


Abstract
Many dsDNA viruses encode DNA-packaging terminases, each containing a nuclease domain that resolves concatemeric DNA into genome-length units. Terminase nucleases resemble the RNase H-superfamily nucleotidyltransferases in folds, and share a two-metal-ion catalytic mechanism. Here we show that residue K428 of a bacteriophage terminase gp2 nuclease domain mediates binding of the metal cofactor Mg2+. A K428A mutation allows visualization, at high resolution, of a metal ion binding mode with a coupled-octahedral configuration at the active site, exhibiting an unusually short metal-metal distance of 2.42 Å. Such proximity of the two metal ions may play an essential role in catalysis by generating a highly positive electrostatic niche to enable formation of the negatively charged pentacovalent phosphate transition state, and provides the structural basis for distinguishing Mg2+ from Ca2+. Using a metal ion chelator β-thujaplicinol as a molecular probe, we observed a second mode of metal ion binding at the active site, mimicking the DNA binding state. Arrangement of the active site residues differs drastically from those in RNase H-like nucleases, suggesting a drifting of the active site configuration during evolution. The two distinct metal ion binding modes unveiled mechanistic details of the two-metal-ion catalysis at atomic resolution.
INTRODUCTION
Terminases are virally encoded multi-component, multi-functional molecular machines employed by many tailed double-stranded DNA (dsDNA) bacteriophages and herpesviruses to resolve newly synthesized concatemeric viral DNA in the infected host cells into genome-length units and package each into a preformed capsid precursor termed procapsid (1–3). Phage terminases contain two fundamental proteinaceous components: a DNA-recognition subunit and a catalytic subunit, also known as the ‘small’ and ‘large’ subunit, respectively. The terminase small subunit (TerS) specifically binds to concatemeric viral DNA, and the terminase catalytic subunit (TerL) cleaves the DNA to generate a new terminus, which is then threaded into the procapsid in an ATP-dependent manner. The terminase catalytic subunit consists of an N-terminal ATPase domain that powers DNA translocation by translating the chemical energy from ATP hydrolysis into physical movement of DNA, and a C-terminal nuclease domain that processes the DNA (4,5). Nuclease activity of the terminase catalytic subunit nuclease domain is precisely controlled. The DNA cleavage by TerL occurs only at certain steps of the DNA-packaging process, typically at the beginning and completion of each DNA-packaging cycle, to avoid aberrant DNA cutting. During DNA translocation, the nuclease activity must be turned off. TerL on its own typically exhibits only weak nuclease activity as observed in full-length proteins (4,6) or isolated domains (7,8), consistent with the physiological requirement that it may not randomly cut DNA in infected host cells, and the nuclease activity may be stimulated by the N-terminal ATPase domains (4,8).
TerL nuclease activity may be either sequence-specific, e.g. in phages such as lambda which cuts at a sequence-specific site in the cos region of the phage genome (9), or none sequence-specific, e.g. for phages that use headful packaging mechanisms such as Sf6, T4, P22 and SPP1 (10). In those ‘headful’ phages, there is typically an initiation cleavage as the first step of the concatemeric DNA packaging process, followed by a series of ‘headful’ cleavage events, each occurring upon the completion of a packaging cycle. The initiation cleavage shows some degree of site specificity as it usually occurs within a specified region in phage genome, but such site specificity is putatively conferred by TerS. The headful cleavage is not sequence specific, resulting in variable sequence and length of the DNA packaged into each capsid that is slightly longer than the unit-length genome (10). In herpes simplex virus type I, the terminase catalytic subunit pUL15 may cut at a sequence-specific site within the DR motif in the ‘a’ sequence of the viral genome, but this specificity is perhaps related to spatial positioning of another terminase component pUL28.
Structural studies of terminase catalytic subunits in full length (4,5) and their nuclease domains (7,11–13) revealed core folds resembling those of RNase H-like endonucleases, whose active site architecture is built around several conserved acidic residues. RNase H-like nucleases are a large class of enzymes that play diverse roles in nucleic acid metabolism (14). Members of this class of enzymes, such as RNases H, reverse transcriptases, integrases, topoisomerases, DNA and RNA polymerases, transposases, Holliday-junction resolvases, RNAi slicer Argonaute, CRISPR Cas nucleases and viral terminases, are fundamental to an array of biological processes ranging from DNA replication, recombination and repair, RNA maturation, processing and interferencing, to host defense, cell death (14) and virus DNA packaging. RNase H-like nucleases share a common two-metal-ion catalytic mechanism (14). In the mechanism proposed by Steitz and Steitz (15) and well studied in RNase H (16–19), the two metal ions are jointly coordinated by the scissile phosphate and two active-site carboxylates, bisecting the scissile phosphate. Metal ion A was thought to deprotonate a water molecule to form OH, which is aligned for a nucleophilic attack on the scissile phosphate. Metal ion B may play a role in stabilization of the transition state pentacovalent phosphate (14,16). However, the detailed catalytic mechanism and the precise roles of these metal ions in pentacovalent phosphate formation remain to be established experimentally. The two metal ions were ∼3.4–4 Å apart in previous structures of RNase H-like nucleases, and were thought to have to move closer allowing the nucleophile to access the scissile phosphate for phosphodiester bond cleavage (16,19–21). However, such a closer metal-metal distance has not been experimentally observed. The two-metal-ion nucleases typically require Mg2+ or Mn2+, and in most cases Ca2+ does not support the catalysis although it supports nucleic acid substrate binding and is abundant in cells. The mechanism underlying differentiation against Ca2+ remains to be established, although it was attributed to different geometry of the Ca2+ coordination system.
Bacteriophage Sf6 belongs to the Podoviridae family of tailed dsDNA bacteriophages (22,23). Sf6 infects Shigella flexneri, an important human pathogen that causes acute diarrhea and bacillary dysentery. Sf6 can alter the host's serotype and virulence through changing the host lipopolysaccharide structure using a horizontal gene transfer mechanism (24). The Sf6 terminase is composed of the small subunit gp1 (25,26) and the large subunit gp2 (4). Like many other phages such as T4, P22 and SPP1, Sf6 uses a headful DNA-packaging mechanism (22). Here we show that residue K428 of the C-terminal nuclease domain of the dsDNA bacteriophage Sf6 terminase catalytic subunit gp2 (gp2C) mediates the binding of its metal cofactor, which likely serves to regulate terminase nuclease activity in a timely manner during viral genome packaging. A K428A mutation has allowed us to capture two distinct metal ion binding modes in the gp2C nuclease active site. High resolution structures reveal a metal ion binding mode with a unique coupled-octahedral configuration, exhibiting an ultra-short metal-metal distance of 2.42 Å. Such a distance is unusual given the 1.6 Å atomic radius of Mg2+ and is among the shortest metal-metal distances in biological systems thus far documented. This provides a structural basis for requirement of Mg2+ or Mn2+ in RNase H-like nucleases and the well-known inability of Ca2+ to support catalysis. Such an ultra-short metal:metal distance may generate a highly positive electrostatic niche that drives formation of the pentacovalent phosphate transition state. Using a divalent metal-sequestering α-hydroxytropolone as a molecular probe, we have observed a second distinct mode of metal ion binding, which reflects the DNA binding state. The arrangement of the active site residues is altered drastically compared to those in other RNase H-like nucleases, suggesting a drifting of the active site configuration during evolution despite of conserved protein folds. These results unveil the atomic detail of the two-metal-ion catalytic mechanism.
MATERIALS AND METHODS
Protein expression, purification and crystallization
The coding sequence for the Sf6 gp2 C-terminal domain (residue 213–470) was amplified by PCR from the phage genomic DNA with the following primers: 5′- CCA CAT ATG GCA ATC ATC AAA CGT GAA -3′, which contains a Nde1 site followed by the gp2C coding sequence and 5′- GGC CTC GAG TTA CCA ACC GGA GGA TGA GGG -3′, the latter of which harbors the gp2 C-terminus, a stop codon and a Xho1 site. The PCR product was digested with the appropriate restriction enzymes, gel purified and cloned into the vector pET28b (Novagen). The resulting construct contained the gp2C DNA sequence with an N-terminal His-tag and a thrombin cleavage site. The K428A mutation was generated by site-directed mutagenesis using the QuickChange Site-Directed Mutagenesis Kit (Stratagene). Plasmids encoding the wild type gp2C and K428A mutant were separately transformed in E. coli strain BL21(DE3) (Novagen). The cells were grown at 37°C in LB media supplemented with 30 μg/ml Kanamycin until OD600 reached around 0.6, after which IPTG was added to a final concentration of 1 mM, the medium temperature was subsequently reduced to 16°C, and cells were grown overnight. Cells were harvested by centrifugation for 10 min at 5000 rpm. Cell pellets were resuspended in buffer A (20 mM Tris-HCl pH 8.5, 500 mM NaCl and 10 mM β-mercaptoethanol) and frozen at −20°C. The cells were thawed at room temperature, lysed by French press and centrifuged at 15 000 rpm with a Sorvall SS-34 rotor for 60 min at 4°C. The supernatant was loaded on a Ni-NTA-Sepharose column equilibrated in buffer B (buffer A with 20 mM imidazole), washed with buffer B and eluted with buffer C (buffer A with 200 mM imidazole). Fractions containing gp2C were pooled and loaded onto a size exclusion chromatography column (Sephacryl S-300, GE healthcare) pre-equilibrated with a buffer containing 20 mM Tris-HCl pH 8.5, 150 mM NaCl, 1 mM EDTA and 1 mM DTT. The gp2C peak fractions were pooled and concentrated with an Ultrafree concentrator (Milipore, MW cutoff 10 000) to ∼24.5 mg/ml and stored at −80°C. The Se-Met gp2C was expressed in E. coli B834(DE3) in the M9 minimal medium with 30mg/l L-selenomethionine. Purification was as described above.
