


Abstract
Following DNA double-strand breaks, poly(ADP-ribose) (PAR) is quickly and heavily synthesized to mediate fast and early recruitment of a number of DNA damage response factors to the sites of DNA lesions and facilitates DNA damage repair. Here, we found that EXO1, an exonuclease for DNA damage repair, is quickly recruited to the sites of DNA damage via PAR-binding. With further dissection of the functional domains of EXO1, we report that the PIN domain of EXO1 recognizes PAR both in vitro and in vivo and the interaction between the PIN domain and PAR is sufficient for the recruitment. We also found that the R93G variant of EXO1, generated by a single nucleotide polymorphism, abolishes the interaction and the early recruitment. Moreover, our study suggests that the PAR-mediated fast recruitment of EXO1 facilities early DNA end resection, the first step of homologous recombination repair. We observed that other PIN domains could also recognize DNA damage-induced PAR. Taken together, our study demonstrates a novel class of PAR-binding module that plays an important role in DNA damage response.
INTRODUCTION
Maintenance of genome integrity is essential to prevent the accumulation of mutations that leads to cancer, ageing and other diseases. The integrity of our genome is constantly challenged by DNA replication errors, environmental hazards and other genotoxic stresses. In response to such stresses, cells activate an evolutionarily conserved pathway termed DNA damage response (DDR), to orchestrate various cellular responses (1–3). It has been shown that poly (ADP-ribosyl)ation (PARylation), a unique posttranslational modification, participates in sensing DNA lesions and facilitates DNA damage repair (4–6). PARylation by PARP1, the founding member of the PARP family, is one of the earliest responses to DNA damage, including single-strand breaks (SSBs) and double-strand breaks (DSBs). Upon DNA damage, the N-terminal zinc finger domains of PARP1 directly recognize the ends of DNA breaks and activate PAR synthesis on substrates including PARP1 itself and adjacent histones (7,8). Accumulated evidence shows that DNA damage-induced PAR may serve as docking signals to recruit DNA damage response factors to DNA lesions (7–17). Some PAR-binding modules, such as PAR-binding motif, PBZ domains, Macro domain, WWE domain and RRM motif, have been characterized in DNA damage response and other cellular processes (4,9,10,12,18–29). Our recent studies also show that a set of BRCT domain, FHA domain and OB-fold domain recognize PAR, and facilitate the rapid recruitment of proteins containing these domains to the sites of DNA damage (14,17,30). These studies reveal novel “readers” of PARylation as well as novel functions of PARylation. Besides these PAR-binding modules, other PAR-binding domains may exist and facilitate different steps of DNA damage repair. Thus, it is important to identify other “readers” and elucidate the molecular mechanism of PAR in DNA damage response.
EXO1 is a member of the RAD2 family nuclease that possesses 5′-3′ exonuclease and flap structure-specific endonuclease activity (31). EXO1 exonuclease plays an important role in the repair of mismatches and DSBs (32). Besides nuclease activity for excising the nicked strand during mismatch repair (MMR), EXO1 also interacts with a number of MMR proteins, such as MSH2, MSH3 and MLH1, which are subunits of the mismatch repair heterodimer complex of MutSα (MSH2-MSH6), MutSβ (MSH2-MSH3) and MutLα (MLH1-PMS2), respectively (33–36). The functional interaction between EXO1 and other MMR proteins suggests that EXO1 plays a structural role in stabilizing multiprotein complexes containing MMR proteins. In DSB repair, EXO1 fulfills 5′-to-3′ DNA resection to produce single-stranded DNA that leads to the formation of a Rad51 filament required to initiate homologous recombination (HR) (32,37–39). Depletion of EXO1 results in cellular chromosomal instability and hypersensitivity to ionizing radiation (IR) (40). Recent studies also indicate that MMR proteins participate in HR (41–44). Thus, it is possible that EXO1-associated MMR machinery has crosstalk with HR for DSB repair.
It has been shown that EXO1 is rapidly co-localized with BRCA1 at laser microirradiation-induced DNA lesions (40). However, the molecular mechanism of EXO1's recruitment to these DNA damage sites is unclear. Here, we show that the PIN domain of EXO1 is a PAR-binding domain and DNA damage-induced PAR targets EXO1 to the sites of DNA damage. Moreover, the interaction between PAR and EXO1 regulates EXO1-dependent DNA damage response.
MATERIALS AND METHODS
GFP-EXOI plasmids were a gift from Zhongsheng You. N-terminus (a.a. 1–352), middle region (a.a. 353–549) and C-terminus (a.a .550–846) of EXO1, SMG5-PIN (a.a. 831–1016), GEN1-PIN (a.a. 1–210) were cloned into pEGFP-C1 or pGEX-4T vector. The EXO1 natural variants and siRNA resistant forms were generated using the QuikChange site-directed mutagenesis kit (Stratagene).
The siRNA sequences targeting PARP1, PARP2, MSH3 and EXO1 are 5′-CAAAGUAUCCCAAGAAGUUdTdT-3′, 5′-GGAGAAGGAUGGUGAGAAAdTdT-3′, 5′-GAAGAACAAUAUCCUACUAdTdT-3′ and 5′-CAAGCCUAUUCUCGUAUUUdTdT-3′ respectively. siRNAs were transfected into cells using oligofectamine (Invitrogen) according to manufacturer's instructions.
Anti β-actin, anti-biotin, anti-EXO1 and anti-GFP antibodies were purchased from Sigma. Anti-PARP1 and anti-PARP2 antibodies were purchased from Millipore. Anti-PAR antibody was purchased from Trevigen. Anti- RPA32 and phospho-H2AX (γ-H2AX) were purchased from Cell Signaling.
Synthesis, purification and fractionation of PAR
His-tagged human poly (ADP-ribose) polymerase1 (PARP1) was expressed in bacteria and purified by Ni-NTA affinity resin. PAR was synthesized and purified as described previously except for the following modifications (45). PAR was synthesized in a 20 ml incubation mixture containing 100 mM Tris-HCl pH7.8, 10 mM MgCl2, 1 mM NAD+, 10 mM DTT, 60 μg calf thymus histone, 50 μg octameric oligonucleotide GGAATTCC and 2 mg PARP1. To generate biotinyl-PAR, 10 μM biotinyl-NAD+ (Trevigen) was included in the reaction. The mixture was incubated at 30°C for 60 min and stopped by an addition of 20 ml ice-cold 20% TCA. Oligo DNA was removed by DNase I and proteins were digested by proteinase K. Purified PAR was fractionated according to chain length by anion exchange HPLC protocol as described previously (45).
Laser micro-irradiation and imaging of cells
U2OS cells and MEFs with or without transfection of indicated plasmids were plated on glass-bottomed culture dishes (Mat Tek Corporation). Laser micro-irradiation was performed using an IX 71 microscope (Olympus) coupled with the MicroPoint Laser Illumination and Ablation System (Photonic Instruments, Inc.). A 337.1 nm laser diode (3.4 mW) transmits through a specific Dye Cell and then yields 365 nm wavelength laser beam that is focused through X60 UPlanSApo/1.35 oil objective to yield a spot size of 0.5–1 μm. Cells were exposed to the laser beam for about 3.5 ns. The pulse energy is 170 μJ at 10 Hz. Images were taken by the same microscope with CellSens software (Olympus). GFP fluorescence at the laser line was converted into a numerical value using Axiovision software (version 4.5). Normalized fluorescent curves from 20 cells from three independent experiments were averaged. Error bars represent the standard deviation.
