


Abstract
Many archaea and bacteria have an adaptive immune system known as CRISPR which allows them to recognize and destroy foreign nucleic acid that they have previously encountered. Two CRISPR-associated proteins, Cas1 and Cas2, are required for the acquisition step of adaptation, in which fragments of foreign DNA are incorporated into the host CRISPR locus. Cas1 genes have also been found scattered in several archaeal and bacterial genomes, unassociated with CRISPR loci or other cas proteins. Rather, they are flanked by nearly identical inverted repeats and enclosed within direct repeats, suggesting that these genetic regions might be mobile elements (‘casposons’). To investigate this possibility, we have characterized the in vitro activities of the putative Cas1 transposase (‘casposase’) from Aciduliprofundum boonei. The purified Cas1 casposase can integrate both short oligonucleotides with inverted repeat sequences and a 2.8 kb excised mini-casposon into target DNA. Casposon integration occurs without target specificity and generates 14–15 basepair target site duplications, consistent with those found in casposon host genomes. Thus, Cas1 casposases carry out similar biochemical reactions as the CRISPR Cas1-Cas2 complex but with opposite substrate specificities: casposases integrate specific sequences into random target sites, whereas CRISPR Cas1-Cas2 integrates essentially random sequences into a specific site in the CRISPR locus.
INTRODUCTION
Bacteria and archaea have evolved several mechanisms to protect against invading viruses and plasmids (1,2). For example, they can secure their perimeters by synthesizing capsules or by down-regulating or mutating the surface receptors that bacteriophages need for binding and entry. In addition, the well-known restriction-modification systems rely on enzyme pairs, one to modify ‘self’ DNA when it is synthesized—e.g. by methylation—and the other to degrade any DNA that has entered the cell but which is not appropriately modified. It has only recently become clear that many bacteria and archaea also have an adaptive immune system, the so-called CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-Cas system, in which they maintain a genetic record of nucleic acid they have previously encountered and which can be subsequently used to specifically target foreign genetic material for degradation (reviewed in 3–5).
At a CRISPR locus, identical palindromic repeats (generally 25–50 bp in length) alternate with ‘spacers’ that correspond to short stretches derived from foreign DNA. The CRISPR locus is then used as a template for the synthesis of a long RNA transcript which is subsequently processed into shorter RNA molecules, crRNAs, that serve as guides to target foreign nucleic acid for destruction. There are many different types of CRISPR systems, with different collections of Cas (‘CRISPR-associated’) proteins (6). Common to all active CRISPR systems are two proteins Cas1 and Cas2 that are required for the initial step of spacer acquisition (7–9). Both proteins are reported to be endonucleases (10–15), although the active site residues of Cas2 are not needed for acquisition in E. coli (9). It was recently demonstrated that the purified E. coli CRISPR Cas1–Cas2 complex can integrate protospacers into the E. coli CRISPR locus (16), indicating that the two proteins together constitute a DNA integrase, although only Cas1 is needed for the reverse reaction, disintegration (16,17). How protospacers are generated is not yet known.
Although the vast majority of cas1 genes identified in sequenced genomes are associated with CRISPR-Cas systems, phylogenetic studies revealed that there are two small families (Cas1-solo group 1 and group 2) that are not affiliated with CRISPR repeats or other cas genes (8). Rather, the Cas1-solo genes of group 2 are found in the proximity of several other non-cas genes including genes encoding a PolB family DNA polymerase and usually an HNH endonuclease (Figure 1A). These clusters of genes are located between short terminal inverted repeat (TIR) sequences (18), a hallmark of DNA transposons (19,20). This observation led to the intriguing suggestion (18) that these genetic neighborhoods are mobile genetic elements, designated ‘casposons’, in which the TIRs delineate a region of DNA that can be mobilized from one location and integrated into another. It was further proposed (18) that the casposon-encoded Cas1 protein is the DNA transposase (or ‘casposase’).
Figure 1.

Features of casposon-encoded Cas1 family 2 casposons. (A) The 9075 bp Aciduliprofundum boonei casposon encodes six predicted proteins annotated by NCBI and (18) as: a DNA polymerase type B (red; bp 380 359–383 058); an HNH endonuclease (green; bp 383 051–383 485); a hypothetical protein (beige; bp 383 492–384 064) containing an HTH domain; a transcriptional regulator containing an HTH domain (orange; bp 384 136–385 170); a Cas1 protein (blue; bp 385 171–386 385); and an N6-methyltransferase (yellow; bp 386 408–389 311). The bp numbering is that of the complete genome of Aciduliprofundum boonei T469 (Accession number NC_013926.1). (B) DNA sequence alignment of the TSD-TIR junctions for seven members of casposon-encoded Cas1 family 2. The abbreviations are: MetFor, Methanoregula formicicum SMSP (Accession number NC_019943); MetMaz, Methanosarcina mazei Goe1 (NC_003901); MetMah, Methanohalophilus mahii DSM 5219 (NC_014002); MetTin, Methanolobus tindarius DSM 2278 (NZ_AZAJ01000001); MetLum, Methanomassiliicoccus luminyensis B10 (NZ_CAJE01000015); AciBoo, Aciduliprofundum boonei T469 (NC_013926.1); MetArv, Methanocella arvoryzae MRE50 (NC_009464). Only the top strand sequence is shown. The conserved G/C motif at each end is boxed in grey, the target site duplications (TSDs) are shown in green, and notable sequence patterns are highlighted in red or blue. Bases shown in bold are those that differ between the LE and RE TIRs of a given casposon. The perfect palindrome in the terminal 20 bp of the AciBoo RE is underlined (there is a single bp difference compared to the LE). (C) Amino acid sequence alignment (using T-Coffee) of the Cas1 C-terminal helix-turn-helix (HTH) domains of seven members of the casposon-encoded Cas1 family 2. Only six residues are completely conserved within this alignment (boxed in grey, asterisk below the alignment), yet there are notable regions of identity between subsets of proteins, highlighted in red or blue. The double dots below the alignment represent conservative mutations; single dots are semi-conservative mutations. The α-helices of the HTH domain were predicted using Jpred4 (49). The number at the end of each sequence indicates the number of amino acids in each protein.
The phylogenetic relationship between CRISPR-associated and casposon-encoded cas1 genes has suggested that casposons may be the evolutionary ancestors of CRISPR-Cas systems (18,21). If so, this would elegantly parallel the development of the V(D)J recombination system of jawed vertebrates that originated from an ancient Transib DNA transposon (22,23), and would suggest that mobile genetic elements are ancestors of adaptive immune systems across the three domains of life. However, to date, there is no experimental evidence that casposons are active mobile elements or that their Cas1 proteins possess any catalytic activity. Thus, to investigate the possibility that casposons are mobilizable by casposases, we have studied the in vitro properties of one representative example, and here show that the casposon-encoded Cas1 protein from the archaeal thermoacidophile Aciduliprofundum boonei is an active DNA integrase that acts specifically on its casposon ends.