Crystals were obtained by hanging drop vapor diffusion, in which 1 ul protein was mixed with 1 ul well solution containing 100 mM HEPES pH 7.5, 50 mM NaCl and 8% PEG8000. Crystals appeared after a few days and grew to 0.4 × 0.05 × 0.01 mm3 within a week. Crystals were flash-frozen in 30% ethylene glycol prior to X-ray data collection. For structures complexed with metal ions or β-thujaplicinol (BTP) (27), crystals were soaked with MgCl2 or MnCl2 at various concentrations for various lengths of time prior to flash freezing (Table 1).
Table 1. X-ray data collection and structure refinement statistics.
| Se-Met gp2C | gp2C | gp2C-K428A:Mg2+ | gp2C-K428A:Mn2+ | gp2C-K428A: Mn2+:BTPe | gp2C-K428A:Ca2+ | |
|---|---|---|---|---|---|---|
| Crystal ID | C1 | A10 | D8 | D1 | F12 | E8 |
| Radiation source | NSLS | APS 23ID-B | APS 23ID-B | APS 23ID-B | APS 23ID-D | APS 23ID-B |
| Wavelength (Å) | 0.97920 | 1.03316 | 1.03316 | 1.03316 | 1.03330 | 1.03319 |
| Resolution (Å) | 64.9–2.44 (2.57–2.44)a | 29.69–1.55 (1.57–1.55) | 29.54–1.52 (1.56–1.52) | 50–1.57 (1.60–1.57) | 28.79–1.86 (1.90–1.86) | 29.6–1.59 (1.59–1.62) |
| Completeness(%) | 98.7(97.2) | 97.7(90.8) | 93.1(97.6) | 97.6(84.5) | 95.4(85.2) | 98.6(90.7) |
| Rmergeb(%) | 14.9(80.4) | 3.3(49.7) | 7.3(38.5) | 4.1(77.6) | 4.1(52.9) | 5.2(81.4) |
| I/σ | 11.0(3.3) | 17.7(1.9) | 9.6(1.8) | 27.0(1.0) | 17.1(2.0) | 16.1(1.2) |
| Redundancy | 7.2 | 3.7 | 3.3 | 3.1 | 3.5 | 3.0 |
| Space group | C2 | C2 | C2 | C2 | P21 | C2 |
| Unit cell (a, b, c, β) | 131.82, 59.5, 46.61, 100.02° | 133.683, 57.734, 46.654, 98.865° | 132.645, 57.519, 46.598, 98.761° | 132.766, 57.365, 46.646, 98.710° | 46.59, 58.42, 53.38, 96.43° | 133.078, 57.437, 46.568, 98.993° |
| Refinement statistics | ||||||
| Rwork/Rfreec | - | 0.14/0.16 | 0.16/0.18 | 0.16/0.19 | 0.17/0.21 | 0.16/0.19 |
| Rmsd bond length/Å | - | 0.009 | 0.009 | 0.009 | 0.007 | 0.006 |
| Rmsd bond angle/° | - | 1.179 | 1.208 | 1.173 | 1.063 | 0.983 |
| Metal | - | 10 mM MgCl2 | 100 mM MgCl2 | 100 mM MnCl2 | 100 mM MnCl2 and 1mM BTP | 10 mM CaCl2 |
| Soaking time | - | 16 h | 5 min | 5 min | 2 h | 1.5 h |
| Metal ions/ligand in the active site | - | None | Two Mg2+ | Two Mn2+ | Two Mn2+ and one BTP | One Ca2+ |
| Metal ion(s) occupancy | - | - | 0.88/0.96 | 0.89/0.68 | 1.0/1.0d | 1.0d |
| B-factor(Å2) | - | - | 19.6/45.5 | 31.9/65.0 | 25.7/27.4 | 33.1 |
aValues in the parentheses are for the outermost resolution shells.
bRmerge = ΣhklΣi|Ii(hkl) − <I(hkl)>| / ΣhklΣiIi(hkl), where Ii(hkl) is the observed intensity of reflection hkl and <I(hkl)> is the averaged intensity of symmetry-equivalent measurements
cRwork = Σhkl∥Fobs| − |Fcalc∥ / Σhkl|Fobs|, where Fobs and Fcalc are structure factors of the observed reflections and those calculated from the refined model, respectively. Rfree has the same formula as Rwork, except that it was calculated against a test set of the data that was not included in the refinement.
dOccupancy not refined.
eBTP, β-thujaplicinol.
X-ray data collection and structure determination
X-ray data for the Se-Met-gp2C were collected at the National Synchrotron Light Source (NSLS), and were processed with the program HKL2000 (28) (Table 1). The structure was determined at 2.44 Å resolution using the single-wavelength anomalous dispersion method with the program SOLVE/RESOLVE (29). Automated model building with the program PHENIX (30) resulted in a model covering ∼80% of the protein (205 out of 258 residues). This model was used in further model building and refinement with the program COOT (31) and PHENIX.
X-ray data for the wild-type and mutants gp2C were collected at the Advanced Photon Source (APS) and Stanford Synchrotron Radiation Light source (SSRL), respectively. Data were processed with the program HKL2000 (28). Structures were determined with the molecular replacement method with PHENIX, and models were manually rebuilt with COOT and refined with the program PHENIX using standard approaches. Refinement statistics are summarized in Table 1. Residues 338–349 are disordered and not modeled in structures of gp2C, gp2C-K428A:Mg2+ and gp2C-K428A:Mn2+:BTP. Residues 337–350 are disordered and not modeled in the structure of gp2C-K428A:Ca2+. Additionally, residues 249–251 and 337–350 in the structure of gp2C-K428A:Mn2+ are disordered thus not modeled.
Fluorescence-based thermal shift assay
Thermal shift assays were performed with purified proteins of gp2C or gp2C-K428A at a concentration of 3.5 μM alone or in the presence of MgCl2 in a buffer containing 20 mM HEPES pH 7.5, 100 mM NaCl and SYPRO Orange (800-folded diluted from the 5000X stock solution, Invitrogen) using procedures essentially as described (32). Solution mixtures were prepared and pipetted into 96-well PCR plates at a final volume of 50 μl. Melting curves were measured in the temperature range of 20°C to 80°C at 1°C/min on a StepOnePlus real-time PCR machine (Applied Biosystems). Melting temperatures were obtained at the maxima of the derivatives of SYPRO Orange fluorescence curves with the StepOne software v2.3. To test the effect of BTP on protein thermal stability, assays were performed in the presence of 2%(v/v) dimethyl sulfoxide (DMSO), 2% DMSO plus 10 mM MgCl2 or 10 mM MgCl2 plus 20 μM BTP (BTP was added from a DMSO stock solution, resulting in a final DMSO concentration of 2%).
RESULTS
The side chain of residue K428 extends into the active site, obstructing metal ion binding
The X-ray structure of gp2C was solved at 2.44 Å using the single-wavelength anomalous diffraction method and refined at 1.55 Å resolution (Table 1). Superimposition with the B. halodurans RNase H structure (17) shows that, despite lack of apparent sequence identity, the gp2C overall fold resembles those of the RNase H-family endonucleases (14), containing a characteristic five-stranded β-sheet sandwiched between α-helices (Figure 1). The five-stranded β-sheet and the two α-helices on one side superimpose well with the RNase H fold. There are seven α-helices on the other side of the gp2C molecule, representing structural elements that may be unique to terminase nucleases (Figure 1). The loop harboring an active site residue D192 in B. halodurans RNase H (17) is substituted by an α-helix near the C-terminus in gp2C, and such an α-helix is also present in human RNase H1 (18).
Figure 1.
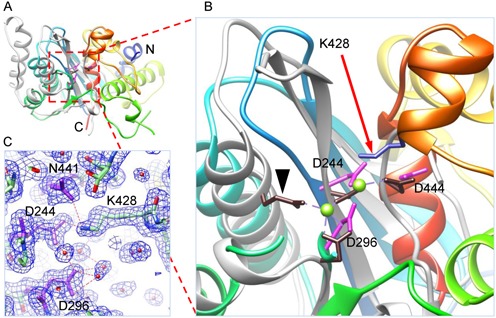
Structure of wild-type gp2C. (A) and (B) The gp2C structure (ribbon diagram rainbow-colored from the N-terminus to the C-terminus) is superimposed with that of B. halodurans RNase H (grey; RCSB PDB code 2G8H). Side chains of acidic residues in the active site of gp2C and K428 are shown as stick models in magenta and blue, respectively. The active site residues of RNase H are shown as stick models in brown. One of the active site residue of RNase H, E109, is indicated with a black arrow head. The two bound Mg2+ in RNase H are shown as green spheres. (C) The 1.55 Å resolution 2mFo–DFc electron density map of gp2C contoured at 1.0 sigma superimposed with the refined model. Water molecules, spheres in magenta. H-bonds, dashed lines in red.
Three acidic residues, D244, D296 and D444, in gp2C superimpose well with the active site residues D71, D132 and D192N in B. halodurans RNase H, respectively (D145, D210 and D274 in human RNase H1) (Figure 1), although the fourth active site residue E109 that coordinates the metal ion B in B. halodurans RNase H is replaced by residue A178 in gp2C. However, no metal ion was observed in the structures of the native gp2C or gp2C soaked or co-crystallized with MgCl2 or MnCl2. Closer inspection of the gp2C structure shows that the K428 side chain extends into the active site. The K428 side chain is well defined in the electron density map with strong density, whose conformation is immobilized by H-bonds to side chain oxygen atoms of D244 and N441 (Figure 1). Such a K428 side chain conformation is essentially identical to that in the previously reported full-length gp2 structure (4), whose nuclease domain is virtually superimposable to the present wild-type gp2C structure and did not show metal ions bound to the nuclease active site either. The gp2C K428 side chain amino group NZ atom is within 2.5 Å of the metal ion A in the superimposed B. halodurans RNase H structure, which would apparently generate steric hindrance for any bound metal ion in addition to the expellant electrostatic interaction.
