


Abstract
CRISPR/Cas9 has emerged as a versatile genome-engineering tool that relies on a single guide RNA (sgRNA) and the Cas9 enzyme for genome editing. Simple, fast and economical methods to generate sgRNAs have made targeted mutagenesis routine in cultured cells, mice, zebrafish and other model systems. Pre-screening of sgRNAs for target efficacy is desirable both for successful mutagenesis and minimizing wasted animal husbandry on targets with poor activity. Here, we describe an easy, quick and cost-effective fluorescent polymerase chain reaction (PCR)-based method, CRISPR Somatic Tissue Activity Test (CRISPR-STAT), to determine target-specific efficiency of sgRNA. As a proof of principle, we validated our method using 28 sgRNAs with known and varied levels of germline transmission efficiency in zebrafish by analysis of their somatic activity in injected embryos. Our data revealed a strong positive correlation between the fluorescent PCR profiles of the injected embryos and the germline transmission efficiency. Furthermore, the assay was sensitive enough to evaluate multiplex gene targeting. This method is easy to implement by laboratories with access to a capillary sequencer. Although we validated the method using CRISPR/Cas9 and zebrafish, it can be applied to other model systems and other genome targeting nucleases.
INTRODUCTION
Recent advances in genome editing by zinc finger nucleases (ZFNs), TAL effector nucleases (TALENs) and clustered regularly interspersed palindromic repeat/Cas9 nucleases (CRISPRs) have made it possible to perform targeted mutagenesis in many systems, including zebrafish (1–7). While assembly of target-specific ZFNs and TALENs have limitations due to design constraints, cost and/or laborious protocols (8–11), CRISPR/Cas9 requires design of a single guide RNA (sgRNA) that can be synthesized quickly and at very low cost (12,13). Furthermore, the target sequence for CRISPR/Cas9 is flexible, limited only by the requirement of a protospacer adjacent motif (PAM site) (14,15). Thus it is straightforward to rapidly design and generate sgRNAs for multiple targets. However, all sgRNAs do not exhibit similar levels of activity in generating mutations at the target site (16). Prediction of activity by computational methods may not be reliable for all applications as the criteria are derived from limited, context specific data (17,18). Thus a rapid experimental validation for the level of any given sgRNA activity is essential.
A number of methods have been developed to evaluate activity of targeting nucleases including CRISPR/Cas9 (19–23). The ‘gold standard’ method to determine the level of somatic activity is by polymerase chain reaction (PCR) of the target region followed by cloning and sequencing of a large number of clones (21,22). Although this method has high sensitivity and specificity to determine the activity level, it is both expensive and labor-intensive. Simple mismatch-based assays such as high resolution melt analysis (HRMA) and DNA cleavage by Surveyor or T7 endonuclease have poor specificity in organisms such as zebrafish that possess a high rate of polymorphism in the genome that cause false positive results (20,24–25) and anecdotally in our hands has a tendency to underestimate activity. Similarly, a restriction fragment length polymorphism assay is limited by the choice of target sites that contain an appropriate restriction enzyme site. The quantitative PCR based protocol developed by Yu et al. (23) is a feasible method to detect somatic activity. However, it might be difficult to design robust primers overlapping with the predicted cut site in every case. Recently two sequence-based methods, termed TIDE and CRISPR-GA, were developed for quantitative assessment of sgRNA activity in the cell culture experiments (26,27). However, both methods have limitations in their application, such as TIDE requiring high quality sequence reads of 500–1500 bp in length that may be feasible for only a small subset of targets. For the majority of targets, the exon sizes are small (<300 bp) and the flanking intronic sequences are highly polymorphic containing both SNPs and insertions or deletions (indels). CRISPR-GA requires next generation sequencing data that can be both expensive and out of reach for many small laboratories aiming to generate mutants in a small number of genes. Currently there are no assays that can be easily used for all model systems.
In zebrafish, the experimental procedure for targeted mutagenesis requires microinjections of the nuclease of choice (ZFNs, TALENS or CRISPR/Cas9) in one-cell stage embryos, growing them to adulthood (∼three months) and screening their progeny for indels at the target site (22,28). Pre-screening for activity saves time and costs associated with the animal husbandry and founder screening. In our experience with ZFNs and TALENs, the somatic activity in injected embryos has been a good indicator of the success of generating genetic mutants (22). These factors prompted us to explore the utility of using the fluorescent PCR method we developed previously (22) to evaluate somatic activity of the injected nuclease. We evaluated 28 sgRNAs with known germline transmission efficiency in zebrafish by fluorescent PCR of the injected embryos at 48 hours post fertilization (hpf) (13) and could accurately determine their potential for generating indels at the target site.
