


Abstract
In this study, we show that silencing of CITED2 using small-hairpin RNA (shCITED2) induced DNA damage and reduction of ERCC1 gene expression in HEK293, HeLa and H1299 cells, even in the absence of cisplatin. In contrast, ectopic expression of ERCC1 significantly reduced intrinsic and induced DNA damage levels, and rescued the effects of CITED2 silencing on cell viability. The effects of CITED2 silencing on DNA repair and cell death were associated with p53 activity. Furthermore, CITED2 silencing caused severe elimination of the p300 protein and markers of relaxed chromatin (acetylated H3 and H4, i.e. H3K9Ac and H3K14Ac) in HEK293 cells. Chromatin immunoprecipitation assays further revealed that DNA damage induced binding of p53 along with H3K9Ac or H3K14Ac at the ERCC1 promoter, an effect which was almost entirely abrogated by silencing of CITED2 or p300. Moreover, lentivirus-based CITED2 silencing sensitized HeLa cell line-derived tumor xenografts to cisplatin in immune-deficient mice. These results demonstrate that CITED2/p300 can be recruited by p53 at the promoter of the repair gene ERCC1 in response to cisplatin-induced DNA damage. The CITED2/p300/p53/ERCC1 pathway is thus involved in the cell response to cisplatin and represents a potential target for cancer therapy.
INTRODUCTION
Cisplatin-based therapy is one of the most effective chemotherapeutic treatments for ovarian, testicular, head and neck, and non-small cell lung cancer (NSCLC). The mechanism of action of cisplatin involves induction of DNA damage and apoptosis. Cisplatin cross-links to DNA, leading to unwinding of the double helix and attraction of various protein factors, including high-mobility-group (HMG) proteins. Presumably due to a shielding effect caused by these proteins, cisplatin-modified DNA is poorly repaired (1,2), a phenomenon which leads to cell cycle arrest and apoptosis. The resulting crosslinks consist of guanine–guanine and guanine–adenine intra-strand crosslinks (70–78%), intra-strand crosslinks of two non-adjacent guanines (8–10%) and other minor crosslink lesions (3,4). Intra-strand crosslinks are usually repaired by nucleotide excision repair (NER) while other lesions are repaired by complex mechanisms, which make use of NER, double-strand break (DSB) repair, and trans-lesion synthesis (TLS) components (5). Ataxia telangiectasia mutated (ATM) protein kinase and ATM-related (ATR) protein kinase are activated in cells during the early response to DNA damage. While ATM is activated by DSBs, ATR is activated by stalled DNA replication forks. Coupling of cisplatin damage to apoptosis also requires mismatch repair (MMR), and abortive attempts to repair DNA lesions play a key role in the cytotoxicity induced by the drug. Recent observations further suggest the involvement of DNA repair by homologous recombination (HR) in this process (2).
Increased DNA repair has been proposed to represent a major mechanism underlying cisplatin resistance. Studies performed on a series of cisplatin-resistant ovarian and cervical cancer cell lines show a clear relationship between DNA repair and reduced cisplatin cytotoxicity (1–2,6). While intra-strand DNA lesions (the major cisplatin-induced DNA adducts) are repaired by NER, the exact mechanism and events occurring during inter-strand crosslinks repair are poorly understood (7,8). Cisplatin-induced inter-strand crosslinks can obstruct DNA replication fork progression in dividing cells, resulting in the formation of DSBs as indicated by the presence of γ-H2AX, a phosphorylated form of histone H2AX (9). DNA damage response (DDR) proteins that co-localize with γ-H2AX foci include the MRE11/RAD50/NBS1 (MRN) complex, BRCA1, RAD51, MDC1 and FANCD2, which represent major components of HR DNA repair (10,11). ICLs induced by cisplatin, mitomycin C, and the combination of psoralen and ultraviolet (UV) light have also been reported to induce the formation of γ-H2AX foci (12–15). This observation raises the possibility that persistence of γ-H2AX foci after treatment with inter-strand crosslinks-inducing agents could reflect a defective HR system, either as a direct inability to repair inter-strand crosslinks or replication-associated DSBs. The formation of γ-H2AX-associated DSBs following cisplatin treatment indicates critical DNA damage that, if not repaired, may be responsible for cisplatin-induced cytotoxicity.
The excision repair cross-complementing group 1 protein (ERCC1), an important mediator of NER, forms a heterodimer with the xeroderma pigmentosum complementation group F protein (XPF), forming a complex that performs a critical incision step during the NER reaction (16,17). The XPF–ERCC1 complex also plays specific roles in inter-strand crosslinks repair (18,19) and in completion of HR during inter-strand crosslinks repair (20), and it facilitates the repair of DSBs induced by cisplatin- inter-strand crosslinks processing (19). Thus, the XPF–ERCC1 complex participates in repair functions beyond NER. Furthermore, ERCC1 expression levels positively correlate with DNA repair capacity, and are associated with cellular and clinical resistance to platinum-based chemotherapy (21–24). Studies that analyzed the role of ERCC1 as an NER component, using both fresh and formalin-fixed paraffin-embedded NSCLC, ovarian and gastric cancer tissues, have been conducted on large numbers of patients (see ref. (25) for a recent review). ERCC1 expression can be used as a prognostic marker for chemoresistance, normal tissue tolerance and patient outcome during platinum-based chemotherapy (26). For example, ERCC1 expression was found to be predictive of patient outcome for NSCLC (27) and gastric cancer (28) treated with cisplatin-based chemotherapy. Early prospective validation studies in patients with NSCLC showed promising therapeutic results (29–31) and several large prospective studies evaluating ERCC1 expression as a biomarker for cisplatin-based therapy response have recently been completed (reviewed in ref. (25)).
The tumor suppressor p53 is stabilized and activated in cells in response to different forms of cellular stress, such as ionizing radiation (IR), which involves ATM activity, as well as UV light, which requires the participation of ATR. These stimuli may induce downregulation of MDM2 expression or changes in the subcellular localization of p53 and MDM2 (32,33). The p53 protein is also involved in modulating cisplatin sensitivity and the NER pathway in response to cisplatin-induced DNA damage (34–36). p53 activates several downstream effector genes, including p21 which leads to cell cycle arrest, as well as PUMA/BAX which induce apoptosis. Transcriptional activity of p53 toward the expression of either apoptotic or cell cycle arrest genes can be determined by post-translational modifications of the protein via phosphorylation at Ser46 (37,38) or through mono-ubiquitination at Lys320 (39), respectively. p53 can also be acetylated by histone acetyltransferases (HAT) which were initially found to modify histones. CBP/p300 and the associated protein P/CAF bind to and acetylate p53 in response to DNA damage, and are needed for full p53 transactivation, as well as for producing downstream p53 effects on growth arrest and apoptosis (40,41). CBP/p300 can both positively and negatively regulate p53 transactivation, as well as p53 protein turnover, depending on cellular context and environmental stimuli. For example, acetylation of p53 at Lys-373 by HAT enzymes plays an important role in stabilizing p53 and in inducing apoptosis via translocation of Bax to the mitochondria (42). In contrast, phosphorylation of p53 N-terminal residues allows interaction of p53 with CBP/p300, which leads to acetylation of p53 at the C-terminus (43), followed by an increase of p53 stability and sequence-specific DNA-binding activity (43–46).
CITED2—a CBP/P300-interacting transactivator protein containing a Glu/Asp-rich C-terminal domain—is critical for the regulation of various genes involved in cell growth and oncogenesis (47). CITED2 has been shown to inhibit the activity of the transcription factor hypoxia-induced factor-1 (HIF-1) (48), and to regulate FOXO3a, which inhibits HIF-1-induced apoptosis (49). In addition, the activity of CITED2 and HIF-1 is affected by the tumor suppressor TSG101 in ovarian carcinoma cells (50). CITED2 also functions as an activator of peroxisome proliferator-activated receptors (PPAR) α and γ (51) and MMP9 (52). In addition, upregulation of CITED2 and activation of PPARα are responsible for neuronal cell death induced by the DNA-damaging agent camptothecin (53), suggesting that CITED2 may have pro-apoptotic properties. In contrast, CITED2 was found to be overexpressed in cisplatin-resistant ovarian and cervical cancer cells, and knockdown of CITED2 using siRNA enhanced the cytotoxic effects of cisplatin in these cells (54,55). These findings suggest that CITED2 may have both apoptotic and anti-apoptotic properties depending on the cellular context.
We previously reported that CITED2 expression level and p53 modifications are implicated in cisplatin resistance in various cancer cell lines (55,56). Knockdown of CITED2 sensitized cancer cells to cisplatin. However, the mechanism underlying CITED2's effect on apoptosis following cisplatin treatment remains unclear. It is unclear whether the interaction between CBP/p300 and CITED2 may affect p53 activity and regulation of ERCC1 expression in response to cisplatin. In the present study, we found that silencing of CITED2 decreased ERCC1 expression via prevention of chromatin relaxation and p53 targeting to the ERCC1 promoter. CITED2 silencing also impaired DNA repair and sensitized cells to cisplatin, and these effects could be rescued by ectopic expression of ERCC1. In addition, silencing of CITED2 sensitized cancer cells and cell line-derived xenografts (CDX) to cisplatin, leading to enhanced cancer cells apoptosis in response to the drug.
MATERIALS AND METHODS
Cell lines and chemical reagents
The cell lines used in this study included human embryonic kidney cells (HEK293) and tumorigenic cells (cervix HeLa and lung H1299; American Tissue Type Collection, Manassas, VA, USA). Cells were cultured as described before (55). Cisplatin, vincristine and taxol (paclitaxel) were purchased from Bristol-Myers Squibb (New York, NY, USA). PFTα and histone acetyltransferase inhibitor (HATi II) were obtained from Biomol Research Laboratories (Farmingdale, NY, USA) and Calbiochem (San Diego, CA, USA), respectively. Other chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA). All reagents were used according to the instructions provided by the supplier.
Gene knockdown using short-hairpin RNA
Lentivirus-based pLKO.1 plasmids expressing short-hairpin RNA (shRNA) that target either CITED2 or p300 were purchased from the National RNAi Core Facility (Academia Sinica, Taipei, Taiwan). A plasmid expressing luciferase shRNA (TRCN0000072244) was used as a negative control. Five plasmid clones for CITED2 or p300 gene were tested for gene knockdown efficiency in HEK293 cells as before (55). The most effective shCITED2 (TRCN0000015653) and shp300 (TRCN0000009882) plasmids were used in the present study. CITED2 and p53 mRNA levels in HEK293 cells were monitored by polymerase chain reaction (PCR) three days following plasmid transfection. Stable clones expressing shRNA plasmids via a lentivirus were established in HeLa and H1299 cells. Recombinant lentivirus constructs were incubated with the cells for 10 days in puromycin-containing selection medium according to the procedures described by the supplier (National RNAi Core Facility).
Quantitative RT-PCR (qRT-PCR)
Quantitative reverse transcription-PCR (in short qRT-PCR) was performed on total cellular RNA extracted with the Trizol reagent (Invitrogen, Waltham, MA, USA) as described before (57). GenBank sequences NM 006079 and NM 000996 were used to design PCR primers for CITED2 and GAPDH, respectively. PCR primers were designed using Primer Express 2.0.0 (Applied Biosystems, Foster City, CA, USA). The primers, which were used at a concentration of 200 nM, were: CITED2, forward, 5′-CCTACCCCCACAACCACTACA-3′; CITED2, reverse, 5′-GCAATCTCGGAAGTGCTGGT-3′; ERCC1, forward, 5′-GCCTATGAGCAGAAACCAGC-3′; reverse, 5′-AATGTGGTCAGGAGGGTCTG-3′; GAPDH, forward, 5′-TCCTGCACCACCAACTGCTT-3′; GAPDH, reverse, 5′-GAGGGGGCCATCCACGTCTT-3′; p300, forward, 5′-CAGATTGATCCCAGCTCCAT-3′; reverse, 5′-GAAAGAAGACTCGGCGTTTG-3′. Relative quantification was performed using the ΔΔCt method with normalization to GAPDH. Namely, the ΔCt for each candidate gene was calculated as ΔCt (candidate) = [Ct (candidate) − Ct (GAPDH)]. The relative abundance of tested mRNA was shown as 2ΔCt(candidate) − ΔCt(GAPDH).
Plasmid expression, cell extracts and immunoblot analysis
Construction and expression of plasmids (CITED2 or p53 cDNA inserted into the pcDNA3 vector) were performed as described before (55). Following 48 h of incubation with DNA and Lipofectamine (Invitrogen), the cells were treated with cisplatin and incubated for an additional 24 h. Fifty microgram or the quantity indicated of protein extract was prepared for immunoblotting as before (58). Proteins were separated using a 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis, transferred onto PVDF membranes and incubated with primary antibodies raised against the following proteins: ERCC1 (FL-297), CITED2 (JA-22), GAPDH (FL-335), p53 (DO-1), p300 (C-20) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), phosphorylated p53 (Ser15 and Ser46; Abcam, Cambridge, MA, USA), acetylated p53 (Lys373; Upstate Biotechnology, Lake Placid, NY, USA); Histone 3 (H3), H3K4Me, H3K9Me2, H4Ac (Active Motif, Carlsbad, CA, USA), H3Ac, H3K9Ac, H3K14Ac, GCN5L2 (Cell Signaling Technology, Beverly, MA, USA), 53BP1 (A3-272A) (Bethyl Laboratories, Montgomery, TX, USA) and γ-H2AX (05-636-I) (Millipore, Billerica, MA, USA). Membranes were incubated with the following secondary antibodies: goat anti-mouse or goat anti-rabbit-horseradish peroxidase (Amersham, Buckinghamshire, UK). Signal was visualized using enhanced chemiluminescence (Amersham).