Electrophoretic mobility shift assay (EMSA)
The affinity between the PIN of EXO1 and PAR was measured by electrophoretic mobility shift assay (EMSA). Briefly, the EXO1 PIN was incubated with 32P-labeled PAR (20 000 cpm, 0.5 nM) in a 30 μl reaction containing buffer A (50 mM Tris–HCl, pH 8.0, 50 mM NaCl, 1 mM DTT, 0.05% NP-40 and 6% glycerol) for 30 min at 4°C. The reaction mixtures were electrophoresed at 4°C on 5% non-denaturing polyacrylamide gel in a buffer containing 7 mM Tris–HCl (pH 7.4), 3 mM boric acid and 1 mM EDTA. Gels were dried and autoradiographed. The apparent dissociation constant (Kd) was estimated as the protein concentration at which half of the radiolabeled DNA probe was shifted in an EMSA essentially as described (46).
GST fusion protein expression and pull-down assay
GST fusion proteins were expressed in E. coli and purified using standard procedures. Purified GST fusion proteins (1 pmol) were incubated with biotin-labeled PAR (5 pmol) and streptavidin beads for 2 h at 4°C. After washing with NETN-100 buffer four times, samples were boiled in SDS-sample buffer and elutes were analyzed by Western blotting with anti-GST antibody.
Cell culture, cell lysis, immunoprecipitation and Western blotting
Human cancer cell lines were maintained in RPMI 1640 medium with 10% fetal calf serum and cultivated at 37°C in 5% CO2 (v/v). For ionizing radiation, cells were irradiated by using JL Shepherd 137Cs radiation source at indicated doses. Cells were lysed with NETN buffer containing 10 mM NaF and 50 mM β-glycerophosphate. Immunoprecipitation and western blotting were performed following standard protocol as described previously (47).
Poly (ADP-ribose) (PAR) binding assays
Approximately 2 μM (5X) or 0.4 μM (1X) of each recombinant protein were incubated with 10 μM PAR, 30 μl glutathione agarose in the buffer containing 10 mM NaH2PO4 (pH 7.5), 100 mM NaCl. After incubation for 1 h at room temperature, beads were extensively washed with PBS, and bound proteins were released by adding 30 μl sample buffer (150 mM Tris-HCl, 10% SDS, 10 mM EDTA) followed by heating at 80°C for 10 min. 2 μl aliquots of samples were dot-blotted onto nitrocellulose membranes. After incubation for 1 h at 60°C, membranes were subjected to dot blotting analysis with anti-PAR antibodies.
Immunofluorescence staining
Cells grown on coverslips were fixed with 3% paraformaldehyde for 20 min and permeabilized with 0.5% Triton X-100 in PBS for 5 min at room temperature. Samples were blocked with 5% goat serum and then incubated with primary antibody for 60 min. Samples were washed three times and incubated with secondary antibody for 30 min. Coverslips were mounted onto glass slides and visualized by a fluorescence microscope.
DNA damage end resection assay
Cells were grown on 15 mm glass bottom cell culture dish such that they are rapidly dividing (usually 50% confluent). Cells were exposed to laser beam for about 3.5 ns. The pulse energy is 170 μJ at 10 Hz. Cells were immediately pretreated with ice-cold NETN-300 buffer (0.5% NP-40, 2 mM EDTA, 100 mM Tris-HCl pH 7.5, 300 mM NaCl) and fixed with 3% paraformaldehyde for 30 min. Immunostaining was performed as described before (47).
Isothermal titration calorimetry (ITC)
ITC measurements were performed using an ITC200 calorimeter (Northampton, MA). The concentration of GST-EXO-PIN was measured by UV absorption at 280 and the concentration of PAR was measured by UV absorption at 258 as described previously (48). Protein and PAR were extensively dialyzed against the buffer containing 10 mM NaH2PO4 (pH 7.5), 100 mM NaCl, and degassed before use. Titrations were carried out in 10 mM NaH2PO4 buffer, (pH 7.5), containing 100 mM NaCl at 16°C. The reaction cell contained protein at 20 μM, and the syringe contained PAR at 250 μM. 20 injections of 2 μl were performed at intervals of 120 s while stirring at 1000 rpm. Binding isotherms were integrated and analyzed using the software Origin 7.0 provided by the manufacturer. Data were fit by a one-site model.
Homologous recombination assay
The assay was established and modified by Dr. Jasin's group (49). Briefly, U2OS cells stably with a single copy of DR-GFP were transfected with siRNA as indicated. siRNA treated cells were transfected with indicated plasmid and infected by adenovirus encoded I-SecI (adeno-I-SecI). Cells were harvested two days after infection and subjected to flow cytometry analysis and GFP positive cell population was measured.
RESULTS
The PIN domain mediates the fast recruitment of EXO1 to DNA lesions
EXO1 has been shown to relocate to DNA lesions in response to DSBs, however, the kinetics of the recruitment of EXO1 to DNA lesions is unclear. Here, we expressed N-terminal GFP-tagged EXO1 (GFP-EXO1) in mouse embryonic fibroblasts (MEFs). Cells were treated with laser microirradiation, and the localization of GFP-EXO1 was monitored with live cell imaging. Interestingly, we found that EXO1 was recruited to DNA lesions within 30 s, which is much faster than we expected (Figure 1A). EXO1 was also retained at DNA lesions for prolonged time (Figure 1A). These results suggest that EXO1 quickly relocates to DNA lesions in response to DNA damage.
Figure 1.
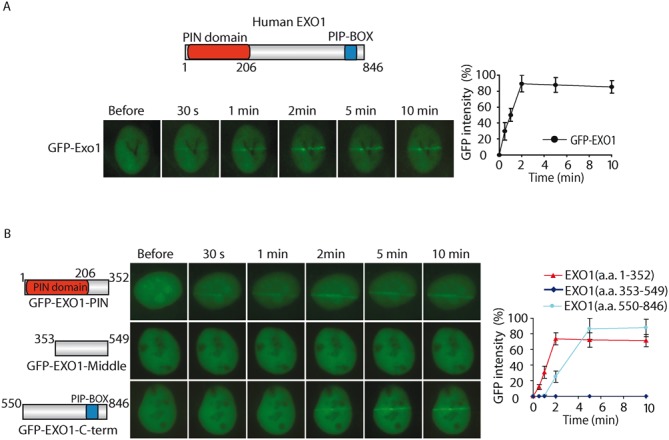
The PIN domain mediates the fast recruitment of EXO1 to DNA lesions. (A)The relocation kinetics of GFP-EXO1 to DNA damage sites. GFP-tagged EXO1 was expressed in MEFs, and the relocation kinetics was monitored in a time course following laser microirradiation. Upper panel: Schematic representation of the domain structure of EXO1. (B) The relocation kinetics of EXO1 truncated mutations to DNA damage sites. GFP-tagged EXO1 truncated mutation was expressed in MEFs, and the relocation kinetics as measured as in (A). For quantitative and comparative imaging (A and B), signal intensities at the laser line were converted into a numerical value using Axiovision software (version 4.5). Normalized fluorescent curves from 20 cells were averaged. The error bars represent the standard deviation. Signal intensities were plotted using Excel.
EXO1 contains several domains including an N-terminal PIN domain and a C-terminal PIP-Box motif. To explore which domain(s) of EXO1 is responsible for the recruitment, we divided EXO1 to N- terminus, C-terminus and the middle regions. Each part was fused with a GFP tag and a nuclear localization sequence (NLS). We found that both N and C-terminal regions but not the middle region of EXO1, were able to relocate to the sites of DNA damage (Figure 1B). We also measured the kinetics of this recruitment. Interestingly, similar to the full length of EXO1, the N-terminal PIN domain was quickly recruited to DNA lesions within 30 s and stayed for prolonged time (Figure 1B). The C-terminal region fused to GFP-NLS was also recruited to DNA damage sites, but was much slower compared to N-terminal PIN domain region. It has been shown that the C-terminal PIP-Box interacts with PCNA, which is likely to mediate the slow accumulation or retention of EXO1 at the sites of DNA damage (50), whereas the N-terminal PIN domain region is responsible for the fast recruitment of EXO1 to DNA lesions.
