


Abstract
Fluctuations in mRNA levels only partially contribute to determine variations in mRNA availability for translation, producing the well-known poor correlation between transcriptome and proteome data. Recent advances in microscopy now enable researchers to obtain high resolution images of ribosomes on transcripts, providing precious snapshots of translation in vivo. Here we propose RiboAbacus, a mathematical model that for the first time incorporates imaging data in a predictive model of transcript-specific ribosome densities and translational efficiencies. RiboAbacus uses a mechanistic model of ribosome dynamics, enabling the quantification of the relative importance of different features (such as codon usage and the 5′ ramp effect) in determining the accuracy of predictions. The model has been optimized in the human Hek-293 cell line to fit thousands of images of human polysomes obtained by atomic force microscopy, from which we could get a reference distribution of the number of ribosomes per mRNA with unmatched resolution. After validation, we applied RiboAbacus to three case studies of known transcriptome-proteome datasets for estimating the translational efficiencies, resulting in an increased correlation with corresponding proteomes. RiboAbacus is an intuitive tool that allows an immediate estimation of crucial translation properties for entire transcriptomes, based on easily obtainable transcript expression levels.
INTRODUCTION
Translation, the synthesis of proteins by ribosomes using an mRNA template, is a fundamental process in biology. It relies upon complex interactions between molecular actors that modulate this process at a number of translation check-points: initiation (1–3), elongation (4–6), termination and ribosome recycling (7,8). Moreover, mRNA determinants such as codon usage bias (9), GC content (10), 5′ mRNA structures (11,12), cis regulatory elements (13), protein–protein interaction (14,15), ribosome pausing (16–18), alternative termination (19) and drop off (20,21) influence translational efficiencies or translation rates in vivo. In cells, several ribosomes translate the same mRNA forming the so-called polyribosome or polysome (22–24). At steady state, the total number of ribosomes per transcript are the result of an equilibrium among initiation, elongation and termination events. The precise contribution of the number of ribosomes per transcript to the final protein production remains elusive and unexplored because of the challenge posed by obtaining experimental genome-wide distributions of ribosome number per transcript.
Translation has been the subject of intense modelling efforts in the last five decades, using various mathematical and computational approaches (18,25–32). These models aimed at predicting protein production rates and understanding the role of mRNA features or contributions of translation stages. Several models purely deal with biophysical theoretical descriptions of ribosome fluxes along mRNAs (29,31,33), while recent experimental methods to study translation using ribosome footprinting (17,34) or polysome profiling (32) motivated new mathematical modelling approaches based on genome-wide maps of ribosome occupancy and/or ribosome density along transcripts (18,31,35). Despite the many insights afforded by these modelling studies, a consensus model remains elusive, as different modelling approaches/assumptions often lead to contradictory conclusions concerning the role of mRNA determinants (in particular the contribution of codon usage), the interplay between initiation and elongation, translational rates and efficiencies. Employing ribosome profiling data to develop mathematical models is undoubtedly promising, but several problems have been encountered. For example, biases determined by alignment of ambiguous RNA reads to mRNA isoforms, artefacts caused by missing normalization (36), fragment bias that depends on the length of the sequenced fragments (37–39) can introduce errors that may affect the robustness of translation efficiencies (TEs) calculated using these data. Ribosome profiling has been extensively used for obtaining estimates of ribosome occupancy per transcript. These estimates are essential for parameterizing mechanistic models of translation, however their reliability is questionable, as they are computed by collapsing ribosome positional information from thousands of copies of the very same transcript. Another technique for obtaining ribosome occupancy, ribosome density and the number of ribosomes per transcript could be the employment of polysomal profiling followed by microarray or RNA-seq (40–43). Unfortunately, this approach provides an indirect estimation of the number of ribosomes per transcript. A more precise way for obtaining this information is the employment of imaging techniques, followed by ribosome counting (44). In principle this approach allows to determine the exact number of ribosomes with a single transcript resolution, if a polysome can be univocally identified.
Recently, much effort has been directed at elucidating by imaging the three-dimensional (3D) structure of polysomes in bacteria (45) and eukaryotes using Cryo-ET and atomic force microscopy (AFM) (44,46–48). The emerging model describes polysomes as groups of tightly interacting ribosomes. In addition, independent groups of ribosomes, or ribo-cliques, spaced by naked mRNA can be observed along the same transcript, as demonstrated by AFM (44). Despite the unique advantages of Cryo-ET for obtaining high-resolution information about ribosome–ribosome interactions (48), it cannot be employed to identify coding mRNA filaments uncovered by ribosomes, precluding the possibility to precisely count the number of ribosomes per transcript. Therefore, AFM is of major help for precisely and univocally counting the number of ribosomes in thousands of transcripts purified from cells or tissues.
Ribosome profiling studies introduced the concept of ‘5′ ramp’, identified as a region of about 50 codons (34). This region immediately follows the start codon, where ribosomes display on average an increased density, probably moving with a reduced elongation speed (36) with respect to the remaining coding sequence (CDS). Although definitive molecular evidences and mechanistic explanation are still missing, a body of clues indicates the existence of the ramp effect (49), that has been identified in bacteria (35,50), yeast (18,34,35) and mammals (31,36,51,52). While existing mathematical models of translation have often included a heuristical ramp effect, to our knowledge the ramp parameters have never been systematically explored or optimized.
Here, for the first time, we exploit the rich data provided by AFM images to calibrate a mechanistic model of translation. We develop RiboAbacus, a new mathematical model of translation calibrated using thousands of single-polysome AFM images. The output of RiboAbacus is the prediction of transcript-specific ribosome numbers and ribosome occupancy from transcriptome data. The model takes into account the main steps of the elongation phase to predict in a transcript specific fashion the number of ribosomes per transcript and derive the corresponding translational efficiency (TE). The proposed method has also been compared with polysome profiling in yeast, showing an increased resolution in determining the number of ribosomes per transcript, and a general agreement for single transcript predictions. We took advantage of the experimental distribution of the number of ribosome per transcript in one human cell line (HeK-293) to tune RiboAbacus parameters (ramp length and slowdown) during the training of the model. A second genome-wide dataset (human MCF-7) and one enriched in a single transcript (rabbit globin from in vitro translation system) were used for validation. Finally, the predicted number of ribosomes per transcript was employed to calculate the TE of mRNAs expressed in three additional biological systems: the human medulloblastoma cell line DAOY (53), primary mouse motoneurons from stem cells (54) and NIH3T3 mouse fibroblasts (55), significantly increasing the experimental correlation between transcript and protein abundances. This application illustrates the effectiveness of model-based predictions in estimating proteome abundances from transcriptome data. In synthesis, RiboAbacus is an intuitive tool that allows an almost immediate estimation of crucial translation properties for entire transcriptomes, based on easily obtainable transcript expression levels.
MATERIALS AND METHODS
Chemicals
All solution used for polysome purifications has been prepared in RNase-free water containing 100 μg/ml cycloheximide in order to prevent ribosome subunit disassembly. All reagents, unless otherwise cited, were of molecular biological grade and purchased from Sigma.
Cell culture and human polysomal purification
The baker's yeast Saccharomyces cerevisiae wild-type strain BY4741 (MATa, his3D1, leu2D0, met15D0, ura3D0) was obtained from the EUROSCARF repository (EUROpean Saccharomyces Cerevisiae ARchive for Functional analysis, Institute for Molecular Biosciences, Johann Wolfgang Goethe-University Frankfurt, Germany, www.euroscarf.de). A single yeast colony was grown overnight to stationary phase in 5 ml of YPDA growth medium (1% Yeast Extract, 2% Peptone, 2% Dextrose and 200 mg/l Adenine) at 30°C. The day after the culture was diluted 1/10 in 20 ml of fresh YPDA and allowed to reach the mid-log growth phase. Translation was blocked by adding 0.01 mg/ml cycloheximide. Yeast cells were then collected by centrifugation and lysed with little modifications to Arava's protocol (40). Briefly, yeast cells were transferred to 2 ml round bottom tubes with 1 ml of freshly prepared lysis buffer (20 mM Tris–HCl, pH 8.0, 140 mM KCl, 1.5 mM MgCl2, 0.5 mM dithiothreitol (DTT), 0.01 mg/ml of cycloheximide, 1% Sodium DeoxyCholate, 1% Triton X-100, 20U RNAse inhibitor) and washed twice. Cells were then lysed using 0.7 ml of lysis buffer with 0.6 vol of pre-chilled acid-washed glass beads (0.45–0.55 mm, Sigma-Aldrich). Complete lysis was performed through six cycles of vortexing (30 s) followed by incubation in ice (1 min). Lysates were harvested by collecting supernatants from two subsequent rounds of cold centrifugation with increasing speed (2600 and 7200 g, respectively). Lysates were then diluted to 0.8 ml with lysis buffer and stored at −80°C. Polysomes were purified as described below for human cellular lysates.
Hek-293 and MCF-7 cells were seeded at a density of 2.5 × 104 cells/cm2 and maintained for 3 days in growth medium (Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, 100 units/ml penicillin and 100 mg/ml streptomycin at 37°C, 5% CO2). At 80% confluence, cells were incubated for 3 min with cycloheximide (100 μg/ml) at 37°C to interfere with the translocation step during protein synthesis, blocking translational elongation and trapping ribosomes on the mRNA. Cells were washed with phosphate buffered saline (PBS + cycloheximide 100 μg/ml) and scraped directly on the plate with 300 μl lysis buffer (10 mM NaCl, 10 mM MgCl2, 10 mM Tris–HCl, pH 7.5, 1% Triton X-100, 1% sodium deoxycholate, 0.2 U/μl RNase inhibitor (Fermentas), cycloheximide 10 μg/ml and 1 mM DTT). After a nuclei and cellular debris removal by centrifugation (5 min at 12 000 g at 4°C), the supernatant was directly transferred onto a 15–50% linear sucrose gradient containing 30 mM Tris–HCl, pH 7.5, 100 mM NaCl, 10 mM MgCl2 and centrifuged in a Sorvall ultracentrifuge on a swinging rotor for 100 min at 180 000 g at 4°C. The fractions corresponding to the 80S peak and to the polysomes were collected monitoring the absorbance at 254 nm. Each fraction was aliquoted, flash frozen in liquid N2 and stored at −80°C before AFM imaging.