MATERIALS AND METHODS
DNA and plasmids
Oligonucleotides were either purchased from IDT (Coralville, IA) or synthesized by the NIH Facility for Biotechnology Resources, Center for Biologics Evaluation and Research (FBR, CBER). Synthetic genes codon-optimized for expression in E. coli for casposon-encoded Aciduliprofundum boonei Cas1 (ABOO_RS01975) and HNH nuclease (ABOO_RS01960) were purchased from Bio Basic Inc. (Markham, Ontario, Canada). The gene encoding casposon-encoded Ab Cas1 was cloned into a modified pBAD vector which contained the thioredoxin (Trx) tag and multiple cloning site of a modified pET-32b plasmid (gift of D. Ronning). The resulting plasmid encodes Ab Cas1 as an N-terminally-tagged Trx fusion protein with a C-terminal histidine tag, both of which are cleavable with TEV protease. The gene encoding the Ab HNH nuclease was cloned into a similar modified pBAD vector such that the resulting fusion protein had an N-terminal Trx tag and a linker containing a 6xhistidine tag followed by a thrombin cleavage site. The Cas1 active site mutant H242A was introduced by PCR mutagenesis (24), and all constructs were fully sequenced.
A plasmid (designated pAbLE30RE30) containing a 2840 bp mini-Ab casposon enclosing a kanamycin-resistance gene was generated using PCR mutagenesis to insert the bps corresponding to the terminal 30 bp of the Ab casposon Left End (LE) and Right End (RE) immediately adjacent to the Eag1 and XhoI sites, respectively, of pHL2577 (25). PCR mutagenesis using pAbLE30RE30 as a template was then carried out to introduce two different 15 bp direct repeats (DR1, DR2) flanking the mini-casposon (pAbLE30RE30DR1 with the direct repeat observed in A. boonei: 5′-CCCCACTACGAGGAG; and pAbLE30RE30DR2, corresponding to 5′-ACGGTCACAGCTTGT). PCR mutagenesis was used to subsequently delete 15 bp of casposon TIR sequence to leave only 15 bp TIRs on each end, generating pAbLE15RE15DR1 and pAbLE15RE15DR2.
Protein expression and purification
Ab Cas1 was expressed in Top10 cells by growth at 42°C until OD600nm 0.4–0.6, at which point the cells were cooled to 19°C, arabinose added to a final concentration of 0.012% (w/v), followed by growth overnight. Cells were harvested, resuspended in 25 mM Tris pH 7.5, and stored at -80°C until use. Upon thawing, cells were lysed by sonication in buffer containing 25 mM Tris pH 7.5, 0.5 M NaCl, 5 mM imidazole (Im), 0.4 mM MgCl2, and 1 mg DNase/100 ml. Following centrifugation at 55,000 x g, the soluble material was loaded onto two 5 ml HiTrap chelating columns (GE Healthcare) connected in tandem which had been preloaded with NiCl2. The columns were washed with Buffer A (20 mM Tris pH 7.5, 0.5 M NaCl, 70 mM Im), followed by a linear gradient over 30 column volumes from Buffer A to Buffer A containing 0.4 M Im and 10% (w/v) glycerol. Fractions containing full-length Ab Cas1 were dialyzed at 4°C against buffer containing 25 mM Tris pH 7.5, 0.5 M NaCl, 10% (w/v) glycerol, and 2 mM dithiothreitol (DTT) in the presence of purified TEV protease. Once cleavage was complete as monitored by SDS-PAGE, the protein was concentrated and subjected to size exclusion chromatography on a Superdex 200 16/60 column (GE Healthcare), where it eluted at the position of a dimer (data not shown). Fractions containing Ab Cas1 were concentrated to ∼4 mg/ml, dialyzed against 25 mM Tris pH 7.5, 0.5 M NaCl, 15% (w/v) glycerol and stored at −80°C until use. An active site mutant, Ab Cas1H242A, was expressed and purified in the identical manner. Ab HNH nuclease was expressed, purified, and stored as described above except that cleavage was with thrombin (Sigma; 300U per protein from one liter harvested cells) and the storage buffer contained 0.2 mM TCEP. All purified proteins were at least 95% pure as judged by SDS-PAGE analysis (data not shown).
In vitro DNA strand transfer assay
Various oligonucleotide substrates were used to assay the ability of Ab Cas1 to catalyze insertion into a pUC19 plasmid substrate. Standard assay conditions consisted of 75 nM Ab Cas1, 200 nM oligonucleotide substrate, and 150 ng pUC19 incubated in 25 mM Tris pH 7.5, 150 mM KCl, 50 μg/ml BSA, 5 mM MnCl2 for 1 h at 37°C unless otherwise stated in the figure legends. Reactions were quenched by the addition of EDTA to a final concentration of 25 mM, followed by proteinase K treatment (NEB; 40 U per reaction tube) for 30 min at 37°C. After addition of glycogen, reaction products were ethanol-precipitated and the tubes air-dried. Reaction products were run on a 1.5% agarose gel in 1X TAE at 100V for 70 min, and visualized using ethidium bromide. For the 6-FAM-labelled LE26 substrate, reaction products were visualized using a GE Typhoon FLA 9500 imager.
In vitro mini-transposon integration assay
To examine the ability of Ab Cas1 to catalyze the insertion of an excised Ab casposon into target DNA in vitro, a blunt-ended 2840 bp LE30/RE30 mini-casposon and a similarly blunt-ended 2810 bp LE15/RE15 mini-casposon were generated by PCR using DNA primers representing the casposon ends and pAbLE30RE30 or pAbLE15RE15, respectively, as the template (for sequences of all primers used, see Table 1). To generate a mini-casposon with flanking sequence, primers were chosen such that their use in a PCR reaction with pAbLE30RE30 as the template resulted in 106 bp of flanking DNA on the LE and 72 bp of flanking DNA on the RE. The mini-casposons were separated from the template plasmids by agarose gel electrophoresis, and resuspended in 10 mM Tris pH 8.0 after QiaQuick gel extraction (Qiagen). Integration reactions (100 μl) were carried out by incubating approximately equimolar amounts of each mini-casposon (estimated from the DNA band intensity on a 1% agarose gel after resuspension in 10 mM Tris) with 150 ng of the pUC19 target plasmid and 75 nM Ab Cas1 in the strand transfer assay buffer indicated above overnight at 37°C. After proteinase K digestion, the DNA products were resuspended in 8 μl 10 mM Tris pH 8.0, and 2 μl used to electroporate 20 μl ElectroMAX E. coli DH10B cells (Invitrogen). After 1 h growth at 37°C in SOC medium, 0.4 ml was plated onto AmpR+KanR LB plates overnight at 37°C, and the number of colonies counted the following day. To analyze the plasmid products by restriction analysis, digests were performed under standard conditions and the products run on a 1.0% agarose gel in 1X TAE and visualized by ethidium bromide staining.