Fast recruitment of EXO1 to DNA lesions is mediated by PAR
It is known that PAR is synthesized by PARPs at DNA lesions within a few seconds following DNA damage. The rapid recruitment of EXO1 within a few seconds raises the possibility that the fast recruitment of EXO1 to DNA lesions could be mediated by PAR. To test this hypothesis, we next examined whether DNA damage-induced PARylation mediates the fast recruitment EXO1. We treated MEFs with olaparib, a potent PARP1/2 inhibitor to suppress PAR synthesis, and found that the recruitment of EXO1 is significantly delayed (Figure 2A). Interestingly, the recruitment of the N-terminal PIN domain, but not the C-terminal region, was significantly delayed with the treatment of olaparib (Figure 2B). These results suggest that the rapid recruitment of EXO1 may be regulated by DNA damage-induced PARylation. Since most DNA damage-induced PARylation is mediated by PARP1 and PARP2 (7,8), whose activities are suppressed by olaparib. We also knocked down PARP1 and PARP2, respectively. In the absence of PARP1 but not PARP2, the early recruitment of EXO1 was significantly suppressed (Figure 2C), suggesting that PARP1-mediated PARylation facilitates the early recruitment of EXO1.
Figure 2.
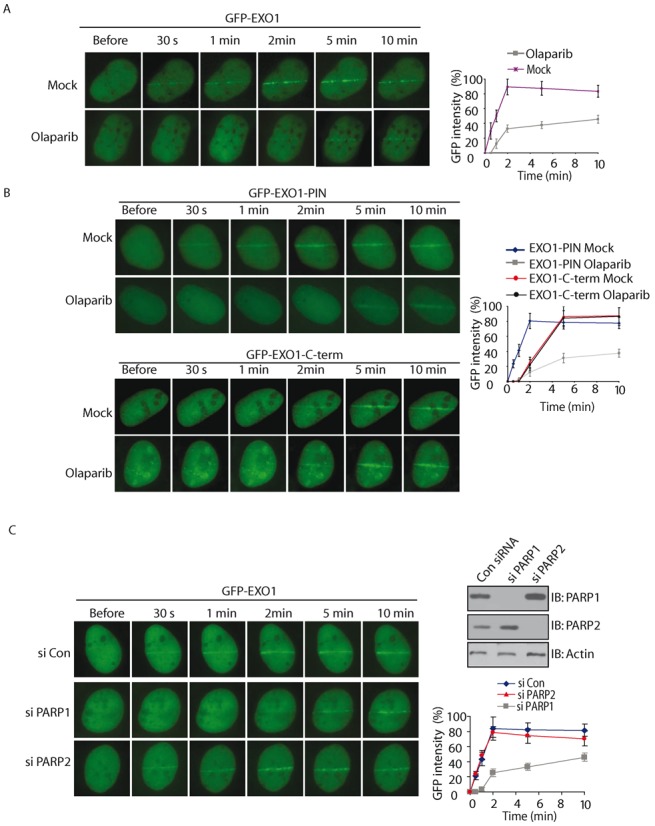
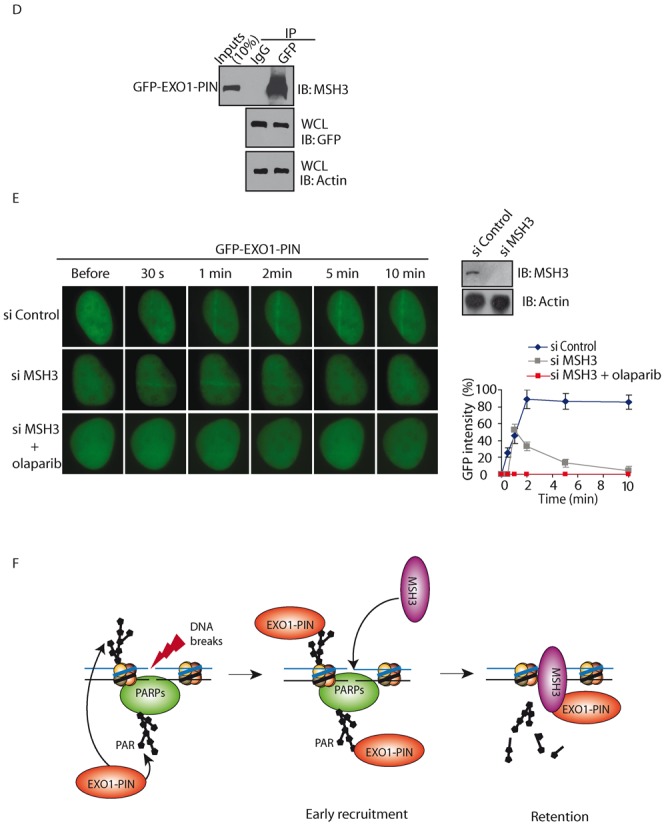
Fast recruitment of EXO1 to DNA lesions is mediated by PAR. (A–B) The effect of olaparib treatment on the recruitment of GFP-EXO1 (A), GFP-EXO1-PIN and GFP-EXO1-C-term (B) to DNA damage sites. GFP-EXO1, GFP-EXO1-PIN or GFP-EXO1-C-term was expressed in MEFs and treated with olaparib. The relocation was monitored in a time course following laser microirradiation. Scale bar = 10 μm. (C) The effect of depletion of PARP1 or PARP2 on the recruitment of GFP-EXO1 to DNA damage sites. GFP-EXO1 was expressed in U2OS cells depletion of PARP1 or PARP2 with siRNA. The relocation was monitored in a time course following laser microirradiation. Depletion of endogenous PARP1 and PARP2 by siRNA was examined by western blotting. Scale bar = 10 μm. (D) MSH3 interacts with EXO-PIN. GFP-EXO1-PIN was expressed in U2OS cells. The interaction between EXO1-PIN and MSH3 was examined with indicated antibodies. (E) Depletion of MSH3 suppresses the retention of EXO-PIN at DNA damage sites. U2OS cells were treated with siMSH3 or olaparib, and the relocation kinetics of GFP-EXO-PIN to DNA damage sites were examined. Depletion of endogenous MSH3 by siRNA was examined by western blotting. (F) PAR mediates the early recruitment of EXO1-PIN to DNA damage sites, whereas MSH3 stabilizes EXO-PIN at DNA damage sites after PAR. For quantitative and comparative imaging (A–C, E), signal intensities at the laser line were converted into a numerical value using Axiovision software (version 4.5). Normalized fluorescent curves from 20 cells were averaged. The error bars represent the standard deviation. Signal intensities were plotted using Excel.
Of note, even lacking of PAR, the N-terminal region of EXO1 is still able to slowly reach to the sites of DNA damage. It has been shown that MSH3 interacts with the N-terminal region (a.a. 129–387) of EXO1 (34). In this study, we confirmed that MSH3 interacted with GPF-EXO1-PIN (a.a. 1–352) (Figure 2D). To assess whether MSH3 regulates the recruitment of EXO to DNA damage sites, we depleted MSH3 by siRNA. As shown in Figure 2E, EXO1-PIN could not be stabilized, but was still recruited to the sites of DNA damage at the sites of DNA damage in the absence of MSH3. However, without both PAR and MSH3, the PIN domain could not be recruited to the sites of DNA damage (Figure 2E). Collectively, these results demonstrate that PAR mediates the fast recruitment of EXO1 to DNA lesions through N-terminal PIN domain, whereas MSH3 retains EXO-PIN for prolonged time at DNA lesions (Figure 2F).