Residue K428 mediates the metal cofactor binding to gp2C
To test if residue K428 affects binding of Mg2+ to gp2C, we constructed and purified the gp2C K428A mutant (gp2C–K428A). Fluorescence-based thermal shift assay was used to monitor protein stability at various concentrations of Mg2+. The gp2C–K428A exhibited reduced thermal stability as evidenced by the drop of Tm from 49.3°C for the wild-type protein to 45.4°C for the K428A mutant (Figure 2A). This would be consistent with loss of interactions of the K428 side chain with neighboring residues such as D244 and N441 as described above (Figure 1C) caused by the K428A mutation. Addition of 10 mM Mg2+ increased the Tm of gp2C–K428A by 3.9°C, but only a ∼0.5°C increase for the wild-type gp2C (Figure 2A). Dose-dependent experiments with Mg2+ concentrations ranging from 0.04 mM to 10 mM showed steady increase of Tm from 45.4°C at 0.2 mM Mg2+ to 49.3°C at 10 mM Mg2+ (Figure 2B) for the mutant protein. In contrast, the Tm of gp2C varied only within a narrow range between 49°C and 50°C (Figure 2B). These data indicate that Mg2+ considerably enhances the thermal stability of gp2C–K428A but has only marginal effect on wild-type gp2C. Taken together with the absence of metal ions in the metal-soaked wild-type gp2C structure, this is consistent with the aforementioned plausible obstruction of Mg2+ binding at the gp2C nuclease active site by the K428 side chain.
Figure 2.
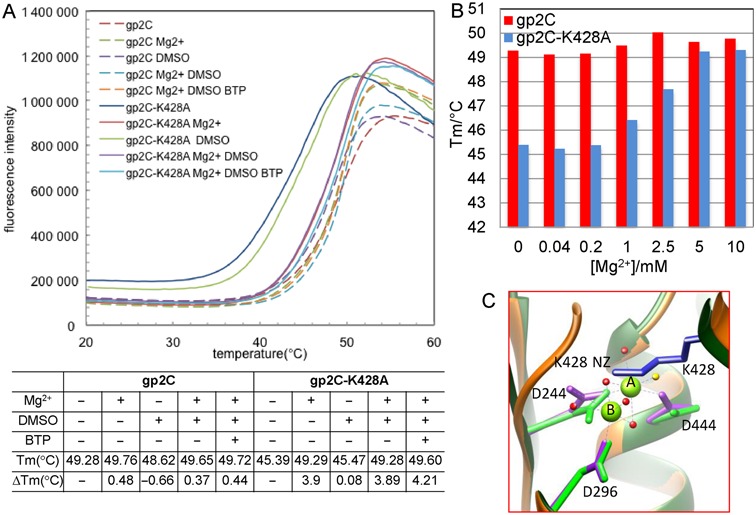
Residue K428 mediates Mg2+ binding to gp2C. (A) Thermal stability of gp2C and gp2C–K428A in the presence and absence of Mg2+, DMSO and/or β-thujaplicinol (BTP). Shown in the upper panel are melting curves for gp2C (dashed curves) and gp2C–K428A (smooth curves). Shown in the lower panel are Tm and ΔTm values compared to proteins alone. (B) A plot of Tm values measured in the presence of various concentrations of Mg2+ for gp2C (red) and gp2C–K428A (blue). (C) Superimposition of the structures of gp2C–K428A:Mg2+ (orange) with the wild-type gp2C (green). Side chains of acidic residues in the active site are shown as stick models in magenta and green for gp2C–K428A:Mg2+ and wt–gp2C, respectively. In the gp2C–K428A:Mg2+ structure, the two Mg2+ ions are shown as green spheres labeled A and B, respectively, and water molecules are shown as red spheres. The water molecule corresponding to the proposed nucleophile is in yellow. Coordinating bonds of the two metal ions are shown as dashed lines in purple.
The K428A mutation allows visualization of metal ions bound to the active site
To test if the K428A mutation eliminates the plausible steric clash thus enables metal ion binding in the active site, we soaked the gp2C-K428A crystal with Mg2+ and solved the structure at 1.52 Å resolution (Table 1). The structure showed superb electron density for the active site, including two Mg2+ ions bound to acidic residue triad D244, D296 and D444 as well as coordinating water molecules (Figures 2C and 3A). Superimposing the Mg2+-complexed and the wild-type gp2C structures shows no significant or global structural alteration across the domain, with a root mean square deviation for Cα atoms of as small as 0.241 Å. Nevertheless, superimposing the Mg2+-complexed structure with the wild-type gp2C structure shows that the nitrogen atom of the K428 side chain is within 1.59 Å and 2.28 Å of the metal ions A and B, respectively (Figure 2C), demonstrating that the K428 side chain abrogates the metal ion binding in the active site. Together with the thermal shift assay results above, these data suggest that K428 mediates metal cofactor binding to gp2C through obstructing the metal ion binding in the active site.
Figure 3.
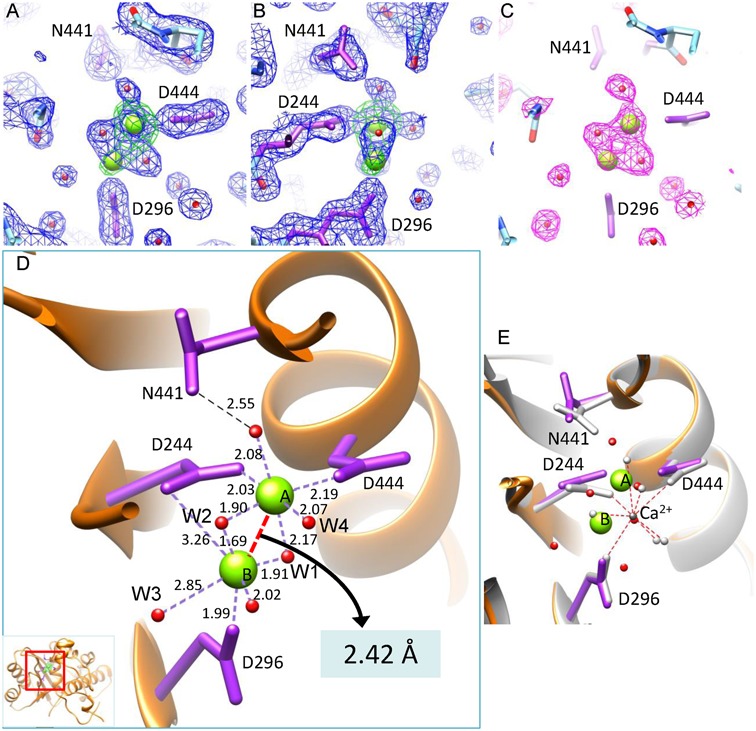
The active site of gp2C–K428A:Mg2+. (A and B) The 1.52 Å resolution 2mFo–DFc electron density map of the gp2C–K428A:Mg2+ structure (blue mesh) contoured at 1.4 σ above background superimposed with the refined model. Green mesh, the anomalous difference map of gp2C–K428A:Mn2+ at 2.47 Å resolution contoured at 8.0 σ above background. Water molecules, spheres in magenta. H-bonds, dashed lines in red. (B) is 90° about the vertical axis from (A). (C) The mFo–DFc difference map (magenta mesh) prior to addition of water and metal ions into the model for refinement, contoured at 3.3 σ above background. The refined structure (stick model) is superimposed. (D) Binding of the two metal ions (green spheres) in the active site of the gp2C–K428A:Mg2+ structure (orange ribbon diagram). Side chains of residues D244, D296, D444 and N441 in the active site are shown as stick models in magenta. Water molecules, red spheres. The coordination bonds of the metal ions are shown as dashed lines in purple with lengths indicated. The distance between the two metal ions is indicated with a red dashed line. The inset shows the location of the figure in the protein. (E) Superimposition of the structures of gp2C–K428A:Mg2+ with gp2C–K428A:Ca2+ (grey). Side chains of residues D244, D296, D444 and N441 in the Ca2+-complexed structure are shown as stick models in grey. The bound Ca2+ is shown as a sphere in dark grey. The coordinating water molecules are shown as grey spheres, with interactions indicated with red dashed lines. The color scheme for the Mg2+-complexed structure is the same as in (D).
Two metal ions concurrently bind to the gp2C–K428A active site
Electron density for two metal ions is observed in the active site of gp2C–K428A soaked with Mg2+ (Figure 3A–C). This is due to physical binding of two metal ions in the active site of a single protein molecule, instead of an averaged effect in the crystal, as evidenced by the following data. First, modeling metal ions into the two electron density peaks is strongly supported by the stereochemistry of the coordination systems including bond lengths and bond angles characteristic of Mg2+ (see description in detail below). Second, in the gp2C–K428A:Mg2+ structure refined at 1.52 Å resolution, Metal ion A has an occupancy of 0.88 and a B-factor of 19.6 Å2, whereas Metal ion B has an occupancy of 0.96 and a B-factor of 45.5 Å2 (Table 1). Third, located at the positions of the two modeled metal ions are density peaks of 11.7 and 5.7 σ above background, respectively, in the Fo–Fc map (3.8 σ and 1.7 σ above background in the 2mFo–DFc map) prior to addition of solvent molecules and metal ions for refinement (5.3 σ and 2.1 σ above background in the 2mFo–DFc map in the final refined structure). Such peak heights are considerably larger than those of water molecules. Finally, the spherical shapes of the two metal ions and the two water molecules are clearly resolved in the electron density map, and there is a hollow space in the electron density map midway between the two water molecules that immediately coordinate with the two metal ions, confirming the high quality of the map (Figure 3A–C).
