became commercially available in the EU (2009), and of the time of data cut-off. Overall, 51 patients (35 %) received miglustat during the observation period (Tables 2–6): 2/8 (25 %) early infantile-onset patients and 17/51 (33 %), 20/42 (48 %) and 12/25 (48 %) patients in the late infantile-, juvenile-, and adolescent/adult-onset subgroups, respectively. No patients with the neonatal form of NP-C and no patients in the ‘non-neurological’ subgroup received miglustat.
The mean ages at treatment start ranged from 0.5 to 49.0 years across the age-at-onset subgroups. Among a total of 38 patients with evaluable information on treatment duration, the overall estimated mean (SD) treatment duration was 2.6 (2.3) years: median (range) 2.0 years (1 week to 8 years). Approximate mean (SD) durations per evaluable age-at-onset subgroup were: 2.6 (2.0) years in late infantile-, 2.8 (2.4) years in the juvenile-, and 2.9 (2.6) years in the adolescent/adult-onset subgroups.
Disease course among related patients
A total of 44 patients from 20 families in this cohort were siblings, most of whom (38 [86 %]) had at least one identified NPC1 mutant allele (see Additional file 1: Table S1). No NP-C gene mutations were identified in three sibling pairs: diagnoses were established by other means based on clinical symptoms and/or laboratory biochemical (filipin testing) and histological methods.
In general, ages at neurological onset and clinical phenotypes appeared congruent between siblings with identical mutant genotypes. Exceptions included patient 6, the sister of patients 25 and 26, all of whom were homozygous for the c.3020C > T (p.Pro1007Leu) mutant allele. Patient 6 had the neonatal-onset form of NP-C, neonatal liver disease and hepatosplenomegaly, and died aged 2 months due to failure to thrive and severe chest infection. Her sisters are twins and have late-infantile onset NP-C. Both twins also had neonatal jaundice and hepatosplenomegaly, with a variety of characteristic neurological signs observed from before the age of 4 years, but both were still alive at the time of data cut-off.
Another sibling pair (patients 80 and 108) were both heterozygotes with c.3182 T > C (p.Ile1061Thr) and c.3019C > G (p.Pro1007Ala) mutant alleles. The older sibling (patient 108) has adolescent/adult-onset NP-C, presented with VSGP, developmental delay and psychiatric disturbances in his late-teens, and survives aged 39 years at last follow up: he had received miglustat therapy for 3 years at data cut-off. His brother (patient 80) had juvenile-onset NP-C and presented with developmental delay and VSGP before 10 years of age. He also had severe seizures that are considered likely to have contributed to his death ‘due to NP-C’ at the age of 18 years.
Finally, patients 139 and 140 both have no neurological manifestations. While patient 139 was diagnosed at an early age following investigations of neonatal liver disease and organomegaly, his older brother has no record of systemic manifestations, and was diagnosed (based on genetic testing) due to mainly to his sibling disease history.
Discussion
It is important to continue gathering knowledge on the natural history of NP-C in order to aid in the clinical management and targeted therapy of affected patients, and to aid in providing counselling and support for their families and caregivers. This summary of data from 146 historical and current UK-based NP-C patients analysed between 1999 and end-2011 provides findings from the largest national NP-C cohort reported to date. Although some patients have minimal data available, they are still included in this report to indicate the full extent of the UK NP-C cohort and provide as full a clinical picture of diagnosed patients as possible.
Based on recent estimates of the total UK population (64.1 million) and national birth rate (12.27/1000) the current birth prevalence of NP-C in the UK is 0.78 cases per 100,000 births, which seems roughly in line with previous estimates of birth prevalence of NP-C in Western Europe (France, Germany and the UK) over the period 1988–2002 [21, 22]. Based on records of all known UK cases diagnosed since 1985, the rate of diagnoses per decade (i.e., the mean number across all years within each decade) has risen steadily over the last three decades, from 3.5 new cases/year during 1985–1994 to 5.1 new cases per year during 1995–2004, and 6.0 new cases per year during 2005–2011. In addition, a total of 21 new cases have been diagnosed between 2012 and 2015, although these most recent cases are not included in this cohort analysis due to non-availability of access to full clinical information after data cut-off. This trend possibly reflects increased awareness of the disease over the last two decades and, in particular, improvements in genetic analysis methods for confirmation of disease-causing mutations.
