


Abstract
The bacterial transposon Tn7 facilitates horizontal transfer by directing transposition into actively replicating DNA with the element-encoded protein TnsE. Structural analysis of the C-terminal domain of wild-type TnsE identified a novel protein fold including a central V-shaped loop that toggles between two distinct conformations. The structure of a robust TnsE gain-of-activity variant has this loop locked in a single conformation, suggesting that conformational flexibility regulates TnsE activity. Structure-based analysis of a series of TnsE mutants relates transposition activity to DNA binding stability. Wild-type TnsE appears to naturally form an unstable complex with a target DNA, whereas mutant combinations required for large changes in transposition frequency and targeting stabilized this interaction. Collectively, our work unveils a unique structural proofreading mechanism where toggling between two conformations regulates target commitment by limiting the stability of target DNA engagement until an appropriate insertion site is identified.
INTRODUCTION
Transposons are genetic elements that can move between sites within a genome. Transposons that move too frequently and/or indiscriminately risk inactivating host genes or altering gene regulation. However, elements that move infrequently risk their own inactivation through the accumulation of mutations. Therefore the frequency and targeting of transposition play an important role in the overall fitness of a transposable element. The process of target site selection has been studied intensively with the bacterial transposon Tn7 that displays tight control of transposition in two separate pathways that recognize different types of targets utilized at different frequencies (1). Recent work suggests that Tn7 is a member of a much larger group of elements that utilize a transposase comprised of two distinct proteins. Elements that utilize this transposase configuration have been referred to as heteromeric transposase elements and appear to have elaborate targeting mechanisms using dedicated target selection proteins (2). These elements have been extremely successful at spreading to diverse bacteria in disparate environments and also facilitate the formation of pathogenicity or fitness islands. Research on heteromeric transposase elements capable of recognizing multiple targets suggest two related questions: How is a specific site of insertion selected and what determines the balance of transposition between multiple pathways?
Tn7 is found in a variety of pathogens in hospital settings where we find all of the Tn7 transposition proteins are identical to canonical Tn7 (3) (Pseudomonas aeruginosa (4), E. coli (5), Acinetobacter baumannii AB300 and A28 (6), Shigella sonnei (7) and Enterobacter cloacae (8)) or nearly identical with a single amino acid change (Klebsiella pneumoniae TnsBG389S(9) and Shigella flexneri TnsEQ177H (10)). Tn7 encodes five proteins that allow the element to carry out the two pathways of transposition; TnsA, TnsB, TnsC, TnsD and TnsE (TnsABCDE). TnsA and TnsB form the heteromeric transposase that is regulated by the AAA+ protein TnsC (11,12). The TnsABC core transposition machinery directs transposition into different types of target sites through the use of the target site selecting proteins, TnsD or TnsE (13,14). The TnsABC+D proteins direct transposition into a single site found in the bacterial chromosome called its attachment site or attTn7. TnsD is a sequence specific DNA binding protein that recognizes a highly conserved sequence in an essential gene in bacteria, glmS, to direct transposition into attTn7 (13,15). The TnsABC+E proteins direct transposition into DNAs facilitating horizontal transfer by recognizing features of DNA replication on the lagging-strand template (16,17).
In the canonical Tn7 element the TnsABC+D pathway of transposition occurs at about a 1000-fold higher frequency than the TnsE pathway that recognizes features found during DNA replication (18). However, previous genetic screens identified TnsE mutants that allowed for much higher levels of TnsE-mediated transposition (19). Some of these TnsE mutants allowed an increase of up to a 1000-fold over wild-type TnsE levels. This indicated that TnsE had the same potential for high frequency transposition as found with the TnsD pathway that recognizes the attTn7 site. Interestingly these TnsE gain-of-activity mutations seemed to show the same target preference found with wild-type TnsE (19). To better understand TnsE-mediated transposition and how Tn7 and other heteromeric transposase elements balance the usage of multiple pathways we dissected and analyzed the mutants genetically, biochemically and structurally.
The structures of the C-terminal domains of TnsE and the TnsE-A453V+D523N variant reveal that gain-of-activity mutations affect at least one of two features on the structure. They either lock the conformation of a V-shaped loop or extend the positively charged groove around the V-shaped loop that likely interacts with target DNA. We suggest that the ability of this surface to interact stably with a target DNA is interlinked with toggling of the V-shaped loop between two alternate conformations. Mutational analysis relates the stability of binding to a target DNA to the ability to control the frequency and selectivity of transposition. Our data suggests that a conformational change within the CTD of TnsE underlies target site selection by regulating stable engagement of the target DNA while providing a signal for activating transposition. We propose that TnsE has an unusual proofreading mechanism where conformational flexibility regulates target commitment by limiting the stability of target DNA engagement until an appropriate insertion site is identified.
MATERIALS AND METHODS
Bacterial strains and plasmids
NLC28 and NLC51 (NLC28 recA51) were described previously (20). BB101 is F’ lacIq lac’ pro’/ara Δ(lac-pro) nalA argE(am) RifR thi-1 slyD::KanR λDE3 was provided by Jennifer Surtees (21). Plasmids pJP103 and pJP104 were based on the pTA106 (pSC101 replicon) vector. TnsE was expressed from its native promoter in plasmid pJP103 and was over-expressed from a Plac promoter in pJP104 (22). All mutant derivatives of the TnsE were constructed by subcloning or synthetic overlap extension PCR using oligonucleotides. All constructs were confirmed by DNA sequencing. Vectors pCW15 (TnsABC) (14) and pCW15* (TnsABCA225V) (23), were described previously.
Crystallization and structure determination
Based on the limited proteolysis results, we subcloned the C-terminal domain of TnsE (residues Leu342-His538) and its A453V+D523N variant in pET22b expression vectors encoding a non-removable C-terminal histidine tag. Plasmids were transformed in BL21 StarTM DE3 cells (Life technologies) supplemented with a plasmid encoding rare codons and incubated at 37°C with orbital agitation up to OD600 ∼0.7. Protein production was then induced by addition of 1 mM IPTG and cell cultures were harvested after 3 h at 37°C. Cell pellets were lysed by sonication and spun at 39 000 g for 40 min. Clarified lysates were loaded onto Ni-chelating HiTrap columns (GE Healthcare) equilibrated with Buffer A (20 mM Tris pH 8, 0.5 M NaCl, 1.4 mM 2-mercaptoethanol, 5% glycerol). Impurities were eliminated with a step gradient and tagged protein was eluted with buffer A supplemented with 240 mM imidazole. Pooled fractions were diluted in buffer B (20 mM Tris pH 8, 1 mM EDTA, 5 mM DTT and 5% glycerol) to drop the NaCl concentration to 50 mM and further purified on a Mono Q 5/50 GL anion exchange column (GE healthcare) using a linear NaCl gradient to 0.5 M. Proteins were concentrated to 6 mg/ml and stored in 20 mM Tris pH 8, 150 mM NaCl, 1 mM EDTA, 5 mM DTT and 5% glycerol. Selenomethionyl protein was produced in B834 (DE3) cells (Novagen) grown in minimal media as described previously (24).