To further verify the concurrent physical existence of two metal ions in the active site, we soaked gp2C–K428A crystals with Mn2+ and solved the structure at 1.57 Å resolution (Table 1). The structure shows excellent electron density for two Mn2+ ions in the active site, occupying essentially the same positions as the Mg2+ ions (Supplementary Figure S1). The two Mn2+ ions show peaks of 23.8 and 9.6 σ above background, respectively, in the Fo–Fc map (8.9 σ and 3.1 σ above background in the 2mFo–DFc map) prior to addition of solvent molecules and metal ions for refinement. We collected anomalous diffraction data for the Mn2+-complexed crystal at the wavelength of 1.89294 Å near the absorption edge of manganese, and the anomalous difference map shows two peaks at 24.0 and 10.2 σ above background at locations corresponding to the two Mn2+, respectively (Figure 3A–B), unambiguously demonstrating that these are indeed bound metal ions. In the Mn2+-complexed gp2C–K428A structure refined at 1.57 Å, the two metal ions have an occupancy of 0.89 and 0.68 and a B-factor of 31.9 and 65.0 Å2, respectively (Table 1).
Taken together, these data demonstrate that two metal ions concurrently occupy the active site.
The structures of gp2C–K428A in presence of Mg2+ or Mn2+ show ultra-short metal:metal distances
The high resolution structures of gp2C–K428A soaked with Mg2+/Mn2+ reveal a metal ion binding mode with a unique coupled-octahedral configuration at the active site comprising the acidic residue triad D244, D296 and D444 (Figure 3D; Supplementary Figure S1D). In the structure of gp2C–K428A soaked with Mg2+ at 1.52 Å resolution, Metal ion A displays a nearly perfect 6-member octahedral coordination system, bonding to a side chain carboxyl oxygen atom from residues D244 and D444, respectively, and four water molecules. The distances between Metal ion A and coordinating atoms range from 1.90 to 2.19 Å, close to the characteristic value of 2.07 Å for the coordinating distances for the inner shell ligands of Mg2+. Metal ion B is situated in the center of a second octahedral coordination system, whose ligands include a side chain carboxyl oxygen atom from residues D244 and D296, respectively and four water molecules. The two octahedral systems are coupled by sharing two water ligands (W1 and W2 in Figure 3D), and these two shared water ligands, together with the two metal ions, assume a planar, tetragonal geometry. The distance from Metal ion B is 1.91 and 1.69 Å for water ligand W1 and W2, respectively, considerably shorter than the characteristic value of ∼2.07 Å for Mg2+. Such short distances are unlikely formed by H-bonds between water molecules, providing additional evidence that Mg2+ occupies position B. The angle between the two bonds formed by Metal ion B and the two water molecules W1 and W2 is 110.1°, which deviates significantly from 90° for an ideal octahedral geometry and is in contrast to 92.9° formed by Metal ion A and the same two water molecules. The distances between Metal ion B and two of its ligands, a water molecule (W3 in Figure 3D) and a carboxyl oxygen of residue D244, are 2.85 and 3.26 Å, respectively. Thus, the coordination system of Metal ion B appears to be slightly deviated from the standard octahedral geometry for Mg2+, perhaps reflecting a stressed, more labile state.
The distance between the two Mg2+ ions is as short as 2.42 Å (2.64 Å between the two manganese ions in the Mn2+-complexed structure), which is unusual given the atomic radii of 1.6 and 1.3 Å for magnesium and manganese, respectively. To the best of our knowledge, such an ultra-short distance may represent the shortest metal:metal distance that has been documented in any biological system such as metallo-enzymes (33,34), and is significantly shorter than those observed in RNase H-family nucleases which typically range from 3.4 to 4.0 Å (14,19). Such an ultra-short metal:metal distance may only be possible for magnesium or manganese ions, but not for calcium ions because of the much larger atomic radius of 1.9 Å. Indeed, the structure of gp2C–K428A mutant soaked with calcium ion shows a totally different active site configuration (Figure 3E). Only a single calcium ion is observed in the active site, which is located differently from magnesium, has an eight-member coordination system with an average metal:ligand distance of 2.48 Å (ranging from 2.19 to 2.79 Å), and is accompanied with different side chain conformations of the active site residues D244 and D444 (Figure 3E). Our results provide a structural basis for distinguishing magnesium from calcium for catalysis in RNase-H-family nucleases (see Discussion).
These results suggest that the two metal ions in the active site exhibits a unique coordinating architecture, which is presumably made possible by the local chemical environment.
The structure of gp2C–K428A in complex with manganese and β-thujaplicinol
The gp2C nuclease activity requires binding of the dsDNA substrate. However, such binding appears transient, as the effort to observe and isolate the gp2C:DNA complex has not been successful thus far. BTP is a natural product isolated from the bark of the Western red cedar and belongs to a group of small molecules called hydroxytropolones (35). These compounds can chelate the two divalent metal ions in the active site of RNase H-like nucleases, were shown to inhibit HIV reverse transcriptase (HIV-RT) through binding to the RNase H domain active site (36,37), and were implicated as candidates for development of antivirals against HIV (27,36–39), herpes simplex viruses (40) and hepatitis B virus (41,42). In a comparative study of the structures of HIV-RT in complex with BTP and human RNase H1 complexed with RNA:DNA hybrid and Ca2+ (18), BTP was shown to occupy the positions of the scissile phosphate of the nucleic acid substrate and the water molecule thought to serve as the nucleophile during catalysis (37). Such binding of hydroxytropolone molecules is also confirmed in the structure of HIV-RT complexed with manicol, a natural product related to BTP (36). We therefore used BTP as a molecular probe as an effort to gain insight into the active site architecture and the DNA-binding state of gp2C.
The crystal of gp2C–K428A was soaked with Mn2+ and BTP, and the structure was determined at 1.86 Å resolution (Table 1). The structure showed superior electron density for BTP and two Mn2+ ions as well as four water ligands (Figure 4; Supplementary Figure S2). The hole in the middle of the tropolone ring is clearly visible, confirming the high quality of the structure. Each Mn2+ ion is both situated at the center of a nearly perfect 6-member octahedral coordination system. Metal ion A is coordinated with two water molecules, BTP oxygen atoms O1 and O2 emanating from the tropolone ring, and side chain oxygen atoms OD1 from residues D244 and N441, respectively. Metal ion B is coordinated with two water molecules, BTP oxygen atoms O7 and O1, and side chain oxygen atoms OD2 from residues D244 and D296, respectively. The BTP tropolone ring exhibits an excellent planar structure, and is nearly coplanar with the two Mn2+ ions, with a small deviation of 7° (Figure 4C). Overall, BTP is nearly planar with the two Mn2+, the side chain carboxyl groups of residues D244 and D296, and the side chain carboxamide group of residue N441 (Figure 4C).
Figure 4.
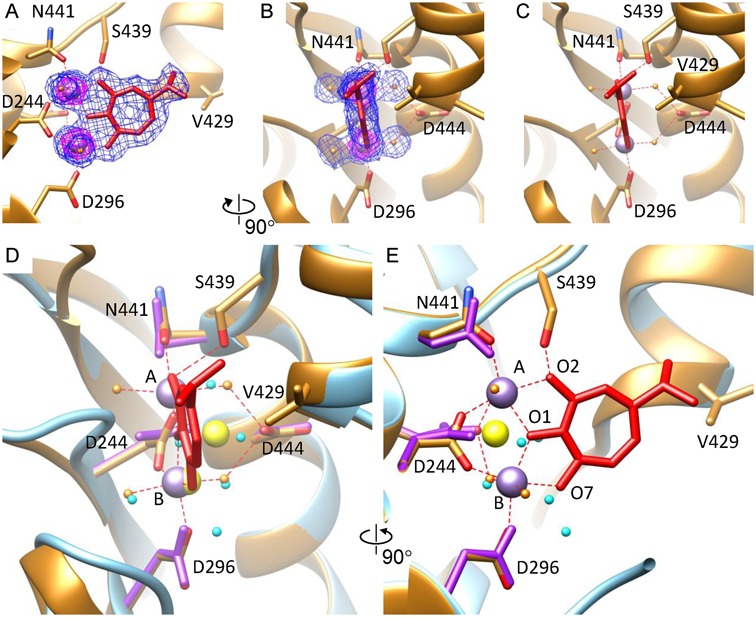
The metal ion binding mode in gp2C–K428A:Mn2+:BTP. (A and B) The gp2C–K428A:Mn2+:BTP structure (ribbon diagram in gold) superimposed with the 1.86 Å resolution 2mFo–DFc electron density map contoured at 1.0 σ (blue mesh) and 5.4 σ (magenta mesh) to show location of the two metal ions. Side chains of the active site residues as well as S439 and V429 are shown as stick models. BTP, red stick model. The two Mn2+, purple spheres. The four coordinating water molecules, gold spheres. (C) The local structure of gp2C–K428A:Mn2+:BTP showing planarity of the BTP tropolone ring and the slight deviation of the BTP plane with respect to the two metal ions. (D) and (E) Superimposition of structures of gp2C–K428A:Mn2+:BTP (gold) and gp2C–K428A:Mg2+ (cyan), where the bound metal ions are shown as purple and yellow spheres and water molecules in gold and cyan, respectively. Metal:ligand bonds in gp2C–K428A:Mn2+:BTP are indicated with red dashed lines. The view in (E) is 90° about the vertical axis from that in (D). Notice the change in position and coordination interactions for Metal ion A.