Table 1. Sequences of oligonucleotides used.
| Name | Sequence (5′-3′ of top strand if duplex) |
|---|---|
| LE30 | GGGGATATATATACATCCCCTCTTAAGTTC |
| ran30 | TAGCCAGCGAGCGAGCGTAGCAGACTCCAT |
| LE26 | GGGGATATATATACATCCCCTCTTAA |
| LE26(A/T) | TGGGATATATATACATCCCCTCTTAA |
| LE21 | GGGGATATATATACATCCCCT |
| LE15 | GGGGATATATATACAT |
| ran26 | TAGCCAGCGAGCGAGCGTAGCAGACT |
| ran26(G/C) | GAGCCAGCGAGCGAGCGTAGCAGACT |
| ran21 | TAGCCAGCGAGCGAGCGTACG |
| ran15 | TAGCCAGCGAGCGAG |
| LE30 + 4fl | GGAGGGGGATATATATACATCCCCTCTTAAGTTC |
| LE30 + 15fl | CCCCACTACGAGGAGGGGGATATATATACATCCCCTCTTAAGTTC |
| LE34 | GGGGATATATATACATCCCCTCTTAAGTTCCCTT |
| LE45 | GGGGATATATATACATCCCCTCTTAAGTTCCCTTTTCACATCACT |
| Primer 1 | CCATGATTACGCCAAGCTCGG |
| Primer 2 (for LE30) | GGGGATATATATACATCCCCTCTTAA |
| Primer 2 (for LE15) | GGGGATATATATACACAGAGAACAACAAC |
| Primer 3 (for RE30) | GGGGATATATATATATCCCCTCTTAA |
| Primer 3 (for RE15) | GGGGATATATATATACAGAGAACTTCAGC |
| Primer 4 | GGGTAACGCCAGGGTTTTCC |
RESULTS
Casposon-encoded Cas1 family 2 members have distinctive terminal inverted repeats
Although the number of casposons identified to date is small, there appear to be three distinct clades (designated families 1–3) (18). When we examined the sequences at or near casposon ends corresponding to members of the largest family 2, we noted a conserved motif of three G/C bps at each end (26). For seven members of the family, this motif is precisely at the junctions with flanking sequences that are direct duplications at each end, and we used these features as guides to manually align casposon ends as shown in Figure 1B. The resulting alignment at both ends suggests that the G/C motif (boxed in grey) is at the tip of the casposon TIRs, and the flanking direct duplications - presumed to be target site duplications (TSDs) (18) - are all between 13–15 bp in length. Furthermore, the subterminal regions of the ∼40 bp TIRs have patterns of conserved bases indicating a bifurcation of family 2 members into those with distinguishing stretches of A/T bps (red, top sequences) or C/G and T/A bps (blue, bottom sequences) within their TIRs.
The same bifurcation is observed in the corresponding amino acid sequences of the C-terminal region of casposon-encoded Cas1 family 2 proteins (Figure 1C), predicted to contain a helix-turn-helix (HTH) domain (18). This apparent division of Cas1 casposons into two distinct sub-types is in accord with the phylogenetic tree of Cas1 proteins, in which those from Aciduliprofundum boonei and Methanocella arvoryzae (the two bottom entries in Figure 1B and C) form a distinct branch (18). The identification and correspondence of two sub-types of TIRs and two sub-types of HTH domains supports the notion that the casposon-encoded Cas1 proteins, at least from family 2, might be active transposases in which the HTH domains play a role in specific recognition of casposon TIRs.
Aciduliprofundum boonei T469 casposon-encoded Cas1 is a DNA integrase
To test the hypothesis that Cas1 family 2 proteins are active DNA transposases, we attempted to express and purify several family members as recombinant fusion proteins expressed in E. coli. Among these, the Cas1 protein from Aciduliprofundum boonei T469 (‘Ab Cas1’) proved to be readily purifiable. The casposon identified in A. boonei contains six ORFs (Figure 1A) and, according to the alignment in Figure 1B, has 36-bp long TIRs that differ by only 1 bp at each end. This assignment of the TIR sequences differs from that of Krupovic et al. (2014) who concluded that the TIRs are 40-bp long, extended by 4 bps into what we identify as the TSDs. The terminal 20 bps of the A. boonei TIRs consist of a palindromic sequence, a property previously noted for several members of the Cas-1 solo family (18). Ab Cas1 shares only 13% amino acid identity with E. coli Cas1 but, like other casposon-encoded Cas1 family 2 members (18), has all of the important active site residues previously identified for CRISPR-associated Cas1 proteins (Figure 2).
Figure 2.

Predicted secondary structure elements of A. boonei Cas1 and alignment with three structurally characterized CRISPR-Cas1 proteins. The β-strands (red arrows) and α-helices (blue cylinders) for Ab Cas1 were predicted using JPred4 (49). The structure-based alignment of Cas1 proteins from E. coli (PDB ID 4P6I), P. aeruginosa (3GOD), and A. fulgidus (4N06) does not differ substantively from those previously reported (9,12,18). The grey secondary structure elements shown below the alignment are those of A. fulgidus. Strictly conserved residues in this alignment are highlighted in grey, and active site residues are boxed. Active site residue numbers are those of Ab Cas1.
We used short dsDNA oligonucleotide substrates to test the ability of Ab Cas1 to integrate casposon TIR ends into target DNA (Figure 3A). In the presence of 150 mM KCl and at 37°C, Ab Cas1 readily integrates a dsDNA 30-mer oligonucleotide representing the Ab casposon LE TIR into a supercoiled pUC19 plasmid substrate, producing products consistent with single-end and double-end insertion events (Figure 3A, lane 5). The reaction requires a divalent metal ion cofactor, and is much more efficient in the presence of 5 mM Mn2+ than 1–10 mM Mg2+ (compare lanes 1–5). Higher activity in Mn2+ than in Mg2+ is consistent with the previously reported activity of the CRISPR Cas1 protein from Pseudomonas aeruginosa (10). Formation of the products requires the oligonucleotide (lane 6), indicating that they are not simply the consequence of Mn2+-catalyzed plasmid nicking. Furthermore, products were not observed when an active site mutant, Cas1H242A (see alignment in Figure 2), was used in place of the wild-type protein (lane 7). As would be expected for a DNA transposase but not for a CRISPR-associated Cas1 protein, DNA integration of oligonucleotide substrates is TIR-sequence specific, as indicated by the lack of activity when an oligonucleotide of random sequence but identical length was used (lane 8).
Figure 3.

Characterization of the in vitro strand transfer activity of A. boonei Cas1. (A) Strand transfer assay using 30-mer oligonucleotides representing the LE TIR (LE30) or a random sequence (ran30). Insertion of a single-end (SE) results in a relaxed target plasmid, whereas double-end insertion (DE) results in a linearized plasmid. Standard reaction conditions were used with the variations as indicated. In all figures, ‘M’ refers to lanes containing DNA markers, ‘sc’ indicates supercoiled pUC19. (B) Strand transfer assay using a fluorescently-labelled oligonucleotide to confirm integration. Oligonucleotide used: Lane1 = none; Lane2 = ran30. Lane3: 6-FAM-LE26. The reactions were run in duplicate and the samples loaded onto the same 1.5% agarose gel which was cut in half for analysis by ethidium bromide staining (left) or fluorescence (right). (C) Strand transfer assay with modified LE substrates (sequences are listed in Table 1). LE26, LE21, LE15 represent the length of the LE TIR oligonucleotide used, and ran26, ran21, ran15 are length-matched random oligonucleotides. (D) Time course of SE and DE product formation using a LE30 oligonucleotide substrate.