Data on visceral symptoms in this cohort were in line with baseline data from 163 patients included in the International NP-C Registry [19]. Neonatal liver disease and/or prolonged jaundice, or a history of just neonatal jaundice, was a feature in all age subgroups in this cohort. As could be expected, neonatal jaundice was recorded in many patients (50–100 %) in the infantile-onset subgroups, and in far lower proportions of patients (0–33 %) in the juvenile- and adolescent/adult-onset subgroups. There was a clear trend for decreased occurrence of organomegaly with increasing age at neurological onset, although organomegaly was not as common in the neonatal-onset subgroup as it was among early-infantile onset patients due to its incremental nature. Nevertheless, splenomegaly is generally considered a consistent indicator of possible NP-C in neonates, and has often served as a signal leading to early diagnosis.
NP-C has been reported as the second most common genetic cause of liver disease during infancy in the UK, after alpha-1-antitrypsin deficiency [23, 24], and investigations to exclude NP-C are common practice in liver disease/neonatal units. A large proportion of early-onset patients in this cohort underwent extensive liver investigations during the neonatal period, but in many cases NP-C was subsequently diagnosed years later, after the appearance of neurological signs. This seems typical of the NP-C population as a whole, as reflected by substantial delays to diagnosis in many cases, globally, and reflects the need for clinicians in all specialities to take a full clinical history when seeing new patients.
Based on previous clinical observations, neonatal jaundice without other overt signs of liver disease can herald a more aggressive clinical course of NP-C, particularly if neurological abnormalities appear during the first 4 years of life [15, 24]. However, the degree of neonatal liver disease does not appear to be a reliable indicator of future disease progression, as illustrated by cases where neurological manifestations became apparent during adulthood despite a history of neonatal liver disease. It is important to consider that early visceral symptoms lack prognostic value in NP-C during family counselling for NP-C cases diagnosed during the neonatal period, providing advice for planning ahead for the emotional and financial burdens of the disease [25]. Of particular note are the sibling patients 132 and 141, who share identical novel heterozygous NPC1 mutations. Both had very severe neonatal liver disease necessitating intensive care, and were followed up over a course of years by their hepatologist. Patient 141 had non-neurological NP-C and died at the age of 36 due to a severe infection in late pregnancy: post-mortem examination showed no storage or neurological problems. Her brother (patient 132) was still alive and self-caring at data cut off, and displayed mild cognitive impairment at last follow up.
The profile of neurological manifestations recorded in this cohort was consistent with other large-cohort NP-C studies [2, 4, 16, 19, 26], with over half of patients displaying one or more of: ataxia; VSGP; dysarthria; dysphagia; and seizures/cataplexy. In terms of age at neurological onset, two-thirds of patients fell into the late infantile-onset and juvenile-onset subgroups. Given the frequency of childhood-onset forms of NP-C in this UK cohort up to the end of 2011, it is not surprising that developmental delay or cognitive deterioration were the most frequently recorded neurological manifestations. Similarly, the frequency of seizures and/or cataplexy was reflective of the overall young age at onset among UK patients, as seizure activity has generally been reported most frequently among patients with late-infantile and juvenile-onset disease [1].
It is notable that ataxia, which is a central component of the recently developed NP-C suspicion index (SI) for the detection of NP-C among patients with suggestive clinical symptoms [9], was the second-most common neurological manifestation in this cohort. VSGP is recognised as one of the earliest signs of neurological deterioration in NP-C [1, 9, 27, 28], so it is unsurprising that this neurological sign was also common. Excluding neonatal cases, VSGP was observed in over half of all UK patients, and was most frequent in those with the juvenile-onset (classical) form of NP-C.