The PIN domain of EXO1 binds to PAR in vitro and in vivo
To examine whether the PIN domain of EXO1 recognizes PAR, we first performed an in vitro binding assay by incubating recombinant GST-tagged PIN domain of EXO1 with PAR. Purified recombinant proteins were dot-blotted on nitrocellulose after incubation with PAR. We found that like CHFR, a known PAR-binding protein, wild type EXO1 could also bind PAR (Figure 3A). Moreover, the PIN domain itself was sufficient to interact with PAR, and deletion of this domain (i.e. ΔPIN a.a. 1–300) abolished the interaction (Figure 3A). Similar results were obtained by using a reverse pull-down assay (Figure 3B, Supplementary Figure S1), suggesting that the PIN domain of EXO1 is required for PAR-binding. Moreover, using EMSA and ITC, we quantitatively measured the affinities between the PIN domain of EXO1 and PAR. The dissociation constant of the interaction is between 200–300 nM (Figure 3C, Supplementary Figure S2A). Taken together, these results suggest that the PIN domain of EXO1 binds to PAR with relative high affinity.
Figure 3.
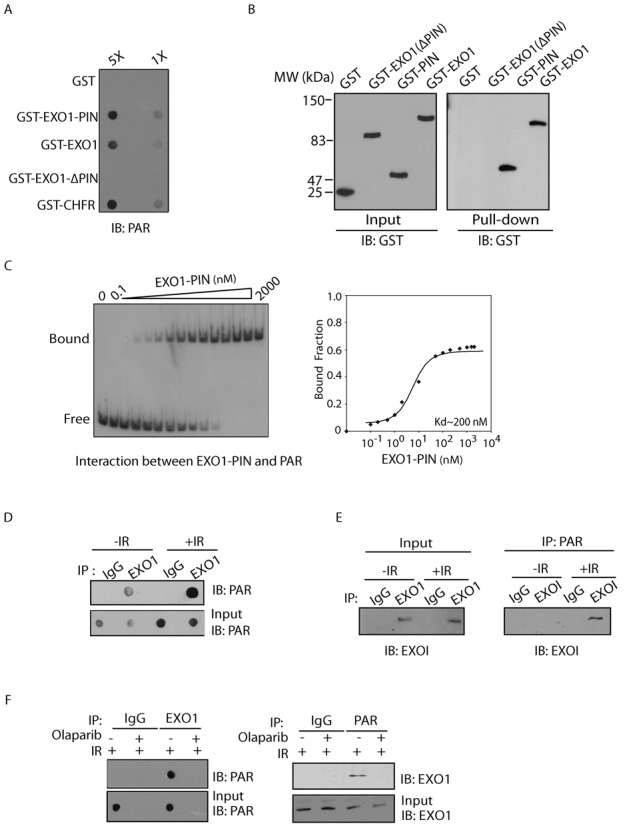
The PIN domain of EXO1 binds to PAR in vitro and in vivo. (A) The recombinant GST-fusion proteins were incubated with PAR. Protein-associated PAR was examined by glutathione agarose beads pull down and dot blotting with anti-PAR antibody. Recombinant GST and GST-CHFR were used as the negative and positive controls, respectively. (B) The PIN domain of EXO1 interacts with biotin-PAR. Left panel: the recombinant GST-EXO1-PIN was incubated with or without biotin-PAR. The interaction was examined by streptavidin beads pull-down assay and Western blotting with anti-GST antibody. Right panel: indicated GST fusion proteins were incubated with biotin-PAR. The interaction was examined by streptavidin beads pull-down assay and Western blotting with anti-GST antibody. GST and GST-CHFR were used as the negative and positive controls, respectively. (C) Analysis of the affinity of the EXO1-PIN toPAR by EMSA. EMSA reactions were carried out in the presence of 0.5 nM radiolabeled oligo(dA) or PAR and varying amounts of EXO1-PIN as indicated. To calculate dissociation constant (Kd), the percentage of radiolabeled DNA-protein complexes was quantified and plotted against the quantity of protein. The data are the average of two independent saturation binding experiments. (D and E) The in vivo interaction between EXO1 and PAR was examined by co-IP (D) and reciprocal co-IP (E). U2OS cells were treated with 0 or 10 Gy of IR. 5 min after IR, cells were lysed and analyzed with indicated antibodies. Input or IPed samples were analyzed by dot blotting (D) or Western blotting (E) with the indicated antibodies. (F) The in vivo interaction between EXO1 and PAR was examined by co-IP and reciprocal co-IP in the presence or absence of olaparib (100 nM) and IR treatment (10 Gy) with the indicated antibodies. Irrelevant IgG was included as the IP control.
Next, we asked if EXO1 interacts with PAR in vivo. Using immunoprecipitation (IP) with anti-EXO1 antibody and dot blotting with anti-PAR antibody, we found that EXO1 associated with PAR in vivo (Figure 3D). Moreover, since DNA damage induces PAR synthesis at the sites of DNA damage (7,8), the interaction between PAR and EXO1 was remarkably increased following IR treatment (Figure 3D and E). This interaction was further confirmed by reciprocal IP (Figure 3E). To exclude the possibility that EXO1 itself was PARylated, we treated the precipitates with 1% SDS to remove the non-covalent interactions. In the presence of SDS, EXO1 was dissociated with PAR (Supplementary Figure S3), suggesting that EXO1 itself is not PARylated in response to DNA damage. We also treated cells with olaparib to suppress PAR synthesis and found that EXO1 no longer interacted with PAR following DNA damage (Figure 3F). Taken together, these results suggest that EXO1 recognizes PAR in vivo, especially when PAR is massively synthesized following DNA damage.
Moreover, IR treatment mainly induces DSBs, whereas PAR can be induced by both DSBs and SSBs. We treated cells with H2O2 to induce SSBs and PARylation. However, the interaction between H2O2-induced PAR and EXO1 is much weaker than that between IR-induced PAR and EXO1. It indicates that specific PARylated proteins might be recognized by EXO1 in response to DSBs. Since EXO1 has several functional partners, such as MSH3 and PCNA (34,51), it is possible that one of its partners could be specifically PARylated in response to DSBs and mediates the early recruitment of EXO1.
The R93G variant of EXO1 abolishes PAR-binding
The PIN domain of EXO1 has 5′-3′ exonuclease activity and cleans DNA lesions. Interestingly, several natural variants of EXO1 have been identified in the PIN domain, without affecting the enzymatic activity. Since the PIN recognizes PAR, we ask if these variants regulate the interaction with PAR. We analyzed a total of five common variants and found that among these variants, the R93G variant largely disrupted the interaction with PAR in vivo (Figure 4A).
Figure 4.
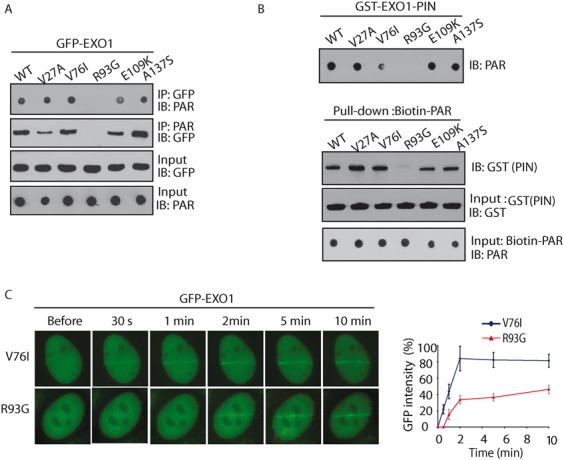
The R93G variant of EXO1 abolishes PAR-binding. (A) GFP-tagged EXO1 and the indicated mutants were expressed in 293T cells. Cell lysates were analyzed by the indicated antibodies. The expression levels of exogenous EXO1 proteins were examined by Western blotting with anti-GFP antibody. (B) The in vitro interaction between EXO1-PIN and PAR was examined by co-IP (upper panel) and reciprocal co-IP (lower panel). Indicated GST fusion proteins were incubated with biotin-PAR. The interaction was examined by streptavidin beads pull-down assay and Western blotting with anti-GST. (C) Indicated GFP-tagged EXO1 variants were expressed in MEFs, and the relocation kinetics as measured as in Figure 1A. GFP fluorescence intensities at the laser line were converted into a numerical value using Axiovision software (version 4.5). Normalized fluorescent curves from 20 cells were averaged. The error bars represent the standard deviation. Signal intensities were plotted using Excel.