Crystals of the C-terminal domain of TnsE (TnsE-CTD) were grown in 27% (weight/volume) PEG 3350, 0.2 M NaCl, 0.1 M Tris pH 8 and belonged to the P21 space group. Three complete data sets using seleno-methionine derived crystals were collected at the selenium peak on the X25 beamline at NSLS (Brookhaven National Laboratory) using a Dectris Pilatus 6M detector. Data were indexed, processed and merged using HKL2000 (25). None of the individual data sets yielded interpretable experimental phases using AutoSol (Phenix). However, a merged data set derived from the three crystals resulted in good quality experimental maps where the 12 molecules of TnsE contained in the asymmetric unit could be readily traced. Despite the high number of molecules in the asymmetric unit, refinement and model building were done using standard protocols in Phenix and COOT (26,27). Although non-crystallographic averaging was not used for phasing in AutoSol, we used non-crystallographic symmetry restraints during the refinement in Phenix. The final model has over 97.4% of the residues within the most favored regions in the Ramachandran plot and none in disallowed regions (see Table 1).
Table 1. Data collection and refinement statistics.
| TnsE-CTD(a) | TnsE-CTDA453V+D523N (b) | |
|---|---|---|
| Data Collection | ||
| Wavelength | 0.979 | 0.979 |
| Space Group | P21 | P21 |
| Cell dimensions | ||
| a, b, c (Å) | 110.3, 87.1, 141.6 | 47.0, 71.3, 50.4 |
| α, β, γ (°) | 90, 95, 90 | 90, 97.3, 90 |
| No. mol asu | 12 | 2 |
| Resolution (Å)c | 50–2.85 (2.9–2.85) | 32.5–1.76 (1.82–1.76) |
| Rmeasc | 0.12 (1.00) | 0.038 (1.13) |
| I/σ(I) | 20.5 (1.9) | 20.5 (1.4) |
| Completeness (%)c | 100 (100) | 99.8 (99.9) |
| Redundancy | 10.6 (10.1) | 3.8 (3.8) |
| Refinement | ||
| Resolution (Å) | 43.2–2.85 | 29.3–1.76 |
| Completeness (%) | 99.9 | 99.7 |
| No. Reflections | 122 621 | 32 706 |
| Rwork / Rfree (%) | 21.1 / 24.2 | 19.6 / 22.3 |
| Atoms refined | 15 020 | 2650 |
| Solvent atoms | 139 | 202 |
| Rmsd in bonds (Å) | 0.005 | 0.004 |
| Rmsd in angles (°) | 0.97 | 0.76 |
| Mean B values (Å) | 84.6 | 47.7 |
(a)Data derived from three merged crystals.
(b)Data derived from single crystal.
(c)Data in the highest resolution shell is shown in parentheses.
Crystals of the TnsE-CTDA453V+D523N double mutant were grown in 25% (weight/volume) PEG 3350, 0.1 M Bis-Tris pH 5.5 and belonged to the P21 space group. A complete data set was collected on the 08B1–1 beamline at the Canadian Light Source (28). Data were reduced and scaled using XDS and SCALA and the structure was determined by molecular replacement using the TnsE-CTD structure lacking the variable arm of the V-shaped loop as the search model. Refinement and model building were done using standard protocols in Phenix and COOT (26,27). The final model has over 97.2% of the residues within the most favored regions in the Ramachandran plot and none in disallowed regions (see Table 1). All figures were generated using PyMol.
Transposition assays
The frequency and targeting of transposition was monitored using a λ vector (λKK1 780 hisG9424::Tn10 del16 del17::attTn7::miniTn7::KanR) as described previously (19,29). To statistically test if the insertions occurred randomly, paired chi-squared tests were used to compare ‘expected’ versus ‘found’ insertions per 100 kb across the E. coli chromosome in each of the genetic backgrounds as done previously (22).
in vitro DNA binding assays and in vitro DNA binding competition assays
Full length proteins were purified using a HIS6 affinity tag as described previously (19) using the slyD− strain BB101 and additional ion exchange chromatography. Electrophoretic mobility shift assays (EMSA) were performed with purified wild-type TnsE and mutant proteins using 3′ recessed DNA templates prepared by annealing oligonucleotides (5′-CCA TTA GCA AGG CCG GAA ACG TCA CCA ATG CAA CGA TCA GCC AAC TAA ACT AGG ACA TCT TTG CC-3′ and 5′-GGC AAA GAT GTC CTA GTT TAG TTG GCT GAT-3′) in the presence of a 50-fold excess of linear blunt-ended competitor DNA as described (19). DNA substrates were labeled with 32P using T4 polynucleotide kinase (NEB) and separated from nucleotide using a G-50 micro spin column (GE healthcare).
For in vitro DNA binding competition assays 0.16 pmol of TnsE (wild type or mutant) was incubated with 0.08 pmol 32P-labeled 3′ recessed DNA substrate for 10 min at 30°C. A 20-fold excess of unlabeled 3′ recessed DNA (1.6 pmol) (5′- CCA TTA GCA AGG CCG GAA ACG TCA CCA ATG CAA CGA TCA GCC AAC TAA ACT AGG ACA TCT TTG CCT TTT TGG CAA AGA TGT CCT AGT TTA GTT GGC TGA T -3′, prepared by annealing) was then added to the reaction and incubated for 10, 20 or 30 min before loading on 6% native gel. 0 min samples were only incubated with 32P-labeled substrate for 10 min at 30°C before loading on the gel.
For quantification, free DNA and super shifted DNA band(s) of each sample were marked, translated into histograms and peak areas were integrated to obtain numerical values using Image J software. For each sample lane the percentage of bound DNA compared to free DNA was calculated using Microsoft Excel. Values for bound DNA at time point 0 min were set as 100% and percentages of bound DNA of the corresponding samples at 10, 20 and 30 min were calculated relative to the 0 min time point. Measurements and calculations were performed for three independent experiments and the average was plotted on the graph including the standard error of the mean.
RESULTS
Gain-of-activity mutations cluster in the C-terminal region of TnsE
A series of TnsE mutants that allowed for very high levels of transposition (M37I+D523N, A453V+G483R, A453V+G515R, E516K+D523N, E522K+D523N and A453V+D523N) have been previously isolated in genetic screens (19). All tested mutants showed a targeting behavior similar to the wild type protein where transposition events occur in a single orientation associated with the direction of DNA synthesis on the lagging-stand template with a preference for the region where DNA replication terminates (19,22). In order to gain mechanistic understanding of these TnsE mutations we constructed and tested the alleles as single mutations. The results indicated that the M37I, G483R, G515R, E516K and E522K residue changes are essentially dependent on the A453V and D523N mutations to allow very high levels of transposition (Figure 1 and Supplementary Table S1), confirming the previous finding that the C-terminal region of TnsE is important for regulating the frequency of TnsE-mediated transposition.
Figure 1.
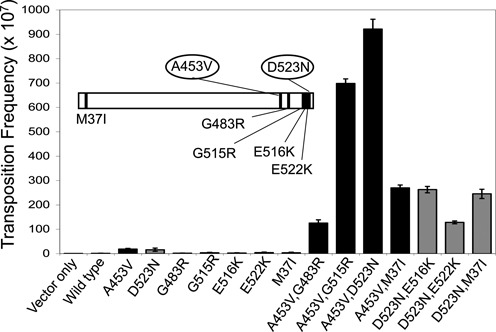
in vivo transposition activity of TnsE and various mutants. Transposition was monitored with cells expressing TnsABC (pCW15) and mutant or wild-type TnsE, (pJP103) (n = 3). Error bars indicate the standard error of the mean. The insert shows the relative positions of the mutations in TnsE.