Psychiatric manifestations were only recorded in patients in the juvenile and adolescent/adult-onset subgroups, which is in agreement with previous screening data [29] and numerous other published findings [1, 2, 30–34]. While little information was available on the precise types of psychiatric disorders for this analysis, patients with juvenile-onset NP-C tend to manifest behavioural problems, impaired learning, and expressive language disorder, which often culminate in failure at school [1]. Patients with adolescent/adult-onset NP-C and psychiatric disorders often exhibit schizophrenia-like psychosis (in up to 25 % of cases), but can also be diagnosed with bipolar disorder, depression and obsessive-compulsive disorder [1, 34].
While the severe neonatal and early-infantile onset forms of NP-C resulted in rapid deterioration and early mortality in the current cohort, total mortality decreased with increasing age at neurological onset among the late-infantile (59 %), juvenile (45 %) and adolescent/adult-onset (32 %) subgroups. Mean age at death values reflected this trend. Some adolescent/adult-onset patients have survived into their fifth and sixth decade of life, which confirms the more insidious, mild course of neurological deterioration that has been reported before among adults with NP-C [1, 26]. Indeed, a recent case has been reported of a 66 year-old female who remains free of any neurological or psychiatric manifestations 18 years after initial presentation [35].
Wide genetic variability coupled with a high degree of phenotypic heterogeneity make genotype–phenotype correlations difficult in NP-C. Variable clinical phenotypes have even been observed in monozygotic twins with the same genetic mutation [36]. Overall, 53/194 (27 %) mutations identified in this cohort were classified as novel. The exon 21 (p.I1061Thr) mutant allele of NPC1 has previously been associated with the common juvenile-onset form of the disease, and is associated with a relatively well characterised cellular function and biochemical phenotype [37–39]. The overall prevalence of this allele has been quoted as approximately 15 % [21], although more recently it has been reported as particularly frequent (in 20–25 % of diagnosed cases) in France and the UK [2]. Based on this cohort update, the p.I1061Thr mutant allele was present in 38 % of patients overall, occurring in approximately equal proportions of patients in the late-infantile and juvenile-onset subgroups. The incidence of the second most frequent known NPC1 mutation – p.1007Ala – appeared relatively much lower, occurring in the heterozygous state in only eight patients overall. The prevalence range for p.1007Ala alleles among the late-infantile, juvenile and adolescent/adult-onset subgroups was 4–10 %.
Interestingly, the prevalence of p.I1061Thr mutations was particularly high in the non-neurological subgroup, where it occurred in 50 % of patients. This begs the question of whether some patients currently in the non-neurological group might progress to develop neurological manifestations in the future. For instance, ‘non-neurological’ patient 136 had no neurological symptoms at last documented follow up aged 2.5 years. However, other patients with homozygous p.I1061Thr mutations (patients 46 and 81–84) all had late-infantile or juvenile onset neurological symptoms. In the same way, many patients with heterozygous p.I1061Thr mutations developed neurological symptoms during the late-infantile period (up to 6 years of age, n = 21), the juvenile period (n = 14), or during adolescence/adulthood (n = 7). Further follow-up might therefore result in re-categorisation of some or all of the ‘non-neurological’ p.I1061Thr patients as having symptomatic neurological disease. Nevertheless, the same cannot be said for eight patients in the non-neurological group in whom p.I1061Thr mutations have been excluded, and/or in whom no known NPC1 mutant alleles have been detected.
Substrate reduction therapy (SRT) using miglustat has been shown to be effective in treating adult patients with Gaucher disease [40–44], and subsequently in the treatment of progressive neurological manifestations in children and adults with NP-C [1, 28, 45–48]. In total 34 % of UK patients have received miglustat therapy for any period of time. Longitudinal analyses of serial clinical status assessments are required to determine the impact of miglustat on neurological disease progression in this cohort.
Conclusions
In summary, the prevalence of NP-C in the UK is in line with previous observational data from other European cohorts, and it is notable that the annual rate of diagnosis of the condition has increased over the past 30 years, possibly due to increasing awareness and improvements in molecular diagnostic methods. The wide phenotypic variability and the overall profile of genotypes detected among UK patients are also consistent with data from other Western populations. In particular, age at onset of neurological manifestations once more appeared associated with more rapid disease progression and a lower age at death. Further analyses are required to assess the impact of miglustat therapy on neurological disease progression.