We further performed an in vitro binding assay by incubating recombinant PIN domain variants of EXO1 with PAR, and further confirmed that the interaction between PAR and PIN domain was abolished by the R93G mutation (Figure 4B). We also found that the interaction between the PIN domain of EXO1 and MSH3 is not affected by the R93G mutation (Supplementary Figure S4). Moreover, this mutation abolished the fast recruitment of the EXO1 to DNA lesions (Figure 4C). In addition, although the R93G mutation abolishes the interaction with PAR, it does not affect the interaction with MSH3 (Supplementary Figure S4). Thus, the R93G mutant could still be recruited to the sites of DNA damage, albeit at much slower rate (Figure 4B). In summary, these results suggest that the R93 in the PIN domain of EXO1 is important for the interaction with PAR.
EXO1 and PARylation regulate the early recruitment of RPA at DNA damage sites
One major function of EXO1 is to start DNA end resection of DSB repair. Following DSBs, the DSB end is processed by EXO1 and other nucleases to generate 3′ overhang, which is first recognized by RPA and subsequently by RAD51 for strand invasion. Here, we further explored the biological function of the PAR-dependent recruitment of EXO1 in DNA end resection by examining the loading of RPA. Consistent with previous studies, in cells depletion of EXO1, the recruitment of RPA at DNA lesions was significantly delayed (Figure 5), suggesting that early DNA end resection is mediated by EXO1. When EXO1-depleted cells were reconstituted with wild type EXO1, the kinetics of RPA recruitment to DNA lesions was restored. However, when cells were reconstituted with the R93G mutant, the recruitment of RPA was not restored (Figure 5). Collectively, these results suggest that the interaction between EXO1 and PAR mediates early DNA end resection.
Figure 5.
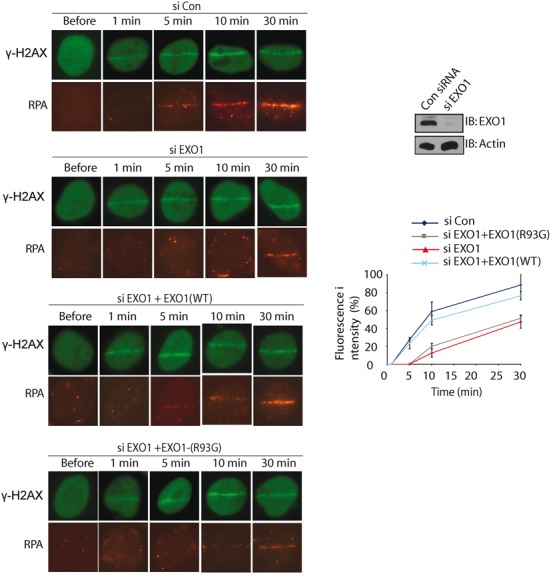
Depletion of EXO1 abolishes the early recruitment of RPA to DNA lesions. U2OS cells were transfected with an EXO1 siRNA or a control siRNA twice, and then co-transfected with siRNA-resistant wildtype EXO1 vector, or a vector expressing the R93G EXO1 mutant. Cells were examined with laser microirradiation and stained with anti-RPA and anti-γH2AX at indicated time points. Scale bar = 10 μm. Depletion of endogenous EXO1 by siRNA was examined by western blotting. Signal intensities at the laser line was converted into numerical value (relative fluorescence intensity) using Axiovision software (version 4.5). Normalized fluorescent curves from 20 cells from three independent experiments were averaged. Signal intensities were plotted using Excel. Error bars represent the standard deviation.
To further examine the functional relevance of the interaction between PAR and EXO1 in the homologous recombination (HR), we used an I-SceI-dependent GFP reporter assay to measure HR in EXO1-depleted cells reconstituted with wild type EXO1 or the R93G variant. Compared to EXO1 wild type, reconstituted with the R93G mutant in EXO1-depleted cells only partially restored the function of wild type EXO1, suggesting that the interaction between PAR and EXO1 is involved in HR (Supplementary Figure S5). However, the functional defect of the R93G in HR is relatively mild. It is because PARylation only mediates the early recruitment of EXO1. Even loss of PARylation, EXO1 could still be recruited to the sites of DNA damage, albeit at much slower rate. Loss of the early recruitment only mildly affects partial HR repair. Alternatively, other enzymes may play redundant function with EXO1 during HR repair. Lacking EXO1 only mildly affects HR repair (37,52–55).
The PIN domain is a PAR-binding domain
Besides EXO1, other DNA damage response factors also contain the PIN domain. Thus, we examined whether the interaction between the PIN domain and PAR is a general phenomenon. We screened seven other PIN domains, and found that the PIN domains of GEN1 and SMG5 could interact with PAR (Figure 6A). This interaction was further confirmed using a reverse pull-down assay (Figure 6B). GEN1 is a flap endonuclease that processes Holliday junction during HR repair, whereas SMG5 forms a complex with SMG7 to process mRNA decay. Thus, it is likely that the interactions between the PIN domains and PAR participate in other steps of DNA damage response.
Figure 6.
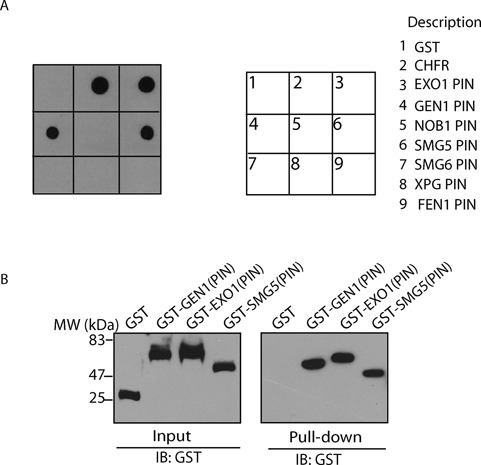
Other PIN domains interact with PAR. (A) Indicated GST fusion proteins were incubated with PAR. The interaction was examined by glutathione agarose bead pull-down and dot blotting with anti-PAR antibody. (B) Indicated GST fusion proteins were incubated with biotin-PAR, the interaction was examined by streptavidin bead pull-down assay and Western blot with anti-GST antibody.
DISCUSSION
Poly (ADP-ribosyl)ation is one of the earliest DNA damage response signals (4–6). Here, we reported that EXO1 recognizes PAR and is quickly recruited to the sites of DNA damage. It has been shown that EXO1 participates in DNA end resection during HR repair (32,37–39). The quick recruitment of EXO1 mediates early DNA end resection. Besides EXO1, the MRN complex also participates in DNA end resection (56–60). Interestingly, the N-terminal BRCT fold of NBS1 also recognizes PAR and is recruited to the sites of DNA damage by PAR (14). Thus, it is likely that DNA damage-induced PARylation recruits both the MRN complex and EXO1 to function together for the initiation of DNA end resection.