Preparation of polysomes from rabbit reticulocytes (RRL)
Briefly, 1 ml of untreated rabbit reticulocytes (RRL) prepared according to Jackson and Hunt (56) was complemented with 20 μM hemin (Fluka), 50 μg/ml creatine phosphokinase, 10 mg/ml creatine phosphate (Fluka), 50 μg/ml of bovine liver tRNAs and 5 mM of D-glucose. Endogenous RNAs were translated in 80 μl reactions containing 40 μl of the complemented, untreated RRL in the presence of 75 mM KCl, 0.5 mM MgCl2, amino acids (20 μM each), 5 mM DTT and 0.1 U/μl RiboLock RNase (Fermentas) for 10 min at 30°C. Reactions were stopped by cooling the tube on ice for 1 min and adding 320 μl of ice-cold, low salt buffer (15 mM NaCl, 1 mM MgCl2, 10 mM Tris pH 7.4, 1 mM DTT, 0.12 mg/ml cycloheximide). Polysome purification following the above-mentioned protocol.
qPCR from RRL polysomal fractions
Nine fractions were collected monitoring the absorbance at 254 nm. From 0.5 ml of each fraction, total RNA was isolated after proteinase K treatment, phenol–chloroform extraction and isopropanol precipitation and resuspended in 20 μl of RNase free water. For each fraction, 4 μl of total RNA was reverse-transcribed using the iScrip™ cDNA Synthesis Kit (Biorad) in a final volume of 20 μl. One microlitre of cDNA and 400 nM of each primer were used in combination with the KAPA SYBR Green kit (KAPA Biosystems) in a final volume of 10 μl. Forty amplification cycles (95°C for 15 s, 55°C for 20 s, 72°C for 25 s) were run in a CFX-96 C1000 thermal cycler (Biorad) using primers specific to rabbit beta-globin (forward: 5′-TTTGCTAAGCTGAGTGAACTGC; reverse: 5′-CCAGCCACCACCTTCTGATA), rabbit 15-lipoxigenase (forward: 5′-TTCTGTCCCCCTGACGATCT; reverse: 5′-GATCTCTCGGCACCAGCTCT) and rabbit 18S rRNA (forward: 5′-ACGGCCGGTACAGTGAAACT; reverse: 5′-GACCGGGTTGGTTTTGATCTG). qPCR amplification efficiency was calculated for each gene using a relative standard curve derived from a cDNA of total RNA isolated from RRL. The Ct values were determined by the CFX Manager 2.1 (Biorad) applying multi-variable, non-linear regression model to individual well fluorescence traces. The amount of each target gene was quantified relative to the fraction n° 14 and normalized to the level 18S gene, according to Pfaffl equation (57). qPCR reactions were carried out in triplicates.
Atomic force microscopy imaging
For AFM imaging a 20 μl of Hek-293 or RRL polysomal fraction were adsorbed for 3 min on freshly cleaved mica pretreated with Ni2 + for 3 min. The samples were then covered with 100 mM Hepes, pH 7.4, 10 mM NaCl, 10 mM MgCl2, 100 μg/ml cycloheximide and 3% (w/v) sucrose. After 1 h of incubation at 4°C, the sample was extensively and gently washed with DEPC-water containing 100 μg/ml cycloheximide and dried at 20°C for at least 1 h.
Imaging was performed using a Cypher AFM (Asylum Research, Santa Barbara, CA, USA) in AC mode, using Asylum routines for the IGOR software environment (WaveMetrics, Portland, OR, USA). Scans have been acquired using OMCL-AC240TS tips (Olympus) with nominal spring constant of 2 N/m. The scanning parameters were as follows: typical driving frequency 70 kHz in air, scanning rate 1-2 Hz. AFM images were levelled line by line and rendered using the Gwyddion (gwyddion.net) software package. Images were analysed in ImageJ (58) to count ribosomes in polysomes, manually picking the ribosomal particles and assigning them to their respective polysomes using a custom ImageJ macro. Thousand polysomes were analysed (♯ objects = 3300 for yeast; ♯ objects = 2251 for Hek-293; ♯ objects = 696 for MCF-7, ♯ objects = 901 for polysomes from RRL) picking more than 20 000 ribosomes.
Model
In order to provide the modelling of translation, the elongation phase was divided in nine different steps, each of them linked to a flux (measured in ribosomes/sec) representing the transition of ribosomes from one stage to the next, similarly to what was done in (29).
The model assigns to each codon of an mRNA nine ordinary differential equations describing the rate of change in the number of ribosomes at position n (referred to the position of the P site of ribosomes), Sn, at the different stages (Equations (1)-(11)) ; note that all the equations are transcript specific). We then set all fluxes equal to 0 to compute the steady-state values of the variables, which is obtained by solving the resulting algebraic system. This procedure, with the addition of a set of initial conditions i.e. of the hypothesis that at time 0 there are no ribosomes along the transcript, allows to compute the steady-state number of ribosomes bound to an mRNA in this condition. See also ‘Results’ section for the assumptions of the model and Supplementary File 1 for further information on fluxes, notations and all parameters involved in the model.
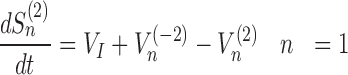 |
(1) |
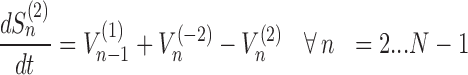 |
(2) |
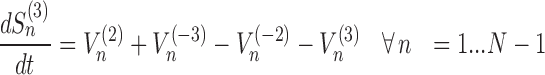 |
(3) |
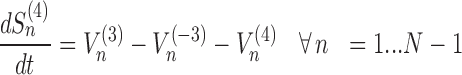 |
(4) |
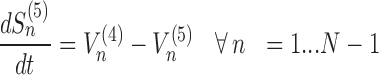 |
(5) |
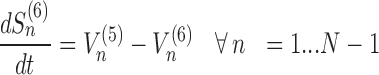 |
(6) |
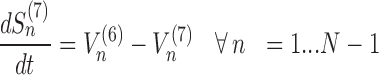 |
(7) |
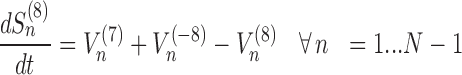 |
(8) |
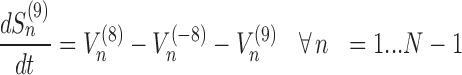 |
(9) |
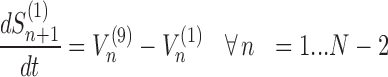 |
(10) |
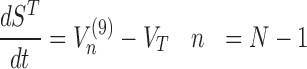 |
(11) |
To be able to solve the system described above, two other equations are necessary: (i) the formula representing the number of codons at position n for transcript r that are not covered by the tail of the preceding ribosomes (Cn, r)
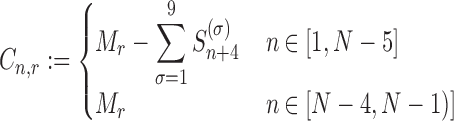 |
(12) |
where Mr is the total number of transcript of species r, and (ii) the probability for a ribosome at position n to move to the next codon (Un)
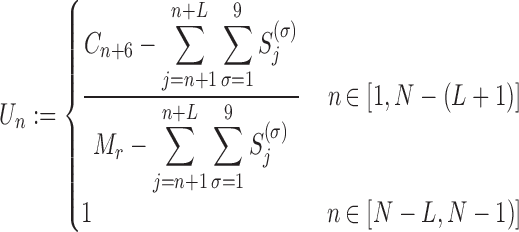 |
(13) |
Equation (12) arises from the assumptions made on the ribosome footprint (see ‘Results’ section and Figure 2A) and the position of the ribosome site with respect to the covered portion of mRNA. Indeed, to obtain the number of free codons at position n, we have to subtract to the maximum amount of codons in that position of the transcript (coincident with the total number of transcripts of species r i.e. Mr) the number of codons occupied by the tail of a ribosome at position n + 4. Since ribosomes leave mRNAs when they reach position N, the number of codons not covered by any ribosome tail is equal to Mr for the last 4 codons. As introduced before, Equation (13) is related to the probability of ribosomes at position n to move forward (Un): since RiboAbacus considers Mr copies of the mRNA species r, the number of ribosomes bound at a specific codon (Sn) ranges from 0 (if all mRNA copies have that position empty) to Mr (if all mRNA copies have that position occupied by ribosomes). (Sn) changes codon to codon and the probability to move forward (Un) depends on the number of free codons close to the tail of ribosomes. In this way the presence of ribosomes along the transcript influences the translocation probability of ribosomes positioned on upstream codons. Basically, (13) coincides with the probability of having a free codon at position n + 5 for only the transcripts not presenting ribosomes between the triplets n + 1 and n + 9, avoiding in this way overestimations of Un. Note that even if Equation (12) is needed exclusively to compute VI (translation initiation) and calculate the probability Un, both the equations are crucial to allow the maintenance of correct distances between the head of each ribosome and the tail of the next one. More precisely, they are necessary to properly compute the only flux related to ribosomes translocation, i.e. 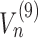 , whereas all the other steps of the process are not affected by them.
, whereas all the other steps of the process are not affected by them.
Figure 2.
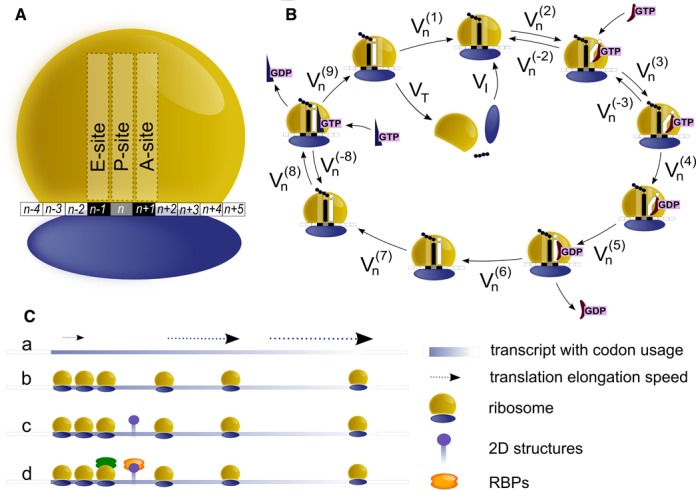
Model description and assumptions. (A) Schematic representation of a ribosome and the portion of the transcript covered. The length of the ribosome footprint is 10 codons. (B) Scheme of the elongation phase, illustrating the chemical reactions considered by the model. Reversible and irreversible reactions during the elongation phase are a simplified version of (64,65) in accordance to (29,66). The kinetic constants for such reactions are taken from (67–70). (C) Schematic representation of the ramp hypotheses: (a) elongation speed is reduced while ribosomes are located on the ramp; (b) the ramp region displays a higher density of ribosomes with respect to the average observed along the remaining transcript; (c) the slowdown effect of the ribosomes along the ramp region could be the consequence of mRNA complex secondary structures; or (d) the presence of RNA binding proteins bound to the region.