Chromatin immunoprecipitation (ChIP) assay
Formaldehyde cross-linking and chromatin immunoprecipitation (ChIP) assays of tissue culture cells were performed using a modified protocol (59) as described before (60) using a commercial kit (Upstate Biotechnology, Lake Placid, NY, USA). Chromatin was sonicated on ice to obtain DNA fragments of appropriate size, averaging 600 bp. Twenty percent of total supernatant was used as a total input control. Following removal of bound proteins, immunoprecipitated DNA was subjected to PCR. The products amplified by regular PCR were separated on a 1.5%-agarose gel and visualized using ethidium bromide staining. p53-binding sites and other possible transcription factor-binding sites on the endogenous ERCC1 promoter and exon 4 region on chromosome 19 (NC_000019.10) were predicted using TFSearch (http://www.cbrc.jp/research/db/TFSEARCH.html; Japan). The primer sequences were designed using Primer 3.0 (Life Technologies, Carlsbad, CA, USA). The primers included: primer pair 1 for the p53-binding site in the promoter region, forward, 5′-GCACAGACACAGGGAATGACTT-3′ and reverse, 5′-ACTGAACCGAAGTCAAAGGAG-3′, which yielded a 169-bp product; primer pair 2 for the negative control-binding site in exon 4, forward, 5′-GGGCCCTGTGGTTATCAAG-3′ and reverse, 5′-ACACTGGGACATGACCCTCC-3′, which yielded a 237-bp product. ChIP-quantitative PCR (qPCR) was performed using a qPCR kit and the same primer pairs. Relative quantification of IP product was performed using the ΔCt method with normalization to control IgG. Namely, the ΔCt for each IP gene was calculated as 2−Δ[Ct(IP)-Ct(input)]-2−Δ[Ct(control IgG)-Ct(input)], which normalized the relative level of DNA (in relation to the input) specifically immunoprecipitated by the p53, H3K9Ac or H3K14Ac antibody to that immunoprecipitated by the control IgG.
Immunofluorescence staining
Transfected cells cultured on glass coverslips were washed with phosphate-buffered saline (PBS) and then fixed with 1% formaldehyde for 15 min at room temperature. Cells were incubated with agitation in 2% BSA (dissolved in PBS) for 1 h followed by incubation with primary mouse anti-γ-H2AX antibody (1 μg/ml) overnight at 4°C and with secondary antibodies conjugated with Alexa-488 (1:200) (Invitrogen) in PBS for 1 h at room temperature. In some experiments, incubations were performed with primary rabbit anti-53BP1 antibody (1 μg/ml) overnight at 4°C and with secondary antibodies conjugated with Alexa-568 (1:200) (Invitrogen) in PBS for 1 h at room temperature. Slides were counterstained with 10 μg/ml of DAPI in 60% glycerol (prepared in PBS). Confocal laser-scanning immunofluorescence microscopy (CLSM) was carried out with a Zeiss LSM510 META confocal microscope (Carl Zeiss, Oberkochen, Germany). Image analysis was done using the LSM510 META Software, and images were assembled using Adobe Photoshop 6.0. Quantitative analysis was performed by inspecting cells (n = 100 per sample) in three separate experiments. Values were expressed as mean ± s.d. The colocalization factor was defined as follows: [(fraction of cells having colocalization) × (fraction of foci colocalized per cell)] × 100.
Cell death and FACS analysis
Cells treated with chemotherapeutic agents (cisplatin, vincristine or taxol) in serum-free medium for 2 h were maintained in drug-containing normal medium for 3 days. Cell viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay (61). The percentage of viable cells was calculated as the ratio of viable cells over the total amount of cells counted. The sensitization factor (SF50) was calculated as the dose which kills 50% of cells in the negative control (e.g. shLuc) divided by the dose which kills 50% of cells in the treatment group (e.g. shCITED2). The resistance factor (RF50) was calculated as the dose which kills 50% of cells in the rescue group (e.g. CITED2) divided by the dose which kills 50% of cells in the treatment group (e.g. shCITED2). To confirm the extent of cell death, apoptotic cells produced by the drug were determined by flow cytometry analysis of sub-G1 cells (62). The LYSYS II software was used to assess cell cycle distribution. Three independent experiments were performed. The data were reported as means ± standard deviation (SD). Statistical significance (P-value) was assessed using a two-tailed Student's t-test for single comparison. The symbol * denotes P < 0.05; ** denotes P < 0.01.
Luciferase ERCC1 promoter analysis
The p53-responsive luciferase reporter of the ERCC1 gene (pERCC1-luc, provided by Dr Muh-Hwa Yang, National Yang-Ming University, Taipei, Taiwan) and wild-type pcep4-p53 (p53 WT) or mutant p53 (p53 Mut) expression plasmids (63) were used. Healthy, growing cells were co-transfected with control or test expression vector and pERCC1-luc gene constructs with Lipofectamine following the procedures provided by the supplier (Invitrogen). A plasmid expressing the bacterial β-galactosidase gene (pCMV-β-gal) was co-transfected in each experiment as an internal control for transfection efficiency. Cells were harvested after 48 h of transfection and luciferase activity was monitored. Transcriptional activity was monitored using a dual luciferase reporter assay (Promega) and a luminescence reader (LMaxII384, Molecular Devices, Sunnyvale, CA, USA) according to the instructions provided by the manufacturers. Promoter activity was assessed using normalization of the luciferase activity by β–galactosidase activity.
Cell-derived tumor xenografts in RAG2-deficient mice
Animal handling and experimental procedures were approved by the Animal Experimental Ethics Committee of Chang Gung University. Female RAG2-deficient C57BL/6 mice of 6–8 weeks of age were used. Tumors were produced by subcutaneous injection of 1 × 107 HeLa cells into RAG2-deficient mice (12 mice per group). Tumor size was measured as described before (64). Three days post-inoculation (tumor diameter was ∼0.5 cm), mice were divided into two groups (group A and group B, six mice per group). Mice from each group were injected intra-tumorally with 5 × 107 copies (in 5 μl of medium) of lenti-shLuc or lenti-shCITED2.
Immunohistochemistry
Tumors were removed and processed with 4% paraformaldehyde at 4°C overnight and 5-μm thick sections were cut on slides using a cryostat. Immunohistochemistry was conducted by incubating the sections in 2% normal goat serum (Vector Laboratories, Burlingame, CA, USA) solution at 37°C for 1 h, after which a first antibody for ERCC1 or γ-H2AX was applied to sections at a 1:200 dilution overnight at 4°C. On the following day, ERCC1 or γ-H2AX was detected by using a goat antibody IgG conjugated to horseradish peroxidase (HRP; Santa Cruz Biotechnology) and diaminobenzidine (DAB). Slides were dehydrated by using a graded series of alcohol and xylene, after which they were mounted in VectaMount (Vector Laboratories) and covered with glass coverslips. All sections were analyzed with an Olympus optical microscope and images were obtained with a digital camera (Pixera, Santa Clara, CA, USA).
Statistical analysis
Data were reported as mean values ± SD. Three independent experiments were performed unless indicated otherwise. Statistical significance (P-value) was calculated with a two-tailed Student's t-test for single comparison.
RESULTS
CITED2 silencing downregulates ERCC1 expression and reduces cell viability in cisplatin-treated cells
Our previous study indicated that CITED2 overexpression is associated with cisplatin resistance in HeLa cells (55). To assess the effect of CITED2 silencing on DNA repair, we examined the expression of ERCC1, a major protein involved in the NER pathway where it recognizes DNA damage and performs the excision process (65). CITED2 silencing in HEK293 cells (which reached over 80% efficiency) significantly reduced ERCC1 mRNA (Figure 1A, P < 0.01) and protein levels (Figure 1B and C, P < 0.01). CITED2 silencing also sensitized HEK293 cells to cisplatin (Figure 1D). Sensitization to the drug was assessed using the sensitization factor SF50, calculated as the IC50 of shControl cells divided by the IC50 of shCITED2 cells (Figure 1D, SF50 = 3.26). CITED2 silencing did not affect the viability of cells treated with the mitotoxins vincristine (SF50 = 1.18) or taxol (SF50 = 1.06) (Table 1). shCITED2 expression significantly increased cisplatin-induced sub-G1 cells (Figure 1E), which represent cells undergoing apoptosis (62). These results suggest that silencing of CITED2 may regulate cell response to DNA-damaging agents.
Figure 1.
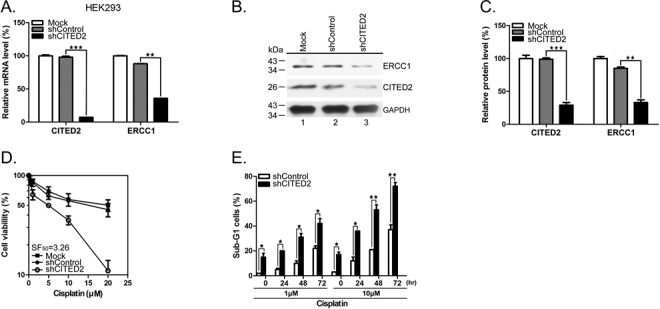
Silencing of CITED2 downregulates ERCC1 expression and cell viability to cisplatin. (A) mRNA levels of the repair gene ERCC1 are reduced in HEK293 cells following CITED2 silencing using shRNA. (B and C) Protein levels of the repair gene ERCC1 are reduced in HEK293 cells following CITED2 silencing. (D) Sensitization of HEK293 cells to cisplatin by shCITED2 compared to shControl. (E) Apoptotic sub-G1 cells in response to cisplatin. The experiments shown in this study were performed in triplicate. Mock control which has no exogenous shRNA was also included for comparison in (A–D).
Table 1. Modification of drug sensitization by CITED2 knockdown in HEK293 cells.
| shControl (IC50) | shCITED2 (IC50) | sensitization factor (SF) | |
|---|---|---|---|
| Cisplatin (μM) | 16.3 ± 0.24 | 5.0 ± 0.13 | 3.26 |
| Vincristine (nM) | 5.3 ± 0.52 | 4.5 ± 0.33 | 1.18 |
| Taxol (nM) | 5.1 ± 0.13 | 4.8 ± 0.84 | 1.06 |
CITED2 silencing reduces DNA repair
To assess whether CITED2 silencing impairs DNA repair, we examined DNA damage status in cisplatin-treated cells. We detected γ-H2AX, which correspond to phosphorylated histone H2AX, a marker of DNA DSBs. Previous studies have shown that γ-H2AX reflects the extent of DSBs and may be used to detect pre-cancerous cells and to monitor the effectiveness of cancer therapy (66). Interestingly, a 10-fold accumulation of γ-H2AX was detected in shCITED2-expressing cells in the absence of cisplatin (1 h: Figure 2A and B, P < 0.005; 24 h: Figure 2C and D, P < 0.005). CITED2 silencing caused a 12- to 14-fold increase of γ-H2AX level in cells treated with 1 and 10 μM of cisplatin for 1 h, respectively. Initial DNA lesions (cisplatin treatment for 1 h) were slightly increased in cisplatin-treated cells compared to control cells. On the other hand, CITED2 silencing caused a five- to six-fold increase of γ-H2AX level in cells treated with 1 and 10 μM of cisplatin for 24 h. These results indicate that the level of DNA damage induced by cisplatin in CITED2 silencing cells was reduced after 24 h of inoculation.
Figure 2.
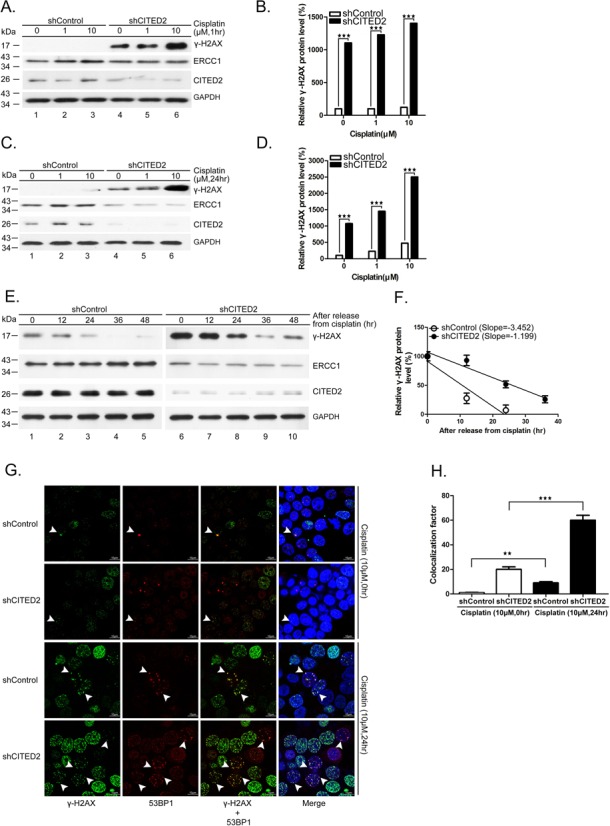
Silencing of CITED2 reduces DNA repair rate. (A and B) Accumulation of DNA damage (γ-H2AX) and reduction of the DNA repair proteins ERCC1 following CITED2 silencing in cisplatin-treated HEK293 cells for 1 h. (B) Quantification of DNA damage shown in (A). (C and D) Accumulation of DNA damage (γ-H2AX) and reduction of the DNA repair proteins ERCC1 following CITED2 silencing in cisplatin-treated HEK293 cells for 24 h. (D) Quantification of DNA damage shown in (C). (E) Reduction of DNA repair rate by shCITED2 expression. HEK293 cells were treated with cisplatin for 24 h, prior to incubation in drug-free medium for the indicated period. (F) Reduced removal of cisplatin-induced γ-H2AX levels in cells expressing CITED2 shRNA. The slopes are indicated. (G) Accumulation of γ-H2AX and 53BP1 double-positive cells following CITED2 silencing in cisplatin-treated HEK293 cells for 24 h. Examples of double-positive cells are indicated with arrowheads. Size reference (10 μm) is indicated. (H) Quantification of γ-H2AX and 53BP1 double-positive cells is shown in (G).