Secondary structure prediction suggested that TnsE has two structured regions connected by a flexible linker. In good agreement, limited trypsin proteolysis revealed that TnsE was readily digested into two fragments of about 48 and 25 kDa (Supplementary Figure S1A). The TnsE protein used in this experiment contained a C-terminal hexa-histidine tag; therefore, we fractionated the digestion products over a Ni-chelating affinity column to identify which of the fragments corresponded to the C-terminal domain. The larger fragment (48 kDa) flowed through the Ni-chelating column, whereas the smaller fragment (25 kDa) was retained in the column indicating that the smaller fragment is the C-terminal domain of the TnsE (Supplementary Figure S1B). These results indicate that the N- and C-terminal regions of TnsE fold independently from each other and can be separated for structural characterization. Therefore, we sought to determine the structure of the C-terminal domain to gain mechanistic understanding of the gain-of-activity mutants identified in this region of TnsE.
TnsE-CTD has a novel fold
Based on the trypsin digestion analysis, we subcloned, produced and crystalized the fragment of TnsE encompassing residues Leu342-His538 (TnsE-CTD) and solved its structure by single-wavelength anomalous diffraction using merged data from three selenomethionyl labeled crystals (see Materials and Methods and Table 1)(30). The structured portion of the domain (residues Asp377-Arg533) adopts a novel α/β fold with a central five-stranded anti-parallel β-sheet surrounded by five α helices; helices α1, α2 and α5 helices cover one face of the β-sheet, while helices α3 and α4 cover the other face (Figure 2A). The secondary structure elements, as well as the extended regions connecting β1-β2 and β3-β4 are well conserved among TnsE proteins from highly divergent organisms (Figure 2 C)(Sequences referenced in (2)). The top (as oriented in Figure 2) region of the domain is highly structured with short loops connecting consecutive secondary structure elements. Conversely, the bottom region of the domain is devoid of structural elements, but extensive hydrogen-bond networks stabilize the long loops connecting secondary structure elements. Importantly, not only the sequence, but also the length of these regions is well conserved (Figure 2 C). The DALI server did not identify any close structural relatives, but TnsE-CTD could be remotely related (8% sequence identity) to the FP domain of the proteasome inhibitor PI31 (31). Comparison of the two structures revealed that the topology similarity is limited to the central antiparallel β-sheet and helices α2 and α5 (Supplementary Figure S2). None of the conserved sequence motifs of PI31 are conserved in TnsE and the interactions stabilizing the catalytic cores of the two proteins are quite different. Furthermore, both the helical and β surfaces of FP domain of PI31 domain mediate protein-protein interactions, confirming the unrelated functions of these domains.
Figure 2.
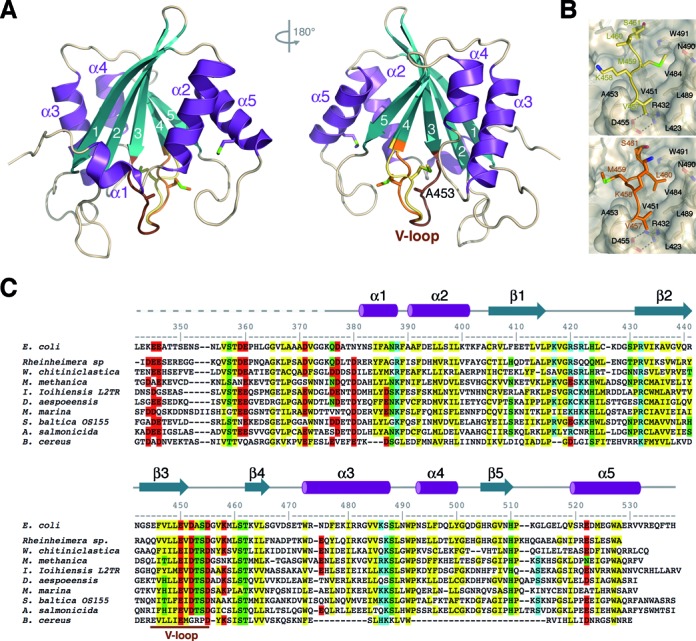
Crystal structure of the C-terminal domain of TnsE. (A) Opposite views of the TnsE-CTD structure shown as a ribbon diagram with strands and helices shown in teal and purple, respectively. The secondary structure elements, as well as the first (Lys375) and last (Arg533) residues visible in the electron density maps are labeled. The V-shaped loop is colored in brown with the alternate conformations for the mobile arm of the loop colored yellow (conformation I) and orange (conformation II), respectively. (B) Detailed views of the alternate conformations of the V-shaped loop. The constant part of TnsE-CTD is shown as a ribbon covered by a semi-transparent surface. The side-chains of residues defining the hydrophobic pocket where the V-shaped loop sits are shown as color-coded sticks and labeled. Mse459 (conformation I, top panel) and Leu460 (conformation II, bottom panel) occupy, respectively, the hydrophobic pocket. (C) Structure based sequence alignment of the C-terminal regions of TnsE from: Escherichia coli, Rheinheimera sp., Wohlfahrtiimonas chitiniclastica, Methylomonas methanica, Idiomarina Ioihienses L2TR, Desulfovibrio aespoeensis, Moritella marina, Shewanella baltica OS155, Aeromonas salmonicida subsp. salmonicica and Bacillus cereus. Conserved residues are highlighted in yellow (hydrophobic), green (polar), red (negatively charged) and blue (positively charged) and the region of the V-shaped loop (V-loop) is indicated.
TnsE-CTD contains a V-shaped loop that toggles between two alternate conformations
The C-terminal domain of TnsE includes two methionine residues (Met459 and Met524), as well as the N-terminal methionine introduced during cloning. N-terminal methionine residues are often processed during protein production and, in this case, it resides in a region of TnsE that it is predicted to be flexible. Therefore, since the asymmetric unit contains twelve copies of TnsE-CTD, we expected to find 24 peaks on the anomalous map. However, 36 strong peaks were found, with 3 peaks per molecule. The strongest peak was located in a terminal α-helix and corresponded to Met524. The other two anomalous peaks were adjacent to each other in all the molecules of the asymmetric unit and corresponded to alternate conformations of Met459 (Supplementary Figure S3). Strands β3 and β4 are connected through a wide mouth loop resembling a letter V where one arm of the ‘V’ is defined by residues Val451-Asp455 and the other arm by residues Val457-Ser461 (Figure 2A). The first arm of the ‘V’ is buried in the hydrophobic core of the protein. The second arm precedes the β4 strand and is exposed to the solvent (Figure 2A). Based on the alternate conformations of Mse459, we modeled this arm of the loop in two alternate conformations (Figure 2 and Supplementary Figure S4). In conformation I, the side chain of Mse459 points toward the β3 strand and sits in a shallow pocket defined by the side chains of residues Leu489, Trp491and Val451 (Figure 2B, top). In conformation II, Mse459 and Leu460 swap places and the side chain of Leu460 fills the hydrophobic pocket, whereas Mse459 sits on the opposite face of the strand (Figure 2B, bottom).
The 12 copies in the asymmetric unit are organized into 2 pseudo-trimers of dimers (Supplementary Figure S5). However, the surface buried by these interactions (both dimers and pseudo-trimers) is below the cutoff for specific interactions as judged using the PISA server (32). Since the apex of the V-shaped loop is part of the dimer interface (Supplementary Figure S5), the packing environment of the flexible arm is identical in the 12 molecules of the asymmetric unit and devoid of any crystallographic contacts. Therefore, the alternate conformations of the V-shaped loop are not due to the diversity of the packing environment. To adopt conformation I, the main chain of Val457 (on the narrow end of the V) gets very close to Ala453. Therefore, we entertained the possibility that the buried arm of the V (residues Val451-Asp455) enables the conformational flexibility of the surface-exposed arm (residues Val457-Ser461). Since mutations in Ala453 greatly enhance transposition frequency when combined with other alleles, we predicted that this V-shaped loop underlies a mechanism to help control target site selection. We hypothesized that interaction with a preferred DNA substrate could drive the mobile arm of the V into a single conformation as a mechanism of control. If that was the case, the Ala453Val mutation in the buried arm of the V-shaped loop may favor the proficient conformation of the exposed arm, thereby enhancing target recognition. To probe this idea, we sought to determine the structure of a variant of the C-terminal domain of TnsE encompassing the double mutation Ala453Val+Asp523Asn (TnsE-CTDA453V+D523N).