Several CRISPR-associated Cas1 proteins have been demonstrated to cleave DNA and RNA substrates (10–12). To rule out the possibility that the products observed in the integration assay are due to cleavage of the plasmid substrate rather than oligonucleotide integration, we repeated the assay using a 26-mer oligonucleotide with a 6-FAM label on nt26 of the 5′ end of the ‘bottom’ strand (indicated by asterisk, Figure 3B). Similar amounts of products were obtained as with the unlabelled oligonucleotide substrate, and the label was incorporated into both the relaxed and linearized plasmid products, indicating that these are indeed single-end and double-end integration products, respectively.
To investigate the TIR sequence requirements for DNA oligonucleotide integration, oligonucleotides of varying TIR length were assayed for integration activity. As shown in Figure 3C, TIRs as short as 15 bp were readily integrated whereas length-matched oligonucleotides with a sequence unrelated to the TIRs were not. This indicates that the palindromic sequence located within the TIRs is not required, and further confirmed that integration is TIR-specific. We also repeated the assay using oligonucleotides in which either the 3′-OH of nt26 of the top strand was replaced by a phosphate group (Figure 3C, ‘LE26_Pi’, lane 9) or the 3′-OH group of nt1 of the bottom strand was replaced with a dideoxy nt (Figure 3C, ‘LE26_ddC’, lane 10). As the latter substitution led to loss of integration activity while blocking the 3′-OH on the top strand had no apparent effect, these results collectively indicate that TIR integration proceeds specifically using the 3′-OH of the bottom strand as the nucleophile.
As the G/C motif at the TIR tip was an important guide in the initial identification of suitable substrates for integration, we wondered if the terminal basepair was important. However, when we replaced the terminal G/C basepair of the TIR with A/T, integration proceeded with wild-type levels of activity (Figure 3C, lane 2). Conversely, appending a G/C bp to an oligonucleotide with unrelated sequence did not confer the ability to be integrated (lane 6). This lack of sensitivity to the exact sequence at the tip of the TIR is consistent with a mode of DNA recognition in which sub-terminal sequences are more important than those at the end, as has been reported for several DNA transposons (27–31).
To investigate the role of target DNA supercoiling in oligonucleotide integration, we performed an integration time course (Figure 3D). When the reaction was permitted to proceed for 18 h at 37°C, a smear of products resulted, consistent with repeated integration of the oligonucleotide substrate into the target plasmid. Thus, integration does not require a supercoiled target plasmid, as every integration event into the plasmid after the initial insertion is into either relaxed or linear DNA, in contrast to the in vitro requirements of E. coli CRISPR Cas1-Cas2 integration (16).
Ab Cas1 catalyzes double-ended integration of a mini-transposon and generates 14–15 bp target site duplications
A target site duplication (TSD) flanking each end of the mobile element is a hallmark of many DNA transposons. These are generated when the mechanism of transposition involves the spatially coordinated insertion of the two transposon ends into opposite strands of the target DNA; the number of bps by which the two insertion sites are offset is identical to the length of the TSDs (20). The number of bps of the TSD is generally a conserved feature of a given transposition system as it is a consequence of the fixed distance between two transposase protomer active sites within the transpososome. To determine if Ab Cas1 generates TSDs upon integration, we created two plasmids in which either 15 bp or 30 bp of the Ab casposon LE and RE sequences flanked a kanamycin resistance gene and were, in turn, enclosed within direct repeats (Figure 4A). Using appropriate primers, we then amplified PCR products corresponding to either a precisely excised LE15/RE15 or LE30/RE30 mini-casposon with blunt ends (2810 and 2840 bp long, respectively), or a LE30/RE30 ‘mini-casposon’ with 106 and 72 bp of flanking sequence on each side, respectively. After agarose gel purification, these mini-casposons (e.g. see Figure 4C, lane 3) were then used as substrates in in vitro integration reactions. The reaction products were electroporated into E. coli, and after overnight growth, plasmids were isolated from single colonies that exhibited combined AmpR + KanR resistance, and their transposon-DNA junctions sequenced.
Figure 4.

In vitro integration of mini-casposons by A. boonei Cas1. (A) Schematic representation of plasmids used as templates for the PCR amplification of mini-casposon substrates. The sequences for Primers 1–4 are listed in Table 1. LE: Left End, RE: Right End, DR: Direct repeat. The restriction site for PmeI is indicated. (B) Number of AmpR + KanR colonies recovered when different mini-casposons were used as a substrate for integration into pUC19. For the mini-casposon with flank present, the results for DR2 are shown. Standard assay conditions were used except the reactions were overnight at 37°C. Reactions were performed in triplicate (indicated in different shades of blue), and the number of colonies for ‘with flank’ were 0, 3 and 2; for ‘no substrate’ were 0, 0 and 0. (C) Restriction digest analysis of plasmid products. One representative plasmid is shown for the LE30/RE30 (Lane 5) and LE15/RE15 (Lane 6) reaction. The mini-casposon (Lane 3; LE15/RE15) is cut by PmeI at pHL2777 nt2753 nt (Lane 2; products marked with arrows on left side) but not by XmnI (Lane 1). pUC19 (Lane 4) is cut by XmnI (Lane 7) but not PmeI (Lane 8). The products of the integration reaction (Lanes 5,6) are cut by both enzymes (Lanes 9–12) generating linearized products of ∼5500 kb. (D) Location of integration sites into pUC19. Direction of the arrows indicate the direction of insertion of the terminal C 3′-OH of the LE bottom strand into the plus strand or the minus strand. The arrow marked with the red asterisk indicates the integration event from which the TSD sequence was subsequently used as DR2. (E) Weblogo (50) for the experimentally obtained TSD sequences. Insertions into the minus strand were reverse complemented before being included in the alignment. Data for the LE30/RE30 and LE15/RE15 mini-casposons were combined.
As shown in Figure 4B, Ab Cas1 integrates pre-excised mini-casposons with either 15 bp and 30 bp ends into a pUC19 target plasmid. Integration was confirmed (Figure 4C) by digesting the resulting plasmids (a representative example of the products obtained for each mini-casposon substrate is shown in lanes 5 and 6, respectively) with XmnI (which cuts pUC19 but does not cut the mini-casposons) and PmeI (which cuts within the mini-casposon but not pUC19). The plasmid products of the integration reactions can be cut with both enzymes yielding a ∼5500 bp linear product (lanes 9–12), indicating the integration of either a 2810 or 2840 bp mini-casposon into pUC19 (2685 bp). Thus, consistent with assays performed using short oligonucleotide substrates, the important elements of protein–DNA recognition lie within the final 15 bp of the TIRs and the palindromic sequences within the TIRs do not play an important role in strand transfer. When the insertion sites were mapped onto the sequence of pUC19 (Figure 4D), the insertion sites were scattered throughout with no evident hot-spots. At the current level of coverage, we have not recovered the same insertion site more than once.