To assess whether the induced DNA damage is due to reduced repair, we treated the cells with 1 μM of cisplatin for 24 h, prior to incubation in drug-free medium. The level of γ-H2AX increased six-fold in shCITED2 cells compared to shControl cells (Figure 2E, compare lanes 1 and 6). While the level of γ-H2AX gradually decreased following incubation (Figure 2E, compare lanes 1–5), the decrease of DNA damage was also observed in shCITED2-expressing cells (lanes 6–10). The levels of γ-H2AX in both types of cells revealed a slower reduction in shCITED2 cells (Figure 2F), indicating a reduced DNA repair rate (slope of −1.199 for shCITED2 cells versus−3.452 for shControl cells). These results suggest that CITED2 silencing may impair DNA repair and result in accumulation of DNA damage.
To confirm these observations, we measured DNA damage by monitoring double immunofluorence of γ-H2AX/53BP1 using confocal analysis in HEK293 cells treated with 10 μM of cisplatin. Following incubation in cisplatin-free medium, the number of γ-H2AX/53BP1 double-positive cells was slightly higher in shCITED2 cells compared to shControl cells (Figure 2G, two upper panels). On the other hand, CITED2 silencing increased the number of γ-H2AX/53BP1 double-positive cells following incubation in cisplatin medium (Figure 2G, compare the two lower panels). While cells with high double-positive stain (>7) were detected in cisplatin-free shCITED2 cells, their number was larger in shCITED2 cells following cisplatin treatment. We noted that the number of cells with low double-positive stain (>7) remained high in shControl cells, even following cisplatin treatment. An equitoxic UV dose, which induces pyrimidine dimers or 6,4-photoproducts, did not elicit similar DNA damage lesions in these cells (data not shown). Statistical analysis indicated a dramatic increase in γ-H2AX/53BP1 double-positive cells (>7 double stain) following CITED2 silencing compared to the shControl (Figure 2H). The levels of γ-H2AX/53BP1 double-positive cells (>7 double stain) increased by about six-fold following cisplatin treatment in shCITED2 cells compared to shControl cells. These values were comparable to that measured earlier for γ-H2AX (Figure 2C and D).
Ectopic expression of ERCC1 rescues shCITED2-induced DNA damage and cell viability
To assess whether ERCC1 overexpression may rescue shCITED2-modified DNA damage and cell viability, we transfected HEK293 cells with Flag-pcDNA3-ERCC1 cDNA. While γ-H2AX levels increased with cisplatin concentration in shControl cells, forced expression of ERCC1 considerably reduced γ-H2AX in cells expressing shControl (Figure 3A, compare lanes 1–3 with 7–9). Ectopic expression of ERCC1 also reduced γ-H2AX levels in shCITED2 cells, with or without cisplatin at 1 μM (Figure 3A, compare lanes 4–5 with 10–11). The level of γ-H2AX induced by a high concentration of cisplatin was not reduced by ERCC1 overexpression in these cells (compare lane 6 with 12). However, statistical analysis of three experiments indicated that ERCC1 overexpression significantly reduced damage level compared to control cells treated with a low dose of cisplatin (Figure 3B). The role of ERCC1 in DSB repair may be limited to specific DSB-repair pathways (such as single strand annealing), and might not be fully representative of global ERCC1 activity. This possibility might explain why ERCC1 overexpression did not reduce γ-H2AX signals in cells treated with a high dose of cisplatin. Furthermore, ectopic expression of ERCC1 rescued cisplatin-induced cell death in both shControl cells (Figure 3C) and shCITED2 cells (Figure 3D). The modulation of cell viability by ERCC1 expression appeared to be significant in the range of cisplatin concentration showing comparable cell viability in both types of cells, namely 5–20 μM in shControl cells and 0.5–5 μM in shCITED2 cells.
Figure 3.
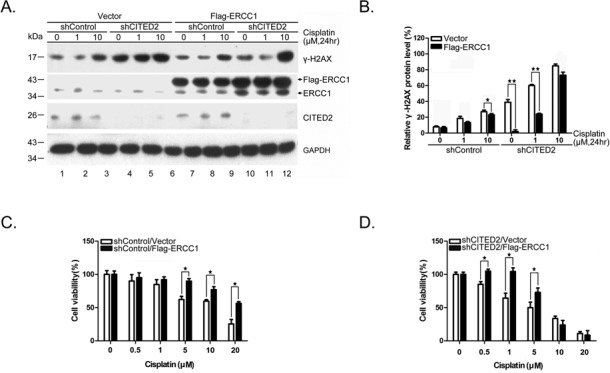
Ectopic expression of ERCC1 rescues shCITED2-induced DNA damage and cell viability. (A) Overexpression of ERCC1 gene reduced γ-H2AX levels. While ERCC1 expression reduced the DNA damage in both control and cisplatin-treated cells (compare lanes 1–3 and 7–9), ERCC1 expression partly reduced shCITED2 potentiated γ-H2AX levels (compare lanes 5 and 11). (B) Quantification of DNA damage shown in (A). (C) Protection of cisplatin-induced cell growth inhibition by ERCC1 overexpression. (D) Protection of cisplatin-induced and/or shCITED2 sensitized cell death by ERCC1 overexpression.
Downregulation of cisplatin-induced ERCC1 mRNA expression by CITED2 silencing is attenuated by p53 inhibitor
To assess the regulatory role of CITED2 silencing on ERCC1 gene expression, we investigated the protein degradation rate of ERCC1. While the level of ERCC1 gradually decreased following cycloheximide treatment in shControl cells, similar protein degradation rates were found in shCITED2 cells (Figure 4A and B). Treatment with 10 μM of cisplatin produced a 2.10-fold decrease of ERCC1 mRNA in shCITED2 cells compared to shControl cells (Figure 4C, P < 0.01). ERCC1 mRNA increased by 3.22- and 2.06-fold in shCITED2 cells following treatment with 1 and 10 μM cisplatin, respectively (Figure 4C). The level of CITED2 silencing in each cell condition was determined (Figure 4D). These results suggest that basal level of ERCC1 mRNA is significantly downregulated by CITED2 silencing.
Figure 4.
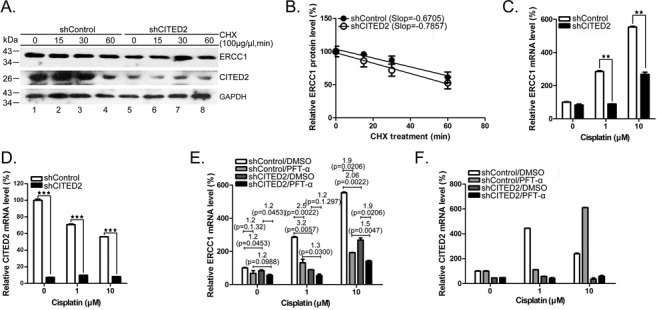
Silencing of CITED2 downregulates cisplatin-induced ERCC1 mRNA levels which were partly suppressed by the p53 inhibitor PFTα. (A) ERCC1 protein instability remained similar in both control and shCITED2-treated cells. Cells were pre-treated with cycloheximide (CHX) for 2 h followed by incubation in CHX-free culture medium for the indicated time. (B) Quantification of ERCC1 protein levels shown in (A). ERCC1 protein level was only minimally affected by shCITED2 as indicated by similar curve slopes. (C) Reduction of ERCC1 mRNA levels by shCITED2. (D) CITED2 mRNA levels remained suppressed by shCITED2. (E) Reduction of cisplatin-induced ERCC1 mRNA levels by shCITED2 is less dramatic in the presence of PFTα. Modulation by PFTα was greater at the low cisplatin concentration used. (F) CITED2 mRNA remained at low levels following shCITED2 expression. mRNA levels of CITED2 appeared to fluctuated in the presence of PFTα. Fold difference of average mRNA levels and P-values are indicated (n = 3).
In response to sub-lethal DNA damage, the cells recruit p53, which is specifically modified (36). To test whether p53 is involved in the regulation of ERCC transcription, we pre-treated the cells with the p53 inhibitor PFTα. While the basal level of ERCC1 mRNA was reduced 1.2-fold in shControl cells following PFTα treatment, there was a 1.4-fold reduction in shCITED2 cells (Figure 4E, P = 0.1032 and P = 0.0162, respectively). ERCC1 mRNA was downregulated 1.2-fold by CITED2 silencing in both control DMSO and PFTα-treated cells. Interestingly, CITED2 silencing induced a significant reduction of ERCC1 mRNA level in cells treated with 1 μM of cisplatin and PFTα: 2.5- and 1.2-fold reductions of mRNA levels were observed in cells treated with 1 μM of cisplatin (Figure 4E, P = 0.0022 and P = 0.1297, respectively). Similarly, there was a reduction of ERCC1 level from 3.0- to 1.9-fold in cells treated with 10 μM of cisplatin (Figure 4E, P = 0.0005 and P = 0.0206, respectively). These results suggest that p53 may be involved in the transcriptional regulation of the ERCC1 gene, under both basal and DNA-damage induced conditions (i.e. cisplatin), with greater effects observed under induced conditions. Furthermore, CITED2 silencing may impair p53-mediated regulation of ERCC1 expression.
Basal and induced p53 binding to the ERCC1 promoter and transactivation of gene expression are reduced by CITED2 silencing
To assess whether p53 regulates ERCC1 gene expression, we performed a ChIP assay. A predicted p53-binding site was identified on the ERCC1 promoter, ranging from 348 to 366 bp upstream of the transcription start site (Figure 5A, parts of the exons and introns are indicated). A 169-bp PCR product containing the p53-binding site of the ERCC1 promoter (the primers used are indicated in Figure 5A) was detected from untreated shControl cells. The p53 binding level was greatly reduced in shCITED2 cells (Figure 5B and C, P < 0.05). No PCR product that included the p53 binding site was detected in the negative control in samples obtained by precipitation with IgG antibody and exon 4 by precipitation with p53 antibody. p53 binding increased in a concentration-dependent manner in cells treated with cisplatin (Figure 5B and C). CITED2 silencing caused a significant reduction of p53 binding to the ERCC1 promoter (Figure 5C, P < 0.01 and P < 0.005 for 1 and 10 μM treatment, respectively). Furthermore, p53 binding to the ERCC1 promoter was reduced by PFTα under both basal and cisplatin-treated conditions (Figure 5D and E). Notably, p53 binding to the ERCC1 promoter was considerably reduced in shCITED2-expressing cells compared to shControl-expressing cells. The level of regulation appeared to be more apparent in cisplatin-treated cells. Furthermore, the ERCC1 promoter assay indicated a dose-dependent induction of luciferase reporter activity (Figure 5F, P < 0.005). While basal activity was significantly reduced by CITED2 silencing (P < 0.01), the inducible activity of the promoter was reduced to a level comparable to basal level following CITED2 silencing. Reduction of CITED2 protein level by shCITED2 was also observed in this assay (Figure 5G). These results indicate that p53 binding to the ERCC1 promoter can be induced by cisplatin and is also regulated by CITED2 silencing. Cisplatin-induced p53 binding to the ERCC1 promoter and transactivation of ERCC1 expression are significantly reduced by CITED2 silencing.
Figure 5.
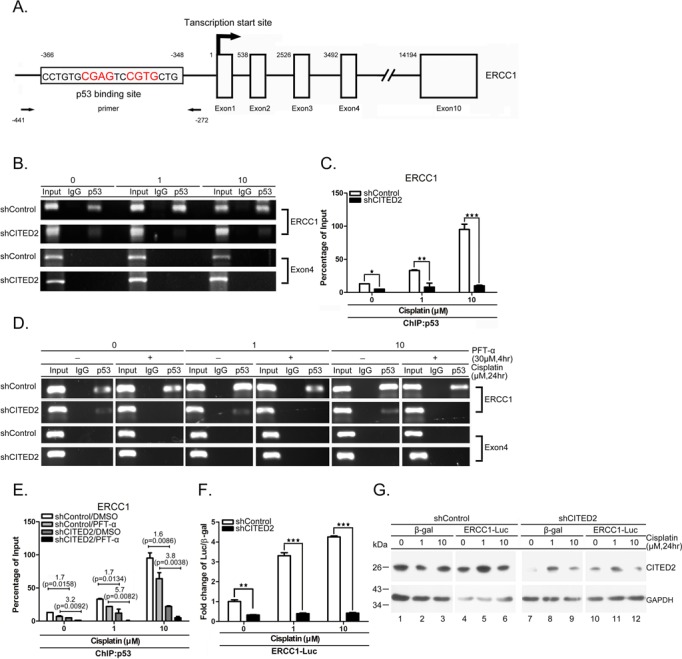
Both basal and induced p53 binding to and transaction of ERCC1 promoter are reduced by shCITED2. (A) Schematic diagram of the promoter and regions of the ERCC1 gene. p53-binding site, transcription initiation site (+1), and exons are indicated. The positions of PCR primers and elements relative to transcription initiation site are indicated with numbers. (B) Representative gel photographs of ChIP PCR products: reduction of p53 binding to ERCC1 the promoter by shCITED2. Chromatin of cells with indicated treatments were immunoprecipitated with p53 antibody or IgG control The co-IP DNA was detected by qRT-PCR for the p53 binding region (ERCC1) or negative control region (Exon 4). (C) Quantification of ChIP DNA from (B) by qRT-PCR: reduction of p53 binding to the ERCC1 promoter following shCITED2 expression. The values of PCR products were normalized to individual input. (D) Representative gel photographs of ChIP PCR products: reduction of p53 binding to ERCC1 promoter by PFTα is partly prevented by shCITED2. Other symbols are the same as in (B). (E) Quantification of ChIP DNA from (D) by qPCR: reduction of p53 binding to ERCC1 promoter by PFTα is partly prevented by shCITED2. Fold difference of average mRNA levels and P-values are indicated (n = 3). (F) Cisplatin-induced ERCC1 promoter activity was reduced by shCITED2 expression. Results were presented as the luciferase activity normalized to β-gal activity as internal control for each treatment. (G) Representative immunoblots of proteins for experiment shown in (F).