MATERIALS AND METHODS
Ethics statement and zebrafish lines used
The zebrafish work was performed under animal study protocol # G05–5 approved by the National Human Genome Research Institute Animal Care and Use Committee at the National Institutes of Health (OLAW Assurance # A-4149–01). Zebrafish were housed in accordance with the Guide for the Care and Use of Laboratory Animals of National Institutes of Health in an AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care) accredited facility. All zebrafish handling, embryo care and microinjections were performed as described in the Zebrafish Book (29). All experiments were performed in the wild-type (WT) fish of genetic strain TAB5.
Selection of sgRNA targets for validation
We used a sgRNA targeting the tyr gene for the proof of principle experiment since its somatic activity can be detected as a pigmentation phenotype in the injected embryos (12). For validation experiments, we selected sgRNAs targeting 28 genes (Table 1, Supplementary Table S1) based on their germline transmission efficiency from our previous study (13).
Table 1. Correlation of CRISPR-STAT data to germline transmission rates for 28 sgRNAs.
| Gene | Percentage of germline transmitting foundersa | Results from CRISPR-STAT | |
|---|---|---|---|
| Fold change | Fold change | ||
| espnlb | 100 | yes | 1500.10 |
| eya4 | 100 | yes | 812.90 |
| grhl2a | 100 | yes | 4623.64 |
| mgea5 | 100 | yes | 5.30 |
| msrb3 | 100 | yes | 1710.94 |
| myo15aa | 100 | yes | 7.16 |
| pou4f3 | 100 | yes | 4000.08 |
| prdx3 | 100 | yes | 4041.19 |
| coch | 77.78 | yes | 8.55 |
| loxhd1b | 75 | yes | 1856.77 |
| man2b1 | 75 | yes | 8.80 |
| tmie | 75 | yes | 1338.47 |
| marveld2a | 50 | yes | 1785.62 |
| mgat5 | 50 | yes | 2056.94 |
| slc26a4 | 50 | yes | 454.96 |
| sh3pxd2aa | 50 | yes | 1165.44 |
| marveld2b | 33.33 | yes | 1.19 |
| myo6a | 30 | yes | 1.34 |
| krt15 | 20 | yes | 3.27 |
| tmc1* | 16.67 | No | 0.99 |
| lhfpl5a | 0 | No | 0.97 |
| myh9a | 0 | No | 1.04 |
| myo7aa* | 0 | yes | 1.94 |
| prps1b | 0 | No | 1.19 |
| ptprq | 0 | No | 0.91 |
| slc26a5 | 0 | No | 1.00 |
| ush1c* | 0 | yes | 1.57 |
| wfs1b | 0 | No | 1.01 |
Notes: a Data from Varshney et al., 2015, asterisks indicate discrepancies between somatic and germline activity based on yes by one method and no by the other method.
sgRNA and Cas9 mRNA synthesis
sgRNAs for each target were generated by the oligonucleotide assembly method as described earlier (13). RNAs were synthesized using the HiScribe™ T7 Quick High Yield RNA Synthesis Kit (New England Biolabs) with an incubation time of 8 h for the in vitro transcription reaction. RNAs were purified with the RNA Clean & Concentrator™-5 kit (Zymo Research) and eluted into 20 μl water. Cas9 mRNA was synthesized from the pT3TS-nCas9n plasmid (Addgene plasmid # 46757) (12) following XbaI digestion using the mMessage mMachine T3 Transcription Kit (Ambion) and recovered by lithium chloride precipitation.
Microinjections and DNA extraction from embryos
Embryos were injected with 300 pg of Cas9 mRNA and 50 pg of each sgRNA at one-cell stage using a PicoPump (World Precision Instruments) and standard microinjection protocol (29). The amount of sgRNA and Cas9 mRNA for each injection was calculated by CRISPR-CALC (Supplementary File S1). For majority of the sgRNAs, we mixed sgRNAs to two genes at 50 pg each to reduce the number of injections. To test for multiplexing, we mixed eight sgRNAs at 25 pg each with 300 pg of Cas9 mRNA (Supplementary File S1). Injected embryos were incubated at 28.5°C and euthanized at 48 hpf for DNA extraction. DNA was extracted from eight uninjected and eight injected embryos for each sgRNA using Extract-N-Amp Tissue PCR Kit (Sigma) with one-fourth of the recommended volumes for each of the solutions. Extracted DNA was diluted at 1:10 ratio with ultra pure water and 1.5 μl was used as template for subsequent PCR reactions.