Acknowledgements
We extend thanks to specialist nurses Alison Cousins, Victoria Crook, Liz Morris, Louise Simmons and Collette Stainforth for their help in updating data on the NP-C patients under their care. We are also indebted to the late Professor Ed Wraith for his expert guidance during the development of the UK NP-C database. Matthew Reilly PhD at InTouch Medical Ltd provided medical writing support in the preparation of this manuscript, paid for by Actelion Pharmaceuticals Ltd.
Abbreviations
- LDL
low-density lipoprotein
- NP-C
Niemann-Pick disease type C
- NPC1/NPC2
genes affected in NP-C
- SD
standard deviation
- SRT
substrate reduction therapy
- VSGP
vertical supranuclear gaze palsy
Additional file
Comparison of sibling profiles. (DOCX 42 kb)
Footnotes
Competing interests
JI has received consultancy fees and travelling expenses from Actelion Pharmaceuticals Ltd. LH, SK and KS have no competing interests to declare.
Authors’ contributions
JI co-ordinated patient care and follow-up, oversaw data entry and management of the NP-C database, co-wrote the first draft of the manuscript and reviewed all successive drafts prior to submission. KS contributed to data entry and database management, co-wrote the first draft manuscript and reviewed all drafts up to manuscript submission. LH and SK performed all genetic analysis and provided critical input during development of the manuscript. All authors approved the final version of the manuscript for submission.
Contributor Information
Jackie Imrie, Phone: 01914150693, Email: jackie@niemann-pick.org.uk.
Lesley Heptinstall, Email: lesley.heptinstall@cmft.nhs.uk.
Stephen Knight, Email: Stephen.Knight@thermofisher.com.
Kate Strong, Email: katestrong@thecareforum.org.uk.
References
- 1.Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F, et al. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab. 2012;106:330–44. doi: 10.1016/j.ymgme.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 2.Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis. 2010;5:16. doi: 10.1186/1750-1172-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walterfang M, Chien YH, Imrie J, Rushton D, Schubiger D, Patterson MC. Dysphagia as a risk factor for mortality in Niemann-Pick disease type C: systematic literature review and evidence from studies with miglustat. Orphanet J Rare Dis. 2012;7:76. doi: 10.1186/1750-1172-7-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jahnova H, Dvorakova L, Vlaskova H, Hulkova H, Poupetova H, Hrebicek M, et al. Observational, retrospective study of a large cohort of patients with Niemann-Pick disease type C in the Czech Republic: a surprisingly stable diagnostic rate spanning almost 40 years. Orphanet J Rare Dis. 2014;9:140. doi: 10.1186/s13023-014-0140-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKay K, Gissen P. Genetic and laboratory diagnostic approach in Niemann Pick disease type C. J Neurol. 2014;261(Suppl 2):569–75. doi: 10.1007/s00415-014-7386-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanier MT, Pentchev P, Rodriguez-Lafrasse C, Rousson R. Niemann-Pick disease type C: an update. J Inherit Metab Dis. 1991;14:580–95. doi: 10.1007/BF01797928. [DOI] [PubMed] [Google Scholar]
- 7.Hendriksz CJ, Pineda M, Fahey M, Walterfang M, Stampfer M, Runz H, et al. The Niemann-Pick disease Type C suspicion index: development of a new tool to aid diagnosis. J Rare Disord Diag Ther. 2015;1:11. [Google Scholar]