Sequencing of integration sites revealed that insertion generated target site duplications (TSDs) of either 14 or 15 bp (Supplementary Table S1). Sequence logos generated from the TSD sequences and the bps preceding and following each within pUC19 are essentially featureless, and there is no obvious pattern to the nucleotide frequency at each position (Figure 4E). Thus, to a first approximation, integration occurs randomly into target DNA.
To see if Ab Cas1 can cleave dsDNA to generate the appropriately processed ends for integration, we performed the in vitro integration reaction using a LE30/RE30 mini-casposon flanked by plasmid sequence. The frequency of recovery of AmpR + KanR resistant colonies was at least 50X lower than with pre-processed ends (Figure 4B), and sequencing of five recovered plasmids revealed that these all contained a direct insertion of the unprocessed mini-casposon, albeit with 14–15 bp TSDs. To determine if some particular feature of the flanking direct repeat sequence observed in A. boonei (i.e. DR1) was responsible for the lack of processing at the casposon ends, we repeated the integration reaction with a direct repeat sequence corresponding to a TSD obtained experimentally from integration of the pre-excised LE30/RE30 casposon into pUC19 (marked with red asterisk in Figure 4D); the results were unchanged. Thus, Ab Cas1 can integrate non-specific DNA with authentic spacing into target DNA but with a substantially lower efficiency relative to DNA that corresponds to its casposon ends.
As there is a second nuclease-encoding gene contained within the A. boonei casposon gene cluster, we investigated the possibility that the A. boonei casposon HNH nuclease might be an active endonuclease responsible for generating free casposon ends, either on its own or in collaboration with Ab Cas1. After expression and purification of Ab HNH nuclease, we repeated the in vitro integration reaction with the unprocessed mini-casposon with Ab Cas1 and the Ab HNH nuclease in equimolar amounts, both at 37°C and at 60°C and for varying reaction times. However, the four recovered AmpR + KanR colonies that we were able to sequence showed no indication of casposon end cleavage or integration, only plasmid rearrangements and religation in other regions, most likely initiated by a non-specific HNH nuclease activity (32).
Ab Cas1 does not cleave TIRs with flanking sequence
As we did not detect coupled cleavage and strand transfer on mini-casposon substrates, we attempted to detect evidence for dsDNA cleavage using oligonucleotide substrates in which flanking DNA corresponding to the A. boonei direct repeat (DR1) was appended to the TIRs. As shown in Figure 5, there was no detectable integration activity in the presence of either 4 or 15 bp of flanking DNA (lanes 2 and 3), indicating that Ab Cas1-catalyzed cleavage at the TIRs does not occur under the specific conditions used here for in vitro integration. We have tested other in vitro conditions and to date have not detected DNA cleavage of oligonucleotides representing combinations of flanking DNA and TIRs (data not shown).
Figure 5.
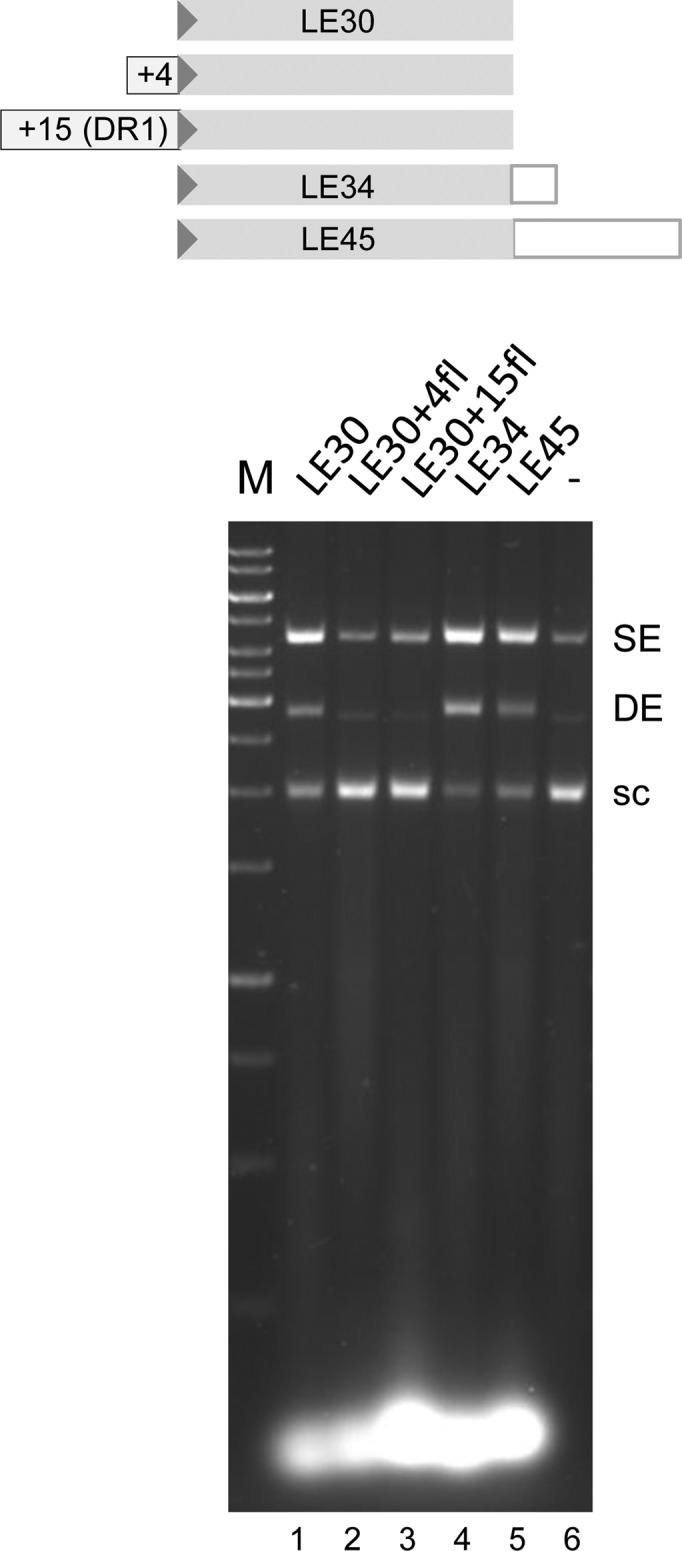
Strand transfer of TIR oligonucleotides with flanking DNA-TIR junctions. The assay was run under standard conditions but for 40 min. The arrowhead represents the casposon end. Nucleotides in the flanking sequence used correspond to those of DR1 (see Materials and Methods), and the oligonucleotide sequences are listed in Table 1.