CITED2 silencing regulates ERCC1 expression and cell viability in p53-null H1299 cells
To further assess the involvement of p53 in ERCC1 transcription and cell viability in response to CITED2 silencing, we monitored cellular response in p53-null H1299 cells. Ectopic expression of p53 upregulated ERCC1 protein expression in H1299 cells (Figure 6A, lanes 2, 4, 6, 8, 10 and 12 versus lanes 1, 3, 5, 7, 9 and 11, respectively). In contrast, γ-H2AX levels were reduced by p53 overexpression in these cells. ERCC1 protein levels were reduced by CITED2 silencing in both untreated and cisplatin-treated H1299 cells, whereas γ-H2AX levels were induced (Figure 6A–C). Furthermore, the viability of H1299 cells was reduced by CITED2 silencing (Figure 6D, SF50 = 3.6). Ectopic expression of p53 further sensitized the cells to cisplatin (SF50 = 5.2). However, expression of mutant p53 did not affect the effects of CITED2 silencing on cell viability in response to the drug (SF50 = 3.0). Unlike HEK293 cells, H1299 cells did not display significant induction of ERCC1 mRNA following cisplatin treatment (Figure 6E). ERCC1 mRNA levels were dramatically induced by cisplatin in H1299 cells following p53 overexpression. Silencing of CITED2 reduced ERCC1 mRNA level slightly more in cisplatin-induced conditions compared to basal levels (Figure 6E, 2.08- and 2.86-fold for induced conditions compared to 1.58-fold for basal conditions). In the presence of p53 overexpression, CITED2 silencing produced an even greater reduction of cisplatin-inducible ERCC1 mRNA levels than the basal level (3.54- and 4.24-fold for induced conditions compared to 1.45-fold for basal levels). The level of CITED2 mRNA following CITED2 silencing was also monitored (Figure 6F). Furthermore, the ERCC1 promoter assay indicated an induction of the promoter activity by wild-type p53 in unstressed cells, but not by mutant p53, and this effect was dramatically reduced by CITED2 silencing (Figure 6G, P < 0.05). Cisplatin-induced promoter activity was also reduced by CITED2 silencing in p53-expressing cells (P < 0.005). Minimal promoter activity which was induced by cisplatin was also reduced by CITED2 silencing. The level of ERCC1 promoter activity regulated by p53 was significantly reduced in cisplatin-treated and untreated H1299 cells in the presence of PFTα (Figure 6H, P < 0.01 and P < 0.005, respectively). These results indicate that basal and inducible expression of ERCC1 can be regulated by exogenous p53, and that silencing of CITED2 impairs the upregulation of ERCC1 in this cell system.
Figure 6.
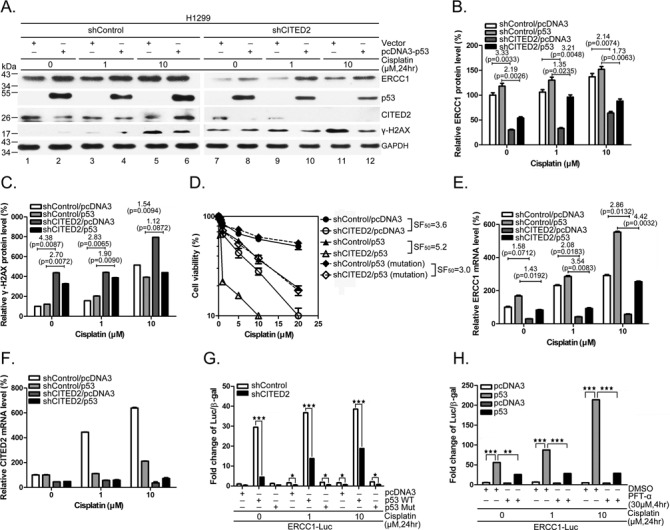
Regulation of ERCC1 expression and cell viability by CITED2 silencing in p53-null H1299 cells. (A) Reduction of ERCC1 protein level and induction of γ-H2AX levels by CITED2 silencing were partly reversed by ectopic expression of p53. (B) Quantification of ERCC1 protein levels in (A). (C) Quantification of γ-H2AX levels of (A). (D) Potentiation of CITED2 silencing-reduced cell viability by ectopic expression of p53. SF50 values are indicated. (E) Reduction of ERCC1 mRNA levels by CITED2 silencing in H1299 cells in the absence or presence of p53. (F) The efficacy of CITED2 silencing of (E). (G) Reduction of p53 activated ERCC1 promoter activity by CITED2 silencing. H1299 cells, with CITED2 silencing or not, were co-transfected with luciferase reporter plasmid and p53 plasmids, in the absence or presence of cisplatin. (H) Reduction of induced ERCC1 promoter activity by p53 inhibitor. P-values are indicated in (B, C and E).
CITED2 silencing reduces both chromatin relaxation and H3Ac/p53 complex binding on the ERCC1 promoter
Previous studies have shown that histone modifications induced by DNA damage, such as acetylation and deacetylation, are involved in both DNA repair and gene transcription (67). To assess whether chromatin relaxation is involved in the regulation of ERCC1 gene expression, we examined the status of representative histone modifications. While overall acetylation of H3 and H4 increased in shControl cells following cisplatin treatment (Figure 7A, lanes 1–3), both basal and cisplatin-induced H3Ac and H4Ac levels were down-regulated by CITED2 silencing (Figure 7A, compare lanes 1–3 with lanes 4–6). Specifically, acetylation of H3K9 and H3K14 was induced following cisplatin treatment. On the other hand, acetylated H3K9 and H3K14 signals were reduced by CITED2 silencing. Nevertheless, methylation of these proteins (H3K4Me and HeK9Me2) remained unchanged. While the level of the acetyltransferase p300 increased in shControl cells following cisplatin treatment, CITED2 silencing moderately reduced p300 levels (Figure 7A, compare lanes 1–3 with 4–6). Reduction of chromatin relaxation, as shown by histone acetylation, is probably caused by loss of p300 activity following CITED2 silencing. Treatment with the HDAC inhibitor trichostatin A (TSA) resulted in increase of basal and cisplatin-induced p53 binding to the ERCC1 promoter (Figure 7B and C). While the basal level of p53 binding to the ERCC1 promoter was slightly reduced by CITED2 silencing (1.86-fold, P = 0.0032), inducible p53 binding was dramatically reduced (3.17- and 5.78-fold for 1 and 10 μM treatment, respectively). Suppression of p53 binding to the ERCC1 promoter (induced by CITED2 silencing) was greatly reduced in cisplatin-treated cells following treatment with TSA (2.81- and 2.42-fold for 1 and 10 μM treatment, respectively, compared to 3.83-fold for untreated cells). These results indicate that reduction of p53 binding to the ERCC1 promoter following CITED2 silencing is less effective in cells in which chromatin is in a relaxed state (induced by TSA treatment). We also noted that DMSO treatment increased p53 binding to the ERCC1 promoter in cisplatin-treated shControl cells. This observation may be explained by a change in p53 conformation or ‘supercharging’ induced by DMSO as described earlier for other proteins (68). Furthermore, while re-ChIP analysis indicated that H3K9Ac/p53 binding to the ERCC1 promoter was induced by cisplatin, silencing of CITED2 dramatically inhibited the binding (Figure 7D, P < 0.01). Similar analysis for H3K14Ac/p53 binding to the ERCC1 promoter indicated that both basal and induced binding were impaired by CITED2 silencing (Figure 7E, P < 0.05 and P < 0.005, respectively). These results indicate that silencing of CITED2 reduces both chromatin relaxation and H3Ac/p53 binding on the ERCC1 promoter.
Figure 7.

Silencing of CITED2 reduces both chromatin relaxation and H3Ac/p53 complex loading on the ERCC1 promoter. (A) Reduction of chromatin relaxation markers following CITED2 silencing. HEK293 cells receiving the indicated treatments were processed for immunoblotting. (B) Enhanced p53 binding to the ERCC1 promoter following treatment with TSA. (C) Quantification of enhanced p53 binding to the ERCC1 promoter by TSA shown in (B). (D) Suppression of H3K9Ac/p53 binding to ERCC1 promoter by shCITED2. (E) Suppression of H3K14Ac/p53 binding to ERCC1 promoter by shCITED2.
Overexpression of CITED2 rescues shCITED2-induced effects on cell viability, ERCC1 and p300 expression, chromatin relaxation, and H3Ac/p53 complex loading on the ERCC1 promoter
To assess whether the sensitizing effects of CITED2 silencing are due to loss of CITED2, we ectopically expressed CITED2 in cisplatin-treated HEK293 cells. Sensitization of HEK293 cells to cisplatin following CITED2 silencing (SF50 = 3.26) was rescued by ectopic expression of CITED2 (RF50 = 1.52) (Figure 8A). Suppression of cisplatin-induced ERCC1 and p300 by CITED2 silencing (Figure 8B, lanes 1–6) was also rescued by ectopic expression of CITED2 (Figure 8B, compare lanes 4–6 with lanes 10–12). Similarly, reduction of chromatin relaxation markers (H3K9Ac and H3K14Ac) following CITED2 silencing was rescued by CITED2 overexpression. On the other hand, accumulation of cisplatin-induced γ-H2AX levels following CITED2 silencing was reduced by ectopic expression of CITED2 (protein level quantification is shown in Supplementary Figure S1A–C). Reduction of ERCC1 and p300 mRNA levels following CITED2 silencing was also rescued by ectopic expression of CITED2 (Figure 8C and D). The changes in the level of CITED2 mRNA are shown in Figure 8E.
Figure 8.
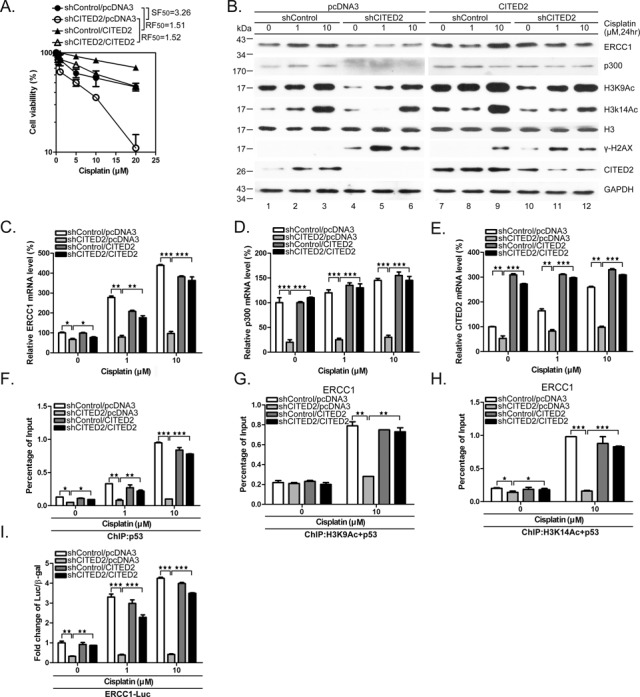
Overexpression of CITED2 rescues shCITED2-induced effects on cell viability, ERCC1 and p300 expression, chromatin relaxation and H3Ac/p53 complex loading on the ERCC1 promoter. (A) Sensitization of HEK293 cells to cisplatin following shCITED2 expression is rescued by ectopic expression of CITED2. (B) Reduction of chromatin-relaxation markers following CITED2 silencing is rescued by ectopic expression of CITED2. HEK293 cells receiving the indicated treatments were processed for immunoblotting. Quantification of protein levels is shown in Supplementary Figure S1A–C. (C) Reduction of ERCC1 mRNA level following CITED2 silencing is rescued by ectopic expression of CITED2. (D) Reduction of p300 mRNA level following CITED2 silencing is rescued by ectopic expression of CITED2. (E) Changes in the CITED2 mRNA level following CITED2 silencing and/or ectopic expression of CITED2. (F) Suppression of p53 binding to ERCC1 promoter by shCITED2 is rescued by ectopic expression of CITED2. (G) Suppression of H3K9Ac/p53 binding to the ERCC1 promoter by shCITED2 is rescued by ectopic expression of CITED2. (H) Suppression of H3K14Ac/p53 binding to the ERCC1 promoter by shCITED2 treatment is rescued by ectopic expression of CITED2. (I) Reduction of cisplatin-induced ERCC1 promoter activity by shCITED2 is rescued by ectopic expression of CITED2. Results are presented as luciferase activity normalized to β-gal activity as internal control for each treatment. The symbols are the same as in Figure 7. Fold difference of average levels and P-values are indicated (n = 3).