- 8.Wraith JE, Sedel F, Pineda M, Wijburg FA, Hendriksz CJ, Fahey M, et al. Niemann-Pick type C Suspicion Index tool: analyses by age and association of manifestations. J Inherit Metab Dis. 2014;37:93–101. doi: 10.1007/s10545-013-9626-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wijburg FA, Sedel F, Pineda M, Hendriksz CJ, Fahey M, Walterfang M, et al. Development of a suspicion index to aid diagnosis of Niemann-Pick disease type C. Neurology. 2012;78:1560–7. doi: 10.1212/WNL.0b013e3182563b82. [DOI] [PubMed] [Google Scholar]
- 10.Boenzi S, Deodato F, Taurisano R, Martinelli D, Verrigni D, Carrozzo R, et al. A new simple and rapid LC-ESI-MS/MS method for quantification of plasma oxysterols as dimethylaminobutyrate esters. Its successful use for the diagnosis of Niemann-Pick type C disease. Clin Chim Acta. 2014;437:93–100. doi: 10.1016/j.cca.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 11.Jiang X, Sidhu R, Porter FD, Yanjanin NM, Speak AO, te Vruchte DT, et al. A sensitive and specific LC-MS/MS method for rapid diagnosis of Niemann-Pick C1 disease from human plasma. J Lipid Res. 2011;52:1435–45. doi: 10.1194/jlr.D015735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porter FD, Scherrer DE, Lanier MH, Langmade SJ, Molugu V, Gale SE, et al. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci Transl Med. 2010;2:56ra81. doi: 10.1126/scitranslmed.3001417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Welford RW, Garzotti M, Marques Lourenco C, Mengel E, Marquardt T, Reunert J, et al. Plasma lysosphingomyelin demonstrates great potential as a diagnostic biomarker for Niemann-Pick disease type C in a retrospective study. PLoS ONE. 2014;9:e114669. doi: 10.1371/journal.pone.0114669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giese AK, Mascher H, Grittner U, Eichler S, Kramp G, Lukas J, et al. A novel, highly sensitive and specific biomarker for Niemann-Pick type C1 disease. Orphanet J Rare Dis. 2015;10:78. doi: 10.1186/s13023-015-0274-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imrie J, Dasgupta S, Besley GT, Harris C, Heptinstall L, Knight S, et al. The natural history of Niemann-Pick disease type C in the UK. J Inherit Metab Dis. 2007;30:51–9. doi: 10.1007/s10545-006-0384-7. [DOI] [PubMed] [Google Scholar]
- 16.Iturriaga C, Pineda M, Fernandez-Valero EM, Vanier MT, Coll MJ. Niemann-Pick C disease in Spain: clinical spectrum and development of a disability scale. J Neurol Sci. 2006;249:1–6. doi: 10.1016/j.jns.2006.05.054. [DOI] [PubMed] [Google Scholar]
- 17.Heron B, Valayannopoulos V, Baruteau J, Chabrol B, Ogier H, Latour P, et al. Miglustat therapy in the French cohort of paediatric patients with Niemann-Pick disease type C. Orphanet J Rare Dis. 2012;7:36. doi: 10.1186/1750-1172-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lachmann RH, te Vruchte D, Lloyd-Evans E, Reinkensmeier G, Sillence DJ, Fernandez-Guillen L, et al. Treatment with miglustat reverses the lipid-trafficking defect in Niemann-Pick disease type C. Neurobiol Dis. 2004;16:654–8. doi: 10.1016/j.nbd.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 19.Patterson MC, Mengel E, Wijburg FA, Muller A, Schwierin B, Drevon H, et al. Disease and patient characteristics in NP-C patients: findings from an international disease registry. Orphanet J Rare Dis. 2013;8:12. doi: 10.1186/1750-1172-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patterson MC, Mengel E, Vanier MT, Schwierin B, Muller A, Cornelisse P, et al. Stable or improved neurological manifestations during miglustat therapy in patients from the international disease registry for Niemann-Pick disease type C: an observational cohort study. Orphanet J Rare Dis. 2015;10:65. doi: 10.1186/s13023-015-0284-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patterson MC, Vanier MT, Suzuki K, Morris JA, Carstea E, Neufeld EB, et al. Niemann-Pick disease type C: a lipid trafficking disorder. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Vogelstein B, et al., editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 3611–33. [Google Scholar]