DISCUSSION
Cas1 proteins of CRISPR-Cas systems have been reported to be metal-dependent nucleases with varying substrate preferences, and their three-dimensional structure appears unrelated to any other characterized proteins (10–12). Together with Cas2, they integrate short protospacers into a specific site within the CRISPR-Cas locus of many bacteria and archaeal species. Our studies here reveal a second biological role for Cas1 proteins: they are active DNA integrases associated with distinct genetic segments designated casposons. In particular, the casposon-encoded Cas1 protein from Aciduliprofundum boonei integrates sequences corresponding to its casposon terminal inverted repeats and generates 14–15 bp target site duplications upon insertion into random DNA.
Although the Cas1–Cas2 complex of CRISPR-Cas systems and the casposon-encoded Cas1 protein both possess DNA integrase activity in vitro, they differ dramatically in their substrate and target requirements, suggesting fundamental differences in how they recognize and act upon DNA. For the well-characterized E. coli CRISPR-Cas system, sequence specificity for spacers is restricted to the 3 bp protospacer-adjacent motif (PAM) from foreign DNA; after processing, only a single nucleotide originating from the PAM is left on the end of the mature protospacer (33–36). Thus, this appears to be the only sequence requirement of the protospacer that becomes integrated into the site of the first repeat following the CRISPR leader sequence (16,17). In contrast, Ab Cas1 specifically integrates oligonucleotides corresponding to the sequence of its casposon ends (Figure 3), and recognition of its TIRs does not appear to involve the terminal bp as changing the terminal C/G bp to A/T does not affect strand transfer and replacing the terminal bp of a random oligonucleotide with C/G does not restore strand transfer activity (Figure 3C). Many characterized DNA transposases specifically recognize sequences subterminal to the very ends of their TIRs rather than the tips of the transposon, and it seems likely that the highly basic C-terminal domain appended to casposon-encoded Cas1 proteins—with its predicted helix-turn-helix motif—confers the property of site-specific binding to casposon ends. Many characterized DNA transposases such as those of IS911, bacteriophage Mu and the Tc1/mariner family use HTH domains (or their variants such as winged helix domains) to recognize specific subterminal sequences within their TIRs (29,37–43).
A characteristic property of CRISPR-Cas systems is that, when new spacers are acquired, they are overwhelmingly integrated into the leader-end of the CRISPR array (16,44). How this integration selectivity is achieved is not yet clear. For Ab Cas1, we observed no evident site-specificity to integration: there was no evidence for hotspots of integration into a pUC19 target plasmid and we could not detect any pattern or specific properties of the integration sites (Figure 4). It is possible because our sequenced sample size is limited, as DNA transposases are often reported to possess a preferred target site in terms of either specific sequence, palindromic nature or basepair content (45). However, if Ab Cas1 has such an underlying preference, it clearly does not dominate target site selection.
It has been reported that protospacer integration in vitro by the E. coli Cas1–Cas2 complex requires target DNA supercoiling (16). Ab Cas1 does not have a similar target requirement as revealed by the time course of SE and DE integration (Figure 3D) in which eventual plasmid fragmentation suggested the repeated integration of short oligonucleotide ends into pUC19. Ab Cas1 also readily integrates oligonucleotide TIRs into short oligonucleotides (data not shown). It is possible that this difference reflects the targeting of protospacer integration to a CRISPR repeat where plasmid supercoiling would favor hairpin extrusion/cruciform formation, whereas Ab Cas1 does not appear to possess such a target preference.
Cas1-mediated integration of protospacers into the CRISPR locus and ∼2.8 kb-mini-casposons into a target plasmid both result in target site duplications. In the case of protospacer integration, it is the CRISPR repeat that becomes duplicated. This is the basis of the signature mechanism by which the CRISPR locus is expanded to become an array of identical CRISPR repeats alternating with random spacers. For casposons, we observed that for each integration event, a randomly selected target site becomes duplicated with a fixed length of 14–15 bp. This suggests that 14–15 bp represents the distance between two Ab Cas1 active sites within the active protein–DNA assembly. The structures of several CRISPR-associated Cas1 proteins have been determined in the absence of bound DNA, and within these dimeric complexes, the active sites are on opposite faces of the dimer (10–12). Whether this arrangement is compatible with integration of two casposon ends into target DNA with a separation of 14–15 bp remains to be established. It is possible that the Ab Cas1 active assembly is a higher-order multimer than a dimer just as the Cas1–Cas2 complex of the E. coli CRISPR-Cas system is reported to be a heterohexamer in which a central Cas2 dimer bridges two Cas1 dimers (9). Furthermore, Cas1 proteins might multimerize in different ways, and the dimer of Ab Cas1 may differ from those observed for CRISPR-associated Cas1 proteins.
Our observations that Ab Cas1 has undetectable cleavage activity on the DNA substrates we tested could have several implications. We note that while endonucleolytic cleavage by CRISPR-Cas1 proteins has been reported, the results are quite variable. In those studies where Cas1 active site mutants have also been examined (as negative controls against potentially co-purifying cellular nucleases), it has been shown that CRISPR-Cas1 from P. aeruginosa, when in ∼300X molar excess, degraded kb-long supercoiled and linear dsDNA and ssDNA, but not ∼500 bp dsRNA or ssRNA (10). The CRISPR-Cas1 from E. coli readily degraded 34-mer ssDNA, ssRNA and dsDNA oligomers, but cannot degrade a 61-mer dsDNA substrate; it was far more active on branched DNA substrates (11). Finally, the CRISPR-Cas1 from A. fulgidus cleaves ssRNA in a Ca2+-dependent manner, but not dsDNA (12). Thus, a unified view of the cleavage activity of CRISPR-associated Cas1 proteins, and what role this might play in spacer acquisition, has not yet emerged. Rather, there is evidence that RecBCD might play a role in generating CRISPR spacers during recovery from stalled replication forks (46), and it has been suggested that a CRISPR-Cas1 nuclease activity might further process the products of the RecBCD complex to generate protospacers of the appropriate length (17). However, this hypothetical step in the adaptation process has not yet been demonstrated.
In the assays described here, we probed linear and supercoiled dsDNA as a substrate, and it is possible that Ab Cas1 recognizes some other form of DNA. For example, the replication-dependent transposition mechanism proposed by Krupovic et al. (18) invokes the formation of a branched DNA substrate when two uncleaved ssDNA casposon ends pair; upon cleavage of both strands at the branch point, the excised ssDNA casposon is then replicated by the casposon-encoded DNA polymerase. We have been unable to detect cleavage on oligonucleotides that mimic the proposed substrate or on single-stranded TIR substrates (data not shown). Very low cleavage activity—if it exists—might explain why so few casposon copies have been identified to date (18). By analogy to the role of RecBCD in the generation of CRISPR spacers, it is more likely that another protein, either encoded by the casposon itself or elsewhere in the genome, is responsible for cleavage at the casposon termini. If another nuclease is involved, it seems likely that the second protein is directed to the casposon ends through an interaction with Cas1. The division of labor at transposon ends between two separate proteins within a heteromeric transposase is rarely seen, but has been observed for the transposase encoded by the cut-and-paste Tn7 transposon (47,48). In this case, the two proteins that constitute the Tn7 transposase are both nucleases, and each cuts one strand at the transposon ends so that a double-strand break is the result of their combined action. The separation of cleavage and integration activities between two subunits of a heteromeric transposase where one generates a 3′-OH group and the other uses it for transesterification implies that the substrate would have to be transferred from one active site to the other during the reaction, which would require some gymnastics. Although we have investigated the possibility that the only other identifiable nuclease within the A. boonei casposon, the HNH nuclease, is responsible for cleavage at the casposon end, to date we have no evidence that its nuclease activity is targeted to the casposon ends, either on its own or in the presence of Ab Cas1. We have also been unable to detect a direct interaction between the two proteins (data not shown).
