

Abstract
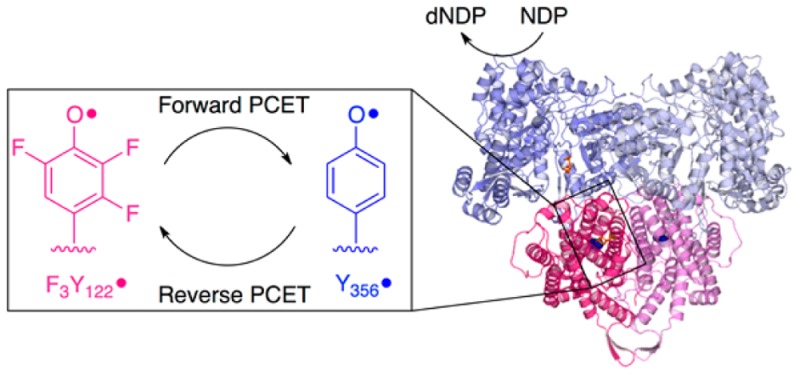
Escherichia coli class Ia ribonucleotide reductase is composed of two subunits (α and β), which form an α2β2 complex that catalyzes the conversion of nucleoside 5′-diphosphates to deoxynucleotides (dNDPs). β2 contains the essential tyrosyl radical (Y122•) that generates a thiyl radical (C439•) in α2 where dNDPs are made. This oxidation occurs over 35 Å through a pathway of amino acid radical intermediates (Y122 → [W48] → Y356 in β2 to Y731 → Y730 → C439 in α2). However, chemistry is preceded by a slow protein conformational change(s) that prevents observation of these intermediates. 2,3,5-Trifluorotyrosine site-specifically inserted at position 122 of β2 (F3Y•-β2) perturbs its conformation and the driving force for radical propagation, while maintaining catalytic activity (1.7 s–1). Rapid freeze–quench electron paramagnetic resonance spectroscopy and rapid chemical-quench analysis of the F3Y•-β2, α2, CDP, and ATP (effector) reaction show generation of 0.5 equiv of Y356• and 0.5 equiv of dCDP, both at 30 s–1. In the absence of an external reducing system, Y356• reduction occurs concomitant with F3Y reoxidation (0.4 s–1) and subsequent to oxidation of all α2s. In the presence of a reducing system, a burst of dCDP (0.4 equiv at 22 s–1) is observed prior to steady-state turnover (1.7 s–1). The [Y356•] does not change, consistent with rate-limiting F3Y reoxidation. The data support a mechanism where Y122• is reduced and reoxidized on each turnover and demonstrate for the first time the ability of a pathway radical in an active α2β2 complex to complete the catalytic cycle.
Introduction
Ribonucleotide reductases (RNRs) catalyze the formation of deoxynucleotides from their corresponding ribonucleotides (Scheme 1) in almost all organisms; allosteric regulation of substrate specificity and activity contributes to fidelity of both DNA replication and repair.1,2 The class Ia RNRs contain two homodimeric subunits, α2 and β2, which form an active α2β2 complex in the case of the E. coli enzyme.3 The β2 subunit houses a diferric-tyrosyl radical (Y122•) cofactor that reversibly oxidizes C439 in the active site of α2 to a thiyl radical.4,5 The C439• initiates nucleotide reduction by H atom abstraction from the 3′ position of the substrate (Scheme 1).6,7 On the basis of in silico docking of the individual X-ray structures of α2 and β2,8,9 the distance between Y122• and C439 is estimated to be >35 Å. This radical transport (RT) process occurs through a specific pathway that involves at least three transient aromatic amino acid radical intermediates (proton-coupled electron transfer or PCET through Y122• → [W48] → Y356 in β2 to Y731 → Y730 → C439 in α2, Figure 1).9,10 During turnover of wild-type (wt) RNR, only the resting state Y122• is observed. In this paper, we describe the perturbation of PCET kinetics by site-specific incorporation of 2,3,5-trifluorotyrosine (F3Y) at position 122 in β2 resulting in accumulation of a pathway tyrosyl radical intermediate (Y356•) that is kinetically and chemically competent to complete the catalytic cycle of RNR.
Scheme 1. Reaction Catalyzed by RNR.

Turnover requires reducing equivalents which are provided by a pair of cysteines in the active site of the enzyme.
Figure 1.