BTP is located in the pocket of the nuclease active site, but does not have extensive direct interactions with the protein, except that its O2 atom is H-bonded to the side chain oxygen of residue S439 and the isopropyl group at the C4 atom of BTP forms non-polar interaction with residue V429 (Figure 4C). Instead, abundant indirect interactions exist between BTP and the protein via the two metal ions and coordinating water molecules. These coordinating water molecules are H-bonded with a side chain carboxyl oxygen of residue D444, the main chain oxygen atom of A252, and the main chain oxygen atom of P245, respectively. Thus, binding of BTP is mainly mediated by interactions with the metal ions but involves minimal direct compound:protein interactions. This is in agreement with the thermal shift data (Figure 2A). For gp2C–K428A in the presence of DMSO, addition of 10 mM Mg2+ resulted in an increase of 3.8°C in Tm, while addition of Mg2+:BTP led to a slightly larger increase of 4.1°C (Figure 2A; compare melting curves and Tm values for proteins with DMSO, with Mg2+ plus DMSO, and with Mg2+ plus BTP), suggesting that the contribution of BTP to the increased thermal stability is much less than that of Mg2+ and that BTP binding largely depends on Mg2+ binding. For wild-type gp2C, addition of Mg2+ and Mg2+:BTP caused only a small increase in Tm by approximately 1.0 and 1.1°C, respectively (Figure 2A; compare Tm values for proteins with DMSO, with Mg2+ plus DMSO and with Mg2+ plus BTP). This is consistent with unsuccessful, or at least much less efficient, binding of BTP and Mg2+ to wild-type gp2C. It is worth pointing out that these data do not rule out the possibility that Mg2+:BTP can displace the K428 side chain and bind to the active site in the wild-type gp2C, but the chance of such a binding mode is low for the case of BTP because (i) BTP has minimal direct interactions with the protein as shown in the gp2C–K428A:Mn2+:BTP structure, which may not be energetically sufficient to drive such a binding mode, and (ii) the space in the active site is limited and may not be adequate to accommodate the displaced K428 side chain, which is further restricted by rotamer conformations. For such a binding mode to occur, significant conformational change in the protein might be required, e.g. potential movement of the hairpin structure where K428 is located (see Discussion), in order to move the K428 side chain away to make room for metal ions. Further studies are needed to test such possibility by using chemically modified forms of BTP (35,36), which might provide more profound compound:protein interaction that is sufficient to drive the required conformational change thus enable such a binding mode.
The metal ion binding mode in gp2C–K428A:Mn2+:BTP is distinct from that in the gp2C–K428A:metal structures
The metal ion binding mode in the gp2C–K428A:Mn2+:BTP ternary complex structure is remarkably distinct from that observed in the gp2C–K428A complexed with metal ions alone described above (Figure 4D–E; Supplementary Figure S1D). Firstly, Metal ion B in both cases occupies approximately the same position, but Metal ion A displays a positional difference of 2.43 Å between the two states. As a result, the distance between the two metal ions is 3.75 Å in the gp2C–K428A:Mn2+:BTP structure, a value that is far more than the ultra-short distance observed in the gp2C–K428A complexed with metal ions alone and falls in the 3.4–4.0 Å range frequently observed in other RNase H-like nucleases (19,20). Secondly, in the BTP-complex structure, D444 is not coordinated with any of the two metal ions, whereas in the gp2C–K428A:metal complex structures it coordinates with Metal ion A (Figure 4D–E; Supplementary Figure S1D), although the conformation of D444 remains virtually unchanged. Third, in the gp2C–K428A:Mn2+:BTP structure, the side chain carboxyl group of D244 undergoes a ∼70° rotation from the conformation in the gp2C–K428A:metal complex structures (Figure 4D–E). As a result, the two carboxyl oxygen atoms are coordinated with the two Mn2+ ions respectively with approximately equal distance.
Side chain flipping of residue N441
The side chain of residue N441 adopts different rotamer conformations in the gp2C–K428A:Mn2+, gp2C–K428A:Mg2+ and gp2C–K428A:Mn2+:BTP structures (Figure 4D–E; Supplementary Figure S1D). In gp2C–K428A:Mg2+, the N441 side chain oxygen atom is H-bonded to a water molecule with a distance of 2.55 Å (Figure 3D). This water molecule is coordinated with Metal ion A with a distance of 2.08 Å, which precisely matches the standard value for the octahedral coordination system of Mg2+ (Figure 3D). In contrast, in gp2C–K428A:Mn2+, the N441 side chain flips toward Metal ion A and the oxygen atom is coordinated to Metal ion A with a distance of 2.59 Å, and the metal:water bond deviates from perpendicular to the central plane of the octahedron (Supplementary Figure S1D). The presence and absence of a water ligand between N441 and Metal ion A in gp2C–K428A complexed with Mg2+ and Mn2+, respectively suggests the less stringent requirement for standard coordinating distances and angles in Mn2+ than in Mg2+. In the gp2C–K428A:Mn2+:BTP structure, the N441 side chain oxygen is coordinated with Metal ion A with a distance of 2.26 Å, and its rotamer conformation is nearly identical to that in gp2C–K428A:Mg2+ (Figure 4D). These observations suggest that the N441 side chain can adopt different conformations as needed to accommodate different configurations and types of the two bound metal ions.
DISCUSSION
A regulatory role of residue K428 in the TerL function through mediating metal cofactor binding
Our structural analysis shows that the gp2C K428 side chain extends into the nuclease active site, obstructing the metal ion binding, while thermal shift assay results support the notion that K428 mediates the metal cofactor binding to gp2C. As metal cofactors are required for the enzymatic activities of RNase H-like nucleases and terminase nuclease domains, these data suggest that residue K428 could play a regulatory role in gp2 nuclease function. This is consistent with the physiological requirement that the TerL nuclease activity must be carefully regulated so that it does not cut random DNA in the infected cells which includes viral DNA but instead only catalyzes DNA processing at specified steps during packaging. It is likely that the K428 side chain may restrict or turn off the nuclease activity by adopting the conformation observed in the wild-type gp2C structure, that is, by extending into the nuclease activity site and thus preventing metal ion binding. This would render the nuclease inactive, thus avoiding aberrant cutting of DNA. When nuclease activity is needed, e.g. at the initiation cleavage step or upon completion of a single packaging cycle, the K428 side chain may be retracted from the active site to allow metal ion binding, thus enabling catalysis. Such an activation mechanism for the gp2 nuclease activity may be accomplished through specific molecular interactions involved in assembly with TerS during the initiation cleavage or in oligomerization of TerL and/or assembly of TerL with the procapsid during the headful cleavage.
Primary sequence identity among TerL nucleases is low, typically below 15%. However, comparative analysis at the 3D structure level shows that a lysine residue of TerL proteins from phage P22 and SPP1 occupies a similar position in a hairpin structure that is reasonably close to the nuclease active site (Supplementary Figure S3). These data support the idea that such a lysine-mediated mechanism might be common among TerL proteins. In phage T4 TerL, no lysine residue exists at a same position, except for a lysine residue K529 that is located in the similar hairpin structure but is less close to the active site and is not known to play such a role (Supplementary Figure S3). These observations are consistent with the fact that phage P22 is a close relative to Sf6 while T4 is more distantly related as shown in a recent comparative structural study (11). The distances of those lysine residues to the active site vary in these phage proteins, with that in Sf6 gp2 the shortest and that in SPP1 G2P the longest, and, interestingly, these appear to coincide with the distances of the hairpins from the active site (Supplementary Figure S3). It was shown that the hairpin structure was conformationally dynamic or disordered in SPP1 G2P (7) and was completely disordered in the gp17 nuclease structure from phage RB49, a close relative to T4 (5) and in herpesvirus TerL nucleases (11,12). These hairpins were suggested to interact with the DNA substrate (4,6,7,11–13), and were proposed to undergo conformational motion that contributed to control of nuclease activity and/or DNA translocation (6,7). It is tempting to speculate that the hairpin may undergo large-scale movement to allow it to swing close to or away from the nuclease core, which would position the lysine residue close to or away from the active site, thus down- or up-regulating the nuclease activity, respectively. Such movement could be triggered by, e.g. interaction with the ATPase domain. This model could explain previous observations that the N-terminal ATPase domain can stimulate the nuclease activity in the context of the full-length TerL proteins (4,6), as the hairpins are located between the nuclease and ATPase domains as observed in the structures of full-length TerL from Sf6 and T4 although the details of domain arrangements differ (4,5). Nevertheless, the hairpins in the Sf6 gp2C structures in the present study and in previous structures of full-length TerL from Sf6 and T4 are well ordered (4,5), and it has not been possible to experimentally observe two conformations for the hairpin in any single TerL nuclease. Further studies are needed to test the viability of this hypothesis.
An ultra-short metal:metal distance in the active site
Formation of the pentacovalent phosphate transition state is the critical step during the two-metal-ion catalytic mechanism shared by RNase H-like nucleases. However, how the two metal ions drive formation of the pentacovalent phosphate transition state remained unclear. It was thought that Metal ion A deprotonates a water molecule, which serves as the nucleophile that attacks on the scissile phosphate, while Metal ion B may contribute to stabilization of the transition state pentacovalent phosphate (14,16). To form the pentacovalent phosphate transition state, the oxygen atom from the nucleophile must move by >1 Å so that it would be within <2 Å of the phosphorous atom for formation of a covalent linkage (16,19). However, the nucleophilic water is unlikely to move over 1Å by itself because this would require it to dissociate from Metal ion A, representing removal of an inner-shell ligand from the metal ion, which is highly costly in energy. Moreover, repulsion between the negatively charged nucleophile and the scissile phosphate also increases energy cost. Thus, it was thought that Metal ion A might move toward Metal ion B within a very short distance, thus bringing the nucleophile closer enough to the phosphorous (16,19).