Being a probability, Un has to satisfy the following condition:
 |
(14) |
To avoid any physical overlap between two consecutive ribosomes, the total number of ribosomes bound to transcripts r has to satisfy the following condition:
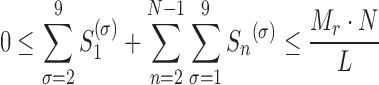 |
(15) |
Solving directly such a complex system is potentially problematic due to the heavy computational load. This can be alleviated by observing that the equations for the last codon are considerably simpler, since 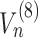 and
and 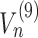 are related to the presence of ribosomes on subsequent codons and hence are trivially zero for the final codon. This allows to devise an efficient backward solution, by fixing a range of values for the exit flux and then computing for each of them the number of ribosomes bound to the mRNA. At this point, we choose the maximum exit flux such that conditions (14)-(15) are both satisfied.
are related to the presence of ribosomes on subsequent codons and hence are trivially zero for the final codon. This allows to devise an efficient backward solution, by fixing a range of values for the exit flux and then computing for each of them the number of ribosomes bound to the mRNA. At this point, we choose the maximum exit flux such that conditions (14)-(15) are both satisfied.
Since the precise nature of the ramp is still controversial (see Figure 2C) , we model the ramp effect by enforcing a lower speed of ribosomes along the first n codons of the transcripts, where n represents the ramp length. Thus, for this portion of the mRNA, we simply multiply the fluxes 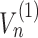 ...
...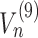 by a constant (ranging from 0 to 1) corresponding to the ribosome slow down rate we want to test and then we proceed as described before.
by a constant (ranging from 0 to 1) corresponding to the ribosome slow down rate we want to test and then we proceed as described before.
Assignment of images to transcripts and calculation of translation efficiency
The output provided by RiboAbacus contains three values for each transcript: (i) the number of ribosomes per transcript, (ii) ribosome occupancy and (iii) TE. The transcriptome-wide distribution of the number of ribosomes per transcript was compared with the experimental distribution obtained from AFM images, both for the training and the validation of the model. As the identity of the individual transcripts imaged by AFM is not known, we couldn't connect directly specific mRNAs to AFM images. We reasonably assumed AFM images to be representative of the distributions of polysomes and transcripts in cells, meaning that the probability of finding the polysome of a certain transcript in an AFM image is proportional to its abundance, easily measurable by experimental approaches. For this reason, in the distributions of the number of ribosomes per transcript obtained with RiboAbacus, we included transcriptome-wide measurements of mRNA levels, given by FPKM (fragments per kilobases per million mapped reads) measurements retrieved from RNA-seq experiments available in literature. RNA-seq provides an empirical distribution of abundance of individual transcripts in a population of cells; the predicted distribution of ribosome counts per transcript was obtained by weighing the predicted number of ribosome on a specific transcript (obtained from RiboAbacus) by its relative frequency (measured by RNA-seq). This marginal distribution can then be directly compared with the distribution of number of ribosomes per transcript measured by AFM. The distance between the experimental and the predicted distribution was calculated constructing two vectors containing at the n-position the experimental and the predicted number of transcripts with exactly n ribosomes attached to them respectively, exploiting then the Euclidean metric to obtain the distance of interest. More precisely, we used the following formula:
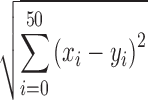 |
(16) |
where xi and yi represent the frequency of mRNAs associated with the number of ribosomes per transcript i respectively for the experimental and the predicted distribution. This distance ranges from 0 (if the two distributions are identical) and 1.
In our study the ribosome occupancy for transript r (ROr) represents the percentage of nucleotides covered by ribosomes for each mRNA and is computed multiplying the predicted number of ribosomes per transcript for the ribosome footprint (L) and normalizing the result for the length of the transcript (N):
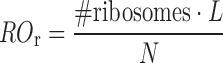 |
(17) |
Translation efficiency (TEr) is obtained by multiplying the ribosome occupancy by the transcript expression levels (Mr):
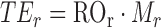 |
(18) |
In the paragraph ‘RiboAbacus improves predictions of proteome data from transcriptome’ the predicted protein abundances were then obtained fitting the length versus ribosome occupancy plot with a negative exponential and using that curve to compute a set of length-specific correction factors, as suggested in (34). We finally used these values in the TE formula obtaining a corrected translation efficiency (cTE).
Statistical analysis
Cross-validation was performed by splitting the Hek-293 transcriptome dataset in two halves. The first was used as training set to optimize the two ramp parameters, the second was used as test set to evaluate the fit of the model. The procedure was repeated 100 times. In parallel, we also approached cross-validation by splitting in two halves the experimental AFM data and we calculated the distance between the two splitted experimental distributions. Also this procedure was repeated 100 times.
To compare the experimental and predicted distributions of the number of ribosomes per transcript, two analytical approaches are used: the first measures the Euclidian distance between the discrete distributions (see the previous subsection), while the second is based on the Kullback-Leibler divergence. In this latter case, using the chi-square minimization method, we first fitted the two distributions with the number of Gaussian curves corresponding to the best fit (usually two or three). Then we computed the Kullback-Leibler divergence between the related curves from experimental and predicted data. After weighting the divergence value to the area under the Gaussian curves that fit the predicted distribution, we finally summed the obtained values.
The Wilcoxon–Mann–Whitney test was used to compare similarity distributions in the paragraph ‘Feature analysis’.
Williams's test was used to analyse differences between two Pearson coefficients in paragraph ‘RiboAbacus improves predictions of proteome data from transcriptome’. The test determines if two dependent correlations are significantly different (59). Williams's test only requires the sample size value and the two correlation values to be compared, and it is the optimal choice for our purposes since it properly works with dependent correlations (60).
RESULTS
Obtaining the distribution of ribosomes per transcripts by atomic force microscopy and comparison with polysome profiling
AFM has been proven to be a powerful approach for studying polysomes and obtaining a great amount of data and information concerning the overall organization of polysomes and the distribution of the number of ribosomes per transcript from thousands of native human polysomes (44). With respect to other methods (40), this technique allows to count the number of ribosomes per transcript at single ribosome resolution and to obtain genome-wide distributions.
To demonstrate the advantages of AFM method, we compared our approach with polysomal profiling coupled to microarray (40) in yeast. Yeast polysomes were isolated from cellular lysates by sucrose gradient sedimentation (Figure 1A). Then, polysomes were imaged by AFM (Figure 1B, left panel) and the number of ribosomes per polysome (i.e. per transcript) was obtained (Figure 1B, right panel). The transcriptome-wide distribution of the number of ribosomes per transcript was determined and shown in Figure 1C. For the analysis, we took into consideration polysomes with high and medium molecular weights that reflect the steady state distribution of ribosomes per transcript for mRNAs with different lengths (40). Given the fact that it is impossible to obtain pure all-steady state polysomes from a cell lysate, we cannot exclude that in the polysome fraction corresponding to medium molecular weight polysomes, some growing polysomes with long transcripts could be possibly present. Next, we employed the dataset of the number of ribosomes per transcript from (40) and compared this distribution (Figure 1D) with ours (Figure 1C). It is clear that AFM provides a much higher resolution of the distribution of ribosomes per transcript. This is to be expected, as AFM enables a direct measurement at single ribosome resolution of the number of ribosomes per transcript. On the other hand, polysome profiling returns this number indirectly employing the absorbance profiles of a sucrose-gradient separation, followed by logarithmic extrapolation of the number of ribosomes in each fraction, microarray analysis (40) or hybridization/blotting (61) and bootstrapping methods for assigning the number or ribosomes to specific transcripts. Transcripts have to be grouped according to the sucrose fraction that corresponds to a fixed number of ribosomes per transcript. This approach leads to the low resolution of the distribution shown in Figure 1D that would be unsatisfactory as training dataset for RiboAbacus. To date no methods exist to assign with single-ribosome resolution the number of ribosomes per transcript in a genome-wide manner. Thus AFM and polysome profiling appear as complementary techniques because AFM has the advantage of ribosome-resolution and polysome profiling of assigning a specific number of ribosome per transcript. For the purpose of modelling the AFM distribution is the most apt technique, because it is reasonable to assume that a big sample size of single-polysomes AFM images is representative of polysomes and transcripts in cells. This means that the probability of finding the polysome of a certain transcript in an AFM image is proportional to the abundance of its mRNA, i.e its transcript expression level. For this reason, RiboAbacus takes into account the level of the transcript, as measured by RNA-seq.
Figure 1.
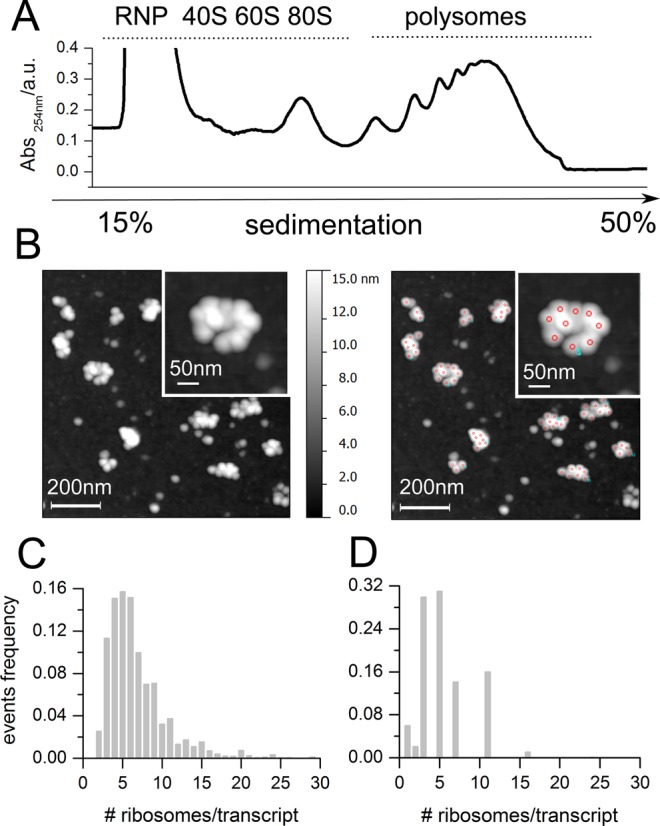
Distribution of the number of ribosomes per transcript by atomic force microscopy (AFM). (A) Representative absorbance profile for sucrose gradient sedimentation of yeast. (B) Example of AFM image of yeast polysomes after absorption on mica (left panel) and example of ribosome detection and counting (right panel, red circles). (C and D) Comparison between the distribution of the number of ribosomes per transcript in yeast obtained using AFM (C) and obtained by Arava and collaborators ((40), D). The number of polysomes considered for counting the number of ribosomes per transcript by AFM is 3300, obtained from 40 independent images.
Assumptions and model development
Before developing the model, we made some preliminary assumptions. RiboAbacus aims at estimating ribosomal densities for an entire transcriptome, without need to know the exact position of ribosomes along the mRNA, an information that could be provided by stochastic (32,62) or probabilistic (27) models. Although some authors considered in their models the availability of ribosomes or their concentrations in the cytoplasm (30,32), our approach does not need to evaluate ribosome competition effects because each transcript is analysed independently. Moreover, we can consider the number of free ribosomes as not limiting. This latter assumption is reasonable given the conditions used for obtaining the training dataset (see next section). In fact under our experimental condition, the number of free ribosomes was calculated to be 2.5·105 ribosomes/cell. This value is similar to what observed in rapidly growing cells of S. cerevisiae (63) as a not limiting condition.