Proposed PCET pathway in E. coli class Ia RNR.10 The pink and blue arrows indicate the movement of electrons and protons through conserved aromatic amino acids (Y356 in β2 and Y731 and Y730 in α2). W48 and its putative proton acceptor D237 are shown in gray, as there is no evidence for their participation in RT. The positions of Y356 and E350 are unknown, as these residues are disordered in all crystal structures of β2.
In wt RNR, PCET steps are preceded by a rate-limiting protein conformational change(s) (5–10 s–1) that occur(s) upon association of α2, β2, substrate (S, CDP), and allosteric effector (E, ATP).11 Forward RT steps, active site chemistry, and reverse RT steps occur on a much more rapid time scale than the protein conformational change(s) preventing observation of radical intermediates either during steady-state turnover or using rapid kinetic methods.11 An approach adopted by our lab has been to change the rate-limiting step of the reaction by site-specifically incorporating tyrosine analogues with perturbed pKa’s and reduction potentials in an effort to accumulate and characterize the proposed radical intermediates (Figure 1). Previously, we described the insertion of 3-nitrotyrosine (NO2Y) at residue 122 in β2.12 The reaction of NO2Y•-β2, α2, CDP, and ATP rapidly generates 0.5 equiv of Y356• and dCDP (>100 s–1). We have proposed that this stoichiometry is a consequence of half-sites reactivity in RNR where only 50% of Y122• in the α2β2 complex reacts at a time.13−17 We have recently shown that, upon radical initiation in wt RNR, a proton is transferred from a water molecule on the diferric cluster to generate the corresponding Y122 phenol (Figure 1).18 In contrast, use of NO2Y•-β2 to initiate RT uncouples this proton transfer (PT) and electron transfer (ET); the conjugate base NO2Y– is generated instead of the anticipated phenol NO2Y. Furthermore, Y356• generated during reverse RT is unable to reoxidize NO2Y–, and thus, this mutant could only perform a single turnover.12 This prevented us from establishing the chemical competence of Y356• to complete the catalytic cycle and determining if it is a true intermediate on the PCET pathway.
In an attempt to engineer a smaller perturbation to the driving force, we inserted F3Y at position 122 in β2 and investigated the reaction of F3Y•-β2, α2, CDP, and ATP.19 Steady-state assays revealed that, unlike the NO2Y•-β2 mutant, F3Y•-β2 can catalyze multiple turnovers at 25% the steady-state wt activity. Hand-quench EPR experiments showed formation of a new tyrosyl radical, assigned to residue Y356 in β2 based on multiple lines of evidence. First, the new radical is observed when the redox inert F is inserted at position 731 but not at position 356.19 Second, the 9 GHz EPR spectrum of the new radical is remarkably similar to that of the radical observed in the NO2Y•-β2 studies.12,19 In NO2Y•-β2, pulsed electron–electron double resonance (PELDOR) spectroscopy experiments measured a distance of 30 Å between the new radical in one α/β pair and NO2Y• in the second α/β pair.20 Similar experiments with other un-natural amino acids inserted at position 356 (either 3,4-dihydroxyphenylalanine21 or 3-aminotyrosine22) provide an identical distance measurement (30 Å). In preliminary PELDOR experiments, a similar distance is also observed between F3Y• and the new radical (Nick, Bennati, unpublished results). These data together support that the radical observed in F3Y•-β2 is located at position 356. Studies with NO2Y•-β2, however, have shown that while the predominant location of the radical is at position 356 (85–90%), Y356• is in equilibrium with Y731• and Y730• in α2 (15–10%).20 These initial studies laid the foundation for the work described herein and gave us an opportunity to investigate the importance of Y356• and reverse RT in an active RNR complex.
In this work, we carry out thorough kinetic analyses of the reaction of F3Y•-β2, α2, CDP, and ATP in the absence and presence of an external reducing system composed of thioredoxin (TR), thioredoxin reductase (TRR), and NADPH. Rapid freeze–quench (RFQ) EPR spectroscopy and rapid chemical-quench (RCQ) studies in the absence of a reducing system demonstrate that Y356• is kinetically and chemically competent; similar amounts of Y356• and dCDP are produced at similar rate constants. Furthermore, subsequent to complete oxidation of α2, Y356• reduction occurs concomitant with F3Y reoxidation. RCQ analysis in the presence of a reducing system shows a burst of dCDP prior to steady-state turnover suggesting that the rate-limiting step occurs after product formation. EPR studies detect no changes in Y356• concentration during steady-state turnover. Together, the data support the conclusion that reverse RT within β2 leading to regeneration of F3Y• is rate-limiting during steady-state turnover of F3Y•-β2.