The early recruitment of EXO1 is mainly dependent on PARylation. But besides PAR, EXO1 also interacts with other DNA damage response factors such as MSH3 and PCNA. Both MSH3 and PCNA are recruited to the site of DNA damage, which is PARylation independent (61–64). It has been shown that PCNA may stabilize EXO1 at the sites of DNA damage via the interaction with the C-terminal PIP-Box motif of EXO1 (50,51). Consistently, in the absence of PAR-binding, the C-terminal PIP-Box alone is able to be slowly accumulated at DNA lesions. In this study, we demonstrated that MSH3 plays a role to retain EXO1 at the sites of DNA damage. MSH3 interacts with the PIN domain of EXO1, but does not affect the interaction between EXO1 and PAR. Besides these two, EXO1 also interacts with MSH2 and MLH1 (33–36). These functional partners of EXO1 may act together to stably retain EXO1 once EXO1 reaches the sites of DNA damage. MSH2, MSH3 and MLH1 all participate in mismatch repair. However, recent studies indicate that the mismatch machinery is involved in HR repair, especially RAD51 loading (65). Thus, it is possible that EXO1 functions together with its binding partners to process DNA ends for the loading of RAD51 during HR repair.
The binding between EXO1 and PAR is through the PIN domain. Strikingly, we found that the R93G mutant, a natural variant caused by a SNP (A277G), abolishes the interaction with PAR. The R93G mutation occurs far away from activation site so that it does not affect the nuclease activity of the PIN domain of EXO1. However, loss of the interaction with PAR delays DNA end resection during HR repair. Thus, it is likely that the R93G variant affects DSB repair and might be a pathogenic mutation. Since DNA damage repair pathway maintains genomic stability and suppresses tumorigenesis, many DNA damage repair factors are important tumor suppressors. Further cancer etiology analysis may reveal the significance of this missense mutation in tumorigenesis.
Because the PIN domain of EXO1 is an exonuclease domain, we also ask whether the PAR-binding regulates the nuclease activity. However, by incubating PAR with the PIN domain of EXO1, we did not observe any change of the nuclease activity. EXO1 has several other functional partners, therefore it is possible that the regulation of the nuclease is very complicated. Recent study shows that supplement of PARP1 into a sophisticated in vitro system, including MSH2, MSH3, the RPA complex, PCNA, EXO1 and DNA, could enhance the nuclease activity of EXO1 (66). PARP1 is very easily activated by DNA, so it is possible that PARyated PARP1 may activate the nuclease activity of EXO1. Thus, PARylation may not only mediate the early recruitment but also facilitate the activation of EXO1.
The PIN domain is an evolutionarily conserved module that is originally thought to recognize and digest nucleic acids. However, not all the PIN domains can recognize and digest nucleic acids (67,68). Although the secondary structure and tertiary folding are similar among the PIN domains, the primary sequence is not conserved, which might provide the biochemical basis for the diversified functions of the PIN domains. Similar phenomena have been observed in the BRCT, FHA and OB-fold domains, three other poly (ADP-ribose) binding modules (13,14,17,30). Both the BRCT and FHA domains have been found as the phospho-amino acid binding modules (69–73). However, due to the various primary sequences, not all the BRCT and FHA recognize phospho-amino acids. A set of BRCT and FHA bind to poly (ADP-ribose). It is likely that these BRCT and FHA recognize the phosphate groups in ADP-ribose. Moreover, the OB-fold domain is thought as a nucleic acid-binding module (74,75). However, not all the OB-folds recognize DNA/RNA because of the unconserved primary sequence. A set of OB-folds interact with poly (ADP-ribose) (17). Similarly, we found that a set of the PIN domain are poly (ADP-ribose) binding domains. Here, we found that the PIN domains of GEN1 and SMG5 also recognize PAR. Since GEN1 participates in Holliday junction processing (76,77), it is likely that PARylation plays a key in this process by either mediating the recruitment or regulating the activity of the PIN domain. The PIN domain of GEN1 is an endonuclease and it is possible that the interaction with PAR may regulate the endonuclease activity of GEN1. Different from EXO1 and GEN1, the PIN domain of SMG5 lacks key residues for enzyme activity and is unlikely to be a nuclease (68). SMG5 forms a heterodimer with SMG7, which is required for efficient nonsense-mediated mRNA decay (78,79). It is possible that the interaction between PAR and SMG5 is to mediate the recruitment of the SMG5/7 complex for mRNA substrate degradation. Further functional analysis is needed to dissect the significance of these interactions. Nevertheless, we have identified a novel class of PAR-binding module.
Supplementary Material
Acknowledgments
This work was supported in part by grants from National Institutes of Health (CA132755, CA130899 and CA187209 to X.Y.), National Natural Science Foundation of China (81572775 to F.Z.) and the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (to F.Z.). X.Y. is a recipient of Era of Hope Scholar Award from the Department of Defense and Research Scholar Award from Leukemia and Lymphoma Society.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [CA132755, CA130899 and CA187209 to X.Y.]; National Natural Science Foundation of China [81572775 to F.Z.]; Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning [to F.Z.]. Funding for open access charge: NIH [R01 CA132755, CA130899, CA187209].
Conflict of interest statement. The authors declare that they have no conflict of interest.
REFERENCES
- 1.Harper J.W., Elledge S.J. The DNA damage response: ten years after. Mol. Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 2.Rouse J., Jackson S.P. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297:547–551. doi: 10.1126/science.1074740. [DOI] [PubMed] [Google Scholar]
- 3.Sancar A., Lindsey-Boltz L.A., Unsal-Kacmaz K., Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 4.Gibson B.A., Kraus W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012;13:411–424. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 5.Hassa P.O., Hottiger M.O. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci. 2008;13:3046–3082. doi: 10.2741/2909. [DOI] [PubMed] [Google Scholar]
- 6.Schreiber V., Dantzer F., Ame J.C., de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat. Rev. Mol. Cell. Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 7.Kim M.Y., Zhang T., Kraus W.L. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev. 2005;19:1951–1967. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 8.Luo X., Kraus W.L. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012;26:417–432. doi: 10.1101/gad.183509.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahel D., Horejsi Z., Wiechens N., Polo S.E., Garcia-Wilson E., Ahel I., Flynn H., Skehel M., West S.C., Jackson S.P., et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science. 2009;325:1240–1243. doi: 10.1126/science.1177321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahel I., Ahel D., Matsusaka T., Clark A.J., Pines J., Boulton S.J., West S.C. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature. 2008;451:81–85. doi: 10.1038/nature06420. [DOI] [PubMed] [Google Scholar]