Primer design and fluorescent PCR
Primer sequences for all sgRNAs are listed in Supplementary Table S1 and also described previously (13). In general, the primers were designed to amplify 180–300 bp fragments with the target site roughly in the middle of the amplicon and avoiding intronic sequences as much as possible. M13F adapter (5′-TGTAAAACGACGGCCAGT-3′) and PIGtail adapter (5′-GTGTCTT-3′) sequences were added to the forward and reverse primers respectively (Supplementary Table S1). PCR reactions were set-up using AmpliTaq-Gold (Life Technologies) and a three primer mix with equimolar ratios of target-specific primers and fluorescently labeled M13F (6-FAM) primer as illustrated in Figure 1A with a final reaction volume of 6.5 μl (22). The PCR conditions were as follows: 12 min denaturation at 94°C; 40 cycles of 94°C for 30 s, 57°C for 30 s and 72°C for 30 s; and final extension at 72°C for 10 min followed by a hold at 4°C.
Figure 1.
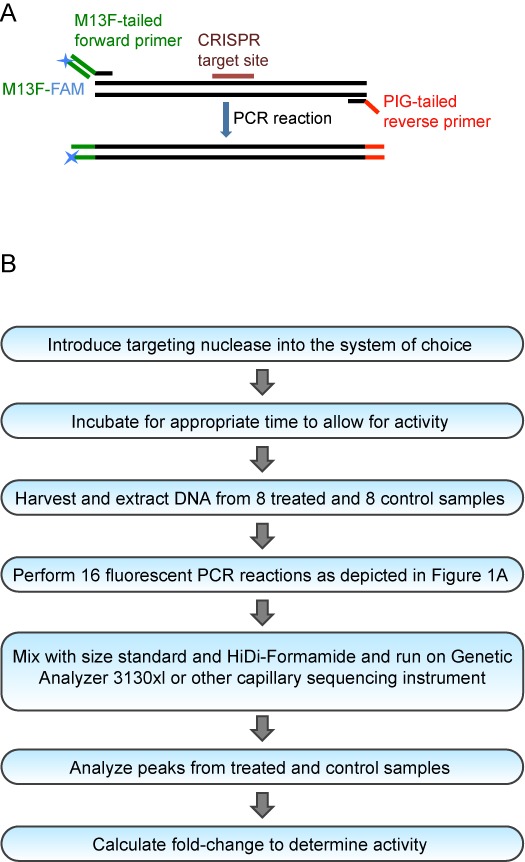
Overview of the CRISPR-STAT Method. (A) Schematic of the primer design strategy for fluorescent PCR. (B) Flowchart of step-by-step experimental procedures for the CRISPR-STAT method.
Fragment separation by capillary electrophoresis
PCR products were mixed with the GeneScan 400HD ROX dye size standard (Life Technologies) by adding 10 μl of 1:50 mix of 400HD ROX and HiDi-Formamide (Life Technologies) to 2 μl of PCR product. Samples were denatured at 95°C for 5 min and run on the Genetic Analyzer 3130xl using POP-7 polymer and a 36 cm array. The default module was used and the injection time of 23 seconds was modified to 60 seconds.
Titration experiment with known levels of mosaicism
DNA from WT and homozygous mutant fish with a known 7 bp insertion mutation in pus3 (unpublished data) were prepared as 50 ng/μl stock solutions and mixed at known ratios ranging from 0 to 100% of the mutant DNA as follows: 100 + 0, 90 + 10, 80 + 20, 70 + 30, 60 + 40, 50 + 50, 40 + 60, 30 + 70, 20 + 80, 10 + 90, 0 + 100 μl of WT and mutant DNA respectively. To account for pipetting errors, fluorescent PCR reactions were set-up in triplicates using 2 μl of mixed DNA as template.
Activity level calculations
Data were analyzed for sizes and heights of all observed peaks using GeneMapper or Genotyper (Life Technologies) by setting the Peak Amplitude Threshold at 100 as the level for background peaks. The expected sizes of the WT peaks were determined from the size of the amplicon and M13F and PIGtail adapters (25 bp) as listed in Supplementary Table S1. We first calculated the ratio of total peak height to the peak height of the WT peak plus 1 for each sample (∑heightTotal/ height WT + 1). The ‘+1’ was used so that we always had a denominator since some injected embryos had no detectable WT peak. We then calculated the average for each group (uninjected and injected) from a total of eight embryos analyzed. Fold change reduction of the WT peak was determined by dividing the averaged ratio for the injected embryos by the averaged ratio for the uninjected embryos. A fold change of 1 indicates no sgRNA activity. For sgRNAs with somatic activity, the higher the fold change, the higher the target-specific efficiency of sgRNA.