Moreover, we considered a ribosome coverage of 10 codons (34) (Figure 2A). The choice of this parameter is of utmost importance to avoid collisions between neighbouring ribosomes. To allow the maintenance of correct distances between the head of each ribosome and the tail of the next one, we calculated the probability of any ribosome to move forward and to start a new cycle of translation (i.e the probability for the first 6 codons of the transcript to be uncovered). To do so we defined the codons occupied by E, P and A sites. From now on we will refer to the position of a ribosome as the position of its P site: for example, if a ribosome is at codon position n, this means that its E, P and A sites cover the n − 1th, nth and n + 1th codons, respectively (Figure 2A). The 3 codons upstream the A site and the 4 codons downstream the E site are therefore also covered by the same ribosome given the ribosome coverage of 30 nt. The ribosome coverage length and the position of the ribosome centered in n position (as in Figure 2A) allow to precisely define the overall occupancy of the ribosome and the probability of a ribosome to bind the transcript, start a new cycle of translation and move forward.
The core of the model is based on the elongation phase of translation that was divided into nine steps (Figure 2B) and modelled as nine ordinary differential equations, similarly to what was done in (29). Since the release of the tRNA from the E site is the first reaction that takes place once the ribosome has reached this site and is positioned on a new codon, we considered this reaction as the first step of the elongation phase for each triplet (Figure 2B). In fact, when a ribosome translocates from the codon at position n to the position n + 1 it becomes ready to accept a new tRNA but its E site is still occupied by the old tRNA. Therefore the tRNA release is the very first reaction related to the n + 1th codon. The nine steps of the elongation phase can be described as follows: (i) tRNA release from the E site; (ii) binding of the tRNA along with the elongation factor eEF1A and the GTP (the so-called ternary complex) at the A site in a codon-independent process; (iii) binding of the ternary complex at the A site (codon-dependent process); (iv) GTP hydrolysis; (v) eEF1A·GDP position change; (vi) eEF1A·GDP release; (vii) accommodation of the tRNA in the A site and transpeptidation; (viii) eEF2·GTP binding; (ix) ribosome translocation. With respect to the others, ribosomes placed at start and stop codons present some differences, leading to slightly different formulations of the equations for these positions (see ‘Materials and Methods’ section for further details). Ribosomes starting a new cycle of translation do not translocate from previous codons rather entering a new cycle from the tail of the transcript. Ribosomes that reach the end of the transcript and leave the last codon, release the completed polypeptide chain and temporarily detach from the transcript. Since these ribosomes do not translocate to next triplets, this step can be considered the last one of the process and the flux of ribosomes that leave the stop codon was used as starting point to infer the total number of ribosomes per transcript.
As general input parameters, we considered the organism specific codon usage bias values (downloaded from http://www.kazusa.or.jp/codon) and the kinetic constants of translation elongation (Supplementary File 1). As transcript specific input we used the following information: (i) the transcript sequence (from ENSEMBL 73) and (ii) the transcript expression level (from RNA-Seq data).
As mentioned in the ‘Introduction’ section, independently of the possible mechanisms giving rise to the ramp (Figure 2C), RiboAbacus includes the ramp effect with two tunable parameters: the ramp length and the ribosome slowdown rate. These parameters were optimized to minimize the distance between the distribution of ribosomes per transcript predicted by the model and the distribution experimentally obtained by AFM (see the following section).
See the ‘Use of RiboAbacus’ section for more details on how to use the software.
Training the model with Hek-293 transcriptome
Given the assumptions of the previous section, we optimized the unknown parameters linked to the ramp (length and slowdown rate), using the experimental distribution of the number of ribosomes per transcript obtained from AFM images of polysomes purified from human Hek-293 cells (Figure 3A and B). The experimental distribution of Hek-293 polysomes was hypothesized to be the sum of normal distributions that we fitted with Gaussian curves (Figure 3C). We obtained as best fit of the experimental dataset three curves with values 5.0 ± 1.3, 9.0 ± 2.4 and 15.0 ± 5.7 ribosomes per transcript (R2 = 0.997). This experimental distribution was taken as reference for training RiboAbacus.
Figure 3.
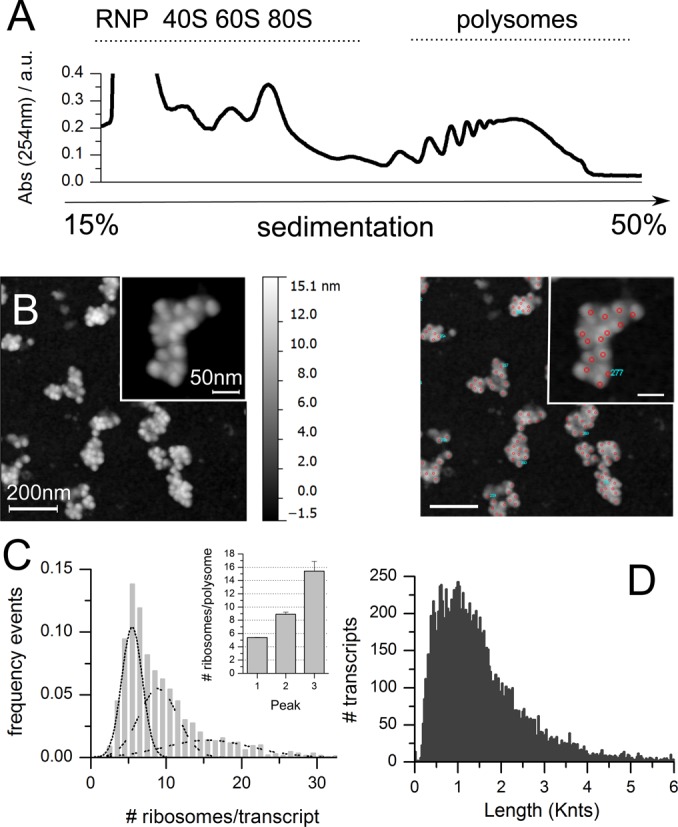
Training dataset: experimental determination of the number of ribosomes per transcript in Hek-293. (A) Example of polysomal profile of Hek-293 lysates. (B) Example of AFM image of Hek-293 polysomes after absorption on mica (left panel) and example of ribosome counting (right panel). (C) Distribution of the number of ribosomes per transcript for Hek-293 transcriptome, as determined from experimental AFM data. The distribution was fitted with three Gaussian curves, with means plotted in the inset (R2 = 0.997). The total number of polysomes considered is 2446, obtained from 20 independent images. (D) Nucleotide length distribution of transcript coding sequences (CDSs) in Hek-293, based on expressed transcripts (GEO ID: GSM936076).
To predict the distribution of ribosomes per transcript of the Hek-293 transcriptome, we used as input the expression levels of Hek-293 mRNAs determined by RNA-seq (GSM936076) and the corresponding transcript sequences. The transcriptome of Hek-293 consists of 14230 transcripts, whose distribution of the CDS lengths is shown in Figure 3D. To optimize the ramp parameters, we adopted a grid approach and selected 91 different combinations of the two parameters. We run the model for each combination, obtained the corresponding distribution of the number of ribosomes per transcript and compared it with the experimental one. Two examples of these comparisons are displayed in Figure 4A, without ramp (left panel) and with slowdown rate 60% and ramp length of 40 codons (right panel). The distance between experimental and predicted distributions was estimated as described in ‘Materials and Methods’ section. The procedure was repeated 100 times with 50–50 cross-validation (see ‘Materials and Methods’ section and Supplementary Figure S1).
Figure 4.
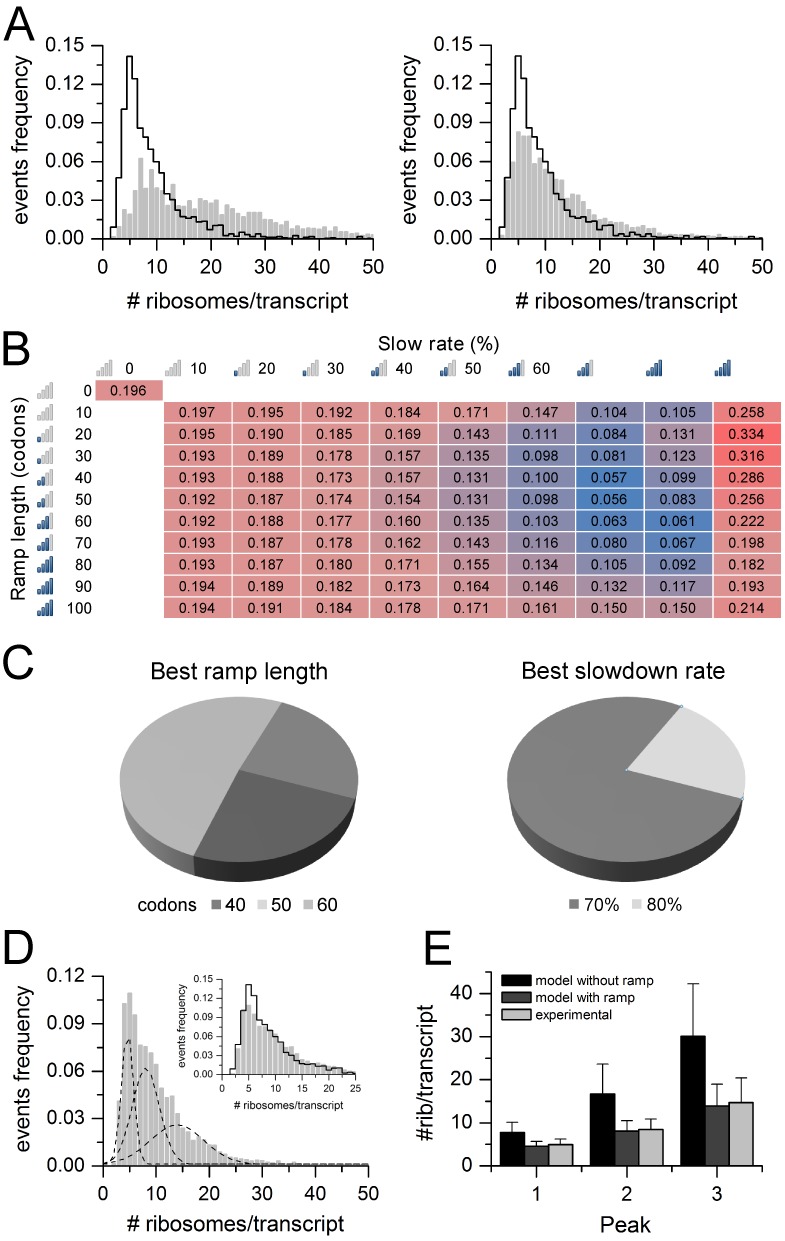
Optimization of model parameters in Hek-293. (A) Comparison between the experimental distribution (black line) and the predicted distribution (grey bars) of the number of ribosomes per transcript, setting the ramp length parameter to 0 (left panel) and to 40 codons with 60% slowdown rate (right panel). (B) Heatmap showing the average distance (100 cross validations) between the experimental and the predicted distribution of the number of ribosomes per transcript, varying the ramp length parameter (from 0 to 100 codons) and the ribosome slowdown rate parameter (from 0 to 90%). Higher distances are highlighted in red gradient, smaller distances in blue gradient. The minimum distance value is obtained with ramp length of 50 codons and ribosome slowdown rate of 70%. (C) Pie charts showing the results of 100 ramp parameters optimizations performed with 50–50 cross validations on the Hek-293 transcriptome. (D) Predicted distribution of the number of ribosomes per transcript, determined by RiboAbacus with optimized ramp parameters (ramp length 50 codons and ribosome slowdown 70%) fitted with three Gaussian curves. The inset shows the comparison with the experimental distribution (black line). (E) Bar plot showing the estimated means of the three Gaussian curves that fit the distribution of the number of ribosomes per transcript, according to experimental data (light grey), predictions from RiboAbacus with ramp length equal to 0 (black), predictions from RiboAbacus with optimized ramp parameters (dark grey).