Furthermore, suppression of p53 binding to the ERCC1 promoter following silencing of CITED2 was rescued by ectopic expression of CITED2 (Figure 8F shows quantified data; representative gels of ChIP PCR products are shown in Supplementary Figure S1D). Suppression of H3K9Ac/p53 or H3K14Ac/p53 binding to the ERCC1 promoter following shCITED2 treatment was rescued by ectopic expression of CITED2 (Figure 8G and H). Further supporting these data, we also found that reduction of cisplatin-induced ERCC1 promoter activity following CITED2 silencing was rescued by CITED2 overexpression (Figure 8I). Cell immunofluorescence assays also indicated that accumulation of cisplatin-induced γ-H2AX/53BP1 double-positive cells following CITED2 silencing was rescued by ectopic expression of CITED2 (Supplementary Figure S3). These results suggest that CITED2 silencing caused sensitization to cisplatin and that this result may be due to reduction of ERCC1 expression, especially at the transactivation level. Moreover, reduction of ERCC1 expression appears to be associated with impairment of chromatin relaxation and p53 binding to the gene promoter.
Reduction of p300 following CITED2 silencing impairs H3KAc/p53 complex binding on the ERCC1 promoter
To assess the role of p300 which is reduced following silencing of CITED2, we treated HEK293 cells with shCITED2 in the presence or absence of cisplatin, and observed a dramatic reduction of p300 mRNA and protein (Figures 7A and 9A). Specific knockdown of p300 with shRNA caused a moderate inhibition of ERCC1 protein, especially in cisplatin-treated cells (Figure 9B). We observed that p300 protein level increased in cisplatin-treated cells (Figure 9B, lanes 1–3). As seen for CITED2 silencing, the mRNA level of ERCC1 was dramatically suppressed by silencing of p300, in both untreated and cisplatin-treated cells (Figure 9C). More than 50% of p53 binding to the ERCC1 promoter was inhibited in unstressed cells, as revealed by ChIP assay, and an even larger inhibition was found in cisplatin-treated cells (Figure 9D). Under the same conditions, binding of acetylated histone 3 (H3K9Ac and H3K14Ac) to the ERCC1 promoter was dramatically inhibited by p300 silencing (Figure 9E and F). We further assessed the modified H3/p53 complex on binding to the ERCC1 promoter by re-ChIP assay. Notably, while H3K9Ac/p53 complex binding to the ERCC1 promoter was greatly reduced by p300 silencing in cisplatin-treated cells, it was not affected in untreated cells (Figure 9G). In contrast, H3K14Ac/p53 binding to the ERCC1 promoter was dramatically reduced by p300 silencing in the presence or absence of cisplatin (Figure 9H). The inhibition effects produced by p300 silencing were similar as that seen following CITED2 silencing (Figure 7D and E).
Figure 9.
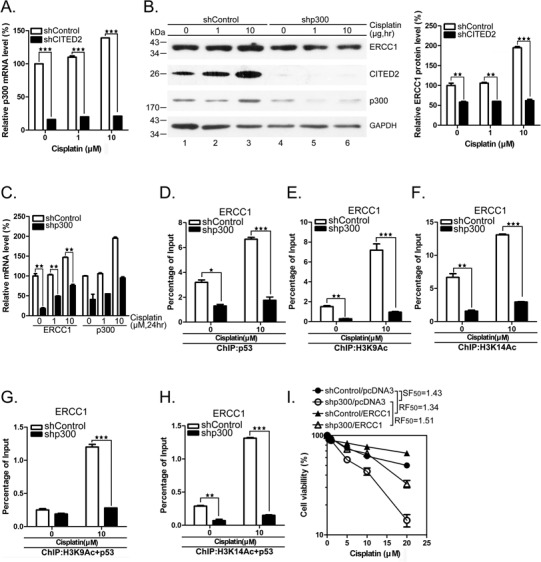
Silencing of p300 reduces chromatin relaxation, H3Ac/p53 complex loading on the ERCC1 promoter and sensitizes cells to cisplatin. (A) Silencing efficacy of CITED2 by shRNA. p300 mRNA levels were determined in untreated or cisplatin-treated HEK293 cells. (B) Reduction of ERCC1 protein levels following silencing of p300. Quantification of p300 protein levels of (A) is shown. (C) Reduction of ERCC1 mRNA levels following p300 silencing. Quantification of p300 mRNA levels of (A) is shown in the right panel. (D) ChIP data indicating reduced binding of p53 to the ERCC1 gene following p300 silencing. Representative PCR product levels are shown on top. The binding level of triplicates was presented as described in Figure 5. (E) Reduced binding of H3K9Ac to ERCC1 gene following p300 silencing. (F) Reduced binding of H3K14Ac to ERCC1 gene following p300 silencing. (G) Reduced binding of H3K9Ac/p53 to ERCC1 gene following p300 silencing. (H) Reduced binding of H3K14Ac/p53 to ERCC1 gene following p300 silencing. (I) Sensitization of HEK293 cells to cisplatin following shp300 expression is rescued by ectopic expression of ERCC1.
To further assess the role of p300 in modulating cisplatin-induced cell death, we treated HEK293 cells with shp300, with or without transfection of the ERCC1 expression plasmid. Silencing of p300 sensitized the cells to cisplatin (Figure 9I, SF50 = 1.43). This effect was rescued by overexpression of ERCC1 (resistance factor, RF50 = 1.51). Taken together, these results indicate that p300 which is regulated by CITED2 plays a role in regulating chromatin relaxation, through selective modification of histones, formation of p53 complex binding to the ERCC1 promoter and regulation of ERCC gene expression and cell sensitivity to genotoxic agents like cisplatin.
CITED2 silencing impairs ERCC1 expression, DNA repair and cell viability in cancer HeLa cells
To assess whether ERCC1 expression, DNA repair and cell viability are also impaired by CITED2 silencing in cancer cells, we used HeLa cells since CITED2 was initially identified as a cisplatin resistance gene in this cell model (56). As in HEK293 cells, HeLa cells in which CITED2 was silenced (Figure 10A) displayed a dramatic increase of γ-H2AX (Figure 10B, compare lanes 1 and 4; Figure 10C). Meanwhile, the level of ERCC1 was moderately reduced by CITED2 silencing in these cells. DNA damage level significantly increased following CITED2 silencing in cells exposed to either 1 or 10 μM of cisplatin (Figure 10B and C, P < 0.01 and P < 0.001, respectively). While DNA damage level slightly increased following cisplatin treatment in shControl cells, ERCC1 protein level was reduced by about 50% in cells expressing shCITED2 (Figure 10B and D, P < 0.01). p300 protein level was also reduced in cells expressing shCITED2 (Figure 10B and E). Furthermore, ERCC1 and p300 mRNA levels (both basal and cisplatin-induced) were considerably downregulated by shCITED2 in HeLa cells (Figure 10F and G, respectively). Unlike HEK293 cells, HeLa cells showed slightly induced CITED2 mRNA levels in the presence of cisplatin (compare Figure 4D and 10A). Nevertheless, CITED2 mRNA levels were reduced by CITED2 silencing in both cell lines. CITED2 silencing also caused a dramatic reduction in cell viability in HeLa cells (Figure 10H, SF50 = 1.43). Induction of resistance of the cells to cisplatin following shCITED2 expression was partly rescued by ERCC1 overexpression (Figure 10H, RF50 = 1.41). Furthermore, ERCC1 and p300 protein levels (both basal and cisplatin-induced) were considerably down-regulated by shp300 in HeLa cells (Figure 10I and J). Silencing of p300 caused a dramatic reduction in cell viability in HeLa cells (Figure 10K, SF = 1.23). Cell viability was rescued by overexpression of ERCC1 (RF50 = 1.32) (Figure 10K). These results indicate that the cisplatin-sensitizing effects of shCITED2 occur through loss of p300 and ERCC1. Furthermore, suppression of ERCC1 and p300 protein levels and chromatin-relaxation markers following CITED2 silencing (Figure 10L, lanes 1–6) was rescued by ectopic expression of CITED2 in HeLa cells (Figure 10L, compare lanes 4–6 with lanes 10–12). As seen in HEK293 cells, CITED2 silencing induced accumulation of γ-H2AX in HeLa cells (Figure 10, compare lanes 1–3 with lanes 4–6). Also, the ectopic expression of CITED2 was rescued the changes in γ-H2AX levels (compare lanes 4–6 with lanes 10–12; quantification of these proteins is shown in Supplementary Figure S2A–C). Reduction of ERCC1 and p300 mRNA levels following shCITED2 treatment was rescued by ectopic expression of CITED2 (Supplementary Figure S2D and E). Cell immunofluorescence assays also indicated that accumulation of cisplatin-induced γ-H2AX/53BP1 double-positive cells by CITED2 silencing was rescued by ectopic expression of CITED2 in HeLa cells (Supplementary Figure S4). These results suggest that CITED2 silencing causes sensitization of HeLa cells to cisplatin by reducing ERCC1 expression and by impairing DNA repair, a mechanism also seen in HEK293 cells.
Figure 10.
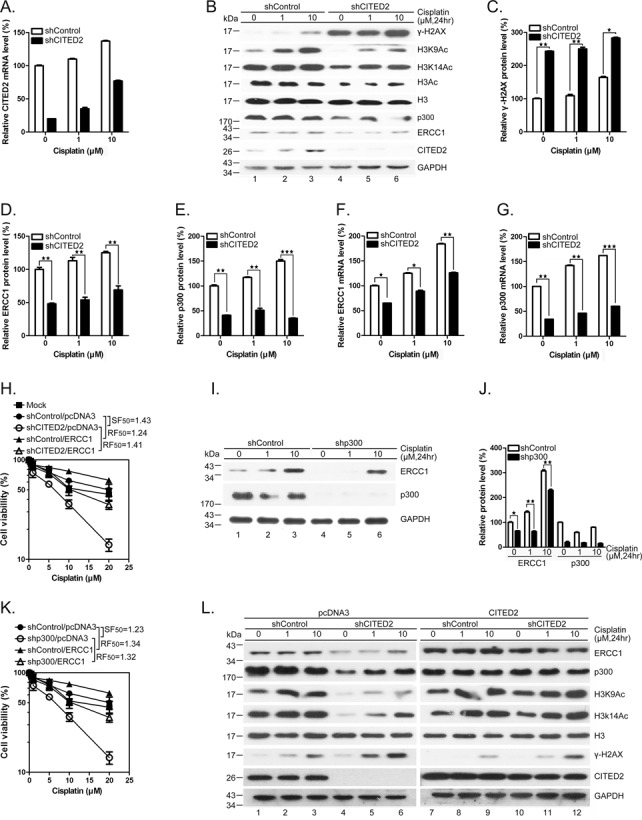
Silencing of CITED2 impairs p300 and ERCC1 protein levels, increases DNA damage and reduces cell viability in cancer HeLa cells. (A) Silencing efficacy of CITED2 by shRNA. (B) Inhibition of p300 and ERCC1 protein levels and accumulation of γ-H2AX levels following CITED2 silencing. (C) Quantification of accumulation of γ-H2AX levels following CITED2 silencing as shown in (B). (D) Quantification of reduction of ERCC1 protein levels following CITED2 silencing in (A). (E) Quantification of reduction of p300 protein levels following CITED2 silencing as shown in (A). (F) Inhibition of ERCC1 mRNA levels by CITED2 silencing. (G) Inhibition of p300 mRNA levels following CITED2 silencing. (H) Potentiation of cisplatin induced inhibition of cell viability by CITED2 silencing and rescued by ectopic expression of ERCC1. (I) Inhibition of ERCC1 protein levels following p300 silencing. (J) Quantification of ERCC1 and p300 protein levels following p300 silencing as shown in (I). (K) Potentiation of cisplatin-induced inhibition of cell viability by p300 silencing and rescue by ectopic expression of ERCC1. (L) Reduction of ERCC1, p300 and chromatin-relaxation markers following CITED2 silencing is rescued by ectopic expression of CITED2. HeLa cells receiving the indicated treatments were processed for immunoblotting. Quantification of protein levels is shown in Supplementary Figure S2A–C.
CITED2 silencing potentiates cisplatin-induced inhibition of CDX tumor growth in mice
To assess the inhibitory effect of CITED2 in vivo, we established HeLa cell CDX tumors in RAG2-deficient mice. CITED2 silencing reduced tumor size at day 7 following implantation (Figure 11A shows representative samples; Figure 11B shows statistical analysis of 12 mice). The growth rate of CDX tumors appeared to be same in the two sets of mice. Following cisplatin treatment, CDX tumor size was dramatically reduced in shCITED2 mice (Figure 11C). Immunohistochemistry staining revealed that while ERCC1 was dramatically reduced in shCITED2 tumors compared to shControl tumor, γ-H2AX levels were clearly enhanced in shCITED2 tumors (Figure 11D). Hematoxylin and eosin (H&E) and CITED2 staining was also performed in these representative samples. Although the level of ERCC1 did not increase significantly in cisplatin-treated tumor cells compared to untreated tumor cells, γ-H2AX levels appeared to increase in response to cisplatin. Western blotting also revealed downregulation of ERCC1 following CITED2 silencing (Figure 11E, compare lanes 1–4 with 5–8). Cisplatin-induced γ-H2AX levels further increased in response to CITED2 silencing (Figure 11E, compare lanes 1–2 with 3–4 for induction by cisplatin; compare lanes 3–4 with 7–8 for enhancement by CITED2 silencing). The expression level of these proteins was quantified (Figure 11F), which indicated that CITED2 silencing caused a downregulation of ERCC1 expression, while reduced DNA damage levels were still observed in vivo. These results reveal that sensitization of CDX tumors to cisplatin can be achieved by combination of cisplatin and CITED2 shRNA in mice.
Figure 11.