Ala453 controls the conformation of the toggle
Crystals of TnsE-CTDA453V+D523N also belonged to the P21 space group, but had smaller unit cell dimensions and diffracted to higher resolution than the wild-type crystals of TnsE-CTD (Table 1). We determined the structure of the double mutant by molecular replacement, but we removed the variable portion of the V-loop from the search model to avoid model bias. The two structures are virtually identical with the exception of the mobile arm of the V-shaped loop that adopts a single conformation, conformation II, in the TnsE-CTDA453V+D523N structure (Figure 3A and Supplementary Figure S6). As opposed to the crystal structure of TnsE-CTD, the packing environment is different for the two molecules of TnsE-CTDA453V+D523N in the asymmetric unit. However, the exposed arm of the V-shaped loop is not involved in any crystal interactions and the alternate conformation of the loop can be modeled without clashes with neighboring molecules. Therefore, the single conformation of the V-shaped loop appears to be imposed by the presence of the bulkier side chain at position 453 that precludes the formation of conformation I. The relative position of Val453 is fixed, because hydrogen-bond interactions between Asp452 with Arg388 and Ser454, as well as a salt bridge between Asp455 and Arg432, stabilize the extended region following β3 (Figure 3A). This region of TnsE, especially Asp455 and Arg432, is highly conserved demonstrating the significance of these interactions for the structural integrity of the domain (Figure 2C). Supporting this idea, point mutations in Asp452 or Asp455 decrease transposition frequency indicating that the structural integrity of this region is important for TnsE function (Figure 3B). Interestingly, the interactions mediated by Asp452 and Asp455 are recapitulated in the transposition experiment. D452N, but not D452A, can still form a hydrogen bond and hence this mutant has similar transposition frequency to wild-type TnsE. Conversely, only Asp455 can mediate the formation of the salt bridge with Arg432 and, thus both D455N and D455A mutations decrease transposition frequency to a similar extent (Figure 3A–B).
Figure 3.
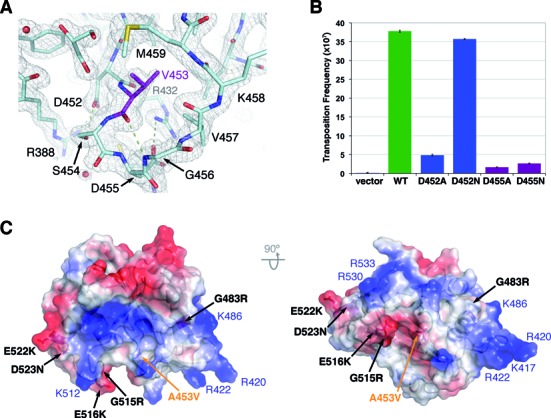
The A453V+D523N mutation locks the conformation of the mobile arm of the V-shaped loop and extends the DNA-binding interface. (A) Detail of the 2Fo-Fc electron density map of the TnsE-CTDA453V+D523N structure around Val453. The model is shown as color-coded sticks, with Val453 shown in purple, and the electron density map (contoured at 1.5σ) is shown as a grey mesh. Val453 is held in place by hydrogen-bond interactions between Arg388, Asp452 and Ser454, as well as a salt bridge between Arg432 and Asp455. (B) In vivo transposition activity of TnsE and TnsE variants affecting the conformation of the loop containing Ala453. Transposition was monitored with cells expressing TnsABC (pCW15) and mutant or wild-type TnsE, (pJP104) (n = 3). Error bars indicate the standard error of the mean. (C) Orthogonal views of the TnsE-CTDA453V+D523N structure shown as a semi-transparent electrostatic potential surface. The location of the point mutations increasing transposition frequency is indicated with black labels. Positive charged residues defining the shallow groove are labeled in blue, and the location of Ala453 is shown in orange.
While the widespread Tn7 element has the Ala453 allele, most related elements (called Tn7-like elements) do not have the Ala453 residue found in the highly conserved 446–455 region found in two conformations in the wild type protein (Figure 2). Most TnsE homologs have a threonine at this position (Figure 2C). Given that the A453V residue change was an important gain-of-activity allele, we constructed and tested a TnsEA453T mutant in the context of the protein from Tn7. We found that the A453T amino acid change was capable of harnessing the secondary mutations like the A453V amino acid change; the TnsEM37I+A453T and TnsEA453T+D523N proteins allowed higher levels of transposition compared to the individually tested constituent, albeit to a lower level than the A453V allele (Supplementary Figure S7). This is probably due to the fact that the side chain of Thr453 (but not Val453) could form a hydrogen bond with the main chain of Lys457, thereby relieving some of the steric constraints imposed by the bulkier side chain. Therefore, while TnsE homologs with a valine or a threonine at position 453 are responsive to mutations that alter the DNA binding surface, the frequency of transposition found with the canonical TnsE protein in Tn7 with an alanine at position 453 is not altered by these same mutations (Supplementary Figure S7).
A two-prong mechanism to control target selection and transposition
In contrast to Ala453, which is buried in the protein core, Asp523 lays on the surface of the domain exposed to the solvent. The D523N mutation removes a negative charge from the surface of the domain and could stabilize DNA binding. Indeed, all the gain-of-activity mutants, but A453V, identified in our screens (Gly483, Gly515, Glu516 and Glu522) are also exposed to the solvent (Figures 1 and 3C). Importantly, these residues lay on the same surface of the domain and are part of a shallow positively charged groove that runs perpendicularly to the V-shaped toggle (Figure 3C). Residues contributing to this surface are generally well conserved (Lys417, Arg420, Arg422, Lys486, Lys512, Arg530 and Arg533). Gain-of-activity mutations extend the positively charged area surrounding this track (i.e. G483R, G515R, E516K, E522K and D523N)(Figure 3C), supporting the notion that they define the DNA-binding interface of TnsE. While speculative, the apparent bend in the positively charged groove in the protein could contribute to preference for a single to double-stranded DNA junction. Interestingly, this surface of the domain is rather flat (Figure 3C), likely explaining why the C-terminal domain does not bind DNA in the absence of the N-terminal domain (data not shown).
Based on the structures of TnsE-CTD and TnsE-CTDA453V+D523N, the A453V mutation, located on the buried arm of the V-shaped loop, locks the mobile arm into a single conformation likely favoring the productive interaction with the preferred substrate, whereas the D523N mutation, located on the surface of the domain, likely stabilizes the TnsE-DNA by extending the DNA-binding interface (Figure 3C). Therefore, gain-of-activity mutations seem to effect TnsE-mediated transposition frequency by two complementary mechanisms. In agreement with this idea, the double mutants that combine the A453V mutation with other surface exposed residues such as G515R or D523N have a highly synergistic effect on DNA transposition whereas double mutants combining two surface-exposed residues do not achieve the same high levels (Figure 1). The A453V+G483R double mutant, however, does not follow this trend (Figure 1). All surface exposed residues increasing transposition frequency, except G483, are located within the β5-α5 loop or the α5 helix. G483 resides on the opposite side of the V-shaped loop and is surrounded by conserved positively charged residues, suggesting that point mutations at this position may have less impact on transposition frequency than mutations on the other end of the groove where positive charged residues are interspersed with negatively charged residues (Figure 3C). To probe whether the different point mutations influenced the stability of the TnsE-DNA interaction, we measured how the various mutants interact with a DNA substrate.