It cannot be ruled out that casposons, although a provocative clustering of genes contained within inverted repeats, are not mobile genetic elements. They are not common and no sequenced genome contains multiple copies of a casposon flanked by TIRs. Alternatively, they may have once been active mobile elements but mutations in Ab Cas1 have inactivated cleavage activity while integration activity remains. If so, this suggests that the CRISPR-Cas1 proteins may have also followed the same evolutionary trajectory: integration activity (and its reversal, disintegration) have been clearly demonstrated (16,17) but whether they are responsible for generating the 3′-OH group on spacers for subsequent integration is not yet known. Nonetheless, it is clear that two distinct families of Cas1 proteins have evolved, both with extant integration activity but with different substrate requirements and integration outcomes. Whether one was the evolutionary ancestor of the other remains a fascinating question.
Supplementary Material
Acknowledgments
We thank Nancy Craig for first bringing casposases to our attention, and Mick Chandler, Ivana Grabundzija and Susu He for helpful suggestions and insights.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Program of the National Institute of Diabetes and Digestive and Kidney Diseases, NIH. Funding for open access charge: The Intramural Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (NIH), Bethesda MD.
Conflict of interest statement. None declared.
REFERENCES
- 1.Abedon S.T. Bacterial ‘immunity’ against bacteriophages. Bacteriophage. 2012;2:50–54. doi: 10.4161/bact.18609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Westra E.R., Swarts D.C., Staals R.H.J., Jore M.M., Brouns S.J.J., van der Oost J. The CRISPRs, they are a-changing: how prokaryotes generate adaptive immunity. Annu. Rev. Genet. 2012;46:311–339. doi: 10.1146/annurev-genet-110711-155447. [DOI] [PubMed] [Google Scholar]
- 3.van der Oost J., Westra E.R., Jackson R.N., Wiedenheft B. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat. Rev. Microbiol. 2014;12:479–492. doi: 10.1038/nrmicro3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heler R., Marraffini L.A., Bikard D. Adapting to new threats: the generation of memory by CRISPR-Cas immune systems. Mol. Microbiol. 2014;93:1–9. doi: 10.1111/mmi.12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fineran P.C., Charpentier E. Memory of viral infections by CRISPR-Cas adaptive immune systems: acquisition of new information. Virology. 2012;434:202–209. doi: 10.1016/j.virol.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Makarova K.S., Haft D.H., Barrangou R., Brouns S.J.J., Charpentier E., Horvath P., Moineau S., Mojica F.J.M., Wolf Y.I., Yakunin A.F., et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yosef I., Goren M.G., Qimron U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012;40:5569–5576. doi: 10.1093/nar/gks216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makarova K.S., Wolf Y.I., Koonin E.V. The basic building blocks and evolution of CRISPR-Cas systems. Biochem. Soc. Trans. 2013;41:1392–1400. doi: 10.1042/BST20130038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nuñez J.K., Kranzusch P.J., Noeske J., Wright A.V., Davies C.W., Doudna J.A. Cas1-Cas2 complex formation mediates spacer acquisition during CRISPR-Cas adaptive immunity. Nat. Struct. Mol. Biol. 2014;21:528–534. doi: 10.1038/nsmb.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiedenheft B., Zhou K., Jinek M., Coyle S.M., Ma W., Doudna J.A. Structural basis for DNase activity of a conserved protein implicated in CRISPR-mediated genome defense. Structure. 2009;17:904–912. doi: 10.1016/j.str.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 11.Babu M., Beloglazova N., Flick R., Graham C., Skarina T., Nocek B., Gagarinova A., Pogoutse O., Brown G., Binkowski A., et al. A dual function of the CRISPR-Cas system in bacterial antivirus immunity and DNA repair. Mol. Microbiol. 2011;79:484–502. doi: 10.1111/j.1365-2958.2010.07465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim T.-Y., Shin M., Yen L.H.T., Kim J.-S. Crystal structure of Cas1 from Archaeoglobus fulgidus and characterization of its nucleolytic activity. Biochem. Biophys. Res. Commun. 2013;441:720–725. doi: 10.1016/j.bbrc.2013.10.122. [DOI] [PubMed] [Google Scholar]
- 13.Beloglazova N., Brown G., Zimmerman M.D., Proudfoot M., Makarova K.S., Kudritska M., Kochinyan S., Wang S., Chruszcz M., Minor W., et al. A novel family of sequence-specific endoribonucleases associated with the clustered regularly interspaced short palindromic repeats. J. Biol. Chem. 2008;283:20361–20371. doi: 10.1074/jbc.M803225200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han D., Krauss G. Characterization of the endonuclease SSO2001 from Sulfolobus solfataricus P2. FEBS Lett. 2009;583:771–776. doi: 10.1016/j.febslet.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 15.Nam K.H., Ding F., Haitjema C., Huang Q., DeLisa M.P., Ke A. Double-stranded endonuclease activity in Bacillus halodurans clustered regularly interspaced short palindromic repeats (CRISPR)-associated Cas2 protein. J. Biol. Chem. 2012;287:35943–35952. doi: 10.1074/jbc.M112.382598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nuñez J.K., Lee A.S.Y., Engelman A., Doudna J.A. Integrase-mediated spacer acquisition during CRISPR-Cas adaptive immunity. Nature. 2015;519:193–198. doi: 10.1038/nature14237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rollie C., Schneider S., Brinkmann A.S., Bolt E.L., White M.F. Intrinsic sequence specificity of the Cas1 integrase directs new spacer acquisition. eLife. 2015;4:e08716. doi: 10.7554/eLife.08716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krupovic M., Makarova K.S., Forterre P., Prangishvili D., Koonin E.V. Casposons: a new superfamily of self-synthesizing DNA transposons at the origin of prokaryotic CRISPR-Cas immunity. BMC Biol. 2014;12:36. doi: 10.1186/1741-7007-12-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Craig N.L. A moveable feast: An introduction to mobile DNA. In: Craig NL, Chandler M, Gellert M, Lambowitz AM, Rice PA, Sandmeyer S, editors. Mobile DNA. 3rd edn. Washington DC: American Society for Microbiology; 2014. pp. 3–39. [Google Scholar]
- 20.Hickman A.B., Dyda F. Mechanisms of DNA transposition. In: Craig NL, Chandler M, Gellert M, Lambowitz AM, Rice PA, Sandmeyer S, editors. Mobile DNA. 3rd edn. Washington DC: American Society for Microbiology; 2014. pp. 531–553. [Google Scholar]