- 11.Chou D.M., Adamson B., Dephoure N.E., Tan X., Nottke A.C., Hurov K.E., Gygi S.P., Colaiacovo M.P., Elledge S.J. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. U.S.A. 2010;107:18475–18480. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li G.Y., McCulloch R.D., Fenton A.L., Cheung M., Meng L., Ikura M., Koch C.A. Structure and identification of ADP-ribose recognition motifs of APLF and role in the DNA damage response. Proc. Natl. Acad. Sci. U.S.A. 2010;107:9129–9134. doi: 10.1073/pnas.1000556107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M., Bian C., Yu X. Poly(ADP-ribosyl)ation is recognized by ECT2 during mitosis. Cell Cycle. 2014;13:2944–2951. doi: 10.4161/15384101.2014.947197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li M., Lu L.Y., Yang C.Y., Wang S., Yu X. The FHA and BRCT domains recognize ADP-ribosylation during DNA damage response. Genes Dev. 2013;27:1752–1768. doi: 10.1101/gad.226357.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masson M., Niedergang C., Schreiber V., Muller S., Menissier-de Murcia J., de Murcia G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell. Biol. 1998;18:3563–3571. doi: 10.1128/mcb.18.6.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okano S., Lan L., Caldecott K.W., Mori T., Yasui A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol. Cell. Biol. 2003;23:3974–3981. doi: 10.1128/MCB.23.11.3974-3981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang F., Chen Y., Li M., Yu X. The oligonucleotide/oligosaccharide-binding fold motif is a poly(ADP-ribose)-binding domain that mediates DNA damage response. Proc. Natl. Acad. Sci. U.S.A. 2014;111:7278–7283. doi: 10.1073/pnas.1318367111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z., Michaud G.A., Cheng Z., Zhang Y., Hinds T.R., Fan E., Cong F., Xu W. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes Dev. 2012;26:235–240. doi: 10.1101/gad.182618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y., Liu S., Mickanin C., Feng Y., Charlat O., Michaud G.A., Schirle M., Shi X., Hild M., Bauer A., et al. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat. Cell Biol. 2011;13:623–629. doi: 10.1038/ncb2222. [DOI] [PubMed] [Google Scholar]
- 20.Aravind L. The WWE domain: a common interaction module in protein ubiquitination and ADP ribosylation. Trends Biochem. Sci. 2001;26:273–275. doi: 10.1016/s0968-0004(01)01787-x. [DOI] [PubMed] [Google Scholar]
- 21.Han W., Li X., Fu X. The macro domain protein family: structure, functions, and their potential therapeutic implications. Mutat. Res. 2011;727:86–103. doi: 10.1016/j.mrrev.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mehrotra P.V., Ahel D., Ryan D.P., Weston R., Wiechens N., Kraehenbuehl R., Owen-Hughes T., Ahel I. DNA repair factor APLF is a histone chaperone. Mol. Cell. 2011;41:46–55. doi: 10.1016/j.molcel.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gottschalk A.J., Timinszky G., Kong S.E., Jin J., Cai Y., Swanson S.K., Washburn M.P., Florens L., Ladurner A.G., Conaway J.W., et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. U.S.A. 2009;106:13770–13774. doi: 10.1073/pnas.0906920106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karras G.I., Kustatscher G., Buhecha H.R., Allen M.D., Pugieux C., Sait F., Bycroft M., Ladurner A.G. The macro domain is an ADP-ribose binding module. EMBO J. 2005;24:1911–1920. doi: 10.1038/sj.emboj.7600664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eustermann S., Brockmann C., Mehrotra P.V., Yang J.C., Loakes D., West S.C., Ahel I., Neuhaus D. Solution structures of the two PBZ domains from human APLF and their interaction with poly(ADP-ribose) Nat. Struct. Mol. Biol. 2010;17:241–243. doi: 10.1038/nsmb.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pleschke J.M., Kleczkowska H.E., Strohm M., Althaus F.R. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J. Biol. Chem. 2000;275:40974–40980. doi: 10.1074/jbc.M006520200. [DOI] [PubMed] [Google Scholar]
- 27.Huambachano O., Herrera F., Rancourt A., Satoh M.S. Double-stranded DNA binding domain of poly(ADP-ribose) polymerase-1 and molecular insight into the regulation of its activity. J. Biol. Chem. 2011;286:7149–7160. doi: 10.1074/jbc.M110.175190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kustatscher G., Hothorn M., Pugieux C., Scheffzek K., Ladurner A.G. Splicing regulates NAD metabolite binding to histone macroH2A. Nat. Struct. Mol. Biol. 2005;12:624–625. doi: 10.1038/nsmb956. [DOI] [PubMed] [Google Scholar]
- 29.Krietsch J., Caron M.C., Gagne J.P., Ethier C., Vignard J., Vincent M., Rouleau M., Hendzel M.J., Poirier G.G., Masson J.Y. PARP activation regulates the RNA-binding protein NONO in the DNA damage response to DNA double-strand breaks. Nucleic Acids Res. 2012;40:10287–10301. doi: 10.1093/nar/gks798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li M., Yu X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell. 2013;23:693–704. doi: 10.1016/j.ccr.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lieber M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 32.Tran P.T., Erdeniz N., Symington L.S., Liskay R.M. EXO1-A multi-tasking eukaryotic nuclease. DNA Repair (Amst) 2004;3:1549–1559. doi: 10.1016/j.dnarep.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 33.Dherin C., Gueneau E., Francin M., Nunez M., Miron S., Liberti S.E., Rasmussen L.J., Zinn-Justin S., Gilquin B., Charbonnier J.B., et al. Characterization of a highly conserved binding site of Mlh1 required for exonuclease I-dependent mismatch repair. Mol. Cell. Biol. 2009;29:907–918. doi: 10.1128/MCB.00945-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmutte C., Sadoff M.M., Shim K.S., Acharya S., Fishel R. The interaction of DNA mismatch repair proteins with human exonuclease I. J. Biol. Chem. 2001;276:33011–33018. doi: 10.1074/jbc.M102670200. [DOI] [PubMed] [Google Scholar]
- 35.Tishkoff D.X., Boerger A.L., Bertrand P., Filosi N., Gaida G.M., Kane M.F., Kolodner R.D. Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proc. Natl. Acad. Sci. U.S.A. 1997;94:7487–7492. doi: 10.1073/pnas.94.14.7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tran P.T., Simon J.A., Liskay R.M. Interactions of Exo1p with components of MutLalpha in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9760–9765. doi: 10.1073/pnas.161175998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mimitou E.P., Symington L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mimitou E.P., Symington L.S. Nucleases and helicases take center stage in homologous recombination. Trends Biochem. Sci. 2009;34:264–272. doi: 10.1016/j.tibs.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 39.Mimitou E.P., Symington L.S. DNA end resection–unraveling the tail. DNA Repair (Amst) 2011;10:344–348. doi: 10.1016/j.dnarep.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bolderson E., Tomimatsu N., Richard D.J., Boucher D., Kumar R., Pandita T.K., Burma S., Khanna K.K. Phosphorylation of Exo1 modulates homologous recombination repair of DNA double-strand breaks. Nucleic Acids Res. 2010;38:1821–1831. doi: 10.1093/nar/gkp1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abuin A., Zhang H., Bradley A. Genetic analysis of mouse embryonic stem cells bearing Msh3 and Msh2 single and compound mutations. Mol. Cell. Biol. 2000;20:149–157. doi: 10.1128/mcb.20.1.149-157.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Wind N., Dekker M., Berns A., Radman M., te Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82:321–330. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- 43.Elliott B., Jasin M. Repair of double-strand breaks by homologous recombination in mismatch repair-defective mammalian cells. Mol. Cell. Biol. 2001;21:2671–2682. doi: 10.1128/MCB.21.8.2671-2682.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y., Rohde L.H., Wu H. Involvement of nucleotide excision and mismatch repair mechanisms in double strand break repair. Curr. Genomics. 2009;10:250–258. doi: 10.2174/138920209788488544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fahrer J., Kranaster R., Altmeyer M., Marx A., Burkle A. Quantitative analysis of the binding affinity of poly(ADP-ribose) to specific binding proteins as a function of chain length. Nucleic Acids Res. 2007;35:e143. doi: 10.1093/nar/gkm944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Y., Sass L.E., Du C., Hsieh P., Erie D.A. Determination of protein-DNA binding constants and specificities from statistical analyses of single molecules: MutS-DNA interactions. Nucleic Acids Res. 2005;33:4322–4334. doi: 10.1093/nar/gki708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang F., Ma J., Wu J., Ye L., Cai H., Xia B., Yu X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr. Biol. 2009;19:524–529. doi: 10.1016/j.cub.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shah G.M., Poirier D., Duchaine C., Brochu G., Desnoyers S., Lagueux J., Verreault A., Hoflack J.C., Kirkland J.B., Poirier G.G. Methods for biochemical study of poly(ADP-ribose) metabolism in vitro and in vivo. Anal. Biochem. 1995;227:1–13. doi: 10.1006/abio.1995.1245. [DOI] [PubMed] [Google Scholar]