Cloning and sequencing of PCR products
A single injected embryo that showed sgRNA activity was selected for loxhd1b, myo6a and sh3pxd2aa. In addition eight injected embryos were pooled for myh9a. DNA extraction and PCR were performed as described above but without the M13F-FAM primer. PCR products were purified using the MinElute PCR purification Kit (Qiagen) and cloned into pCR4-TOPO vector (Life Technologies). Colony PCR was performed as follows: 50 μl of PCR master mix containing everything except template DNA was dispensed into each well of a 96-well plate. Individual colonies were picked with sterile P-20 pipette tips and dipped into the master mix followed by PCR. The PCR conditions were as follows: 12 min denaturation at 94°C; 35 cycles of 94°C for 30 s, 57°C for 30 s and 72°C for 30 s; and final extension at 72°C for 10 min followed by a hold at 4°C. Four microliters of PCR products were sequenced with M13F and big-dye v3.1 sequencing mix (Life Technologies) after removal of unused primers and nucleotides with Exo-SAP-IT (Affymetrix). Sequence analysis was performed using software package Sequencher, version 5.3 (Gene Codes).
RESULTS
Overview and sensitivity of CRISPR-STAT method
The goal of our study was to develop an easy, fast, economical and reliable method for pre-screening of genome-targeting nucleases, in particular, sgRNAs for their target-specific activity. We have been using the fluorescent PCR method for high throughput mutagenesis in zebrafish (13,22). Therefore, we sought to validate its performance for pre-screening of sgRNAs by analysis of somatic tissue from injected embryos that are typically mosaic for a variety of indels. We termed this method CRISPR-STAT for CRISPR—Somatic Tissue Activity Test. The primer design strategy and experimental workflow of CRISPR-STAT are described in Figure 1. Briefly, we injected sgRNA/Cas9 mRNA mix into one-cell stage embryos and collected eight injected and eight uninjected embryos at 48 hpf for fluorescent PCR of the target region. We separated the fluorescent PCR products by capillary electrophoresis using a conventional capillary sequencer, i.e. Genetic analyzer 3100xl. We analyzed the observed peak profiles for each embryo (injected and uninjected) using GeneMapper and calculated the fold change reduction in the WT allele with the following formula:
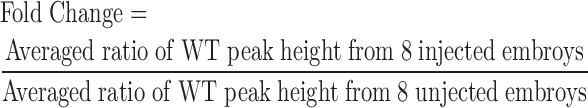 |
To evaluate the sensitivity of the CRISPR-STAT method, we simulated different levels of mosaicism by mixing WT DNA with increasing amounts of mutant DNA (from a known mutant fish homozygous for a 7 bp insertion). As expected, as the amount of WT DNA was decreased and the mutant DNA increased the peak heights of the respective alleles changed accordingly (Figure 2A). Quantification of the data demonstrated a strong correlation (R2 = 0.99) between the percentage of WT allele detected in the mix with the percentage of WT DNA added to the fluorescent PCR reaction (Figure 2B). These data demonstrated the sensitivity of CRISPR-STAT method to the degree of mosaicism in the template DNA, suggesting that it can be used to quantify activity of targeting nucleases.
Figure 2.

Evaluation of sensitivity of CRISPR-STAT method to detect mosaicism. (A) Fluorescent PCR peak profiles showing the changes in peak heights of WT and mutant alleles with varying ratios of WT to mutant DNA. In the fluorescent PCR plots, the X-axis represents the size of the peaks with the red and blue stars denoting WT and mutant alleles respectively, and Y-axis shows the peak heights. Ratios of WT and mutant DNA are shown for each panel. (B) Correlation of the detected WT allele to the known amount of WT allele in the mixture of WT and mutant DNA at various ratios. (C) Representative embryos at 48hpf and corresponding fluorescent PCR peak profiles shown for an uninjected embryo (top panel) and an embryo injected with tyr sgRNA showing reduced pigmentation phenotype (bottom panel). In the fluorescent PCR plots, the X-axis represents the size of the peaks with the red star denoting the position of the WT peak and Y-axis shows the peak heights. Data is shown for peaks observed within 20 nt of the WT peak.
Correlation of somatic activity with germline transmission efficiency
To test whether fluorescent PCR is sensitive enough to identify peaks due to somatic sgRNA activity in mosaic embryos, we selected the sgRNA target for the tyr gene which generates an easy to detect pigmentation phenotype in >90% of the injected embryos (12). We observed a clear reduction of the WT peak (1800-fold change of WT peak height) in the eight injected embryos that were selected by a reduced pigmentation phenotype compared to eight uninjected embryos. Furthermore, as expected we observed multiple secondary peaks within 50 bp of the amplicon size indicative of indels of up to 25 bp in the injected embryos only (Figure 2C). These data suggested that CRISPR-STAT is sensitive enough to detect somatic activity of sgRNAs in mosaic zebrafish embryos.