The matrix of average distance values resulting from the combinations of the ramp parameters is displayed in Figure 4B. It is worth noting that without considering the ramp hypothesis in the model (i.e the ramp and the length parameters are equal to 0), the distance value (0.196) between the predicted and experimental distributions is high (Figure 4A, left panel). Similarly, the model with a slowdown rate of 90% displayed the maximum distance values within the matrix with the worst match with ramp length set at 20 codons (distance value 0.334). On the contrary, lower distance values were observed with slowdown rate ranging between 60 and 80% and ramp length between 20 and 80 codons. The best approximation within experimental data (distance = 0.056) was obtained in the case of ramp length equal to 50 codons and slowdown rate of 70% (Figure 4C and D; see also Supplementary File 2 for the complete RiboAbacus results). After fitting this distribution with three Gaussian curves similarly to what performed for the experimental data, the predictions nicely matched the experimental values. Computing the distance between the mean of the predicted and experimental Gaussian curves, we obtained differences <1 ribosome per transcript (Figure 4E). On the contrary, comparing the experimental means with those derived from the model with ramp length equal to 0 (black bars in Figure 4E), the differences between the predicted and experimental curves are 3 ribosomes per transcript for the first peak and up to 8-16 ribosomes per transcript for the other two. Interestingly, the absence of the ramp clearly overestimates the number of ribosomes per transcript as displayed in Figure 4E and Supplementary Figure S2. Moreover, the model without the ramp predicts a high coverage (∼70%) for both the short and long transcripts, in disagreement with what was observed in yeast in (40), whereas adding the ramp to RiboAbacus the relationship between these two parameters follows an exponential decay trend (Supplementary Figure S3A). It is worth noting that the optimized ramp length value closely matches what experimentally observed by ribosome profiling data (34). Given the results of the training, the two ramp parameters were set to 70% (ramp slowdown) and 50 codons (ramp length) in the following validations.
Feature analysis
To understand the contribution of each transcript feature to RiboAbacus predictions, we started running the model with CDS length as the only feature. Then we progressively added the following features, one at a time: expression level, codon usage bias and optimized ramp parameters. At each step we computed the distance between the resulting predicted distribution of the number of ribosomes per transcript and the experimental distribution. We defined the distribution similarity as 1-distance. In this way the similarity value is 1 if the distributions are identical and close to 0 if huge differences are present. The results of the analysis repeated 100 times with 50–50 cross-validation are displayed in Figure 5, showing a significant improvement in the fit of the model upon addition of each new feature. The inclusion of mRNAs abundances and the codon usage bias leads to an average increment of the similarity of 0.03 and 0.04, respectively. Interestingly, the ramp effect increases the similarity of 0.15, up to 0.95. It is noteworthy that only the combination of all features can properly predict the number of ribosomes. In fact, the removal of one feature leads to lower similarity values (see Supplementary Figure S4). Overall, these results pinpoint the importance of modelling a slowdown mechanisms, such as the ramp and/or initiation rates, and suggest that codon usage bias is not a major determinant of the number of ribosomes per transcript observed in human polysomes, as suggested in (51,52).
Figure 5.
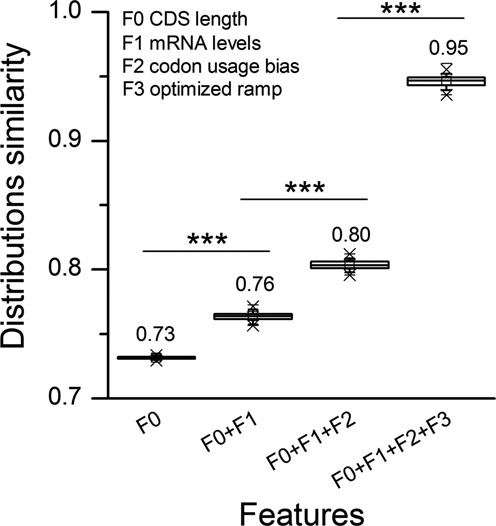
Contribution of transcript features to RiboAbacus predictions. Boxplot showing the similarities (calculated as 1-distance) between the predicted distribution of the number of ribosomes per transcript and the experimental distribution, progressively adding transcript features to the model (F0: CDS length, F1: mRNA level, F2: codon usage bias, F3: optimized ramp parameters). Similarities were calculated in 100 rounds of cross-validation. Statistical significances from Wilcoxon–Mann–Whitney test are shown: (***P-value < 0.001).
Model validation with MCF-7 transcriptome
To validate RiboAbacus, we employed the experimental distribution of the number of ribosomes per transcript obtained from the breast cancer carcinoma cell line MCF-7. The experimental distribution was determined from AFM images by counting the number of ribosomes per polysome after sucrose gradient sedimentation of cell lysates (Figure 6A) in the same way used for Hek-293.
Figure 6.
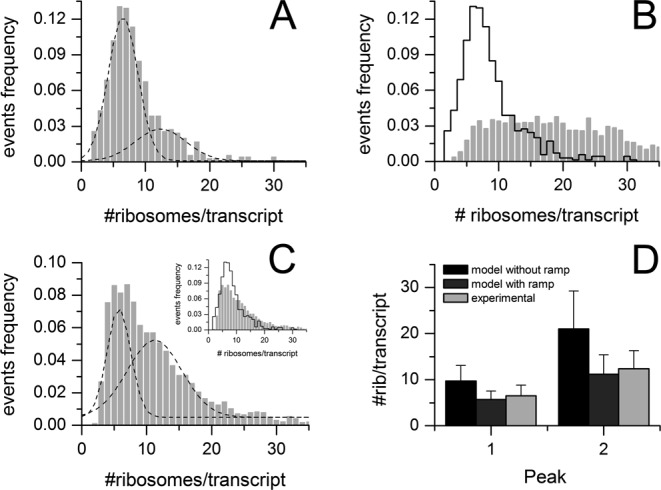
Validation of RiboAbacus in MCF-7. (A) Distribution of the number of ribosomes per transcript for MCF-7 transcriptome, as determined from experimental AFM data. The distribution was best fitted with two Gaussian curves (R2 = 0.996). (B) Comparison between the experimental distribution (black line) and RiboAbacus predicted distribution (grey bars) of the number of ribosomes per transcript, setting the ramp length parameter to 0. (C) Distribution of the number of ribosomes per transcript predicted by RiboAbacus with the previously optimized ramp parameters (length 50 codons and slowdown rate 70%) best fitted with two Gaussian curves. The inset shows the comparison with the experimental distribution (black line). (D) Bar plot showing the estimated means of the two Gaussian curves that fit the distribution of the number of ribosomes per transcript, according to experimental data (light grey), predictions from RiboAbacus with ramp length equal to 0 (black), predictions from RiboAbacus with optimized ramp parameters (dark grey).
We then run RiboAbacus, using the ramp parameters previously optimized in Hek-293 and the abundances of MCF-7 transcripts obtained from RNA-seq data (GSE48213, 29 087 transcripts) and the corresponding transcript sequences (Ensembl 73). Similarly to what observed during the training, the predicted distribution of the number of ribosomes per transcript without ramp (grey bars in Figure 6B) poorly matches the experimental distribution (black line Figure 6B). In this case, the distance between the two distributions is 0.234 (Supplementary Figure S5). The introduction of the optimized ramp parameters (length 50 codons and slowdown rate 70%, Figure 6C and Supplementary File 2) leads to a clear improvement of the prediction and a consequent decrease of the distance value to 0.099.
In the case of MCF-7, the experimental distribution was best fitted with two Gaussian curves (Figure 6A), with means of 6.5 ± 2.3 and 12.4 ± 3.9 ribosomes per transcript (R2 = 0.996). Comparing these values with the means of the two Gaussian curves obtained with the optimized parameters, we found a good agreement (Figure 6D). In fact, fitting the data obtained without the ramp, we observed a difference between the experimental and the predicted mean of ∼3 ribosomes per transcript for the first curve and 8 for the second. Similarly to what observed in Hek-293, the mean values of the optimized model better approximate the experimental means, with differences of around 1 ribosome per transcript. Noteworthy, the optimal ramp slowdown region is cell line independent and characterized by a length between 30 and 70 codons, with a slowdown rate ranging from 60 to 90% (see Figure 4B and Supplementary Figure S5). Overall, these results confirm the ability of the model to consistently estimate the number of ribosomes per transcript.
Model validation in the rabbit reticulocyte system
The use of AFM allowed us to precisely describe the composition, in term of ribosomes per transcript, of thousands of polysomes, i.e. to precisely count with single transcript resolution how many ribosomes are engaged in translation for transcripts expressed in cells. Even though this prediction originates from the most extensive census of ribosome numbers available in literature, AFM cannot recognize the identity of the corresponding transcripts when using a cell lysate. This means that we cannot associate to one specific transcript a specific number of ribosomes using a cell lysate, nor measure the abundance of each transcript in the images. To overcome this problem, we took advantage of the well-known in vitro translation system based on RRL lysates. In this system, unless treated with micrococcal nucleases, two proteins are preferentially produced: globin and lipoxygenase. Indeed, globin represents the great majority of synthesized proteins (71). In addition, given the difference between the length of the two transcripts, it is possible to isolate sucrose fractions that are highly enriched of polysomes formed by the globin transcript. We therefore used this system as additional validation model to count the number of ribosomes per transcript in a population composed of a known transcript. This model has the advantage of offering a single transcript validation of RiboAbacus predictions.