Gain-of-activity mutations stabilize binding of TnsE to target DNA
TnsE has a specific affinity for DNA structures with 3′ recessed ends (19). Indeed, TnsE-mediated transposition is only detectable in a reconstituted in vitro system if a gapped plasmid was used as the target DNA (33). Furthermore, the ability of the A453V+G515R double-mutant to recognize 3′ recessed ends was specifically enhanced suggesting these residues are likely relevant to target site selection (19). Therefore, we constructed affinity tagged derivatives, purified the proteins, and tested the in vitro DNA binding ability of the previously tested double-mutants, three additional double-mutants (TnsEA453V+D523N, TnsEE516K+D523N and TnsEE522K+D523N), and all of the single mutant proteins. We found that all of the TnsE double-mutants displayed enhanced complex formation that is consistent with the previously tested mutants (Figure 4 and data not shown)(19). In good agreement with previously published work (19), the wild-type protein predominantly forms a shifted product while the double-mutants predominantly form slower migrating ‘super shifted’ products when analyzed by EMSA (Figure 4). Super shifted complexes likely arise from multiple TnsE proteins residing on a DNA substrate and/or interaction between protein-DNA complexes, but future work will be required to determine the molecular nature of the various complexes identified by EMSA. Additionally, we found that TnsE proteins with the single A453V, D523N, G483R, G515R, E516K or E522K residue changes in the C-terminus displayed enhanced complex formation with DNA structures with 3′ recessed ends that was essentially the same as the DNA binding displayed by each of the double-mutants (Figure 4 and data not shown).
Figure 4.
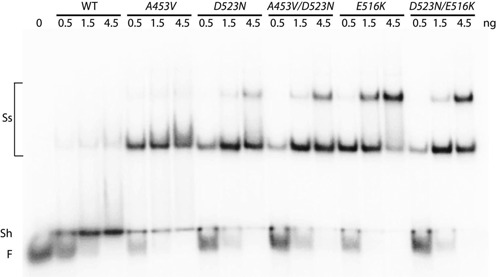
Autoradiogram of a representative gel mobility shift assay with purified mutant and wild-type TnsE proteins. The electophoretic mobility shift assay was carried out with radiolabled substrate DNA (with 0.08 pmoles of a 3′ recessed-end DNA substrate) incubated with buffer alone (Free DNA) or with increasing amounts of TnsE (wild-type or mutant) (0.08, 0.24, 0.72 pmoles) in presence of a 50-fold excess of linear blunt-ended competitor DNA (Material and methods). The position of Free (F) DNA, a shifted product (Sh), and super-shifted (Ss) species are indicated.
However, when carrying out transposition assays with these proteins in vivo frequencies varied over 500-fold (Figure 1), which cannot be explained by the different DNA binding behaviors/affinities in a standard EMSA (Figure 4). To address this unexpected result we examined the ability of the mutant proteins to remain stably bound to the preferred 3′ recessed end DNA substrate with a competition assay where we challenged with excess preferred substrate after pre-binding. TnsE proteins were pre-incubated with a radiolabeled 3′ recessed end substrate for 10 min at 30°C. Excess unlabeled 3′ recessed-end substrate was then added and the amount of protein that remained bound to the radiolabeled substrate was monitored over a 30-min period. Strikingly, under these conditions TnsE proteins with the single mutants behaved clearly different from TnsE proteins with two mutations. With the TnsEA453V and TnsED523N proteins substrate binding was strongly reduced within 10 min, but the TnsEA453V+D523N mutant remained persistently bound over 30 min (Figure 5A). A similar result was found when comparing the E516K and D523N alleles (Figure 5B). Graphing the results from multiple experiments with the single mutants indicates that within 10 min only about 40–60% of the wild-type protein remains bound to the original substrate, whereas with the double-mutants almost 90% remains bound (Figure 5C). Importantly, the amount of protein that remained bound to DNA was proportionally related to the nature of the mutations: double DNA-binding mutant (D523N+E516K) > DNA-binding+V-loop mutant (D523N+A453V) > single DNA-binding mutant (D523N, E516K) > single V-loop mutant (A453V) > wild-type TnsE. These data also support a two-prong mechanism of setting the frequency of transposition, as surface mutants form more stable complexes with DNA than mutations affecting the conformation of the V-shaped loop (Figure 5).
Figure 5.
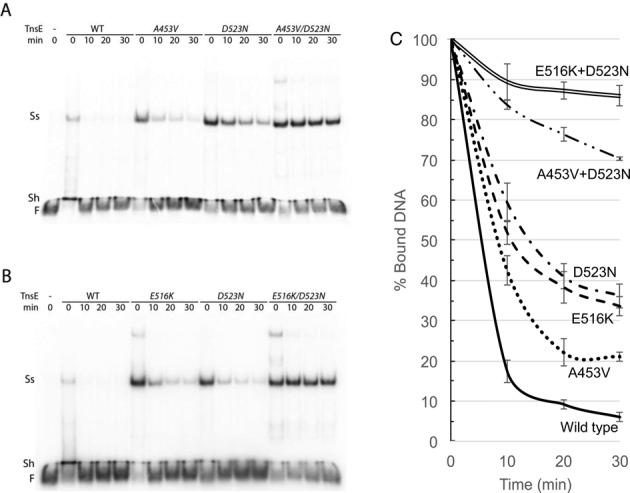
Autoradiogram of a representative gel mobility competition assays with purified mutant and wild-type TnsE proteins. (A–B) To determine binding stability TnsE proteins (0.16 pmoles) were pre-bound with 0.08 pmoles of a radiolabeled 3′ recessed-end DNA substrate for 10 min and then challenged with a 20-fold excess of unlabeled 3′ recessed-end DNA substrate for 10, 20 or 30 min (Materials and Methods). The position of Free (F) DNA, a shifted product (Sh) and prominent super-shifted (Ss) species is indicated. (C) DNA binding stability of wild-type and mutant TnsE proteins. Signal intensities from Figure 4 and repeated experiments were graphed as the percentage of bound DNA at 10, 20 and 30 min relative to the 0 min time point. The Signal Intensity of the super shifted band(s) at time point 0 min was set as 100% and percentages at time points 10, 20 and 30 min were calculated for each sample (Materials and Methods). Values represent the data from three independent experiments. Error bars indicate the standard error of the mean.
Taken together these results indicate that the single amino acid changes allow an enhanced complex formation (Figure 4) and a modest gain-of-activity phenotype in transposition (Supplementary Table S1). However, large increases in the frequency of transposition and increases in DNA binding stability are only found with combinations of two mutations (Figures 1 and 5). Two mutations (i.e. D523N+E516K) extending the positively charged groove in the TnsE-CTD can lead to a substantial increase in TnsE-mediated transposition (Figure 1) and a much more stable interaction with DNA substrate. However the most substantial gain-of-activity phenotype appears to involve double mutants with alleles from each class of mutation (i.e. A453V+G515R or A453V+D523N), thereby supporting the advantage of the two-prong mechanism for setting the frequency of transposition (Figure 1).