- 22.Vanier MT, Millat G. Niemann-Pick disease type C. Clin Genet. 2003;64:269–81. doi: 10.1034/j.1399-0004.2003.00147.x. [DOI] [PubMed] [Google Scholar]
- 23.Mieli-Vergani G, Howard ER, Mowat AP. Liver disease in infancy: a 20 year perspective. Gut. 1991;Suppl:S123–8. doi: 10.1136/gut.32.Suppl.S123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelly DA, Portmann B, Mowat AP, Sherlock S, Lake BD. Niemann-Pick disease type C: diagnosis and outcome in children, with particular reference to liver disease. J Pediatr. 1993;123:242–7. doi: 10.1016/S0022-3476(05)81695-6. [DOI] [PubMed] [Google Scholar]
- 25.Imrie J, Galani C, Gairy K, Lock K, Hunsche E. Cost of illness associated with Niemann-Pick disease type C in the UK. J Med Econ. 2009;12:219–29. doi: 10.3111/13696990903245863. [DOI] [PubMed] [Google Scholar]
- 26.Wraith JE, Baumgartner MR, Bembi B, Covanis A, Levade T, et al. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol Genet Metab. 2009;98:152–65. [DOI] [PubMed]
- 27.Salsano E, Umeh C, Rufa A, Pareyson D, Zee DS. Vertical supranuclear gaze palsy in Niemann-Pick type C disease. Neurol Sci. 2012;33:1225–32. doi: 10.1007/s10072-012-1155-1. [DOI] [PubMed] [Google Scholar]
- 28.Patterson MC, Vecchio D, Prady H, Abel L, Wraith JE. Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 2007;6:765–72. doi: 10.1016/S1474-4422(07)70194-1. [DOI] [PubMed] [Google Scholar]
- 29.Bauer P, Balding DJ, Klunemann HH, Linden DE, Ory DS, Pineda M, et al. Genetic screening for Niemann-Pick disease type C in adults with neurological and psychiatric symptoms: findings from the ZOOM study. Hum Mol Genet. 2013;22:4349–56. doi: 10.1093/hmg/ddt284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shulman LM, David NJ, Weiner WJ. Psychosis as the initial manifestation of adult-onset Niemann-Pick disease type C. Neurology. 1995;45:1739–43. doi: 10.1212/WNL.45.9.1739. [DOI] [PubMed] [Google Scholar]
- 31.Campo JV, Stowe R, Slomka G, Byler D, Gracious B. Psychosis as a presentation of physical disease in adolescence: a case of Niemann-Pick disease, type C. Dev Med Child Neurol. 1998;40:126–9. doi: 10.1111/j.1469-8749.1998.tb15374.x. [DOI] [PubMed] [Google Scholar]
- 32.Sandu S, Jackowski-Dohrmann S, Ladner A, Haberhausen M, Bachmann C. Niemann-Pick disease type C1 presenting with psychosis in an adolescent male. Eur Child Adolesc Psychiatry. 2009;18:583–5. doi: 10.1007/s00787-009-0010-2. [DOI] [PubMed] [Google Scholar]
- 33.Nia S, Geiblinger S, Gruber C, Gallmetzer P, Gerschlager W, Baumgartner C. Psychiatric symptoms as an early sign of the adult form of lysosomal storage disease Niemann-Pick type C. J Neurol Neurochir Psychiatr. 2011;12:94–5. [Google Scholar]
- 34.Walterfang M, Fietz M, Fahey M, Sullivan D, Leane P, Lubman DI, et al. The neuropsychiatry of Niemann-Pick type C disease in adulthood. J Neuropsychiatry Clin Neurosci. 2006;18:158–70. doi: 10.1176/jnp.2006.18.2.158. [DOI] [PubMed] [Google Scholar]
- 35.Greenberg C, Barnes JG, Kogan S, Seargent LE: A rare case of Niemann–Pick disease type C without neurological involvement in a 66-year-old patient. Molec Genet Metab Rep [article in press] DOI: 10.1016/jymgmr201502004 2015. [DOI] [PMC free article] [PubMed]