We purified rabbit reticulocyte polysomes by sucrose gradient fractionation (Figure 7A) and purified RNA along the gradient to identify by qPCR the sucrose fraction enriched in globin polysomes (Figure 7B). The polysome fraction with the peak of globin mRNA (arrow in Figure 7A and B) was analysed by AFM imaging (Figure 7C) to determine the experimental distribution of the number of ribosomes per transcript (Figure 7D). The experimental mean number of ribosomes per transcript (4.7 ± 0.89) was compared to the number predicted by RiboAbacus in absence or presence of the ramp parameters optimized in Hek-293. RiboAbacus predicted 9 ribosomes per globin transcript without the ramp assumption, and 4 ribosomes per transcript with the optimized ramp parameters (Figure 7D), a number very close to the mean of the experimental distribution. This transcript-specific validation further demonstrates that RiboAbacus is a powerful model for accurately predicting the number of ribosomes per transcript.
Figure 7.
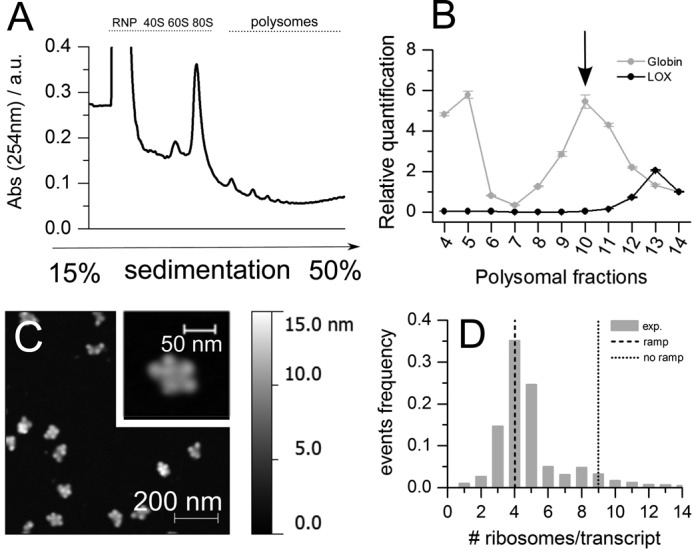
Validation of the model with the globin transcript in rabbit reticulocytes (RRL). (A) Representative absorbance profile for sucrose gradient sedimentation of rabbit reticulocyte lysates after incubation at 37°C for 10 min. (B) PCR quantification of globin and lipoxygenase (LOX) transcripts along the sucrose gradient fractions. The fraction with the highest abundance of the globin transcript is marked with a black arrow. This fraction was chosen for AFM imaging. (C) Example of AFM image of RRL polysomes after absorption on mica. (D) Comparison between the experimentally determined distribution of the number of ribosomes per transcript (♯ counted objects = 901; mean ♯ ribosomes/transcript 4.7 ± 0.8, in agreement with what observed in (22)), the number predicted with ramp length equal to 0 (♯ ribosomes/transcript = 9, dotted line) and with the optimized ramp parameters (♯ ribosomes/transcript = 4, dashed line).
RiboAbacus improves predictions of proteome data from transcriptome data
The great advantages of modelling translation are mainly the possibility to (i) predict protein levels starting from transcript abundances and (ii) obtain information about how mRNA determinants or other parameters can contribute in defining protein production. The quantification of protein levels is sometimes challenging for lowly expressed proteins, difficult samples such as tissues, biopsies, single cells and subcellular compartments such as axons. The experimental detection of transcript levels is far more easy and cost effective, but it has been shown that mRNAs levels poorly correlate with protein levels in several organisms (53,72). We wondered whether this discrepancy between transcriptome and proteome could be reduced by using the number of ribosomes per transcript predicted by RiboAbacus (Supplementary File 2).
To prove this, we selected three studies where protein and transcript abundances have been experimentally determined (53–55) and we checked whether RiboAbacus predictions were able to increase the correlation between experimental transcriptomes and proteomes. For each dataset we computed the predicted number of ribosomes per mRNA and obtained the corresponding ribosome occupancy values using the ramp parameters optimized in Hek-293 (Supplementary File 2). Plotting the ribosome occupancy as a function of the corresponding mRNAs length, we observed that the relationship between these two parameters follows an exponential decay trend (see Supplementary Figure S3A). This means that the shorter the transcript, the higher the ribosome occupancy, supporting previous observations (34,40). To calculate the TE we introduced a correction parameter that takes into account this effect, as previously suggested in (34). In this way we obtained a cTE. For each dataset we then computed the correlation between the predicted cTEs and the experimental protein levels. To measure whether RiboAbacus significantly improved the correlation between transcriptomes and proteomes, we performed the Williams's tests (Table 1). In parallel this comparison was repeated running RiboAbacus without ramp parameters, to understand the role of slowdown effects also in this context.
Table 1. List of transcriptome/proteome and cTE/proteome correlations with and without the ramp hypothesis for three different transcriptome/proteome datasets.
| Cell line | Number of | Transcriptome/ | cTE (ramp)/ | Correlation | cTE (no ramp)/ | Correlation |
|---|---|---|---|---|---|---|
| transcripts | proteome | proteome | increase | proteome | increase | |
| correlation | correlation | P-value (ramp) | correlation | P-value (no ramp) | ||
| DAOY | 904 | 0.425 | 0.531 | 3.50·10−3 | 0.468 | 0.254 |
| Motoneuron | 5600 | 0.473 | 0.532 | 2.92·10−5 | 0.480 | 0.631 |
| NIH3T3 | 5830 | 0.615 | 0.655 | 2.95·10−4 | 0.655 | 6.04·10−4 |
The number of transcripts involved and the P-values from Williams's tests are also reported for each analysis.
The first transcriptome/proteome dataset used was obtained from human medulloblastoma cell line DAOY (53). In this case, the experimentally measured protein quantities better correlate with the predicted cTE (R = 0.531) than with transcript levels (R = 0.425). The increase of the correlation is statistically significant (P-value 3.5·10−3), suggesting that RiboAbacus better approximates protein production (Figure 8A and B). Moreover, the correlation calculated without the ramp hypothesis (R = 0.468, P-value = 0.25) is not significantly higher than the experimental correlation, confirming the important role of slowdown mechanisms in correctly modelling the process of protein production (Figure 8C). Similarly, we applied RiboAbacus to primary mouse motoneurons (54). Again, RiboAbacus significantly increased the correlation between transcript and protein levels using the optimized ramp parameters (R = 0.532 versus R = 0.473, P-value = 2.92·10−5), but not without the ramp hypothesis (R = 0.480, P-value = 0.63). Finally, we run RiboAbacus on a third transcriptome-proteome dataset from NIH3T3 mouse fibroblasts (55). Using this dataset the increase in correlation with optimized ramp parameters is smaller than in previous cases, (R = 0.615 versus R = 0.655, P-value = 2.95·10−4). In contrast to previous examples, the increase in correlation was significant also without the ramp hypothesis (R = 0.653, P-value = 6.01·10−4). This slight increase could be due to the higher initial correlation between the experimental transcriptome and proteome.
Figure 8.
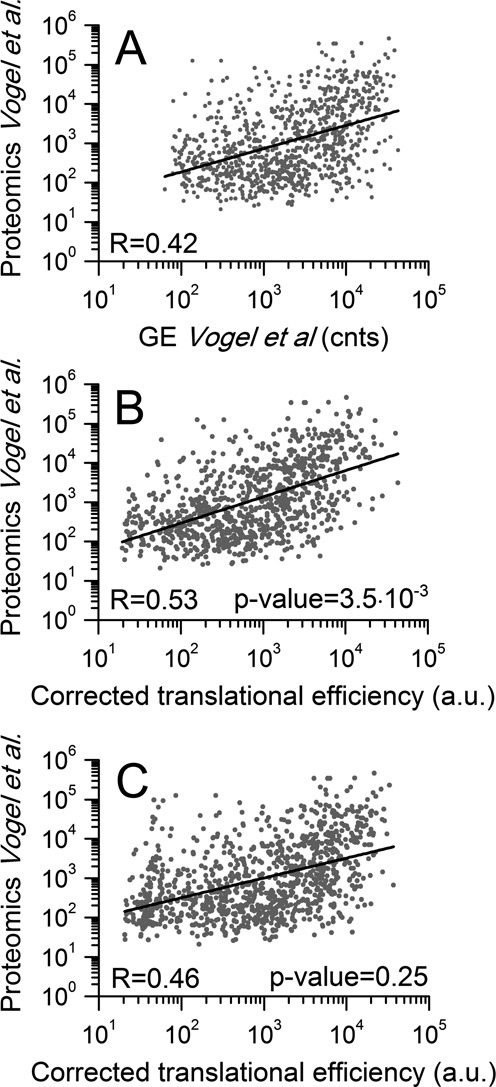
Improved correlation between transcript and protein abundances using translation efficiencies (TEs) calculated by RiboAbacus. (A) Scatterplot of experimental transcript abundances versus protein abundances (53). (B) Scatterplot of cTEs calculated by RiboAbacus with the optimized ramp parameters versus protein abundances. (C) Scatterplot of cTEs calculated by RiboAbacus with ramp equal to 0 versus protein abundances.
Use of RiboAbacus
RiboAbacus is coded in C and available in GitHub at http://fabiolauria.github.io/RiboAbacus/. Two input files are needed: a list of transcript CDSs with related expression levels and a list of organism-specific codon usage bias values. The transcript file must contain for each transcript two lines: the first reporting the expression level, along with general information about the transcripts (gene ID, transcript ID, protein ID and protein level) and the second reporting the CDS. The codon usage file must contain the list of codons and the corresponding codon usage bias values, arranged in two columns. We provide three options for Homo sapiens (default), Mus musculus and S. cerevisiae. Note that the set of kinetic constants is fixed (see Supplementary File 1). RiboAbacus outputs two files: the first contains for each transcript the number of ribosomes, the ribosome occupancy and the TE; the second file contains the frequencies of the number of ribosomes per transcript, that can be used to build the transcriptome-wide distribution. For further information please refer to the Readme file in GitHub.
A run of RiboAbacus on an entire transcriptome takes less than a minute on a standard personal computer.
DISCUSSION
We developed RiboAbacus, a model trained on experimental imaging-derived data, able to quickly and accurately predict the steady state number of ribosomes per transcript in entire transcriptomes.
The number of ribosomes bound to a mRNA directly contributes to the final amount of the corresponding protein in cells, since ribosomes are the molecular machines responsible for protein synthesis. Therefore, understanding the contribution of the number of ribosomes bound to a transcript is of major importance for unravelling the impact of translational controls and possibly using transcriptome data to predict TEs. Nevertheless, measuring numbers of ribosomes bound to transcripts is challenging, leading researchers to neglect this important parameter in the development of mathematical models of translation.