Wild-type and gain-of-activity TnsE mutants show different abilities to redirect transposition events
Control of Tn7 transposition involves the TnsC regulator protein and similar regulatory proteins are found in other transposons (12,34). TnsC is a AAA+ protein and the ADP versus ATP bound state of TnsC determines if the TnsABC complex is competent for initiating transposition (11–13). TnsC mutants, likeTnsCA225V, with slowed ATPase activity show transposition even in the absence of TnsD or TnsE, but can be directed to specific sites by natural target site selecting proteins (like TnsD to attTn7) or by synthetic mechanisms like triplex forming DNA (23,35–36). TnsABCA225V transposition occurs without a requirement for TnsD or TnsE in a standard in vivo transposition assay where the element is delivered into E. coli on a defective lambda phage that cannot replicate or integrate in the host (Figure 6A)(37). We mapped transposition events mediated by TnsABCA225V in E. coli and found they occurred without any obvious bias for position or orientation throughout the 4.6 Mbp chromosome (Figure 6B)(22). For statistical analysis we binned the number of Tn7 insertions per 100 kb interval across the chromosome and graphed and analyzed the distribution (Supplementary Figure S8A). Consistent with previous work (22) the distribution of insertions with TnsABCA225V was not significantly different from what is expected for a random distribution (P = 0.52).
Figure 6.
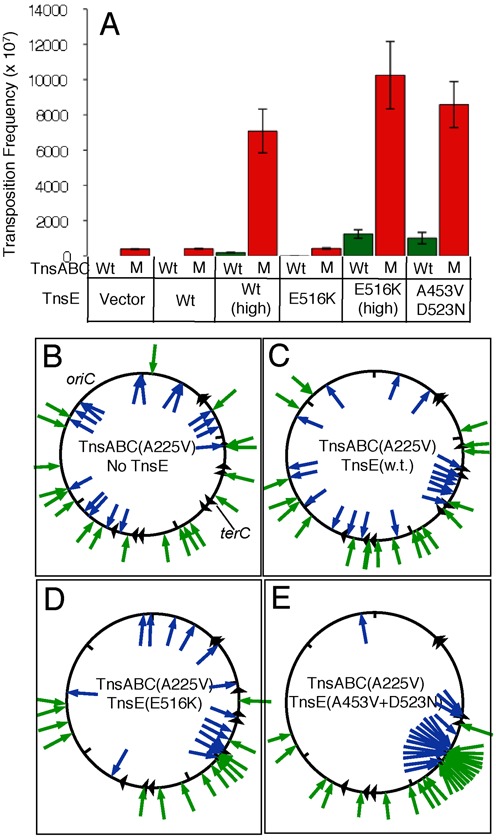
The frequency of Tn7 transposition with various TnsC and TnsE alleles and the location and orientation of transposition events. (A) Transposition frequency found with wild type (WT)(Green) or mutant (M)(Red) TnsABC core machinery (pCW15 and pCW15* respectively). Strains had the empty vector (Vector) or expressed TnsE in various configurations; wild type from its native promoter (WT) or lac promoter (WT(high)), TnsEE516K from its native promoter (E516K) or lac promoter (E516K (high)), or TnsEA453V+D523N from its native promoter (A453V+D523N). Panels (B–E) indicate the position of independently collected transposition events (arrows) in the E. coli chromosome containing TnsABCA225V and an empty vector (pCW15* pTA106)(B), TnsABCA225V +TnsEWT (pCW15* pJP104-TnsEWT)(C), TnsABCA225V +TnsEE516K (pCW15* pJP104-TnsEE516K)(D) and TnsABCA225V +TnsEA453V+D523N (pCW15* pJP103- TnsEA453V+D523N)(E). Blue arrows inside the circle indicate right-to-left insertions; Green arrows outside the circle indicate left-to-right insertions. Triangles indicate the approximate position of ter sites. The approximate position of oriC and terC are given.
Over expression of TnsE synergistically increased the frequency of TnsABCA225V transposition (>100-fold) indicating the TnsC mutant maintained its ability to functionally interact with TnsE (Figure 6A), however, these transposition events did not occur with the expected pattern normally found with TnsE (Figure 6C). This result indicated that in the context of the mutant TnsCA225V regulator protein, wild-type TnsE was able to support transposition without recognizing its normal target features. Statistical analysis also indicated that the distribution found with the TnsABCA225V and wild-type TnsE combination was not significantly different from a random distribution (P = 0.52) as observed with TnsABCA225V alone (Supplementary Figure S8B). Although wild-type TnsE stimulated transposition the result suggests that it did not occur through its capacity to recognize the preferred substrate found during DNA replication. Given our finding that wild-type TnsE binds the preferred substrate weakly, we hypothesized that the ability of TnsCA225V to bind DNA without sequence or structural specificity ‘overrides’ the interactions found with wild-type TnsE. An idea we explore below.
We addressed the role of DNA binding by analyzing the TnsE mutant alleles in the context of TnsCA225V; under the same conditions used to test wild-type TnsE we analyzed the TnsEE516K mutant that shows enhanced complex formation. Like wild-type TnsE, the TnsEE516K mutant stimulated transposition in cells with the TnsABCA225V machinery >100-fold (Figure 6A). However, unlike wild-type TnsE the TnsEE516K mutant was able to redirect transposition events to replication-associated targets; transposition with TnsEE516K favors an orientation bias that correlates with the direction of DNA replication and shows a preference for the region where DNA replication terminates (Figure 6D). For example, while only 8% of the insertions found with wild-type TnsE occur within 100 Kb of terC (Figure 6C), 35% of the insertions occur in this region with the TnsEE516K protein (Figure 6D). In addition, the distribution of insertions found with TnsEE516K was significantly different from random (P = 2.1 × 10−6) (Supplementary Figure S8C). Therefore the increased DNA binding stability identified with TnsEE516K in vitro correlates with the ability of the protein to redirect TnsABCA225V to DNA replication targets.
TnsE proteins with two gain-of-activity mutations bind the preferred DNA substrate with much greater stability than those with a single mutation (Figure 5). We found that under the control of the native promoter even low expression levels of TnsEA453V+D523N very strongly redirects the TnsABCA225V machinery to DNA replication targets; transposition events occur in a single orientation with DNA replication and have a strong preference for the terminus region (Figure 6E). Compared to only 8% of the wild-type TnsE insertions occurring within 100 Kb of terC (Figure 6C), 50% of the insertions occur in this region with the TnsEA453V+D523N protein (Figure 6E). In this case, the distribution of insertions found with TnsABCA225V and TnsEA453V+D523N was strikingly different from random (P = 1.0 × 10−57) (Supplementary Figure S8D).
Taken together, we find that with TnsABCA225V, wild-type TnsE participates in transposition, however, these insertions do not occur at replication targets, but instead at random positions across the chromosome, like those found with TnsABCA225V alone (Figure 6 and Supplementary Figure S8). Conversely, TnsEE516K and TnsEA453V+D523N are capable of recognizing features found during DNA replication, with specific targeting being much more pronounced with TnsEA453V+D523N (Figure 6 and Supplementary Figure S8).