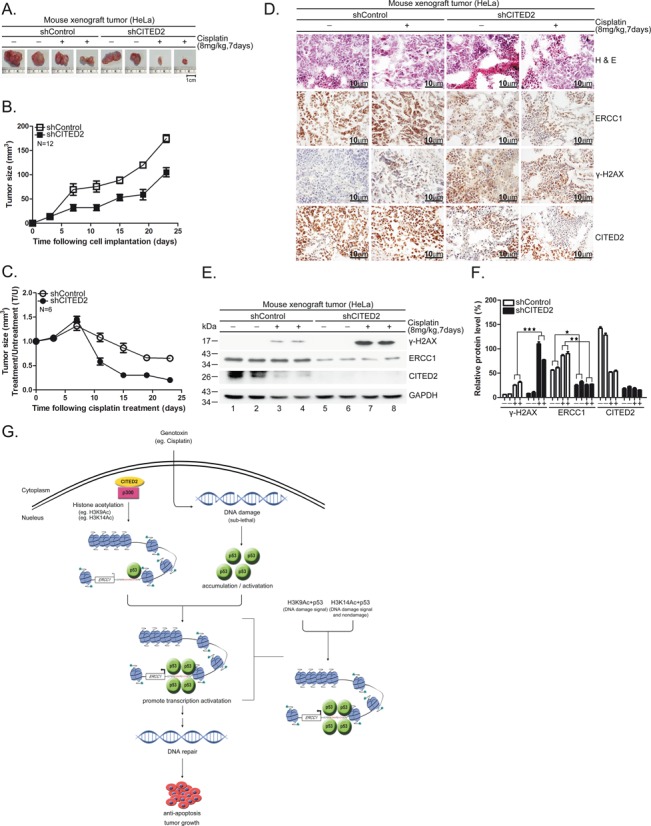
Silencing of CITED2 potentiates inhibition of CDX tumor growth by cisplatin in mice. (A) Representative tumor size excised 7 days after cisplatin treatment. Size reference (cm) is indicated at the bottom. (B) Reduced growth rate of HeLa cells expressing shCITED2. Compared to the tumorigenic cells expressing shControl, the tumorigenic HeLa cells that expressed shCITED2 grew slower, starting 23 days post-inoculation in RAG2-deficient mice. (C) Kinetic changes in tumor size of CDX following repeated cisplatin injections. Average tumor size was calculated from tumors of six mice for each time point. (D) Detection of CITED2 in shControl and shCITED2 mouse tissues by immunohistochemistry. (a, a’) H&E staining of shControl mouse tissues with or without cisplatin treatment. (b, b’) H&E staining of shCITED2 mouse tissues with or without cisplatin treatment. (c, c’) ERCC1 detection of shControl mouse tissues. (d, d’) ERCC1 protein levels of shCITED2 mouse tissues. (e, e’) DNA damage (γ-H2AX) in shControl mouse tissues. (f, f’) DNA damage (γ-H2AX) detection of shCITED2 mouse tissues. (g, g’) CITED2 detection of shControl mouse tissues. (h, h’) CITED2 protein levels of shCITED2 mouse tissues. Positive staining (brown) in tumor tissues is shown. Size reference (10 μm) is indicated. (E) Cisplatin-induced DNA damage (γ-H2AX) levels are dramatically enhanced following silencing of CITED2 as revealed by western blotting of mouse tumors. (F) Quantification of (E). ERCC1 protein levels were reduced by shCITED2. (G) Working model of CITED2/p300/H3ac/p53 in the regulation of ERCC1 expression. CITED2/p300/H3ac/p53 play a positive role in regulating ERCC1 expression. In case of stressed cells, p53 activation by DNA damage may further amplify the regulatory effect depending on the level of damage. In response to sub-lethal damage, activated p53 recruits CITED2/p300 acetylated H3 (H3K9Ac, H3K14Ac) and may target genes like ERCC1 for expression to repair DNA damage, leading to anti-apoptosis effects. Accordingly, cancer cells receiving low levels of cytotoxic agents during chemotherapy may acquire upregulation of CITED2, providing the opportunity of gaining resistance to the drug. This model also illustrates that the H3K14Ac/p53 complex targets ERCC1 gene in response to both non-damage and DNA damage signals. H3K9Ac/p53 complex targets to the ERCC1 gene in response to DNA damage signal.
DISCUSSION
In this study, we found that silencing of CITED2/p300 can downregulate ERCC1 gene expression in both HEK293 and HeLa cells. The downregulation of this DNA repair gene is associated with accumulation of DNA damage in both basal and cisplatin-induced conditions, and with reduced cell viability. The negative regulatory effects of CITED2/p300 silencing can be largely reversed by ectopic expression of ERCC1. These results suggest that CITED2/p300, a chromatin modulator, is involved in cisplatin-induced DDR which requires p53 activation and expression of DNA repair gene. Accordingly, CITED2 plays an important role in bridging chromatin remodulation and p53-mediated transcriptional initiation of DNA repair genes, a process which may be determinant for the cell response to genotoxic agents. In addition, the cisplatin-sensitizing effects of CITED2 silencing could be reproduced in CDX tumors in mice, showing the possible clinical relevance of our observations.
Our results demonstrate that CITED2 silencing by shRNA impairs p53 binding to the ERCC1 promoter, leading to reduced ERCC1 mRNA expression. This observation is supported by the result that impairment of p53 activity by a chemical inhibitor (i.e. PFTα) reduced the expression level of ERCC1 mRNA. It is widely accepted that the activation of p53 can lead to cell-cycle arrest or apoptosis, depending on the genotoxic agent and the subsequent post-translational modifications observed (69). For example, phosphorylation of p53 at the N-terminal region leads to recruitment of CBP/p300 and PCAF to p53-responsive elements on the chromatin, resulting in the acetylation and stabilization of p53 (41,43,44). We have previously found that CITED2 silencing enhanced p53 acetylation (Lys373) in HEK293 cells, leading to a decrease of p53 ubiquitination and subsequent accumulation of p53 protein (55). In this study, we demonstrated that CITED2/p300 modified the binding of the H3/p53 complex to the ERCC1 promoter in order to regulate gene expression and DNA repair, as well as sensitivity to cisplatin. Furthermore, p53-dependent regulation of ERCC1 gene expression by CITED2 silencing was observed in H1299 cells (p53-null) in which exogenous p53 was expressed. These results demonstrated that p53 is involved in mediating the effects of CITED2 silencing on ERCC1 expression. However, the effects of CITED2 silencing on ERCC1 expression were also observed in H1299 cells in the absence of p53, suggesting possible p53-independent modes of regulation.
Previous studies have shown that upregulation of ERCC1 mRNA is dependent on AP-1 in ovarian cancer cells exposed to either cisplatin or phobol ester (70,71). In comparison, upregulation of ERCC1 mRNA requires GATA in EGF-stimulated hepatoma cells (72) while the same process is dependent on p38β activity in cisplatin-treated lung cancer cells (73). These results suggest that the ERCC1 gene may be activated by various transcription factors via multiple pathways that depend on the specific cellular stimuli and the type of cells involved. The involvement of p53 in inducing ERCC1 expression as observed in the present study further extends the list of genetic factors that control expression of this critical DNA repair gene. The p53-independent pathways involved in ERCC1 upregulation (which is often found in cancer cells) may include AP-1 and others.
We have demonstrated that p53-dependent ERCC1 expression plays a vital role in DNA damage and repair, and subsequent in cell viability. Regulation of this protective protein is especially apparent at low level of DNA damage which is found in p53-null cells, suggesting a p53-independent mechanism of expression (Figure 6). Accordingly, inhibition of p53 activity lowers ERCC1 level and sensitizes the cells to DNA damage-induced cell death. However, PFT-α, a p53 inhibitor, protects the cells from DNA damage-induced apoptosis by a p53-independent mechanism that takes place downstream of mitochondrial activity and which might involve cyclin D1 (74). Activation of p53 can lead to cell-cycle arrest or apoptosis, depending on the genotoxic agents, damage level and the subsequent post-translational modifications observed (69). Such response to DNA damage will be impaired in cells that lack p53 activity. Therefore, the role of p53-independent CITED2/p300/ERCC1 expression in DNA damage-induced apoptosis may be more complex than previously thought.
CBP/p300 acts as a protein scaffold upon which a multicomponent transcriptional regulatory complex can be built, thereby connecting different sequence-specific transcription factors such as p53 to the transcription apparatus (75). This protein complex also provides HAT activity, which endows these proteins with the capacity to influence chromatin activity by modulating nucleosomal histones (75). Furthermore, global assessment of the p300 HAT transcriptome in human melanoma identified functional roles in promoting chromatin assembly, activation of DNA repair pathways and cell cycle progression through direct transcriptional regulatory mechanisms (76). In this study, we demonstrated that silencing of CITED2/p300 caused a dramatic reduction of histone relaxation markers, such as H3K9ac and H3K14ac, which were associated with reduced binding of p53 to the ERCC1 promoter. Indeed, silencing of CITED2 exerted similar negative effects on H3K9ac and H3K14ac in targeting the ERCC1 promoter as did p300 silencing (Figure 7D, E and Figure 9G and H). Both CITED2 silencing and p300 silencing caused H3K9ac/p53 complex binding in response to DNA damage and H3K14ac/p53 complex binding in response to both non-damaged and damaged DNA (Figure 11G). These results indicate that the effects of CITED2 silencing on ERCC1 expression are mediated through p300 and p53 activities. Furthermore, p53 binding to ERCC1 promoter was moderately enhanced by TSA (an HDAC inhibitor), and this effect was suppressed by CITED2 silencing. Thus, the regulation of ERCC1 expression by CITED2 involves regional targeting by H3Kac/p53, rather than global expression levels of H3Kac along the genome. Together, these results support the notion that regulation of ERCC1 gene expression in response to cisplatin is tightly regulated by p53 and epigenetic factors.
Though most histone modifications do not change appreciably in response to genotoxic stress, the steady-state levels of H3K9 and H3K56 acetylation reversibly decreased in human osteosarcoma (U2OS) cells following DNA damage induced by hydroxyurea or phleomycin (77). However, we found an increase of H3ac in non-cancer HEK293 cells treated with cisplatin, while H3K9ac remained unchanged in these cells (Figure 7A). The regulatory effects of CITED2/p300 silencing on chromatin relaxation are unlikely to involve changes in H3K56ac levels since H3K56 has been shown to be acetylated by the HAT GCN5/KAT2A (77). These results suggest that acetylation/deacetylation of histones plays an important role in cell response to DNA damage, depending on the types of damage and cells. CITED2 physically interacts with CBP/p300 (48) and silencing of either CITED2 or p300 produced similar effects on H3 acetylation and on the formation of p53-containing protein complex on the ERCC1 promoter. These results suggest that the regulatory effects of CITED2 silencing on ERCC1 expression may require p300 activity.
While H3 level may be slightly reduced by CITED2 silencing, H3K9me2 is increased by cisplatin in shCITED2 cells (Figure 7A). Earlier studies have demonstrated that upregulation of H3K9me2 is associated with inhibition of DSB repair induced by either γ–irradiation or doxorubicin (78,79). Increase of H3K9me2 by cisplatin in shCITED2 cells may be involved in the inhibition of DSB repair and may lead to enhanced cytotoxicity.
Unlike the direct DSBs induced by IR which are predominately repaired by non-homologous end joining mechanisms, cisplatin-induced inter-strand crosslinks and other replication-associated DSBs require HR for repair (80,81). Cells defective in ERCC1, XPF or components of HR are sensitive to cell killing by ICL-inducing chemotherapeutic agents such as cisplatin. It appears that both unrepaired inter-strand crosslinks (and probably their associated DSBs) constitute the major cytotoxic lesions induced by inter-strand crosslinks agents (81–84). In this context, cisplatin induced inter-strand crosslinks which comprise only 8–10% of lesions, and these are relevant for causing apoptosis. In this study, we found high levels of γ-H2Ax in cells following CITED2 silencing even in unstressed cells. Induced inter-strand crosslinks and associated DSBs are unrepaired and probably lead to ROS accumulation in replicating cells. Furthermore, these damages were dramatically reduced and cell viability increased following ectopic expression of ERCC1 in cisplatin-treated cells. ERCC1 may be indirectly involved in repair of DSBs. It has been demonstrated that increased repair of inter-strand crosslinks, as measured by the comet assay, contributes to clinically acquired resistance to melphalan in multiple myeloma and platinum in ovarian tumor cells (85,86). In terms of DNA repair, this method appears to measure the initial rate of inter-strand crosslink unhooking by ERCC1-XPF since cells with defective ERCC1 or XPF show decreased unhooking and increased cellular sensitivity (81–83). Accordingly, impaired removal of DSBs in cisplatin-treated cells following CITED2 silencing is regulated by defects in the early stage of inter-strand crosslinks repair due to ERCC1 deficiency. This observation may partly explain that the expression level of ERCC1 as regulated by CITED2 plays an important role in DDR to cisplatin. γ-H2Ax is only important during repair of DSBs or ROS, and has very limited functional importance during the DDR after cisplatin damage in which DSBs are not the major lesions (87). In this study, however, silencing of CITED2 significantly increased γ-H2Ax levels and impaired its removal in HEK293 and HeLa cells. CITED2 silencing may interfere with cell replication and lead to accumulation of ROS, leading to the formation of DSBs in these cells. Alternatively (or additionally), CITED2 silencing may potentiate accumulation of significant DSBs in response to cisplatin through HR processing of inter-strand crosslinks components, although the quantity of γ-H2Ax directly generated by cisplatin may show little relation to its functional importance.