Materials and Methods
Materials
(His)6 wt-α2 (specific activity of 2500 nmol/min/mg) was expressed from pET28a-nrdA and purified using our standard protocol.23 Wt-α2 was pre-reduced by the addition of DTT and hydroxyurea prior to use.24 Tyrosine phenol lyase (TPL) was expressed and purified as described.25 F3Y was enzymatically synthesized from the corresponding phenol using TPL.26 The pBAD-nrdB122TAG and pEVOL-FnYRS-E3 plasmids were generated and isolated as described.19 Apo F3Y-β2 was expressed, purified, and reconstituted as detailed in the Supporting Information (SI). Yields of 10–15 mg of pure apo protein/g cell paste are routinely obtained. Reconstituted F3Y•-β2 has a specific activity (750–1000 nmol/min/mg) that varies directly with the radical content (0.6–0.8 F3Y•/β2). E. coli TR (40 U/mg) and TRR (1400 U/mg) were purified using established protocols.27,28 [3H] CDP was purchased from ViTrax (Placentia, CA). Hepes, MgSO4, EDTA, 2XYT microbial medium, ampicillin (Amp), chloramphenicol (Cm), ATP, CDP, and carrier deoxycytidine (dC) were obtained from Sigma-Aldrich. Promega provided isopropyl β-d-1-thiogalactopyranoside (IPTG) and DTT. Calf alkaline phosphatase was purchased from Roche. Assay buffer consists of 50 mM Hepes pH 7.6, 15 mM MgSO4, and 1 mM EDTA.
Reaction of F3Y•-β2, wt-α2, CDP, and ATP Monitored by RFQ-EPR Spectroscopy
RFQ experiments were performed on an Update Instruments 1019 syringe ram unit and a model 715 Syringe Ram controller (ram speed 1.25–1.6 cm/s) equipped with a Lauda RM6 circulating water bath set at 5 or 25 °C. F3Y•-β2 (0.4–0.8 F3Y•/β2, 80 μM) and CDP (2 mM) in assay buffer was mixed on a rapid time scale (16 ms–15 s) with an equal volume of wt-α2 (80 μM) and ATP (6 mM) in assay buffer. The reaction was quenched in liquid isopentane (−140 °C), and the crystals were packed into EPR tubes for analysis by EPR spectroscopy. A packing factor of 0.60 ± 0.02 was determined for wt-β2. The reaction at 5 °C was additionally monitored on a longer time scale (20 s–2 min) by mixing all assay ingredients by hand (30 μM wt-α2, 30 μM F3Y•-β2, 1 mM CDP, and 3 mM ATP) and quenching in liquid isopentane. EPR spectroscopy was performed at the Department of Chemistry Instrumentation Facility at MIT using wt-β2 (1.2 Y•/β2) as a standard. The concentration of Y• in the wt-β2 standard was previously estimated using a Cu(II)SO4 standard.29 EPR spectra were recorded at 77 K on a Bruker EMX X-band spectrometer with a quartz finger dewar containing liquid N2. The parameters were as follows: microwave frequency 9.45 GHz, power 30 μW, modulation amplitude 1.50 G, modulation frequency 100 kHz, time constant 5.12 ms, and scan time 41.93 s. From each composite spectrum, residual F3Y• was subtracted by aligning the radical’s distinct features on the high- and low-field sides of the spectrum as previously reported (Figure S1).19 The subtracted spectrum was reintegrated to quantitate the percentage of any observed pathway radical. The complete data sets at 5 and 25 °C were fit to eq 1:
| 1 |
where A1 and A2 are the amplitudes of the two phases and k1 and k2 are the observed rate constants.
Reaction of F3Y•-β2, wt-α2, CDP, and ATP Monitored by the RCQ Method
RCQ experiments were performed on a KinTek RQF-3 instrument equipped with a Lauda RM6 circulating water bath set at 5 or 25 °C. Syringe A containing 20 μM wt-α2 and 6 mM ATP in assay buffer was mixed with an equal volume from syringe B containing 20 μM F3Y•-β2 (0.85 F3Y•/β2) and 1 mM [3H] CDP (22 000 cpm/nmol) in assay buffer. The reaction was aged for varying times (5 ms–100 s) and quenched with 2% HClO4 in syringe C. The reaction was additionally monitored at >100 s by mixing the contents of the two syringes by hand, incubating the reaction mixture in a circulating water bath for the desired period of time, and manually quenching the reaction with 2% HClO4. All samples were neutralized by the addition of 110–160 μL of 0.5 M KOH and worked up as described.11,30 For the measurement of radioactive background from [3H] CDP, an equal volume of the contents of syringe B was mixed with assay buffer, followed by 2% HClO4 and KOH. The reaction was also performed by hand (100 s at 5 or 25 °C) before and after the entire RCQ time course to account for any air oxidation of wt-α2. The 5 °C data set was fit to eq 1, and the 25 °C data set was fit to eq 2:
| 2 |
In eq 2, the first phase is fixed, A2 and k2 represent the amplitude and rate constant of the second phase, and k3 represents the rate constant for the third phase.