An sgRNA has to be extremely efficient to cause biallelic mutations in enough cells that a phenotype can be detected in the injected embryos (as observed for the tyr gene). However, to generate stable germline transmitted mutants, the injected embryos need to be healthy and devoid of lethal phenotypes so that they can be raised to fertile adults. To test whether fluorescent PCR can be used to determine sgRNA activity in the injected embryos regardless of whether the gene has an observable phenotype or not, we selected sgRNAs targeting 28 genes with a range of germline transmission rates (from 0 to 100%) that had been previously determined (13). Two sgRNAs were co-injected with Cas9 RNA and for each target eight embryos were analyzed by fluorescent PCR at 48 hpf. Representative examples of peak profiles for sgRNA with 50% positive founders (sh3pxd2aa, Figure 3A), 30% positive founders (myo6a, Figure 3B) and no positive founders (myh9a, Figure 3C) are shown in Figure 3. We observed a reduction in the WT peak height and presence of multiple secondary peaks in injected embryos for all but one (tmc1) (Table 1, Figure 3 A and B) of the 20 targets with positive germline transmission data. Only three sgRNAs (myo7aa, tmc1 and ush1c) showed somatic activity that incorrectly predicted germline transmission (Table 1). The discrepancies between somatic and germline activity of tmc1, myo7aa and ush1c could be due to technical reasons as mRNA synthesis and injections were carried out by different people in the two experiments or screening of insufficient number of founder fish for germline transmission, especially for myo7aa and ush1c. Overall, our data suggested a strong positive trend between the fold reduction in the WT peak and the percentage of positive founders. The correlation coefficient for these experiments was 0.75 (Figure 3D, Table 1). Thus based on our data a decrease in the WT peak and presence of secondary peaks in the injected embryos can be taken as an indication of target-specific sgRNA activity.
Figure 3.

Correlation of somatic and germline transmission activity of sgRNA. Representative fluorescent PCR plots for sgRNAs with medium (A, sh3pxd2aa), low (B, myo6a) and no (C, myh9a) germline transmission activity are shown. In each case, data from an uninjected embryo is shown in the left panel and an injected embryo in the right panel. In the fluorescent PCR plots, the X-axis represents the size of the peaks with red star denoting the position of the WT peak and Y-axis shows the peak heights. Data is shown for peaks observed within 20 nt of the WT peak. (D) Graph showing the correlation of somatic activity calculated as a log fold change in the WT peak (X-axis) and percentage of founders positive for germline transmission (Y-axis).
Correlation of the secondary peaks with indels at the target site
To verify that the secondary peaks observed in sgRNA injected embryos are due to indel mutations at the target site, we cloned and sequenced amplicons for one gene each with high (loxhd1b), medium (sh3pxd2aa), low (myo6a) and no (myh9a) germline transmission activity. As expected, 100% (39/39) of sequenced clones for myh9a sgRNA (with no secondary peaks), had WT sequence (Figure 3C, Supplementary Figure S1). On the other hand, no WT clones were found for loxhd1b and sh3pxd2aa sgRNAs (Figures 3A and 4, Supplementary Figure S1). Overall, we found sequences correlating by size to predicted indels based on the peak profiles of the embryos (Figure 4, Supplementary Figure S1). For example, in the embryo injected with loxhd1b sgRNA, a 3 bp deletion represented the strongest secondary peak as well as the most frequently observed sequence in the clones (Figure 4). Thus, our data showed that most of the secondary peaks detected by fluorescent PCR are representative of target-specific indels.
Figure 4.
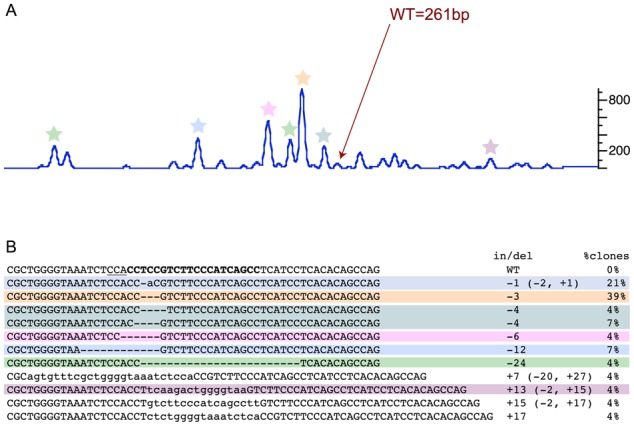
Correlation of fluorescent PCR peak profile and mutations. (A) Peak profile of an embryo injected with loxhd1b sgRNA. The WT peak is marked by an arrow and the secondary peaks for which clones with matching size of indels were detected are marked by colored stars. In the fluorescent PCR plot, X-axis represents the size of the peaks with WT denoting the position of the WT peak and Y-axis shows the peak heights. Data is shown for peaks observed within 30 nt of the WT peak. (B) Sequences of WT and mutant clones with percent of clones for each mutation. The target site is in bold and the PAM site in underlined in the WT sequence. The indels and percentage of clones columns are highlighted to match with the corresponding secondary peaks in (A).