In the present studies we have observed ultra-short metal:metal distances in the active site. Such ultra-short distances are to the best of our knowledge among the shortest documented in biological systems such as metallo-enzymes (33,34), and are close to metal:metal bond lengths in multimetallic complexes in inorganic systems such as the Mo-Mo distance of 2.12 Å (43) and the Co-Zr distances ranging from 2.33 to 2.44 Å (44,45). A Ni-Fe distance of 2.5 Å was reported in the 2.15 Å resolution X-ray structure of the reduced state of a [NiFeSe] hydrogenase (46), and the 1.36 Å structure of a carbon monoxide dehydrogenase showed a Ni-Fe distance of 2.56 Å (47). In the P-clusters and the FeMo cofactors of nitrogenases, metal:metal distances can be as short as 2.50 and 2.58 Å, respectively (48–50). The oxygen-evolving complex of photosystem II is a Mn4CaO5 cluster, in which the shortest Mn-Mn distance was 2.68 Å (51). The ultra-short metal:metal distances observed in the present work are significantly shorter than those reported for RNase H-family nucleases which typically range from 3.4 to 4.0 Å or longer (14,16,19). It was reported that two Mg2+ ions were as close as 3 Å in the 2.88 Å resolution structure of the phage T7 RNA polymerase complexed with substrate, which belongs to the RNase H-family enzymes (52). Previous TerL nuclease structures were at much lower resolution and failed to reveal such an ultra-short metal:metal distance. In the 2.5 Å resolution structure of phage SPP1 G2P soaked with Mn2+, the two metal ions were 3.9 Å apart (7). In the 3.2 Å resolution structure of human cytomegalovirus TerL pUL89 nuclease domain, two Mn2+ were observed in the active site with a metal:metal distance of 3.4 Å (12). The structure of phage P22 TerL gp2 nuclease domain at 2.02 Å resolution was reported to contain an Mg2+ with octahedral coordination and a second Mg2+ with a non-canonical tetrahedral coordination as far as 7.9 Å apart, which were not confirmed with anomalous diffraction (13). Only one Mg2+ was observed in the active site of the isolated nuclease domain of TerL from phage RB49, a close relative of phage T4 (5), and no metal ions were reported in the TerL nuclease active sites of phage T4 (5), Sf6 (4) and herpes simplex virus type 1 (11).
The effect of the ultra-close proximity of the two metal ions is 2-fold. First, it would generate a highly positive electrostatic niche in the active site, which may be essential for destabilizing the scissile phosphate and neutralizing the abundant negative charges in the pentacovalent phosphate, thus promoting the formation of the pentacovalent phosphate transition state. Second, it would bring one of its coordinating water molecules toward the scissile phosphate within a distance from the phosphorous atom that is short enough for formation of a covalent linkage (see below and Figure 6). Hence, the close metal:metal distance observed here may represent a critical state during the two-metal-ion catalysis, that is, a state that is poised for the formation of the pentacovalent phosphate prior to P-O bond breaking.
Figure 6.
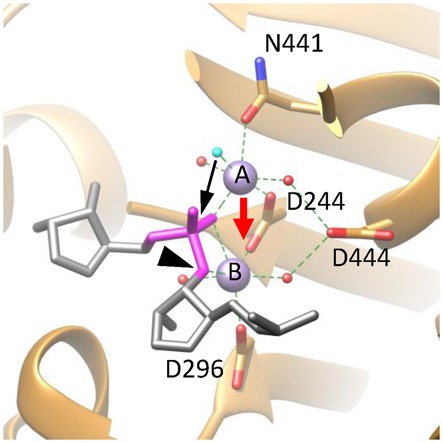
A proposed catalytic mechanism for gp2C. The active site architecture of the DNA-binding state based on the gp2C–K428A:Mn2+:BTP structure (ribbon diagram in gold), in which BTP has been replaced with the DNA substrate fitted as in Figure 5 (stick model in grey) and the nucleophilic water (cyan) that takes the position of the O2 atom of BTP in the gp2C-K428A:Mn2+:BTP structure shown in Figures 4 and 5. For clarity, only the backbone of a strand of the DNA is schematically shown, in which the scissile phosphate is in magenta and the leaving 3′-OH group is indicated with a black arrowhead. Side chains of active side residues are shown as stick models. The two metal ions are shown as purple spheres and are labeled with A and B, respectively. The nucleophilic water is in cyan. Other water molecules, red spheres. Coordination bonds of the metal ions are shown as green dashed lines. The proposed movement of Metal ion A (red arrow) brings the two metal ions closer, generating a positively charged niche that neutralizes, thus helps to stabilize, the highly negatively charged pentacovalent phosphate. The movement of Metal ion A also brings the nucleophilic water (cyan) closer to the scissile phosphate (black arrow) within a distance from the phosphorous that is short enough for formation of a covalent bond. As a result, the nucleophilic attack occurs and the pentacovalent phosphate transition state is formed.
The binding mode of BTP and Mn2+ mimics the DNA-binding state of gp2C
The gp2C–K428A:Mn2+:BTP structure reveals a metal ion binding mode distinct form that observed in the structures complexed with metal ions alone. Comparative studies with the HIV-RT:BTP and the human RNase H1:RNA/DNA:Ca2+ complex structures showed that BTP usurped the positions of the nucleic acid scissile phosphate and the nucleophilic water molecule (37). Thus, the location of BTP would reflect that of bound DNA substrate with respect to the nuclease active site. To gain insight into the DNA binding in gp2C, we first superimposed the HIV-RT:BTP complex structure onto the gp2C–K428A:Mn2+:BTP structure by matching the positions and orientations of the bound BTP and the two metal ions (Supplementary Figure S4A–C). Subsequently, we superimposed the human RNase H1:RNA/DNA:Ca2+ complex structure onto the resultant HIV-RT:BTP complex structure based on highly conserved protein folds and configurations of active site residues between RNase H1 and the HIV-RT RNase H domain (Supplementary Figure S4D). The resultant RNA:DNA hybrid from the superimposed human RNase H1:RNA/DNA:Ca2+ structure thus reflects the approximate location of the DNA substrate with respect to the gp2C nuclease active site (Figure 5). This notion is also supported by the observations that the two Mn2+ ions in gp2C structure occupy essentially the same positions as the two Ca2+ ions in the superimposed human RNase H1 structure, and that the carboxyl/carboxamide groups of D244, N441 and D296 in the gp2C structure superimpose nicely with the carboxyl groups of D145, D274 and E186 in the RNase H1 structure, respectively (Figure 5). Despite the small positional difference, the carboxyl groups of D444 in gp2C and D210N in RNase H1 also have approximately the same locations (Figure 5). This superimposition aligns the RNA strand of the RNA/DNA hybrid along the crevice of the gp2C active site. The water molecule proposed as the nucleophile in the RNase H1 structure (18) is close to the BTP O2 atom in the gp2C structure (Figure 5). The O1 and O7 atoms of BTP are superimposable with a non-bridging oxygen atom of the scissile phosphate and the oxygen atom of the leaving 3′-OH group, respectively (Figure 5). Thus, the structure of gp2C–K428A:Mn2+:BTP mimics the state of DNA substrate binding, shedding light on the architecture of the active site and the spatial arrangement of the scissile phosphate with respect to the two metal ions and the active site residues.
Figure 5.
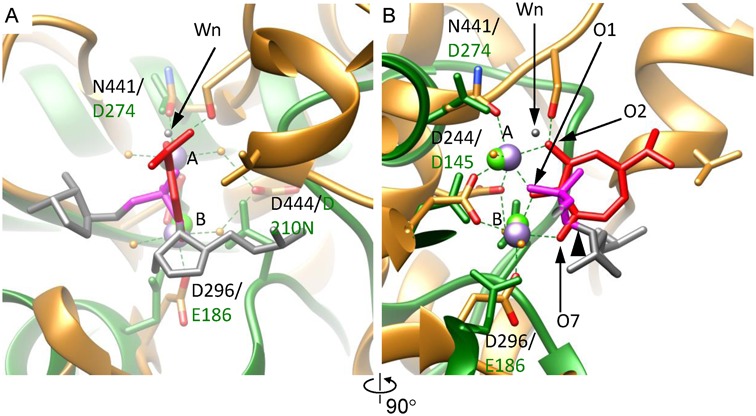
The metal ion binding mode of gp2C–K428A:Mn2+:BTP mimics the DNA-binding state. (A) The gp2C–K428A:Mn2+:BTP structure (gold) is superimposed onto human RNase H1 in complex with an RNA/DNA hybrid and Ca2+ (RCSB PDB code 2G8H) (green) assuming the metal ion binding modes are conserved and the bound BTP reflects the DNA scissile phosphate (see text). Side chains of active side residues are shown as stick models. Each superimposed pair of active site residues are indicated with the residue numbers in gp2C on the top and in RNase H1 on the bottom, respectively. The two Mn2+ in gp2C–K428A structure are shown as purple spheres and labeled with A and B, respectively. The two Ca2+ ions in RNase H1 are shown as green spheres. The RNA strand of the RNA/DNA hybrid in the RNase H1 structure is shown as stick models in grey. For clarity, only two nucleotides are shown. The scissile phosphate is in magenta. The leaving 3′-OH group is indicated with a black arrowhead in (B). The nucleophilic water in the RNase H1 structure is indicated with a black arrow and labeled Wn. The oxygen atoms O1, O2 and O7 of BTP are indicated. (B) 90° about the vertical axis from (A).
A model for the two metal ion catalytic mechanism
Observations of the two distinct metal ion binding modes, one for DNA substrate binding and the other putatively for the pentacovalent phosphate formation, suggest a transition between the two states during catalysis, which involves a ∼2.43 Å positional change of Metal ion A from N441 to D444 (Figure 4D–E). What triggers such a positional change? It is likely that the negatively charged D444 side chain carboxyl favors a charge–charge interaction with Metal ion A, which is unavailable for the uncharged N441 side chain, thus an acidic residue at position 444 may be important for triggering the plausible movement of Metal ion A. D444 in gp2C corresponds to D210 in human RNase H1 and, interestingly, the human RNase H1 D210N mutant lost nuclease activity (18), supporting the importance of the negative charge of the D210 side chain. Additionally, it could be that the energy barrier for such movement is small enough to allow such a movement to occur, owing to the architecture of the active site.