Reaction of F3Y•-β2, wt-α2, CDP, ATP, TR/TRR/NADPH Monitored by Hand-Quench EPR Spectroscopy
Reactions were performed in a total volume of 250 μL containing 10 μM wt-α2, 10 μM F3Y•-β2 (0.6 F3Y•/β2), 1 mM CDP, 3 mM ATP, 40 μM TR, 0.8 μM TRR, and 1 mM NADPH in assay buffer. Samples were incubated in a circulating water bath set at 5 °C and quenched for EPR analysis between 20 and 90 s in liquid isopentane (−140 °C). The reactions were also performed at 25 °C in a final volume of 250 μL containing 30 μM wt-α2, 10 μM F3Y•-β2 (0.6 F3Y•/β2), 2.5 mM CDP, 3 mM ATP, 80 μM TR, 1.6 μM TRR, and 2.5 mM NADPH.
Reaction of F3Y•-β2, wt-α2, CDP, ATP, and TR/TRR/NADPH Monitored by the RCQ Method
The reaction was performed in an identical fashion to that described in the absence of a reducing system with minor modifications. For data collected at 5 °C, syringe A contained 20 μM wt-α2, 6 mM ATP, 80 μM TR, and 1.6 μM TRR in assay buffer, while syringe B contained 20 μM F3Y•-β2 (0.6 F3Y•/β2), 1 mM [3H] CDP (20 000 cpm/nmol), and 2 mM NADPH. For the 25 °C reaction, the amount of [3H] CDP in syringe B was increased to 2 mM. Samples were quenched and worked up as described earlier. The time courses of the reactions were fit to eq 3:
| 3 |
Here A and k1 are the amplitude and rate constant for the burst phase, respectively, and k2 is the rate constant for the linear phase.
Results
“Two or None” Radical Distribution and Half-Sites Reactivity in F3Y•-β2
The diferric-F3Y• cofactor is self-assembled from apo F3Y-β2 by the addition of Fe2+ and O2 to produce ∼0.8 F3Y•/β2 (SI), lower than the 1.2 Y•/wt-β2.31 While the radical distribution in β2 has remained difficult to probe experimentally, evidence collected over the past few years supports that active β2 contains one Y• in each monomer (“two or none”, Figure 2A), suggesting that only ∼40% of β2 is active in our F3Y•-β2 samples.12,13,20−22,32,33 To provide support for this conclusion, we monitored the effect of F3Y• concentration on the amount of Y356• that accumulates. RFQ-EPR experiments were performed at 25 °C with F3Y•-β2 containing 0.4 F3Y•/β2 or 0.85 F3Y•/β2, wt-α2, CDP, and ATP. The kinetic analysis of these studies is discussed subsequently, but the amount of Y356• is 0.43 and 0.5 equiv/F3Y•, respectively (Table S1). This amount can be rationalized by the “two or none” radical distribution model (Figure 2A) as well as half-sites reactivity in RNR (Figure 2B); only one of the 2 F3Y• reacts at a time to generate Y356•.20−22
Figure 2.

(A) “Two or none” model for radical distribution in F3Y•-β2. The amounts of active (40%) and inactive β2 (60%) are shown for a sample containing 0.8 F3Y•. The amount of radical in F3Y•-β2 is lower than that typically seen in wt-β2 (1.2 Y•/β2).31 (B) Half-sites reactivity in F3Y•-β2. The total amount of Y356• that can accumulate on one turnover is 0.5 equiv/F3Y•.