DISCUSSION
While mutants of TnsE enhancing transposition were genetically identified over a decade ago, a mechanistic understanding of these TnsE variants has been lacking. In this study we carried out a structure-function analysis with the TnsE target site selecting protein of Tn7 to understand how insertion sites are selected. The crystal structure of the C-terminal domain of TnsE unveils a novel fold including a built-in two-prong mechanism to regulate commitment to a target DNA. This mechanism explains the modes of action of the gain-of-activity mutations identified in the C-terminal region of TnsE. One class of mutation targets surface exposed residues and enhances TnsE-mediated transposition by increasing DNA-binding affinity. The second type of mutation targets a single residue within the hydrophobic core of the domain and determines the conformation of a mobile V-shaped loop. The latter mechanism is unique to TnsE and likely favors target specificity by sacrificing transposition frequency. Indeed, wild-type TnsE naturally appears to form an unstable complex with target DNA, whereas double mutants of TnsE with high transposition frequency form highly stable interactions with 3′ recessed DNA substrates. Therefore, our work unveils a new type of ‘proofreading’ mechanism in TnsE where conformational flexibility in the CTD regulates target commitment by limiting the stability of target DNA engagement until an appropriate insertion site is identified.
Only one gain-of-activity mutation was identified in the N-terminal domain of TnsE, M37I. Mutations extending the positively charged groove or locking the conformation of the V-shaped loop responded similarly to the M37I gain-of-activity allele (Figure 1). The M37I amino acid change was originally isolated twice with the D523N allele (i.e. M37I+D523N) (19), however, we found the same high level of transposition with the M37I+A453V alleles (Figure 1). Preliminary work suggests that TnsEM37I interacts similarly with DNA or processivity factor to wild-type TnsE, however the TnsEM37I protein is toxic and poorly maintained except under low expression on a low copy number vector (data not shown). A mechanistic understanding of the role of the M37I mutation and the N-terminal domain in target selection awaits further characterization.
Minimally, two interactions guide the ability of TnsE to recognize attributes found during active DNA replication on the lagging-strand template; interaction with the processivity factor and interaction with a 3′ recessed end substrate (19,33). A conserved motif found in the N-terminal domain of TnsE allows interaction with processivity factor, but the mechanism that allows interaction with the 3′ recessed end substrate has been enigmatic. A requirement for two features would help explain why no gain-of-activity mutation was identified that showed relaxed specificity. One model that could account for the behavior of TnsE is that only when both the N-terminal domain interacts with processivity factor and the C-terminal domain of TnsE interacts with a 3′ recessed end do the two domains interact productively to signal the core TnsABC machinery to initiate transposition. Virtually all TnsE-mediated transposition events appear to be targeted into the template replicated by lagging-strand DNA replication, something that can be explained by recognizing two features using distinct domains of the protein.
We suggest that the alternate conformations of the V-shaped loop can provide a form of proofreading where the active conformation of the wild type protein will only be stabilized when engaged with DNAs containing a 3′ recessed end substrate (Supplementary Figure S9). This would help account for the strict targeting of TnsE-mediated transposition to the lagging-strand template during replication. While the gain-of-activity mutants still recognize features found during DNA replication, it was previously shown that the ratio of insertions that occurred into conjugal plasmids dropped considerably with the mutants (19). Therefore, part of the proofreading function found in wild-type TnsE allows more selective transposition into conjugal DNA replication. It has previously been argued that the bias for DNA replication on the lagging-strand template during conjugation may be explained by the coordination of the replication process (33); for chromosomal DNA replication the leading- and lagging-strand templates are processed in a single replisome, however, with conjugal replication these templates are processed in separate donor and recipient cells. The ability to toggle between alternate conformations as a mechanism to fine tune substrate recognition may therefore help by allowing conjugal DNA replication to be targeted in preference to chromosomal DNA replication.
Beyond proofreading, the alternate conformations of the V-shaped loop may also mitigate negative effects on DNA replication. Wild-type TnsE naturally forms a highly unstable complex with the preferred DNA substrate producing primarily a shifted product with little or no super shifted product (Figures 4 and 5). A strong interaction with a potential target DNA during DNA replication could be expected to adversely affect DNA replication. Consistent with this idea, it was previously shown that double mutants were only isolated on low copy vectors under low expression from the native promoter (19). Therefore, the conformational toggling identified in wild-type TnsE could also provide a mechanism to test DNAs with a ‘loose grip’ until the most favorable target for transposition are identified (i.e. DNA replication events in recipient cells during conjugation). This is reminiscent of the TnsE interaction with sliding clamp processivity factor. TnsE interacts weakly with the sliding clamp with a KD of ∼2.44 μM as measured by surface plasmon resonance, presumably limiting the negative effect a stronger interaction would have on DNA replication and other sliding clamp dependent processes. However, aggressive over expression of wild-type TnsE with an arabinose promoter will induce the SOS damage response indicating excessive single-stranded gaps through an interaction with the processivity factor (33).
It has recently been found that other heteromeric transposase elements may utilize distinct target site selection proteins to access attachment sites other than the attTn7 site (2). It will be interesting to determine if stability of target binding also controls the relative frequency for competing transposition pathways in these elements. The ability to recognize the template strand replicated by lagging-strand replication has been implicated in a number of different types of mobile elements (Reviewed in (38)), therefore conformational toggling might be a general mechanism to identify structural features in replicating DNA used by mobile elements.
ACCESSION NUMBERS
Atomic coordinates and structure factors have been deposited with the Protein Data Bank (accession codes 5D16 and 5D17).
Supplementary Material
Acknowledgments
We thank Nancy Craig (Johns Hopkins Medical School, Howard Hughes Medical Institute) for strains and comments on the manuscript.
Footnotes
Present Address: Qiaojuan Shi, Department of Molecular Biology and Genetics, Cornell University.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health, National Science Foundation [R01 GM069508 and MCB-1244227 to J.E.P.]; Canadian Institutes of Health Research [MOP-67189 to A.G.]. Crystallographic data included in this study was measured at beam lines X25 of the National Synchrotron Light Source (NSLS) and 08B1–1 at the Canadian Light Source (CLS). US Department of Energy the National Center for Research Resources [P41RR012408 to NSLS]; National Institute of General Medical Sciences [P41GM103473 to NSLS]; Natural Sciences and Engineering Research Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada and the University of Saskatchewan [to CLS]. Funding for open access charge: National Science Foundation [MCB-1244227 to J.E.P.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Peters J.E. Tn7. Microbiol. Spectr. 2014;2:1–20. doi: 10.1128/microbiolspec.MDNA3-0010-2014. [DOI] [PubMed] [Google Scholar]