The distributions of the number of ribosomes per transcript obtained from AFM images, that underpins RiboAbacus predictions, have been compared with polysome profiling in yeast, using the well known dataset from (40). We were able to show that AFM enables to reach an unparalleled resolution in determining the number of ribosomes per transcript. On the other hand, high-throughput approaches based on hybridization or sequencing allow the identification of transcripts, that is not possible in AFM. Nevertheless, a general agreement for single transcript predictions between arrays and RiboAbacus has been shown (Supplementary Figure S6).
RiboAbacus takes as input a list of transcripts whose sequence and expression levels are known, and the organism codon usage bias and the translational kinetic constants. As experimental reference for tuning the model output, we took advantage of experimental data obtained from AFM images of purified polysomes that uniquely allows the precise count of ribosomes per transcript. Without additional parameters, RiboAbacus predictions overestimate the number of ribosomes per transcript (Figure 4A, left panel). Such overestimation has already been observed in other models, indicating that codon usage alone is not sufficient to account for ribosome dynamics (29,32).
We thus took into consideration the existence of 5′ slowdown mechanisms that may give rise to the so-called ramp described in yeast by ribosome protecting assays (34). The possible biological reasons for the existence of the ramp are still under debate and the conclusions discordant (18,31,34,36,49–52). A hypothesis is that regions rich of rare codons could affect the waiting time for the correct tRNA binding to ribosomes (51,52). In addition, the presence of RNA structures, produced by intramolecular base pairing, could also induce a slowdown movement of mRNA helicases (35) and a consequent stalling of ribosomes. Most probably, these two features contribute simultaneously to final ramp effects (31,50). Regardless of the specific mechanism involved, we decided to model the ramp effect by introducing in RiboAbacus two ramp parameters: ramp length and ramp slowdown rate. We optimized these parameters in Hek-293, computing the best fit with the experimental data. Interestingly, the optimal value of the ramp length (50 codons; Figure 3B) is in agreement with data available in literature (34,35,51,52). Importantly, our results highlight that codon usage bias plays a minor role than the ramp hypothesis in the accuracy of prediction (Figure 5). Therefore, our predictions indicate that the ramp, or any slowdown events taking place at the beginning of the CDS, plays an important role in determining the overall number of ribosomes per transcript.
Another confirmation of the importance of slowdown mechanisms can be observed inspecting the ribosome occupancy or coverage (i.e the percentage of nucleotides covered by ribosomes). Using the optimized ramp parameters we could observe that the ribosome occupancy per mRNA was inversely proportional to the length of the CDS, similarly to what was experimentally observed in other studies (34,40). It is then possible that additional translation mechanisms, such as different initiation rates or ribosomes drop off (20,21), can play a role to avoid the loading of high number of ribosomes on long transcripts keeping the mRNA coverage at a low level. Indeed, we found that ribosome occupancy is almost constant for transcripts longer than 2000 nt even if the total number of ribosomes per mRNA increases with their length.
Using RiboAbacus, we tried to understand the contribution that the number of predicted ribosomes per transcript may give to explain the total protein level in cells. In fact, mRNA abundances, measured by microarray or by next-generation sequencing (NGS) techniques, are widely used as proxies for protein measurements, but a general poor correlation between the experimental measures of mRNA and protein levels has been reported in many works in mammalian cells. For example, (53) showed that the mRNA abundance in cells may account for approximately one-third of the downstream protein production yield (R2 =0.29). Computational approaches have been attempted in order to identify and select mRNA features that could bridge the gap between transcriptome and proteome measurements by employing multivariate linear regression models. In S. cerevisiae, a set of transcript-specific features (including codon usage, transcript length, ribosome density, evolutionary conservation) was selected to maximally increase the prediction of protein levels from mRNA levels (from 0.69 to 0.76 in (73), from 0.76 to 0.86 in (74)). In mammalian systems, Vogel and co-workers (53) identified 25 mRNA features that increased the coefficient of determination from 0.29 to 0.67 on a subset of 512 transcripts. Thus, we asked what could be the overall contribution of the number of ribosomes uploaded on transcripts in determining the proteome. RiboAbacus is able to estimate the cTE for each transcript given its abundance. We found that the number of ribosomes per transcript significantly increased the experimental correlation in three different datasets: from 0.42 to 0.53 in human medulloblastoma cell line DAOY, from 0.47 to 0.53 in mouse motor neurons and from 0.61 to 0.66 in mouse fibroblasts NIH3T3. Interestingly, without the introduction of the ramp parameters, the increase in correlation is considerably lower and, with the exception of the last dataset, not significant. This result is an additional clue that the slowdown of ribosomes at the 5′ of the CDS should play a pivotal role in regulating the final protein abundance. According to the improved correlation provided by RiboAbacus, up to 10% of protein levels can be explained by the number of ribosomes per transcript.
While the improvement in explanatory power afforded by RiboAbacus is both sizeable and statistically significant, there remains a considerable amount of proteomic variability unaccounted for, pointing to the need of additional translational regulation mechanisms. Future work is needed to better understand how additional translational controls could be included in mathematical models to improve the correlation between transcript and protein levels.
RiboAbacus stands as a simple and immediate approach that may be useful to deal with problems that we have with other methods for studying translation. In fact it can predict numbers of bound ribosomes and transcript-specific translation properties solely from global gene expression. As such, RiboAbacus can be applied to any gene-expression dataset, requiring much fewer experimental resources than polysome profiling methods and representing a quick complementary method to more expensive and demanding experimental techniques to study translational control of gene expression. It can also be used to predict protein levels and translational properties in systems (e.g. biopsies, single cells, subcellular compartment etc.) where a proteomic quantification is still challenging.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
AXonomIX research project financed by the Provincia Autonoma di Trento, Italy; European Research Council [MLCS 306999 to G.S.]. Funding for open access charge: International AXonomIX Research Project financed by the Provincia Autonoma di Trento, Italy.
Conflict of interest statement. None declared.
REFERENCES
- 1.Sonenberg N., Hinnebusch A.G. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aitken C.E., Lorsch J.R. A mechanistic overview of translation initiation in eukaryotes. Nat. Struct. Mol. Biol. 2012;19:568–576. doi: 10.1038/nsmb.2303. [DOI] [PubMed] [Google Scholar]
- 3.Jackson R.J., Hellen C.U., Pestova T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010;324:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ingolia N.T., Lareau L.F., Weissman J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stadler M., Fire A. Wobble base-pairing slows in vivo translation elongation in metazoans. RNA. 2011;17:2063–2073. doi: 10.1261/rna.02890211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li G.-W., Oh E., Weissman J.S. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012;484:538–541. doi: 10.1038/nature10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nürenberg E., Tampé R. Tying up loose ends: ribosome recycling in eukaryotes and archaea. Trends Biochem. Sci. 2013;38:64–74. doi: 10.1016/j.tibs.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 8.des Georges A., Hashem Y., Unbehaun A., Grassucci R.A., Taylor D., Hellen C.U., Pestova T.V., Frank J. Structure of the mammalian ribosomal pre-termination complex associated with eRF1·eRF3·GDPNP. Nucleic Acids Res. 2014;42:3409–3418. doi: 10.1093/nar/gkt1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Novoa E.M., Ribas de Pouplana L. Speeding with control: codon usage, tRNAs, and ribosomes. Trends Genet. 2012;28:574–581. doi: 10.1016/j.tig.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 10.Lynn D.J., Singer G.A., Hickey D.A. Synonymous codon usage is subject to selection in thermophilic bacteria. Nucleic Acids Res. 2002;30:4272–4277. doi: 10.1093/nar/gkf546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gray N.K., Hentze M.W. Regulation of protein synthesis by mRNA structure. Mol. Biol. Rep. 1994;19:195–200. doi: 10.1007/BF00986961. [DOI] [PubMed] [Google Scholar]
- 12.Kudla G., Murray A.W., Tollervey D., Plotkin J.B. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324:255–258. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pircher A., Bakowska-Zywicka K., Schneider L., Zywicki M., Polacek N. An mRNA-derived noncoding RNA targets and regulates the ribosome. Mol. Cell. 2014;54:147–155. doi: 10.1016/j.molcel.2014.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shalgi R., Hurt J.A., Krykbaeva I., Taipale M., Lindquist S., Burge C.B. Widespread regulation of translation by elongation pausing in heat shock. Mol. Cell. 2013;49:439–452. doi: 10.1016/j.molcel.2012.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friend K., Campbell Z.T., Cooke A., Kroll-Conner P., Wickens M.P., Kimble J. A conserved PUF-Ago-eEF1A complex attenuates translation elongation. Nat. Struct. Mol. Biol. 2012;19:176–183. doi: 10.1038/nsmb.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolin S.L., Walter P. Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J. 1988;7:3559–3569. doi: 10.1002/j.1460-2075.1988.tb03233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ingolia N.T., Brar G.A., Rouskin S., McGeachy A.M., Weissman J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah P., Ding Y., Niemczyk M., Kudla G., Plotkin J.B. Rate-limiting steps in yeast protein translation. Cell. 2013;153:1589–1601. doi: 10.1016/j.cell.2013.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noller H.F. Evolution of protein synthesis from an RNA World. Cold Spring Harb. Perspect. Biol. 2012;4:a003681. doi: 10.1101/cshperspect.a003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herr A.J., Wills N.M., Nelson C.C., Gesteland R.F., Atkins J.F. Drop-off during ribosome hopping. J. Mol. Biol. 2001;311:445–452. doi: 10.1006/jmbi.2001.4899. [DOI] [PubMed] [Google Scholar]
- 21.Cruz-Vera L.R., Magos-Castro M.A., Zamora-Romo E., Guarneros G. Ribosome stalling and peptidyl-tRNA drop-off during translational delay at AGA codons. Nucleic Acids Res. 2004;32:4462–4468. doi: 10.1093/nar/gkh784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warner J.R., Rich A., Hall C.E. Electron microscope studies of ribosomal clusters synthesizing hemoglobin. Science. 1962;138:1399–1403. doi: 10.1126/science.138.3548.1399. [DOI] [PubMed] [Google Scholar]