Detection of somatic activity in multiplexed injections
It has been demonstrated that multiple sgRNAs can be combined to target multiple genes (13,30–31). The multiplexing approach is of greater importance in generating gene knockouts in zebrafish and mice as it reduces the number of injections, reagents and time to raise animals. Therefore, we evaluated zebrafish embryos co-injected with eight sgRNAs by CRISPR-STAT to determine their somatic activity. We detected somatic sgRNA activity in all eight genes (Table 2, Figure 5) with most injected embryos with activity in at least six genes. Thus, our data strongly supports the use of CRISPR-STAT to quickly screen sgRNA for target-specific activity both for single and multiplexed mutagenesis projects.
Table 2. Somatic activity of individual sgRNAs in multiplexed embryos.
| Gene | % of embryos with secondary peaks (n = 8) | Fold change |
|---|---|---|
| loxhd1b | 100% | 17.69 |
| man2b1 | 87.5% | 1.11 |
| marveld2b | 37.5% | 0.98 |
| myo15aa | 87.5% | 1.89 |
| myo6a | 75.0% | 1.35 |
| prdx3 | 100% | 4.63 |
| slc26a4 | 100% | 1.68 |
| sh3pxd2aa | 100% | 276.07 |
Figure 5.
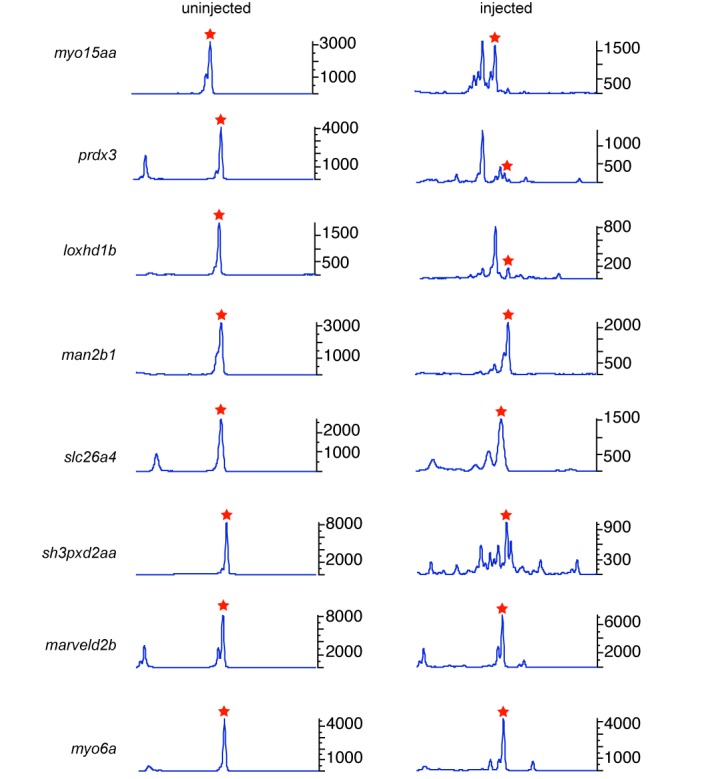
Peak profiles of an embryo injected with pooled sgRNAs to eight genes. The left panel shows an uninjected embryo as control and right panel shows an injected embryo. In the fluorescent PCR plots, X-axis represents the size of the peaks with WT denoting the position of the WT peak and Y-axis shows the peak heights. Data is shown for peaks observed within 20 nt of the WT peak that is denoted by a red star.
DISCUSSION
Successful genome editing by targeting nucleases, i.e. ZFNs, TALENs and CRISPR/Cas9 depends on their target-specific activity. A fast and cost-effective approach to test their efficacy at the target-site would help in developing optimized genome editing strategies for a variety of applications, such as disease modeling and functional genomics using animal models and developing gene therapy approaches using cultured cells. The CRISPR-STAT method presented here can give an accurate assessment of sgRNA activity in 1 day. We used a three-primer strategy to save costs associated with fluorescent labeling of individual target-specific primers. For laboratories performing targeted mutagenesis on a single gene, the M13F-FAM primer and genomic-M13 fused primer can be replaced with a fluorescently labeled gene-specific primer.