- 49.Weinstock D.M., Nakanishi K., Helgadottir H.R., Jasin M. Assaying double-strand break repair pathway choice in mammalian cells using a targeted endonuclease or the RAG recombinase. Methods Enzymol. 2006;409:524–540. doi: 10.1016/S0076-6879(05)09031-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen X., Paudyal S.C., Chin R.I., You Z. PCNA promotes processive DNA end resection by Exo1. Nucleic Acids Res. 2013;41:9325–9338. doi: 10.1093/nar/gkt672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liberti S.E., Andersen S.D., Wang J., May A., Miron S., Perderiset M., Keijzers G., Nielsen F.C., Charbonnier J.B., Bohr V.A., et al. Bi-directional routing of DNA mismatch repair protein human exonuclease 1 to replication foci and DNA double strand breaks. DNA Repair (Amst) 2011;10:73–86. doi: 10.1016/j.dnarep.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu Z., Chung W.H., Shim E.Y., Lee S.E., Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mimitou E.P., Symington L.S. DNA end resection: many nucleases make light work. DNA Repair (Amst) 2009;8:983–995. doi: 10.1016/j.dnarep.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moreau S., Morgan E.A., Symington L.S. Overlapping functions of the Saccharomyces cerevisiae Mre11, Exo1 and Rad27 nucleases in DNA metabolism. Genetics. 2001;159:1423–1433. doi: 10.1093/genetics/159.4.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gravel S., Chapman J.R., Magill C., Jackson S.P. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sartori A.A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., Jackson S.P. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hopkins B.B., Paull T.T. The P. furiosus mre11/rad50 complex promotes 5′ strand resection at a DNA double-strand break. Cell. 2008;135:250–260. doi: 10.1016/j.cell.2008.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nicolette M.L., Lee K., Guo Z., Rani M., Chow J.M., Lee S.E., Paull T.T. Mre11-Rad50-Xrs2 and Sae2 promote 5′ strand resection of DNA double-strand breaks. Nat. Struct. Mol. Biol. 2010;17:1478–1485. doi: 10.1038/nsmb.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langerak P., Mejia-Ramirez E., Limbo O., Russell P. Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet. 2011;7:e1002271. doi: 10.1371/journal.pgen.1002271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nimonkar A.V., Genschel J., Kinoshita E., Polaczek P., Campbell J.L., Wyman C., Modrich P., Kowalczykowski S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25:350–362. doi: 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hong Z., Jiang J., Hashiguchi K., Hoshi M., Lan L., Yasui A. Recruitment of mismatch repair proteins to the site of DNA damage in human cells. J. Cell Sci. 2008;121:3146–3154. doi: 10.1242/jcs.026393. [DOI] [PubMed] [Google Scholar]
- 62.Mortusewicz O., Schermelleh L., Walter J., Cardoso M.C., Leonhardt H. Recruitment of DNA methyltransferase I to DNA repair sites. Proc. Natl. Acad. Sci. U.S.A. 2005;102:8905–8909. doi: 10.1073/pnas.0501034102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hashiguchi K., Matsumoto Y., Yasui A. Recruitment of DNA repair synthesis machinery to sites of DNA damage/repair in living human cells. Nucleic Acids Res. 2007;35:2913–2923. doi: 10.1093/nar/gkm115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Essers J., Theil A.F., Baldeyron C., van Cappellen W.A., Houtsmuller A.B., Kanaar R., Vermeulen W. Nuclear dynamics of PCNA in DNA replication and repair. Mol. Cell. Biol. 2005;25:9350–9359. doi: 10.1128/MCB.25.21.9350-9359.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dietlein F., Thelen L., Jokic M., Jachimowicz R.D., Ivan L., Knittel G., Leeser U., van Oers J., Edelmann W., Heukamp L.C., et al. A functional cancer genomics screen identifies a druggable synthetic lethal interaction between MSH3 and PRKDC. Cancer Discov. 2014;4:592–605. doi: 10.1158/2159-8290.CD-13-0907. [DOI] [PubMed] [Google Scholar]
- 66.Liu Y., Kadyrov F.A., Modrich P. PARP-1 enhances the mismatch-dependence of 5′-directed excision in human mismatch repair in vitro. DNA Repair (Amst) 2011;10:1145–1153. doi: 10.1016/j.dnarep.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arcus V.L., McKenzie J.L., Robson J., Cook G.M. The PIN-domain ribonucleases and the prokaryotic VapBC toxin-antitoxin array. Protein Eng. Des. Sel. 2011;24:33–40. doi: 10.1093/protein/gzq081. [DOI] [PubMed] [Google Scholar]
- 68.Glavan F., Behm-Ansmant I., Izaurralde E., Conti E. Structures of the PIN domains of SMG6 and SMG5 reveal a nuclease within the mRNA surveillance complex. EMBO J. 2006;25:5117–5125. doi: 10.1038/sj.emboj.7601377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu X., Chini C.C., He M., Mer G., Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 70.Durocher D., Smerdon S.J., Yaffe M.B., Jackson S.P. The FHA domain in DNA repair and checkpoint signaling. Cold Spring Harb. Symp. Quant. Biol. 2000;65:423–431. doi: 10.1101/sqb.2000.65.423. [DOI] [PubMed] [Google Scholar]
- 71.Manke I.A., Lowery D.M., Nguyen A., Yaffe M.B. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003;302:636–639. doi: 10.1126/science.1088877. [DOI] [PubMed] [Google Scholar]
- 72.Durocher D., Taylor I.A., Sarbassova D., Haire L.F., Westcott S.L., Jackson S.P., Smerdon S.J., Yaffe M.B. The molecular basis of FHA domain:phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms. Mol. Cell. 2000;6:1169–1182. doi: 10.1016/s1097-2765(00)00114-3. [DOI] [PubMed] [Google Scholar]
- 73.Durocher D., Henckel J., Fersht A.R., Jackson S.P. The FHA domain is a modular phosphopeptide recognition motif. Mol. Cell. 1999;4:387–394. doi: 10.1016/s1097-2765(00)80340-8. [DOI] [PubMed] [Google Scholar]
- 74.Flynn R.L., Zou L. Oligonucleotide/oligosaccharide-binding fold proteins: a growing family of genome guardians. Crit. Rev. Biochem. Mol. Biol. 2010;45:266–275. doi: 10.3109/10409238.2010.488216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arcus V. OB-fold domains: a snapshot of the evolution of sequence, structure and function. Curr. Opin. Struct. Biol. 2002;12:794–801. doi: 10.1016/s0959-440x(02)00392-5. [DOI] [PubMed] [Google Scholar]
- 76.Rass U., Compton S.A., Matos J., Singleton M.R., Ip S.C., Blanco M.G., Griffith J.D., West S.C. Mechanism of Holliday junction resolution by the human GEN1 protein. Genes Dev. 2010;24:1559–1569. doi: 10.1101/gad.585310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ip S.C., Rass U., Blanco M.G., Flynn H.R., Skehel J.M., West S.C. Identification of Holliday junction resolvases from humans and yeast. Nature. 2008;456:357–361. doi: 10.1038/nature07470. [DOI] [PubMed] [Google Scholar]
- 78.Jonas S., Weichenrieder O., Izaurralde E. An unusual arrangement of two 14-3-3-like domains in the SMG5-SMG7 heterodimer is required for efficient nonsense-mediated mRNA decay. Genes Dev. 2013;27:211–225. doi: 10.1101/gad.206672.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ohnishi T., Yamashita A., Kashima I., Schell T., Anders K.R., Grimson A., Hachiya T., Hentze M.W., Anderson P., Ohno S. Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol. Cell. 2003;12:1187–1200. doi: 10.1016/s1097-2765(03)00443-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.