- 23.Palade G.E. A small particulate component of the cytoplasm. J. Biophys. Biochem. Cytol. 1955;1:59–68. doi: 10.1083/jcb.1.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wettstein F.O., Staehelin T., Noll H. Ribosomal aggregate engaged in protein synthesis: characterization of the ergosome. Nature. 1963;197:430–435. doi: 10.1038/197430a0. [DOI] [PubMed] [Google Scholar]
- 25.Gerst I., Levine S.N. Kinetics of protein synthesis by polyribosomes. J. Theor. Biol. 1965;9:16–36. doi: 10.1016/0022-5193(65)90054-8. [DOI] [PubMed] [Google Scholar]
- 26.MacDonald C.T., Gibbs J.H. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Biopolymers. 1969;7:707–725. [Google Scholar]
- 27.Gilchrist M.A., Wagner A. A model of protein translation including codon bias, nonsense errors, and ribosome recycling. J. Theor. Biol. 2006;239:417–434. doi: 10.1016/j.jtbi.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 28.Mitarai N., Sneppen K., Pedersen S. Ribosome collisions and translation efficiency: optimization by codon usage and mRNA destabilization. J. Theor. Biol. 2008;382:236–245. doi: 10.1016/j.jmb.2008.06.068. [DOI] [PubMed] [Google Scholar]
- 29.Zouridis H., Hatzimanikatis V. A model for protein translation: polysome self-organization leads to maximum protein synthesis rates. Biophys. J. 2007;92:717–730. doi: 10.1529/biophysj.106.087825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reuveni S., Meilijson I., Kupiec M., Ruppin E., Tuller T. Genome-scale analysis of translation elongation with a ribosome flow model. PLoS Comput. Biol. 2011;7:e1002127. doi: 10.1371/journal.pcbi.1002127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dana A., Tuller T. Determinants of translation elongation speed and ribosomal profiling biases in mouse embryonic stem cells. PLoS Comput. Biol. 2012;8:e1002755. doi: 10.1371/journal.pcbi.1002755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ciandrini L., Stansfield I., Romano M.C. Ribosome traffic on mRNAs maps to gene ontology: genome-wide quantification of translation initiation rates and polysome size regulation. PLoS Comput. Biol. 2013;9:e1002866. doi: 10.1371/journal.pcbi.1002866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Von der Haar T. Mathematical and Computational Modelling of Ribosomal Movement and Protein Synthesis: an overview. Comput. Struct. Biotechnol. J. 2012;1:e20120400. doi: 10.5936/csbj.201204002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tuller T., Veksler-Lublinsky I., Gazit N., Kupiec M., Ruppin E., Ziv-Ukelson M. Composite effects of gene determinants on the translation speed and density of ribosomes. Genome Biol. 2011;12:R110. doi: 10.1186/gb-2011-12-11-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zupanic A., Meplan C., Grellscheid S.N., Mathers J.C., Kirkwood T.B., Hesketh J.E., Shanley D.P. Detecting translational regulation by change point analysis of ribosome profiling data sets. RNA. 2014;20:1507–1518. doi: 10.1261/rna.045286.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bohnert R., Rätsch G. rQuant. web: a tool for RNA-Seq-based transcript quantitation. Nucleic Acids Res. 2010;38:W348–W351. doi: 10.1093/nar/gkq448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hansen K.D., Brenner S.E., Dudoit S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010;38:e131. doi: 10.1093/nar/gkq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts A., Trapnell C., Donaghey J., Rinn J.L., Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2010;12:R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arava Y., Wang Y., Storey J.D., Liu C.L., Brown P.O., Herschlag D. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3889–3894. doi: 10.1073/pnas.0635171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MacKay V.L., Li X., Flory M.R., Turcott E., Law G.L., Serikawa K.A., Xu X.L., Hookeun L., Goodlett D.R., Aebersold R., et al. Gene expression analyzed by high-resolution state array analysis and quantitative proteomics response of yeast to mating pheromone. Mol. Cell. Proteomics. 2004;3:478–489. doi: 10.1074/mcp.M300129-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Darnell J.C., Van Driesche S.J., Zhang C., Hung K. Y.S., Mele A., Fraser C.E., Stone E.F., Chen C., Fak J.J., Chi S.W., et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qin X., Ahn S., Speed T.P., Rubin G.M. Global analyses of mRNA translational control during early Drosophila embryogenesis. Genome Biol. 2007;8:R63. doi: 10.1186/gb-2007-8-4-r63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Viero G., Lunelli L., Passerini A., Bianchini P., Gilbert R.J., Bernabò P., Tebaldi T., Diaspro A., Pederzolli C., Quattrone A. Three distinct ribosome assemblies modulated by translation are the building blocks of polysomes. J. Cell. Biol. 2015;208:581–596. doi: 10.1083/jcb.201406040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brandt F., Etchells S.A., Ortiz J.O., Elcock A.H., Hartl F.U., Baumeister W. The native 3D organization of bacterial polysomes. Cell. 2009;136:261–271. doi: 10.1016/j.cell.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 46.Brandt F., Carlson L.-A., Hartl F., Baumeister W., Grünewald K. The three-dimensional organization of polyribosomes in intact human cells. Mol. Cell. 2010;39:560–569. doi: 10.1016/j.molcel.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 47.Afonina Z.A., Myasnikov A.G., Shirokov V.A., Klaholz B.P., Spirin A.S. Formation of circular polyribosomes on eukaryotic mRNA without cap-structure and poly (A)-tail: a cryo electron tomography study. Nucleic Acids Res. 2014;42:9461–9469. doi: 10.1093/nar/gku599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Myasnikov A.G., Afonina Z.A., Ménétret J.-F., Shirokov V.A., Spirin A.S., Klaholz B.P. The molecular structure of the left-handed supra-molecular helix of eukaryotic polyribosomes. Nat. Commun. 2014;5 doi: 10.1038/ncomms6294. doi:10.1038/ncomms6294. [DOI] [PubMed] [Google Scholar]
- 49.Tuller T., Zur H. Multiple roles of the coding sequence 5’ end in gene expression regulation. Nucleic Acids Res. 2015;43:13–28. doi: 10.1093/nar/gku1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bentele K., Saffert P., Rauscher R., Ignatova Z., Blüthgen N. Efficient translation initiation dictates codon usage at gene start. Mol. Syst. Biol. 2013;9 doi: 10.1038/msb.2013.32. doi:10.1038/msb.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Q., Qu H.-Q. Human coding synonymous single nucleotide polymorphisms at ramp regions of mRNA translation. PloS One. 2013;8:e59706. doi: 10.1371/journal.pone.0059706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tuller T., Carmi A., Vestsigian K., Navon S., Dorfan Y., Zaborske J., Pan T., Dahan O., Furman I., Pilpel Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010;141:344–354. doi: 10.1016/j.cell.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 53.Vogel C., Abreu Rde S., Ko D., Le S.Y., Shapiro B.A., Burns S.C., Sandhu D., Boutz D.R., Marcotte E.M., Penalva L.O. Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol. Syst. Biol. 2010;6 doi: 10.1038/msb.2010.59. doi:10.1038/msb.2010.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hornburg D., Drepper C., Butter F., Meissner F., Sendtner M., Mann M. Deep proteomic evaluation of primary and cell line motoneuron disease models delineates major differences in neuronal characteristics. Mol. Cell Proteomics. 2014;13:3410–3420. doi: 10.1074/mcp.M113.037291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 56.Jackson R.J., Hunt T. Preparation and use of nuclease-treated rabbit reticulocyte lysates for the translation of eukaryotic messenger RNA. Methods Enzymol. 1983;96:50–74. doi: 10.1016/s0076-6879(83)96008-1. [DOI] [PubMed] [Google Scholar]
- 57.Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Williams E.J. Regression Analysis. NY: Wiley; 1959. [Google Scholar]
- 60.Steiger J.H. Tests for comparing elements of a correlation matrix. Psychol. Bull. 1980;87:245–251. [Google Scholar]
- 61.Arava Y., Boas F.E., Brown P.O., Herschlag D. Dissecting eukaryotic translation and its control by ribosome density mapping. Nucleic Acids Res. 2005;33:2421–2432. doi: 10.1093/nar/gki331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gilchrist M.A. Combining models of protein translation and population genetics to predict protein production rates from codon usage patterns. Mol. Biol. Evol. 2007;24:2362–2372. doi: 10.1093/molbev/msm169. [DOI] [PubMed] [Google Scholar]
- 63.Warner J.R. The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 1999;24:437–440. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- 64.Frank J., Gonzalez R.L. Jr. Structure and dynamics of a processive Brownian motor: the translating ribosome. Annu. Rev. Biochem. 2010;79:381–412. doi: 10.1146/annurev-biochem-060408-173330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wohlgemuth I., Pohl C., Rodnina M.V. Optimization of speed and accuracy of decoding in translation. EMBO J. 2010;29:3701–3709. doi: 10.1038/emboj.2010.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zouridis H., Hatzimanikatis V. Effects of codon distributions and tRNA competition on protein translation. Biophys. J. 2008;95:1018–1033. doi: 10.1529/biophysj.107.126128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bilgin N., Claesens F., Pahverk H., Ehrenberg M. Kinetic properties of Escherichia coli ribosomes with altered forms of S12. J. Mol. Biol. 1992;224:1011–1027. doi: 10.1016/0022-2836(92)90466-w. [DOI] [PubMed] [Google Scholar]
- 68.Rodnina M.V., Pape T., Fricke R., Kuhn L., Wintermeyer W. Initial binding of the elongation factor Tu· GTP· aminoacyl-tRNA complex preceding codon recognition on the ribosome. J. Biol. Chem. 1996;271:646–652. doi: 10.1074/jbc.271.2.646. [DOI] [PubMed] [Google Scholar]
- 69.Pape T., Wintermeyer W., Rodnina M.V. Complete kinetic mechanism of elongation factor Tu-dependent binding of aminoacyl-tRNA to the A site of the E. coli ribosome. EMBO J. 1998;17:7490–7497. doi: 10.1093/emboj/17.24.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Savelsbergh A., Katunin V.I., Mohr D., Peske F., Rodnina M.V., Wintermeyer W. An elongation factor G-induced ribosome rearrangement precedes tRNA-mRNA translocation. Mol. Cell. 2003;11:1517–1523. doi: 10.1016/s1097-2765(03)00230-2. [DOI] [PubMed] [Google Scholar]
- 71.Soto Rifo R., Ricci E.P., Decimo D., Moncorge O., Ohlmann T. Back to basics: the untreated rabbit reticulocyte lysate as a competitive system to recapitulate cap/poly(A) synergy and the selective advantage of IRES-driven translation. Nucleic Acids Res. 2007;35:e121. doi: 10.1093/nar/gkm682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ghazalpour A., Bennet B., Petyuk V.A., Orozco L., Hagopian R., Mungrue I.N., Farber C.R., Sinsheimer J., Kang H.M., Furlotte N., et al. Comparative analysis of proteome and transcriptome variation in Mouse. PLoS Genet. 2011;7:e1001393. doi: 10.1371/journal.pgen.1001393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tuller T., Kupiec M., Ruppin E. Determinants of protein abundance and translation efficiency in S. cerevisiae. PLoS Comput. Biol. 2007;3:e248. doi: 10.1371/journal.pcbi.0030248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gunawardana Y., Niranjan M. Bridging the gap between transcriptome and proteome measurements identifies post-translationally regulated genes. Bioinformatics. 2013;29:3060–3066. doi: 10.1093/bioinformatics/btt537. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.