We demonstrated the utility of CRISPR-STAT as a pre-screening method in zebrafish to evaluate sgRNAs. By combining a cloning-free method of generating sgRNAs with evaluation by CRISPR-STAT, any laboratory should be able to generate mutants in their genes of interest and be confident of the efficiency of targeting before raising the animals. We recommend designing at least two sgRNAs per gene and evaluating a small subset of the injected embryos for their activity. This approach facilitates successful mutagenesis and minimizes time and costs associated with animal husbandry and negative founder screening for targets with poor somatic activity. Efficient founder screening strategies, i.e. reducing the number of founders screened and the number of embryos screened per founder, can be based on the efficiency score of the somatic tests. Furthermore, primers designed for CRISPR-STAT can be recycled in subsequent steps, specifically for identification of germline transmitting founders and genotype-phenotype correlation analysis of the knockout mutants.
Efficient nucleases are a prerequisite for several other applications of genome editing. For example, deletions of entire genes or large chromosomal regions and inversions can be generated by using two targeting nucleases (13,32–33). Targeted knock-in to create a missense mutation or insert a reporter gene by using homology directed repair and a donor template DNA is still a challenge due to low efficiencies and can be significantly improved with highly efficient sgRNAs (34). Thus genome-editing applications can be made more efficient by implementation of the CRISPR-STAT as a pre-screening step in the targeted mutagenesis process.
Although we used zebrafish to demonstrate CRISPR-STAT, it would also work in other model systems and for other targeting nucleases. Similar to the mosaic nature of the injected zebrafish embryos, the cultured cells transfected with CRISPR/Cas9 plasmids are mosaic for a number of mutations and often require picking and growing of single cells for subsequent analysis. Pre-screening of transfected cells with CRISPR-STAT would allow one to determine if the desired activity is seen prior to the tedious colony cloning efforts. Calculating the fold change gives an effective quantitative measure of the sgRNA activity, however, in most cases it will not be necessary to perform these calculations. A quick analysis of the fluorescent PCR profiles of injected embryos or transfected cells for reduction of the WT peak and presence of secondary peaks can be interpreted as good, target-specific activity. In conclusion, CRISPR-STAT is a valuable and versatile tool for assessing target-specific efficacy of genome editing nucleases in a variety of systems.
Supplementary Material
Acknowledgments
We thank MaryPat Jones and Julia Fekecs for help with figures and Niraj Trivedi for assistance with data analysis.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of the National Human Genome Research Institute; National Institutes of Health. Funding for open access charge: Intramural Research Program of the National Human Genome Research Institute; National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1.Bedell V.M., Wang Y., Campbell J.M., Poshusta T.L., Starker C.G., Krug R.G. 2nd, Tan W., Penheiter S.G., Ma A.C., Leung A.Y., et al. In vivo genome editing using a high-efficiency TALEN system. Nature. 2012;491:114–118. doi: 10.1038/nature11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doyon Y., McCammon J.M., Miller J.C., Faraji F., Ngo C., Katibah G.E., Amora R., Hocking T.D., Zhang L., Rebar E.J., et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat. Biotechnol. 2008;26:702–708. doi: 10.1038/nbt1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang P., Xiao A., Zhou M., Zhu Z., Lin S., Zhang B. Heritable gene targeting in zebrafish using customized TALENs. Nat. Biotechnol. 2011;29:699–700. doi: 10.1038/nbt.1939. [DOI] [PubMed] [Google Scholar]
- 4.Hwang W.Y., Fu Y., Reyon D., Maeder M.L., Tsai S.Q., Sander J.D., Peterson R.T., Yeh J.R., Joung J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaj T., Gersbach C.A., Barbas C.F. III. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gratz S.J., Cummings A.M., Nguyen J.N., Hamm D.C., Donohue L.K., Harrison M.M., Wildonger J., O'Connor-Giles K.M. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 2013;194:1029–1035. doi: 10.1534/genetics.113.152710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cermak T., Doyle E.L., Christian M., Wang L., Zhang Y., Schmidt C., Baller J.A., Somia N.V., Bogdanove A.J., Voytas D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foley J.E., Yeh J.R., Maeder M.L., Reyon D., Sander J.D., Peterson R.T., Joung J.K. Rapid mutation of endogenous zebrafish genes using zinc finger nucleases made by Oligomerized Pool ENgineering (OPEN) PLoS One. 2009;4:e4348. doi: 10.1371/journal.pone.0004348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reyon D., Tsai S.Q., Khayter C., Foden J.A., Sander J.D., Joung J.K. FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 2012;30:460–465. doi: 10.1038/nbt.