We propose a catalytic mechanism for gp2C based on a transition between the two metal ion binding modes observed here (Figure 6). The DNA substrate docks onto the active site adopting an architecture as shown in the gp2C–K428A:Mn2+:BTP structure in which the two metal ions are 3.75 Å apart. The two metal ions are jointly coordinated by a non-bridging oxygen atom of the scissile phosphate, bisecting the scissile phosphate. Metal ion B is also coordinated with the oxygen atom of the leaving 3′-OH group. The presumed nucleophilic water molecule, occupying a position similar to that of the O2 of BTP, is coordinated to Metal ion A. In such a DNA-binding state, Metal ion A is coordinated with N441 side chain, but not D444, as shown in the gp2C-K428A:Mn2+:BTP structure. Metal ion A then shifts to the position as shown in the structures of gp2C–K428A complexed with metal ions alone. Such movement may be triggered by more favorable interactions of Metal ion A with D444 side chain than N441 due to the negative charge of D444, and may be facilitated by flipping of the N441 side chain. This would bring the two metal ions into proximity, generating a positively charged niche that neutralizes and thus stabilizes the highly negatively charged pentacovalent phosphate. Meanwhile, movement of Metal ion A would bring the nucleophilic water closer to the scissile phosphate so that it is within a distance from the phosphorous that is short enough for formation of a covalent linkage. As a result, the nucleophilic attack occurs and the pentacovalent phosphate is formed. This mechanism sheds light on the intricate steps during the catalysis and how the two metal ions cooperate to drive formation of the pentacovalent phosphate transition state.
Distinguishing Mg2+ from Ca2+ in the two-metal-ion catalysis
The two-metal-ion nucleases typically require Mg2+ or Mn2+, and in most cases Ca2+ does not support catalysis although it supports nucleic acid binding. Distinguishing Mg2+ from Ca2+ for catalysis is physiologically necessary, given comparable biological abundance of the two types of metal ions. The ultra-short metal:metal distances observed in the gp2C–K428A:metal structures may only be possible for magnesium or manganese ions due to their small atomic radii of 1.6 and 1.3 Å, respectively, but not for calcium ions because of the much larger atomic radius of 1.9 Å, as evidenced by the gp2C–K428A:Ca2+ structure which shows a totally different metal ion binding in the active site (Figure 3). Only a single calcium ion is observed in the active site, which not only results in non-productive stereochemistry but also insufficient positive charges. Two Ca2+ were observed in the active site of human RNase H1 D210N mutant in complex with an RNA/DNA hybrid (18). However, such a complex was not productive, presumably because the two Ca2+ ions failed to adopt a short enough metal:metal distance. Therefore, the requirement for an ultra-short metal:metal distance provides a structural basis for distinguishing magnesium from calcium for catalysis in these nucleases.
Drifting of the nuclease active sites in viral terminases
Superimposing the gp2C–K428A:Mn2+:BTP structure with those of the human RNase H1:RNA/DNA:Ca2+ and the HIV-RT:BTP complexes according to the bound metal ions and BTP as described above shows that residues D244, N441, D296 and D444 in the gp2C structure overlie residues D145, D274, E186 and D210N in the RNase H1 structure (D443, D549, E478 and D498 in HIV-RT), respectively (Figure 5; Supplementary Figure S4), indicating concordance in their functional roles. However, superimposition according to the overall protein folds shows that residues D244, D296 and D444 in gp2C fit well with the active site residues D145, D210N and D274 in human RNase H1, respectively (Figure 5; Supplementary Figure S4). In gp2C, residue A178 replaces the fourth active site residue E109 that coordinates Metal ion B in RNase H1, while residue N441 in gp2C occupies the position of residue V552 in RNase H1. Overall, it appears as if the configuration of the active site, that is, the spatial arrangement of active residues D145, D274, E186 and D210 in the RNase H1 with respect to the protein fold, underwent a translation into residues D244, N441, D296 and D444 in the gp2C. Such rearrangement of active site residues built upon a conserved protein fold might occur during evolution through, e.g. acquisition of N441 as in gp2C which gains the function of metal ion binding followed by loss of the acidic residue at position 178 (corresponding to E109 in RNase H1), maintaining the ability to bind two metal ions as required for catalysis, although the two bound metal ions adopt different orientations with respect to the protein fold. Therefore, these results suggest a drifting or migration of the active site configuration on a conserved protein fold. Active site drifting may reflect an intrinsic feature of two-metal-ion catalytic nucleases, which may possess structural and functional plasticity that makes it more amenable for these enzymes to undergo such drifting, as the minimal requirement for catalysis would only include the ability to bind to two metal ions in a defined configuration to form the active site and the accessibility of the active site to the nucleic acid substrate.
Previous comparative structural studies of herpes simplex virus Type 1 terminase catalytic subunit pUL15 C-terminal nuclease domain showed an evolutionary lineage for the nuclease domains of virus terminases and RNase H-like nucleases, and the arrangements of the active site acidic residues in terminase nuclease domains exhibit high variability despite retaining a highly conserved protein fold core, i.e. locations of those active site residues on the protein folds differ remarkably (11). Such variability appears to frequently occur in viruses and phages (11), consistent with their fast mutation rates. The present structural analysis has provided further structural evidence to support active site drifting of virus terminase nucleases. Our structural studies show that BTP is capable of chelating the two metal ions in the nuclease active site, thus blocking catalysis, which conceptually suggests that hydroxytropolones can be used to target terminase nuclease domains in human herpesviruses, a group of clinically important human pathogens. A benefit of hydroxytropolones is that they target an essential chemical feature in two-metal-ion catalysis. Nevertheless, given the active site plasticity of those terminase nucleases, it would be interesting to assess if additional active site drifting arises in response to hydroxytropolones, in which case contiguous redesign of the compounds will be needed.
ACCESSION NUMBERS
The coordinates of the structures and reflection data for gp2C, gp2C–K428A:Mg2+, gp2C–K428A:Mn2+, gp2C–K428A: Mn2+:BTP and gp2C–K428A:Ca2+ have been deposited with the RCSB PDB with the entry codes of 5C10, 5C12, 5C15, 5C2F and 5C2D, respectively.
Supplementary Material
Acknowledgments
We thank the staff at the National Synchrotron Light Source for assistance in X-ray data collection of Se-Met gp2C, and the staff at the Advanced Photon Source and the Stanford Synchrotron Radiation Lightsource for beamline access for data collection of gp2C native crystals and soaked with metal ions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of General Medical Sciences of the National Institutes of Health [R01GM090010 to L.T.]; Intramural Research Program of the National Cancer Institute, National Institutes of Health, Department of Health and Human Services [to S.Le.G and J.A.B]; National Institutes of Health [SC2GM099596 to R.P.M.]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1.Baines J.D. Herpes simplex virus capsid assembly and DNA packaging: a present and future antiviral drug target. Trends Microbiol. 2011;19:606–613. doi: 10.1016/j.tim.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Fokine A., Rossmann M.G. Molecular architecture of tailed double-stranded DNA phages. Bacteriophage. 2014;4:e28281. doi: 10.4161/bact.28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casjens S.R. The DNA-packaging nanomotor of tailed bacteriophages. Nat. Rev. Microbiol. 2011;9:647–657. doi: 10.1038/nrmicro2632. [DOI] [PubMed] [Google Scholar]
- 4.Zhao H., Christensen T.E., Kamau Y.N., Tang L. Structures of the phage Sf6 large terminase provide new insights into DNA translocation and cleavage. Proc. Natl. Acad. Sci. U.S.A. 2013;110:8075–8080. doi: 10.1073/pnas.1301133110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun S., Kondabagil K., Draper B., Alam T.I., Bowman V.D., Zhang Z., Hegde S., Fokine A., Rossmann M.G., Rao V.B. The structure of the phage T4 DNA packaging motor suggests a mechanism dependent on electrostatic forces. Cell. 2008;135:1251–1262. doi: 10.1016/j.cell.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 6.Ghosh-Kumar M., Alam T.I., Draper B., Stack J.D., Rao V.B. Regulation by interdomain communication of a headful packaging nuclease from bacteriophage T4. Nucleic Acids Res. 2011;39:2742–2755. doi: 10.1093/nar/gkq1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smits C., Chechik M., Kovalevskiy O.V., Shevtsov M.B., Foster A.W., Alonso J.C., Antson A.A. Structural basis for the nuclease activity of a bacteriophage large terminase. EMBO Rep. 2009;10:592–598. doi: 10.1038/embor.2009.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cornilleau C., Atmane N., Jacquet E., Smits C., Alonso J.C., Tavares P., Oliveira L. The nuclease domain of the SPP1 packaging motor coordinates DNA cleavage and encapsidation. Nucleic Acids Res. 2013;41:340–354. doi: 10.1093/nar/gks974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Catalano C.E. The terminase enzyme from bacteriophage lambda: a DNA-packaging machine. Cell Mol. Life. Sci. 2000;57:128–148. doi: 10.1007/s000180050503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Black L.W. DNA packaging in dsDNA bacteriophages. Annu. Rev. Microbiol. 1989;43:267–292. doi: 10.1146/annurev.mi.43.100189.001411. [DOI] [PubMed] [Google Scholar]