Nevertheless, our results strongly suggest that the CITED2/p300/H3ac/p53 pathway plays an important role in regulating ERCC1 expression. In the case of cisplatin-stressed cells, p53 activation by DNA damage signals may further amplify the regulatory effect depending on the level of damage. In response to sub-lethal DNA damage, CITED2 recruits p53, which is specifically modified (for instance, by phosphorylation at S15; data not shown), and along with chromatin relaxation such as H3K9ac may target genes like ERCC1 to repair DNA damage (Figure 11). Accordingly, cancer cells receiving low levels of cytotoxic agents during chemotherapy may acquire upregulation of CITED2, giving them the opportunity to gain resistance to the drug. Other repair genes such as RAD51, which is also regulated by CITED2 silencing and which participates in DNA repair in our cell system (data not shown), could not be ruled out as another possible protein mediating the effects of CITED2 silencing. Epigenetic regulation of DNA repair is not limited to cisplatin-induced DNA damage as presented in this study. For example, H3K9ac is increased by the acetyltransferase GCN5 in TFTC and STAGA complexes following UV exposure, which stimulate XPC recruitment and leads to repair of damage by NER (88,89). In addition, the N-terminal activation domains of p53 require GCN5 HAT activity to regulate gene expression by influencing chromatin modifications (90,91). Notably, the effects of CITED2 silencing on p53 accumulation and the increase of p53's target Bax were more pronounced after treatment with a high concentration of cisplatin (55). These results support the notion that, through physical interaction with p53, CITED2/p300 and/or modified H3 (acetylation at specific site) as demonstrated here can regulate p53 transactivation on specific genes, depending on the cellular context and environmental stimuli (40). Therefore, the CITED2/p300/H3ac/p53 complexes found here may link cisplatin-induced DNA damage recognition to nucleosome acetylation and may regulate critical growth regulatory pathways in tumor cells. Our results suggest that CITED2 may serve as a potential therapeutic target for resistant malignancies by promoting cellular responses to DNA damaging agents that are currently ineffective against specific cancers. It will be important to examine the status of CITED2/γ-H2AX/ERCC1 expression in clinical samples using immunohistochemistry and other techniques.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the reporter plasmids provided by Dr Muh-Hwa Yang (National Yang-Ming University).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Council, Taiwan (NSC) [NSC100-2320-B-182-026-MY3]; Chang Gung Memorial Hospital (CMRP) [CMRPD190053], [CMRPD1C0191], [CMRPD1C0263]. Funding for open access charge: NSC [100-2320-B-182-026-MY3]; CMRP [CMRPD1C0263].
Conflict of interest statement. None declared.
REFERENCES
- 1.Chao C.C. Molecular basis of cis-diamminedichloroplatinum(II) resistance: a review. J. Formos. Med. Assoc. 1996;95:893–900. [PubMed] [Google Scholar]
- 2.Jordan P., Carmo-Fonseca M. Molecular mechanisms involved in cisplatin cytotoxicity. Cell. Mol. Life Sci. 2000;57:1229–1235. doi: 10.1007/PL00000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fichtinger-Schepman A.M., van der Veer J.L., den Hartog J.H., Lohman P.H., Reedijk J. Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: formation, identification, and quantitation. Biochemistry. 1985;24:707–713. doi: 10.1021/bi00324a025. [DOI] [PubMed] [Google Scholar]
- 4.Fichtinger-Schepman A.M., van Oosterom A.T., Lohman P.H., Berends F. cis-Diamminedichloroplatinum(II)-induced DNA adducts in peripheral leukocytes from seven cancer patients: quantitative immunochemical detection of the adduct induction and removal after a single dose of cis-diamminedichloroplatinum(II) Cancer Res. 1987;47:3000–3004. [PubMed] [Google Scholar]
- 5.Roos W.P., Kaina B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013;332:237–248. doi: 10.1016/j.canlet.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Selvakumaran M., Pisarcik D.A., Bao R., Yeung A.T., Hamilton T.C. Enhanced cisplatin cytotoxicity by disturbing the nucleotide excision repair pathway in ovarian cancer cell lines. Cancer Res. 2003;63:1311–1316. [PubMed] [Google Scholar]
- 7.Chu G. Cellular responses to cisplatin. The roles of DNA-binding proteins and DNA repair. J. Biol. Chem. 1994;269:787–790. [PubMed] [Google Scholar]
- 8.Gillet L.C., Scharer O.D. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 2006;106:253–276. doi: 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- 9.Clingen P.H., Wu J.Y.H., Miller J., Mistry N., Chin F., Wynne P., Prise K.M., Hartley J.A. Histone H2AX phosphorylation as a molecular pharmacological marker for DNA interstrand crosslink cancer chemotherapy. Biochem. Pharmacol. 2008;76:19–27. doi: 10.1016/j.bcp.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 10.Fernandez-Capetillo O., Lee A., Nussenzweig M., Nussenzweig A. H2AX: the histone guardian of the genome. DNA Rep. 2004;3:959–967. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 11.Lyakhovich A., Surralles J. New roads to FA/BRCA pathway: H2AX. Cell Cycle. 2007;6:1019–1023. doi: 10.4161/cc.6.9.4223. [DOI] [PubMed] [Google Scholar]
- 12.Huang X., Okafuji M., Traganos F., Luther E., Holden E., Darzynkiewicz Z. Assessment of histone H2AX phosphorylation induced by DNA topoisomerase I and II inhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin. Cytometry A. 2004;58:99–110. doi: 10.1002/cyto.a.20018. [DOI] [PubMed] [Google Scholar]
- 13.Niedernhofer L., Odijk H., Budzowska M., van Drunen E., Maas A., Theil A.F., de Wit J., Jaspers N.G., Beverloo H.B., Hoeijmakers J.H., Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol. Cell. Biol. 2004;24:5776–5787. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mogi S., Oh D.H. gamma-H2AX formation in response to interstrand crosslinks requires XPF in human cells. DNA Rep. 2006;5:731–740. doi: 10.1016/j.dnarep.2006.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rothfuss A., Grompe M. Repair kinetics of genomic interstrand DNA cross-links: evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway. Mol. Cell. Biol. 2004;24:123–134. doi: 10.1128/MCB.24.1.123-134.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergstralh D.T., Sekelsky J. Interstrand crosslink repair: can XPF-ERCC1 be let off the hook. Trends Genet. 2008;24:70–76. doi: 10.1016/j.tig.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 17.Bessho T., Sancar A., Thompson L.H., Thelen M.P. Reconstitution of human excision nuclease with recombinant XPF-ERCC1 complex. J. Biol. Chem. 1997;272:3833–3837. doi: 10.1074/jbc.272.6.3833. [DOI] [PubMed] [Google Scholar]
- 18.Niedernhofer L.J., Odijk H., Budzowska M., van Drunen E., Maas A., Theil A.F., de Wit J., Jaspers N.G.J., Beverloo H.B., Hoeijmakers J.H.J., et al. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol. Cell. Biol. 2004;24:5776–5787. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuraoka I., Kobertz W.R., Ariza R.R., Biggerstaff M., Essigmann J.M., Wood R.D. Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombination nuclease. J. Biol. Chem. 2000;275:26632–26636. doi: 10.1074/jbc.C000337200. [DOI] [PubMed] [Google Scholar]
- 20.Al-Minawi A.Z., Lee Y.F., Hakansson D., Johansson F., Lundin C., Saleh-Gohari N., Schultz N., Jenssen D., Bryant H.E., Meuth M., et al. The ERCC1/XPF endonuclease is required for completion of homologous recombination at DNA replication forks stalled by inter-strand cross-links. Nucleic Acids Res. 2009;37:6400–6413. doi: 10.1093/nar/gkp705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosell R., Lord R.V., Taron M., Reguart N. DNA repair and cisplatin resistance in non-small-cell lung cancer. Lung Cancer. 2002;38:217–227. doi: 10.1016/s0169-5002(02)00224-6. [DOI] [PubMed] [Google Scholar]
- 22.Li Q.D., Yu J.J., Mu C.J., Yunmbam M.K., Slavsky D., Cross C.L., Bostick-Bruton F., Reed E. Association between the level of ERCC-1 expression and the repair of cisplatin-induced DNA damage in human ovarian cancer cells. Anticancer Res. 2000;20:645–652. [PubMed] [Google Scholar]
- 23.Olaussen K.A., Dunant A., Fouret P., Brambilla E., Andre F., Haddad V., Taranchon E., Filipits M., Pirker R., Popper H.H., et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. New Engl. J. Med. 2006;355:983–991. doi: 10.1056/NEJMoa060570. [DOI] [PubMed] [Google Scholar]
- 24.Arora S., Kothandapani A., Tillison K., Kalman-Maltese V., Patrick S.M. Downregulation of XPF-ERCC1 enhances cisplatin efficacy in cancer cells. DNA Rep. 2010;9:745–753. doi: 10.1016/j.dnarep.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bowden N.A. Nucleotide excision repair: why is it not used to predict response to platinum-based chemotherapy. Cancer Lett. 2014;346:163–171. doi: 10.1016/j.canlet.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 26.Gossage L., Madhusudan S. Current status of excision repair cross complementing-group 1 (ERCC1) in cancer. Cancer Treat. Rev. 2007;33:565–577. doi: 10.1016/j.ctrv.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Chen S., Zhang J., Wang R., Luo X., Chen H. The platinum-based treatments for advanced non-small cell lung cancer, is low/negative ERCC1 expression better than high/positive ERCC1 expression? A meta-analysis. Lung Cancer. 2010;70:63–70. doi: 10.1016/j.lungcan.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 28.De Dosso S., Zanellato E., Nucifora M., Boldorini R., Sonzogni A., Biffi R., Fazio N., Bucci E., Beretta O., Crippa S., et al. ERCC1 predicts outcome in patients with gastric cancer treated with adjuvant cisplatin-based chemotherapy. Cancer Chemother. Pharmacol. 2013;72:159–165. doi: 10.1007/s00280-013-2181-2. [DOI] [PubMed] [Google Scholar]
- 29.Reynolds C., Obasaju C., Schell M.J., Li X., Zheng Z., Boulware D., Caton J.R., Demarco L.C., O'Rourke M.A., Shaw Wright G., et al. Randomized phase III trial of gemcitabine-based chemotherapy with in situ RRM1 and ERCC1 protein levels for response prediction in non-small-cell lung cancer. J Clin. Oncol. 2009;27:5808–5815. doi: 10.1200/JCO.2009.21.9766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simon G., Sharma A., Li X., Hazelton T., Walsh F., Williams C., Chiappori A., Haura E., Tanvetyanon T., Antonia S., et al. Feasibility and efficacy of molecular analysis-directed individualized therapy in advanced non-small-cell lung cancer. J. Clin. Oncol. 2007;25:2741–2746. doi: 10.1200/JCO.2006.08.2099. [DOI] [PubMed] [Google Scholar]
- 31.Cobo M., Isla D., Massuti B., Montes A., Sanchez J.M., Provencio M., Vinolas N., Paz-Ares L., Lopez-Vivanco G., Munoz M.A., et al. Customizing cisplatin based on quantitative excision repair cross-complementing 1 mRNA expression: a phase III trial in non-small-cell lung cancer. J. Clin. Oncol. 2007;25:2747–2754. doi: 10.1200/JCO.2006.09.7915. [DOI] [PubMed] [Google Scholar]
- 32.Ashcroft M., Taya Y., Vousden K.H. Stress signals utilize multiple pathways to stabilize p53. Mol. Cell. Biol. 2000;20:3224–3233. doi: 10.1128/mcb.20.9.3224-3233.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu W., Feng Z., Levine A.J. The regulation of multiple p53 stress responses Is mediated through MDM2. Genes Cancer. 2012;3:199–208. doi: 10.1177/1947601912454734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seo Y.R., Jung H.J. The potential roles of p53 tumor suppressor in nucleotide excision repair (NER) and base excision repair (BER) Exp. Mol. Med. 2004;36:505–509. doi: 10.1038/emm.2004.64. [DOI] [PubMed] [Google Scholar]
- 35.Dempke W., Voigt W., Grothey A., Hill B.T., Schmoll H.J. Cisplatin resistance and oncogenes–a review. Anticancer Drugs. 2000;11:225–236. doi: 10.1097/00001813-200004000-00001. [DOI] [PubMed] [Google Scholar]
- 36.Siddik Z.H. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 37.Le Cam L., Linares L.K., Paul C., Julien E., Lacroix M., Hatchi E., Triboulet R., Bossis G., Shmueli A., Rodriguez M.S., et al. E4F1 is an atypical ubiquitin ligase that modulates p53 effector functions independently of degradation. Cell. 2006;127:775–788. doi: 10.1016/j.cell.2006.09.031. [DOI] [PubMed] [Google Scholar]
- 38.Yoshida K. Role for DYRK family kinases on regulation of apoptosis. Biochem. Pharmacol. 2008;76:1389–1394. doi: 10.1016/j.bcp.2008.05.021. [DOI] [PubMed] [Google Scholar]
- 39.Oda K., Arakawa H., Tanaka T., Matsuda K., Tanikawa C., Mori T., Nishimori H., Tamai K., Tokino T., Nakamura Y., et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 2000;102:849–862. doi: 10.1016/s0092-8674(00)00073-8. [DOI] [PubMed] [Google Scholar]
- 40.Grossman S.R. p300/CBP/p53 interaction and regulation of the p53 response. Eur. J. Biochem. 2001;268:2773–2778. doi: 10.1046/j.1432-1327.2001.02226.x. [DOI] [PubMed] [Google Scholar]