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sander J.D., Dahlborg E.J., Goodwin M.J., Cade L., Zhang F., Cifuentes D., Curtin S.J., Blackburn J.S., Thibodeau-Beganny S., Qi Y., et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA) Nat. Methods. 2011;8:67–69. doi: 10.1038/nmeth.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jao L.E., Wente S.R., Chen W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc. Natl. Acad. Sci. U.S.A. 2013;110:13904–13909. doi: 10.1073/pnas.1308335110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varshney G.K., Pei W., LaFave M.C., Idol J., Xu L., Gallardo V., Carrington B., Bishop K., Jones M., Li M., et al. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res. 2015;7:1030–1042. doi: 10.1101/gr.186379.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu P.D., Lander E.S., Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mali P., Esvelt K.M., Church G.M. Cas9 as a versatile tool for engineering biology. Nat. Methods. 2013;10:957–963. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu Y., Sander J.D., Reyon D., Cascio V.M., Joung J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doench J.G., Hartenian E., Graham D.B., Tothova Z., Hegde M., Smith I., Sullender M., Ebert B.L., Xavier R.J., Root D.E. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 2014;32:1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gagnon J.A., Valen E., Thyme S.B., Huang P., Ahkmetova L., Pauli A., Montague T.G., Zimmerman S., Richter C., Schier A.F. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS One. 2014;9:e98186. doi: 10.1371/journal.pone.0098186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bell C.C., Magor G.W., Gillinder K.R., Perkins A.C. A high-throughput screening strategy for detecting CRISPR-Cas9 induced mutations using next-generation sequencing. BMC Genomics. 2014;15:1002. doi: 10.1186/1471-2164-15-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dahlem T.J., Hoshijima K., Jurynec M.J., Gunther D., Starker C.G., Locke A.S., Weis A.M., Voytas D.F., Grunwald D.J. Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet. 2012;8:e1002861. doi: 10.1371/journal.pgen.1002861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sander J.D., Cade L., Khayter C., Reyon D., Peterson R.T., Joung J.K., Yeh J.R. Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat. Biotechnol. 2011;29:697–698. doi: 10.1038/nbt.1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sood R., Carrington B., Bishop K., Jones M., Rissone A., Candotti F., Chandrasekharappa S.C., Liu P. Efficient methods for targeted mutagenesis in zebrafish using zinc-finger nucleases: data from targeting of nine genes using CompoZr or CoDA ZFNs. PLoS One. 2013;8:e57239. doi: 10.1371/journal.pone.0057239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu C., Zhang Y., Yao S., Wei Y. A PCR based protocol for detecting indel mutations induced by TALENs and CRISPR/Cas9 in zebrafish. PLoS One. 2014;9:e98282. doi: 10.1371/journal.pone.0098282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim H.J., Lee H.J., Kim H., Cho S.W., Kim J.S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res. 2009;19:1279–1288. doi: 10.1101/gr.089417.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiu P., Shandilya H., D'Alessio J.M., O'Connor K., Durocher J., Gerard G.F. Mutation detection using Surveyor nuclease. Biotechniques. 2004;36:702–707. doi: 10.2144/04364PF01. [DOI] [PubMed] [Google Scholar]
- 26.Brinkman E.K., Chen T., Amendola M., van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guell M., Yang L., Church G.M. Genome editing assessment using CRISPR Genome Analyzer (CRISPR-GA) Bioinformatics. 2014;30:2968–2970. doi: 10.1093/bioinformatics/btu427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Talbot J.C., Amacher S.L. A streamlined CRISPR pipeline to reliably generate zebrafish frameshifting alleles. Zebrafish. 2014;11:583–585. doi: 10.1089/zeb.2014.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Westerfield M. THE ZEBRAFISH BOOK, 5th Edition; A guide for the laboratory use of zebrafish (Danio rerio) Eugene: University of Oregon Press; 2007. [Google Scholar]
- 30.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shah A.N., Davey C.F., Whitebirch A.C., Miller A.C., Moens C.B. Rapid reverse genetic screening using CRISPR in zebrafish. Nat. Methods. 2015;6:535–540. doi: 10.1038/nmeth.3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao A., Wang Z., Hu Y., Wu Y., Luo Z., Yang Z., Zu Y., Li W., Huang P., Tong X., et al. Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res. 2013;41:e141. doi: 10.1093/nar/gkt464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Essletzbichler P., Konopka T., Santoro F., Chen D., Gapp B.V., Kralovics R., Brummelkamp T.R., Nijman S.M., Burckstummer T. Megabase-scale deletion using CRISPR/Cas9 to generate a fully haploid human cell line. Genome Res. 2014;24:2059–2065. doi: 10.1101/gr.177220.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hisano Y., Sakuma T., Nakade S., Ohga R., Ota S., Okamoto H., Yamamoto T., Kawahara A. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Sci. Rep. 2015;5:8841. doi: 10.1038/srep08841. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.