- 36.Benussi A, Alberici A, Premi E, Bertasi V, Cotelli MS, Turla M, et al. Phenotypic heterogeneity of Niemann-Pick disease type C in monozygotic twins. J Neurol. 2015;262:642–7. doi: 10.1007/s00415-014-7619-x. [DOI] [PubMed] [Google Scholar]
- 37.Gelsthorpe ME, Baumann N, Millard E, Gale SE, Langmade SJ, Schaffer JE, et al. Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J Biol Chem. 2008;283:8229–36. doi: 10.1074/jbc.M708735200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pentchev PG, Comly ME, Kruth HS, Vanier MT, Wenger DA, Patel S, et al. A defect in cholesterol esterification in Niemann-Pick disease (type C) patients. Proc Natl Acad Sci U S A. 1985;82:8247–51. doi: 10.1073/pnas.82.23.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Millat G, Marcais C, Rafi MA, Yamamoto T, Morris JA, Pentchev PG, et al. Niemann-Pick C1 disease: the I1061T substitution is a frequent mutant allele in patients of Western European descent and correlates with a classic juvenile phenotype. Am J Hum Genet. 1999;65:1321–9. doi: 10.1086/302626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cox TM, Amato D, Hollak CE, Luzy C, Silkey M, Giorgino R, et al. Evaluation of miglustat as maintenance therapy after enzyme therapy in adults with stable type 1 Gaucher disease: a prospective, open-label non-inferiority study. Orphanet J Rare Dis. 2012;7:102. doi: 10.1186/1750-1172-7-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cox T, Lachmann R, Hollak C, Aerts J, van Weely S, Hrebicek M, et al. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet. 2000;355:1481–5. doi: 10.1016/S0140-6736(00)02161-9. [DOI] [PubMed] [Google Scholar]
- 42.Elstein D, Dweck A, Attias D, Hadas-Halpern I, Zevin S, Altarescu G, et al. Oral maintenance clinical trial with miglustat for type I Gaucher disease: switch from or combination with intravenous enzyme replacement. Blood. 2007;110:2296–301. doi: 10.1182/blood-2007-02-075960. [DOI] [PubMed] [Google Scholar]
- 43.Elstein D, Hollak C, Aerts JM, van Weely S, Maas M, Cox TM, et al. Sustained therapeutic effects of oral miglustat (Zavesca, N-butyldeoxynojirimycin, OGT 918) in type I Gaucher disease. J Inherit Metab Dis. 2004;27:757–66. doi: 10.1023/B:BOLI.0000045756.54006.17. [DOI] [PubMed] [Google Scholar]
- 44.Kuter DJ, Mehta A, Hollak CE, Giraldo P, Hughes D, Belmatoug N, et al. Miglustat therapy in type 1 Gaucher disease: clinical and safety outcomes in a multicenter retrospective cohort study. Blood Cells Mol Dis. 2013;51:116–24. doi: 10.1016/j.bcmd.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 45.Wraith JE, Vecchio D, Jacklin E, Abel L, Chadha-Boreham H, Luzy C, et al. Miglustat in adult and juvenile patients with Niemann-Pick disease type C: long-term data from a clinical trial. Mol Genet Metab. 2010;99:351–7. doi: 10.1016/j.ymgme.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 46.Wraith JE, Imrie J. New therapies in the management of Niemann-Pick type C disease: clinical utility of miglustat. Ther Clin Risk Manag. 2009;5:877–87. doi: 10.2147/TCRM.S5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patterson MC, Vecchio D, Jacklin E, Abel L, Chadha-Boreham H, Luzy C, et al. Long-term miglustat therapy in children with Niemann-Pick disease type C. J Child Neurol. 2010;25:300–5. doi: 10.1177/0883073809344222. [DOI] [PubMed] [Google Scholar]
- 48.Pineda M, Wraith JE, Mengel E, Sedel F, Hwu WL, Rohrbach M, et al. Miglustat in patients with Niemann-Pick disease Type C (NP-C): a multicenter observational retrospective cohort study. Mol Genet Metab. 2009;98:243–9. doi: 10.1016/j.ymgme.2009.07.003. [DOI] [PubMed] [Google Scholar]