- 11.Selvarajan Sigamani S., Zhao H., Kamau Y.N., Baines J.D., Tang L. The structure of the herpes simplex virus DNA-packaging terminase pUL15 nuclease domain suggests an evolutionary lineage among eukaryotic and prokaryotic viruses. J. Virol. 2013;87:7140–7148. doi: 10.1128/JVI.00311-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nadal M., Mas P.J., Blanco A.G., Arnan C., Sola M., Hart D.J., Coll M. Structure and inhibition of herpesvirus DNA packaging terminase nuclease domain. Proc. Natl. Acad. Sci. U.S.A. 2010;107:16078–16083. doi: 10.1073/pnas.1007144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roy A., Cingolani G. Structure of p22 headful packaging nuclease. J. Biol. Chem. 2012;287:28196–28205. doi: 10.1074/jbc.M112.349894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang W. Nucleases: diversity of structure, function and mechanism. Q. Rev. Biophys. 2011;44:1–93. doi: 10.1017/S0033583510000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steitz T.A., Steitz J.A. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl. Acad. Sci. U.S.A. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang W., Lee J.Y., Nowotny M. Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol. Cell. 2006;22:5–13. doi: 10.1016/j.molcel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 17.Nowotny M., Gaidamakov S.A., Crouch R.J., Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005;121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 18.Nowotny M., Gaidamakov S.A., Ghirlando R., Cerritelli S.M., Crouch R.J., Yang W. Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription. Mol. Cell. 2007;28:264–276. doi: 10.1016/j.molcel.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 19.Nowotny M., Yang W. Stepwise analyses of metal ions in RNase H catalysis from substrate destabilization to product release. EMBO J. 2006;25:1924–1933. doi: 10.1038/sj.emboj.7601076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang W. An equivalent metal ion in one- and two-metal-ion catalysis. Nat. Struct. Mol. Biol. 2008;15:1228–1231. doi: 10.1038/nsmb.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Batra V.K., Beard W.A., Shock D.D., Krahn J.M., Pedersen L.C., Wilson S.H. Magnesium-induced assembly of a complete DNA polymerase catalytic complex. Structure. 2006;14:757–766. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Casjens S., Winn-Stapley D.A., Gilcrease E.B., Morona R., Kuhlewein C., Chua J.E., Manning P.A., Inwood W., Clark A.J. The chromosome of Shigella flexneri bacteriophage Sf6: complete nucleotide sequence, genetic mosaicism, and DNA packaging. J. Mol. Biol. 2004;339:379–394. doi: 10.1016/j.jmb.2004.03.068. [DOI] [PubMed] [Google Scholar]
- 23.Gemski P. Jr, Koeltzow D.E., Formal S.B. Phage conversion of Shigella flexneri group antigens. Infect. Immun. 1975;11:685–691. doi: 10.1128/iai.11.4.685-691.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindberg A.A., Wollin R., Gemski P., Wohlhieter J.A. Interaction between bacteriophage Sf6 and Shigella flexner. J. Virol. 1978;27:38–44. doi: 10.1128/jvi.27.1.38-44.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao H., Kamau Y.N., Christensen T.E., Tang L. Structural and functional studies of the phage Sf6 terminase small subunit reveal a DNA-spooling device facilitated by structural plasticity. J. Mol. Biol. 2012;423:413–426. doi: 10.1016/j.jmb.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao H., Finch C.J., Sequeira R.D., Johnson B.A., Johnson J.E., Casjens S.R., Tang L. Crystal structure of the DNA-recognition component of the bacterial virus Sf6 genome-packaging machine. Proc. Natl. Acad. Sci. U.S.A. 2010;107:1971–1976. doi: 10.1073/pnas.0908569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Budihas S.R., Gorshkova I., Gaidamakov S., Wamiru A., Bona M.K., Parniak M.A., Crouch R.J., McMahon J.B., Beutler J.A., Le Grice S.F. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones. Nucleic Acids Res. 2005;33:1249–1256. doi: 10.1093/nar/gki268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otwinowski Z., Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 29.Terwillinger T.C., Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 1999;D55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Groftehauge M.K., Hajizadeh N.R., Swann M.J., Pohl E. Protein-ligand interactions investigated by thermal shift assays (TSA) and dual polarization interferometry (DPI) Acta Crystallogr. D Biol. Crystallogr. 2015;71:36–44. doi: 10.1107/S1399004714016617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindahl P.A. Metal-metal bonds in biology. J. Inorg. Biochem. 2012;106:172–178. doi: 10.1016/j.jinorgbio.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rees D.C. Great metalloclusters in enzymology. Annu. Revi. Biochem. 2002;71:221–246. doi: 10.1146/annurev.biochem.71.110601.135406. [DOI] [PubMed] [Google Scholar]
- 35.Meck C., D'Erasmo M.P., Hirsch D.R., Murelli R.P. The biology and synthesis of alpha-hydroxytropolones. MedChemComm. 2014;5:842–852. doi: 10.1039/C4MD00055B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chung S., Himmel D.M., Jiang J.K., Wojtak K., Bauman J.D., Rausch J.W., Wilson J.A., Beutler J.A., Thomas C.J., Arnold E., et al. Synthesis, activity, and structural analysis of novel alpha-hydroxytropolone inhibitors of human immunodeficiency virus reverse transcriptase-associated ribonuclease H. J. Med. Chem. 2011;54:4462–4473. doi: 10.1021/jm2000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Himmel D.M., Maegley K.A., Pauly T.A., Bauman J.D., Das K., Dharia C., Clark A.D. Jr, Ryan K., Hickey M.J., Love R.A., et al. Structure of HIV-1 reverse transcriptase with the inhibitor beta-Thujaplicinol bound at the RNase H active site. Structure. 2009;17:1625–1635. doi: 10.1016/j.str.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beilhartz G.L., Wendeler M., Baichoo N., Rausch J., Le Grice S., Gotte M. HIV-1 reverse transcriptase can simultaneously engage its DNA/RNA substrate at both DNA polymerase and RNase H active sites: implications for RNase H inhibition. J. Mol. Biol. 2009;388:462–474. doi: 10.1016/j.jmb.2009.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Didierjean J., Isel C., Querre F., Mouscadet J.F., Aubertin A.M., Valnot J.Y., Piettre S.R., Marquet R. Inhibition of human immunodeficiency virus type 1 reverse transcriptase, RNase H, and integrase activities by hydroxytropolones. Antimicrob. Agents Chemother. 2005;49:4884–4894. doi: 10.1128/AAC.49.12.4884-4894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tavis J.E., Wang H., Tollefson A.E., Ying B., Korom M., Cheng X., Cao F., Davis K.L., Wold W.S., Morrison L.A. Inhibitors of nucleotidyltransferase superfamily enzymes suppress herpes simplex virus replication. Antimicrob. Agents Chemother. 2014;58:7451–7461. doi: 10.1128/AAC.03875-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu G., Lomonosova E., Cheng X., Moran E.A., Meyers M.J., Le Grice S.F., Thomas C.J., Jiang J.K., Meck C., Hirsch D.R., et al. Hydroxylated tropolones inhibit hepatitis B virus replication by blocking viral ribonuclease h activity. Antimicrob. Agents Chemother. 2015;59:1070–1079. doi: 10.1128/AAC.04617-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu Y., Cheng X., Cao F., Huang A., Tavis J.E. beta-Thujaplicinol inhibits hepatitis B virus replication by blocking the viral ribonuclease H activity. Antiviral Res. 2013;99:221–229. doi: 10.1016/j.antiviral.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 43.Mashima K., Shimoyama Y., Kusumi Y., Fukumoto A., Yamagata T., Ohashi M. Formation of a dative bond between Pt-0 and Mo-II in linear Pt-0-Mo-II-Mo-II-Pt-0 complexes, (Mo2Pt20)-Pt-II(pyphos)(4)(PR3)(2), and unique 1,4-oxidative addition reaction of diaryl disulfides giving (Mo2Pt2I)-Pt-II(pyphos)(4)(SAr)(2) (pyphos = 6-diphenylphosphanyl-2-pyridonato) Eur. J. Inorg. Chem. 2007:235–238. [Google Scholar]
- 44.Greenwood B.P., Forman S.I., Rowe G.T., Chen C.H., Foxman B.M., Thomas C.M. Multielectron redox activity facilitated by metal-metal interactions in early/late heterobimetallics: Co/Zr complexes supported by phosphinoamide ligands. Inorg. Chem. 2009;48:6251–6260. doi: 10.1021/ic900552b. [DOI] [PubMed] [Google Scholar]
- 45.Thomas C.M., Napoline J.W., Rowe G.T., Foxman B.M. Oxidative addition across Zr/Co multiple bonds in early/late heterobimetallic complexes. Chem. Commun. 2010;46:5790–5792. doi: 10.1039/c0cc01272f. [DOI] [PubMed] [Google Scholar]
- 46.Garcin E., Vernede X., Hatchikian E.C., Volbeda A., Frey M., Fontecilla-Camps J.C. The crystal structure of a reduced [NiFeSe] hydrogenase provides an image of the activated catalytic center. Structure. 1999;7:557–566. doi: 10.1016/s0969-2126(99)80072-0. [DOI] [PubMed] [Google Scholar]
- 47.Jeoung J.H., Dobbek H. Structural basis of cyanide inhibition of Ni, Fe-containing carbon monoxide dehydrogenase. J. Am. Chem. Soc. 2009;131:9922–9923. doi: 10.1021/ja9046476. [DOI] [PubMed] [Google Scholar]
- 48.Zhang L.M., Morrison C.N., Kaiser J.T., Rees D.C. Nitrogenase MoFe protein from Clostridium pasteurianum at 1.08 A resolution: comparison with the Azotobacter vinelandii MoFe protein. Acta Crystallogr. D Biol. Crystallogr. 2015;71:274–282. doi: 10.1107/S1399004714025243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spatzal T., Perez K.A., Einsle O., Howard J.B., Rees D.C. Ligand binding to the FeMo-cofactor: structures of CO-bound and reactivated nitrogenase. Science. 2014;345:1620–1623. doi: 10.1126/science.1256679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Einsle O., Tezcan F.A., Andrade S.L., Schmid B., Yoshida M., Howard J.B., Rees D.C. Nitrogenase MoFe-protein at 1.16 A resolution: a central ligand in the FeMo-cofactor. Science. 2002;297:1696–1700. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 51.Suga M., Akita F., Hirata K., Ueno G., Murakami H., Nakajima Y., Shimizu T., Yamashita K., Yamamoto M., Ago H., et al. Native structure of photosystem II at 1.95 A resolution viewed by femtosecond X-ray pulses. Nature. 2015;517:99–103. doi: 10.1038/nature13991. [DOI] [PubMed] [Google Scholar]
- 52.Yin Y.W., Steitz T.A. The structural mechanism of translocation and helicase activity in T7 RNA polymerase. Cell. 2004;116:393–404. doi: 10.1016/s0092-8674(04)00120-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.