- 2.Peters J.E., Fricker A.D., Kapili B.J., Petassi M.T. Heteromeric transposase elements: generators of genomic islands across diverse bacteria. Mol. Microbiol. 2014;93:1084–1092. doi: 10.1111/mmi.12740. [DOI] [PubMed] [Google Scholar]
- 3.Flores C., Qadri M.I., Lichtenstein C. DNA sequence analysis of five genes, tnsA, B, C, D and E, required for Tn7 transposition. Nucleic Acids Res. 1990;18:901–911. doi: 10.1093/nar/18.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haines A.S., Jones K., Batt S.M., Kosheleva I.A., Thomas C.M. Sequence of plasmid pBS228 and reconstruction of the IncP-1alpha phylogeny. Plasmid. 2007;58:76–83. doi: 10.1016/j.plasmid.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Roche D., Flechard M., Lallier N., Reperant M., Bree A., Pascal G., Schouler C., Germon P. ICEEc2, a new integrative and conjugative element belonging to the pKLC102/PAGI-2 family, identified in Escherichia coli strain BEN374. J. Bacteriol. 2010;192:5026–5036. doi: 10.1128/JB.00609-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramirez M.S., Quiroga C., Centron D. Novel rearrangement of a class 2 integron in two non-epidemiologically related isolates of Acinetobacter baumannii. Antimicrob. Agents Chemother. 2005;49:5179–5181. doi: 10.1128/AAC.49.12.5179-5181.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang F., Yang J., Zhang X., Chen L., Jiang Y., Yan Y., Tang X., Wang J., Xiong Z., Dong J., et al. Genome dynamics and diversity of Shigella species, the etiologic agents of bacillary dysentery. Nucleic Acids Res. 2005;33:6445–6458. doi: 10.1093/nar/gki954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramirez M.S., Pineiro S., Argentinian Integron Study G., Centron D. Novel insights about class 2 integrons from experimental and genomic epidemiology. Antimicrob. Agents Chemother. 2010;54:699–706. doi: 10.1128/AAC.01392-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doi Y., Hazen T.H., Boitano M., Tsai Y.C., Clark T.A., Korlach J., Rasko D.A. Whole-genome assembly of Klebsiella pneumoniae coproducing NDM-1 and OXA-232 carbapenemases using single-molecule, real-time sequencing. Antimicrob. Agents Chemother. 2014;58:5947–5953. doi: 10.1128/AAC.03180-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ye C., Lan R., Xia S., Zhang J., Sun Q., Zhang S., Jing H., Wang L., Li Z., Zhou Z., et al. Emergence of a new multidrug-resistant serotype X variant in an epidemic clone of Shigella flexneri. J. Clin. Microbiol. 2010;48:419–426. doi: 10.1128/JCM.00614-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Z., Craig N.L., Peters J.E. In: Bacterial Integrative Mobile Genetic Elements. Roberts AP, Mullany P, editors. Landes Bioscience; 2013. pp. 1–32. [Google Scholar]
- 12.Stellwagen A.E., Craig N.L. Mobile DNA elements: controlling transposition with ATP-dependent molecular switches. Trends Biochem. Sci. 1998;23:486–490. doi: 10.1016/s0968-0004(98)01325-5. [DOI] [PubMed] [Google Scholar]
- 13.Bainton R.J., Kubo K.M., Feng J.N., Craig N.L. Tn7 transposition: target DNA recognition is mediated by multiple Tn7-encoded proteins in a purified in vitro system. Cell. 1993;72:931–943. doi: 10.1016/0092-8674(93)90581-a. [DOI] [PubMed] [Google Scholar]
- 14.Waddell C.S., Craig N.L. Tn7 transposition: two transposition pathways directed by five Tn7-encoded genes. Genes & Dev. 1988;2:137–149. doi: 10.1101/gad.2.2.137. [DOI] [PubMed] [Google Scholar]
- 15.Mitra R., McKenzie G.J., Yi L., Lee C.A., Craig N.L. Characterization of the TnsD-attTn7 complex that promotes site-specific insertion of Tn7. Mobile DNA. 2010;1:1–14. doi: 10.1186/1759-8753-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolkow C.A., DeBoy R.T., Craig N.L. Conjugating plasmids are preferred targets for Tn7. Genes & Dev. 1996;10:2145–2157. doi: 10.1101/gad.10.17.2145. [DOI] [PubMed] [Google Scholar]
- 17.Finn J.A., Parks A.R., Peters J.E. Transposon Tn7 directs transposition into the genome of filamentous bacteriophage M13 using the element-encoded TnsE protein. J. Bacteriol. 2007;189:9122–9125. doi: 10.1128/JB.01451-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waddell C.S., Craig N.L. Tn7 transposition: two transposition pathways directed by five Tn7-encoded genes. Genes & Dev. 1988:137–149. doi: 10.1101/gad.2.2.137. [DOI] [PubMed] [Google Scholar]
- 19.Peters J.E., Craig N.L. Tn7 recognizes target structures associated with DNA replication using the DNA binding protein TnsE. Genes & Dev. 2001;15:737–747. doi: 10.1101/gad.870201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKown R.L., Waddell C.S., Arciszewska L.A., Craig N.L. Identification of a transposon Tn7-dependent DNA-binding activity that recognizes the ends of Tn7. Proc. Natl. Acad. Sci. U.S.A. 1987;84:7807–7811. doi: 10.1073/pnas.84.22.7807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chivers P.T., Sauer R.T. NikR repressor: high-affinity nickel binding to the C-terminal domain regulates binding to operator DNA. Chem. Biol. 2002;9:1141–1148. doi: 10.1016/s1074-5521(02)00241-7. [DOI] [PubMed] [Google Scholar]
- 22.Peters J.E., Craig N.L. Tn7 transposes proximal to DNA double-strand breaks and into regions where chromosomal DNA replication terminates. Mol. Cell. 2000;6:573–582. doi: 10.1016/s1097-2765(00)00056-3. [DOI] [PubMed] [Google Scholar]
- 23.Stellwagen A., Craig N.L. Gain-of-function mutations in TnsC, an ATP-dependent transposition protein which activates the bacterial transposon Tn7. Genetics. 1997;145:573–585. doi: 10.1093/genetics/145.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hendrickson W.A., Horton J.R., LeMaster D.M. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): a vehicle for direct determination of three-dimensional structure. EMBO J. 1990;9:1665–1672. doi: 10.1002/j.1460-2075.1990.tb08287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otwinowski Z., Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. NY: Academic Press; 1997. [DOI] [PubMed] [Google Scholar]
- 26.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 27.Afonine P., Grosse-Kunstleve R., Adam P. The Phenix refinement framework. CCP4 Newsl. 2005;42:1–7. [Google Scholar]
- 28.Fodje M., P G., Janzen K., Labiuk S., Gorin J., Berg R. 08B1–1: an automated beamline for macromolecular crystallography experiments at the Canadian Light Source. J. Synchrotron Radiat. 2014;21:633–637. doi: 10.1107/S1600577514005578. [DOI] [PubMed] [Google Scholar]
- 29.McKown R.L., Orle K.A., Chen T., Craig N.L. Sequence requirements of Escherichia coli attTn7, a specific site of transposon Tn7 insertion. J. Bacteriol. 1988;170:352–358. doi: 10.1128/jb.170.1.352-358.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Q., Zhang Z., Hendrickson W.A. Multi-crystal anomalous diffraction for low-resolution macromolecular phasing. Acta Crystallogr. D Biol. Crystallogr. 2011;67:45–59. doi: 10.1107/S0907444910046573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holm L., Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 33.Parks A.R., Li Z., Shi Q., Owens R.M., Jin M.M., Peters J.E. Transposition into replicating DNA occurs through interaction with the processivity factor. Cell. 2009;138:685–695. doi: 10.1016/j.cell.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamauchi M., Baker T. An ATP-ADP switch in MuB controls progression of the Mu transposition pathway. EMBO J. 1998;17:5509–5518. doi: 10.1093/emboj/17.18.5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rao J.E., Craig N.L. Selective recognition of pyrimidine motif triplexes by a protein encoded by the bacterial transposon Tn7. J. Mol. Biol. 2001;307:1161–1170. doi: 10.1006/jmbi.2001.4553. [DOI] [PubMed] [Google Scholar]
- 36.Rao J.E., Miller P.S., Craig N.L. Recognition of triple-helical DNA structures by transposon Tn7. Proc. Natl. Acad. Sci. U.S.A. 2000;97:3936–3941. doi: 10.1073/pnas.080061497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stellwagen A., Craig N.L. Avoiding Self: Two Tn7-encoded proteins mediate target immunity in Tn7 transposition. EMBO J. 1997;16:6823–6834. doi: 10.1093/emboj/16.22.6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fricker A.D., Peters J.E. Vulnerabilities on the Lagging-Strand Template: Opportunities for Mobile Elements. Annu. Rev. Genet. 2014:167–186. doi: 10.1146/annurev-genet-120213-092046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.