- 41.Liu L., Scolnick D.M., Trievel R.C., Zhang H.B., Marmorstein R., Halazonetis T.D., Berger S.L. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 1999;19:1202–1209. doi: 10.1128/mcb.19.2.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roy S., Packman K., Jeffrey R., Tenniswood M. Histone deacetylase inhibitors differentially stabilize acetylated p53 and induce cell cycle arrest or apoptosis in prostate cancer cells. Cell Death Differ. 2005;12:482–491. doi: 10.1038/sj.cdd.4401581. [DOI] [PubMed] [Google Scholar]
- 43.Sakaguchi K., Herrera J.E., Saito S., Miki T., Bustin M., Vassilev A., Anderson C.W., Appella E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998;12:2831–2841. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gu W., Roeder R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 45.Espinosa J.M., Emerson B.M. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol. Cell. 2001;8:57–69. doi: 10.1016/s1097-2765(01)00283-0. [DOI] [PubMed] [Google Scholar]
- 46.Luo J., Li M., Tang Y., Laszkowska M., Roeder R.G., Gu W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 2004;101:2259–2264. doi: 10.1073/pnas.0308762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun H.B., Zhu Y.X., Yin T.G., Sledge G., Yang Y.C. MRG1, the product of a melanocyte-specific gene related gene, is a cytokine-inducible transcription factor with transformation activity. Proc. Natl. Acad. Sci. U.S.A. 1998;95:13555–13560. doi: 10.1073/pnas.95.23.13555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhattacharya S., Michels C.L., Leung M.K., Arany Z.P., Kung A.L., Livingston D.M. Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev. 1999;13:64–75. doi: 10.1101/gad.13.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bakker W.J., Harris I.S., Mak T.W. FOXO3a is activated in response to hypoxic stress and inhibits HIF1-induced apoptosis via regulation of CITED2. Mol. Cell. 2007;28:941–953. doi: 10.1016/j.molcel.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 50.Young T.W., Mei F.C., Rosen D.G., Yang G., Li N., Liu J., Cheng X. Up-regulation of tumor susceptibility gene 101 protein in ovarian carcinomas revealed by proteomics analyses. Mol. Cell. Proteomics. 2007;6:294–304. doi: 10.1074/mcp.M600305-MCP200. [DOI] [PubMed] [Google Scholar]
- 51.Tien E.S., Davis J.W., Vanden Heuvel J.P. Identification of the CREB-binding protein/p300-interacting protein CITED2 as a peroxisome proliferator-activated receptor alpha coregulator. J. Biol. Chem. 2004;279:24053–24063. doi: 10.1074/jbc.M401489200. [DOI] [PubMed] [Google Scholar]
- 52.Chou Y.T., Wang H., Chen Y., Danielpour D., Yang Y.C. Cited2 modulates TGF-beta-mediated upregulation of MMP9. Oncogene. 2006;25:5547–5560. doi: 10.1038/sj.onc.1209552. [DOI] [PubMed] [Google Scholar]
- 53.Gonzalez Y.R., Zhang Y., Behzadpoor D., Cregan S., Bamforth S., Slack R.S., Park D.S. CITED2 signals through peroxisome proliferator-activated receptor-gamma to regulate death of cortical neurons after DNA damage. J. Neurosci. 2008;28:5559–5569. doi: 10.1523/JNEUROSCI.1014-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yanagie H., Hisa T., Ogata A., Miyazaki A., Nonaka Y., Nishihira T., Osada I., Sairennji T., Sugiyama H., Furuya Y., et al. Improvement of sensitivity to platinum compound with siRNA knockdown of upregulated genes in platinum complex-resistant ovarian cancer cells in vitro. Biomed. Pharmacother. 2009;63:553–560. doi: 10.1016/j.biopha.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 55.Wu Z.Z., Sun N.K., Chao C.C. Knockdown of CITED2 using short-hairpin RNA sensitizes cancer cells to cisplatin through stabilization of p53 and enhancement of p53-dependent apoptosis. J. Cell. Physiol. 2011;226:2415–2428. doi: 10.1002/jcp.22589. [DOI] [PubMed] [Google Scholar]
- 56.Wu Z.Z., Lu H.P., Chao C.C. Identification and functional analysis of genes which confer resistance to cisplatin in tumor cells. Biochem. Pharmacol. 2010;80:262–276. doi: 10.1016/j.bcp.2010.03.029. [DOI] [PubMed] [Google Scholar]
- 57.Sun C.L., Chao C.C. Cross-resistance to death ligand-induced apoptosis in cisplatin-selected HeLa cells associated with overexpression of DDB2 and subsequent induction of cFLIP. Mol. Pharmacol. 2005;67:1307–1314. doi: 10.1124/mol.104.008797. [DOI] [PubMed] [Google Scholar]
- 58.Kamarajan P., Sun N.K., Sun C.L., Chao C.C. Apaf-1 overexpression partially overcomes apoptotic resistance in a cisplatin-selected HeLa cell line. FEBS Lett. 2001;505:206–212. doi: 10.1016/s0014-5793(01)02817-4. [DOI] [PubMed] [Google Scholar]
- 59.Shang Y., Hu X., DiRenzo J., Lazar M.A., Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 60.Wu Z.Z., Chow K.P., Kuo T.C., Chang Y.S., Chao C.C. Latent membrane protein 1 of Epstein-Barr virus sensitizes cancer cells to cisplatin by enhancing NF-kappaB p50 homodimer formation and downregulating NAPA expression. Biochem. Pharmacol. 2011;82:1860–1872. doi: 10.1016/j.bcp.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 61.Chao C.C., Huang S.L., Huang H.M., Lin-Chao S. Cross-resistance to UV radiation of a cisplatin-resistant human cell line: overexpression of cellular factors that recognize UV-modified DNA. Mol. Cell. Biol. 1991;11:2075–2080. doi: 10.1128/mcb.11.4.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsai S.Y., Sun N.K., Lu H.P., Cheng M.L., Chao C.C. Involvement of reactive oxygen species in multidrug resistance of a vincristine-selected lymphoblastoma. Cancer Sci. 2007;98:1206–1214. doi: 10.1111/j.1349-7006.2007.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun N.K., Huang S.L., Chien K.Y., Chao C.C. Golgi-SNARE GS28 potentiates cisplatin-induced apoptosis by forming GS28-MDM2-p53 complexes and by preventing the ubiquitination and degradation of p53. Biochem. J. 2012;444:303–314. doi: 10.1042/BJ20112223. [DOI] [PubMed] [Google Scholar]
- 64.Wu Z.Z., Chao C.C.K. Knockdown of NAPA using short-hairpin RNA sensitizes cancer cells to cisplatin: Implications to overcome chemoresistance. Biochem. Pharmacol. 2010;80:827–837. doi: 10.1016/j.bcp.2010.05.026. [DOI] [PubMed] [Google Scholar]
- 65.Altaha R., Liang X., Yu J.J., Reed E. Excision repair cross complementing-group 1: gene expression and platinum resistance. Int. J. Mol. Med. 2004;14:959–970. [PubMed] [Google Scholar]
- 66.Bonner W.M., Redon C.E., Dickey J.S., Nakamura A.J., Sedelnikova O.A., Solier S., Pommier Y. GammaH2AX and cancer. Nat. Rev. Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luijsterburg M.S., van Attikum H. Chromatin and the DNA damage response: the cancer connection. Mol. Oncol. 2011;5:349–367. doi: 10.1016/j.molonc.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sterling H.J., Prell J.S., Cassou C.A., Williams E.R. Protein conformation and supercharging with DMSO from aqueous solution. J. Am. Soc. Mass Spectrom. 2011;22:1178–1186. doi: 10.1007/s13361-011-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bode A.M., Dong Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 70.Li Q., Gardner K., Zhang L., Tsang B., Bostick-Bruton F., Reed E. Cisplatin induction of ERCC-1 mRNA expression in A2780/CP70 human ovarian cancer cells. J. Biol. Chem. 1998;273:23419–23425. doi: 10.1074/jbc.273.36.23419. [DOI] [PubMed] [Google Scholar]
- 71.Li Q., Zhang L., Tsang B., Gardner K., Bostick-Bruton F., Reed E. Phorbol ester exposure activates an AP-1-mediated increase in ERCC-1 messenger RNA expression in human ovarian tumor cells. Cell. Mol. Life Sci. 1999;55:456–466. doi: 10.1007/s000180050302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Andrieux L.O., Fautrel A., Bessard A., Guillouzo A., Baffet G., Langouet S. GATA-1 is essential in EGF-mediated induction of nucleotide excision repair activity and ERCC1 expression through ERK2 in human hepatoma cells. Cancer Res. 2007;67:2114–2123. doi: 10.1158/0008-5472.CAN-06-3821. [DOI] [PubMed] [Google Scholar]
- 73.Planchard D., Camara-Clayette V., Dorvault N., Soria J.C., Fouret P. p38 Mitogen-activated protein kinase signaling, ERCC1 expression, and viability of lung cancer cells from never or light smoker patients. Cancer. 2012;118:5015–5025. doi: 10.1002/cncr.27510. [DOI] [PubMed] [Google Scholar]
- 74.Sohn D., Graupner V., Neise D., Essmann F., Schulze-Osthoff K., Janicke R.U. Pifithrin-alpha protects against DNA damage-induced apoptosis downstream of mitochondria independent of p53. Cell Death Differ. 2009;16:869–878. doi: 10.1038/cdd.2009.17. [DOI] [PubMed] [Google Scholar]
- 75.Chan H.M., La Thangue N.B. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 2001;114:2363–2373. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- 76.Yan G., Eller M.S., Elm C., Larocca C.A., Ryu B., Panova I.P., Dancy B.M., Bowers E.M., Meyers D., Lareau L., et al. Selective inhibition of p300 HAT blocks cell cycle progression, induces cellular senescence, and inhibits the DNA damage response in melanoma cells. J. Invest. Dermatol. 2013;133:2444–2452. doi: 10.1038/jid.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tjeertes J.V., Miller K.M., Jackson S.P. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009;28:1878–1889. doi: 10.1038/emboj.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Young L.C., McDonald D.W., Hendzel M.J. Kdm4b Histone Demethylase Is a DNA Damage Response Protein and Confers a Survival Advantage following gamma-Irradiation. J. Biol. Chem. 2013;288:21376–21388. doi: 10.1074/jbc.M113.491514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McDonald O.G., Wu H., Timp W., Doi A., Feinberg A.P. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat. Struct. Mol. Biol. 2011;18:867–874. doi: 10.1038/nsmb.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dronkert M.L., Kanaar R. Repair of DNA interstrand cross-links. Mutat. Res. 2001;486:217–247. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- 81.De Silva I.U., McHugh P.J., Clingen P.H., Hartley J.A. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol. Cell. Biol. 2000;20:7980–7990. doi: 10.1128/mcb.20.21.7980-7990.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.De Silva I.U., McHugh P.J., Clingen P.H., Hartley J.A. Defects in interstrand cross-link uncoupling do not account for the extreme sensitivity of ERCC1 and XPF cells to cisplatin. Nucleic Acids Res. 2002;30:3848–3156. doi: 10.1093/nar/gkf479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Clingen P.H., Arlett C.F., Hartley J.A., Parris C.N. Chemosensitivity of primary human fibroblasts with defective unhooking of DNA interstrand cross-links. Exp. Cell Res. 2007;313:753–760. doi: 10.1016/j.yexcr.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 84.Sasaki M.S., Takata M., Sonoda E., Tachibana A., Takeda S. Recombination repair pathway in the maintenance of chromosomal integrity against DNA interstrand crosslinks. Cytogenet. Genome Res. 2004;104:28–34. doi: 10.1159/000077463. [DOI] [PubMed] [Google Scholar]
- 85.Spanswick V.J., Craddock C., Sekhar M., Mahendra P., Shankaranarayana P., Hughes R.G., Hochhauser D., Hartley J.A. Repair of DNA interstrand crosslinks as a mechanism of clinical resistance to melphalan in multiple myeloma. Blood. 2002;100:224–229. doi: 10.1182/blood.v100.1.224. [DOI] [PubMed] [Google Scholar]
- 86.Wynne P., Newton C., Ledermann J., Olaitan A., Mould T.A., Hartley J.A. Enhanced repair of DNA interstrand crosslinking in ovarian cancer cells from patients following treatment with platinum-based chemotherapy. Br. J. Cancer. 2007;97:927–933. doi: 10.1038/sj.bjc.6603973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cleaver J.E. gamma H2Ax: biomarker of damage or functional participant in DNA repair ‘All that Glitters Is not Gold!’. Photochem. Photobiol. 2011;87:1230–1239. doi: 10.1111/j.1751-1097.2011.00995.x. [DOI] [PubMed] [Google Scholar]
- 88.Brand M., Moggs J.G., Oulad-Abdelghani M., Lejeune F., Dilworth F.J., Stevenin J., Almouzni G., Tora L. UV-damaged DNA-binding protein in the TFTC complex links DNA damage recognition to nucleosome acetylation. EMBO J. 2001;20:3187–3196. doi: 10.1093/emboj/20.12.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Martinez E., Palhan V.B., Tjernberg A., Lymar E.S., Gamper A.M., Kundu T.K., Chait B.T., Roeder R.G. Human STAGA complex is a chromatin-acetylating transcription coactivator that interacts with pre-mRNA splicing and DNA damage-binding factors in vivo. Mol. Cell. Biol. 2001;21:6782–6795. doi: 10.1128/MCB.21.20.6782-6795.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Candau R., Scolnick D.M., Darpino P., Ying C.Y., Halazonetis T.D., Berger S.L. Two tandem and independent sub-activation domains in the amino terminus of p53 require the adaptor complex for activity. Oncogene. 1997;15:807–816. doi: 10.1038/sj.onc.1201244. [DOI] [PubMed] [Google Scholar]
- 91.Nagy Z., Tora L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene. 2007;26:5341–5357. doi: 10.1038/sj.onc.1210604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.