- 21.Koonin E.V., Krupovic M. Evolution of adaptive immunity from transposable elements combined with innate immune systems. Nat. Rev. Genet. 2015;16:184–192. doi: 10.1038/nrg3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kapitonov V.V., Jurka J. RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biol. 2005;3:e181. doi: 10.1371/journal.pbio.0030181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kapitonov V.V., Koonin E.V. Evolution of the RAG1-RAG2 locus: both proteins came from the same transposon. Biol. Direct. 2015;10:20. doi: 10.1186/s13062-015-0055-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makarova O., Kamberov E., Margolis B. Generation of deletion and point mutations with one primer in a single cloning step. Biotechniques. 2000;29:970–972. doi: 10.2144/00295bm08. [DOI] [PubMed] [Google Scholar]
- 25.Evertts A.G., Plymire C., Craig N.L., Levin H.L. The Hermes transposon of Musca domestica is an efficient tool for the mutagenesis of Schizosaccharomyces pombe. Genetics. 2007;177:2519–2523. doi: 10.1534/genetics.107.081075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hickman A.B., Dyda F. CRISPR-Cas immunity and mobile DNA: a new superfamily of DNA transposons encoding a Cas1 endonuclease. Mobile DNA. 2014;5:23. doi: 10.1186/1759-8753-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arciszewska L.K., Craig N.L. Interaction of the Tn7-encoded transposition protein TnsB with the ends of the transposon. Nucleic Acids Res. 1991;19:5021–5029. doi: 10.1093/nar/19.18.5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zou A., Leung P.C., Harshey R.M. Transposase contacts with Mu DNA ends. J. Biol. Chem. 1991;266:20476–20482. [PubMed] [Google Scholar]
- 29.Claeys Bouuaert C., Walker N., Liu D., Chalmers R. Crosstalk between transposase subunits during cleavage of the mariner transposon. Nucleic Acids Res. 2014;42:5799–5808. doi: 10.1093/nar/gku172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richardson J.M., Colloms S.D., Finnegan D.J., Walkinshaw M.D. Molecular architecture of the Mos1 paired-end complex: the structural basis of DNA transposition in a eukaryote. Cell. 2009;138:1096–1108. doi: 10.1016/j.cell.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hickman A.B., Ewis H.E., Li X.H., Knapp J.A., Laver T., Doss A.L., Tolun G., Steven A.C., Grishaev A., Bax A., et al. Structural basis of hAT transposon end recognition by Hermes, an octameric DNA transposase from Musca domestica. Cell. 2014;158:353–367. doi: 10.1016/j.cell.2014.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stoddard B.L. Homing endonuclease structure and function. Q. Rev. Biophys. 2006;38:49–95. doi: 10.1017/S0033583505004063. [DOI] [PubMed] [Google Scholar]
- 33.Mojica F.J., Díez-Villaseñor C., García-Martínez J., Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733–740. doi: 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- 34.Datsenko K.A., Pougach K., Tikhonov A., Wanner B.L., Severinov K., Semenova E. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun. 2012;3:945. doi: 10.1038/ncomms1937. [DOI] [PubMed] [Google Scholar]
- 35.Yosef I., Goren M.G., Qimron U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012;40:5569–5576. doi: 10.1093/nar/gks216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goren M.G., Yosef I., Auster O., Qimron U. Experimental definition of a clustered regularly interspaced short palindromic duplicon in Escherichia coli. J. Mol. Biol. 2012;423:14–16. doi: 10.1016/j.jmb.2012.06.037. [DOI] [PubMed] [Google Scholar]
- 37.Chandler M., Fayet O., Rousseau P., Ton Hoang B., Duval-Valentin G. Copy-out-paste-in transposition of IS911: a major transposition pathway. In: Craig NL, Chandler M, Gellert M, Lambowitz AM, Rice PA, Sandmeyer S, editors. Mobile DNA. 3rd edn. Washington DC: American Society for Microbiology; 2014. pp. 591–607. [Google Scholar]
- 38.Clubb R.T., Omichinski J.G., Savilahti H., Mizuuchi K., Gronenborn A.M., Clore G.M. A novel class of winged helix-turn-helix protein: the DNA-binding domain of Mu transposase. Structure. 1994;2:1041–1048. doi: 10.1016/s0969-2126(94)00107-3. [DOI] [PubMed] [Google Scholar]
- 39.Schumacher S., Clubb R.T., Cai M.L., Mizuuchi K., Clore G.M., Gronenborn A.M. Solution structure of the Mu end DNA-binding Iβ subdomain of phage Mu transposase: modular DNA recognition by two tethered domains. EMBO J. 1997;16:7532–7541. doi: 10.1093/emboj/16.24.7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clubb R.T., Schumacher S., Mizuuchi K., Gronenborn A.M., Clore G.M. Solution structure of the Iγ subdomain of the Mu end DNA-binding domain of phage Mu transposase. J. Mol. Biol. 1997;273:19–25. doi: 10.1006/jmbi.1997.1312. [DOI] [PubMed] [Google Scholar]
- 41.Montaño S.P., Pigli Y, Rice P.A. The Mu transpososome structure sheds light on DDE recombinase evolution. Nature. 2012;491:413–417. doi: 10.1038/nature11602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watkins S., van Pouderoyen G., Sixma T.K. Structural analysis of the bipartite DNA-binding domain of Tc3 transposase bound to transposon DNA. Nucleic Acids Res. 2004;32:4306–4312. doi: 10.1093/nar/gkh770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Izsvák Z., Khare D., Behlke J., Heinemann U., Plasterk R.H., Ivics Z. Involvement of a bifunctional, paired-like DNA-binding domain and a transpositional enhancer in Sleeping Beauty transposition. J. Biol. Chem. 2002;277:34581–34588. doi: 10.1074/jbc.M204001200. [DOI] [PubMed] [Google Scholar]
- 44.Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 45.Linheiro R.S., Bergman C.M. Testing the palindromic target site model for DNA transposon insertion using the Drosophila melanogaster P-element. Nucleic Acids Res. 2008;36:6199–6208. doi: 10.1093/nar/gkn563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levy A., Goren M.G., Yosef I., Auster O., Manor M., Amitai G., Edgar R., Qimron U., Sorek R. CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature. 2015;520:505–510. doi: 10.1038/nature14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.May E.W., Craig N.L. Switching from cut-and-paste to replicative Tn7 transposition. Science. 1996;272:401–404. doi: 10.1126/science.272.5260.401. [DOI] [PubMed] [Google Scholar]
- 48.Sarnovsky R.J., May E.W., Craig N.L. The Tn7 transposase is a heteromeric complex in which DNA breakage and joining activities are distributed between different gene products. EMBO J. 1996;15:6348–6361. [PMC free article] [PubMed] [Google Scholar]
- 49.Drozdetskiy A., Cole C., Procter J., Barton G.J. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015;43:W389–W394. doi: 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crooks G.E., Hon G., Chandonia J.M., Brenner S.E. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.