Kinetics in the Absence of a Reducing System
Kinetics of Formation and Disappearance of Y356• at 25 °C
To assess if Y356• is formed faster than the turnover number of the enzyme (1.7 s–1, Table 1), a RFQ-EPR experiment was performed in which wt-α2, F3Y•-β2, CDP, and ATP were mixed rapidly (16 ms to 15 s). EPR analysis of each sample revealed a mixture of F3Y• and Y356•, and spectral subtractions were performed to quantitate the relative fraction of each radical. The unique hyperfine interactions associated with the fluorine nuclei facilitated spectral deconvolution (Figure S1).19 The results of the experiment are shown in Figure 3A, and the data were fit to a biexponential equation. Rapid loss of 0.51 ± 0.02 equiv of F3Y• (not shown) concomitant with formation of identical amounts of Y356• occurs at 30 ± 5 s–1. Subsequently, reduction of the pathway radical with kapp 0.4 ± 0.1 s–1 is accompanied by reformation of F3Y• with the same kapp (not shown). These data show for the first time, accumulation of a pathway radical (Y356•) in an active RNR complex that can regenerate the stable radical at position 122 (F3Y•). We note that between 0.1 and 1 s, the concentration of Y356• varies minimally. As shown subsequently, F3Y•-β2 can make multiple dCDPs in the absence of a reducing system, and the reduction of Y356• and reoxidation of F3Y are only visualized after the last turnover when all α2s are oxidized. Finally, regeneration of F3Y• is incomplete with only 0.25 equiv of Y356• reoxidizing F3Y within 10 s.
Table 1. DeoxyCDP Formation Kinetics in the Absence and Presence of TR/TRR/NADPHa.
| first phase |
second phase |
total dC |
||||||
|---|---|---|---|---|---|---|---|---|
| β2 | T (°C) | R | k1 (s–1) | A/radical | k2 (s–1) | A/radical | dC/α2 | dC/radical |
| F3Y | 25 | N | 30b | 0.5b | 0.5 (1) | 2.9 (1) | 2.8 (4)c | 3.5 (3)c |
| F3Y | 25 | Y | 22 (9) | 0.40 (5) | 1.73 (4) | |||
| F3Y | 5 | N | 3 (1) | 0.3 (1) | 0.08 (1) | 2.9 (1) | 2.7 (1) | 3.4 (1) |
| F3Y | 5 | Y | 6 (3) | 0.26 (5) | 0.20 (1) | |||
All experiments were performed with 10 μM wt-α2 and 10 μM F3Y•-β2. R notes the absence or presence of a reducing system. A represents the amplitude of each phase.
See description in main text and SI for more details regarding fitting.
Numbers reported reflect the total amount of dC generated within the first two phases. Product generated in the third phase is cytosine.
Figure 3.

Reaction of F3Y•-β2, wt-α2, CDP, and ATP at 25 °C monitored by (A) RFQ-EPR spectroscopy and (B) the RCQ method. All data points represent the averages of two independent trials. Data were fit to a (A) two- or (B) three-phase model with the rate constants shown in Table 1. (B) The inset shows dCDP formation during the first 2 s of the reaction. The rate constants measured for Y356• formation and disappearance correlate with the fitted rate constants for dCDP formation in the first two phases.
Kinetics of Formation and Disappearance of Y356• at 5 °C
The rapid formation of Y356• at 25 °C resulted in generation of 0.25 equiv (50% of total Y356•) prior to the first data point (16 ms, Figure 3A) prompting us to switch to lower temperatures to slow down the reaction. RFQ-EPR experiments were set up at 5 °C as described for 25 °C, and the results are shown in Figure 4A. A kapp of 3.8 ± 0.5 s–1 was measured for formation of Y356• concomitant with loss of F3Y• (not shown). In contrast to our observation at 25 °C, only 0.32 ± 0.02 equiv of Y356• is formed at 5 °C reflecting temperature dependent changes in the rates of formation and decay of the pathway radical. Similar to our observation at 25 °C, the concentration of Y356• varies minimally between 0.8 and 5 s supporting the proposal that F3Y•-β2 catalyzes multiple turnovers prior to visualization of reverse RT. Unlike in the 25 °C reaction, Y356• reduction at 5 °C is accompanied by complete reformation of F3Y• (0.3 equiv, 0.06 ± 0.01 s–1) within ∼40 s.
Figure 4.

Reaction of F3Y•-β2, wt-α2, CDP, and ATP at 5 °C monitored by (A) RFQ-EPR spectroscopy and (B) the RCQ method. All data points represent the averages of two independent trials. Black lines represent biexponential fits to the data with the rate constants given in Table 1. (B) The inset shows dCDP formation during the first 5 s of the reaction. The rate constants measured for Y356• formation and disappearance are identical to the rate constants measured for dCDP formation.
Kinetics and dCDP Formation with F3Y•-β2 at 25 °C
Each dCDP generated by RNR is accompanied by the formation of a disulfide bond in the active site of an α monomer (Scheme 1, Figure 5, step A). Re-reduction of the active site disulfide by a C-terminal cysteine pair on each monomer (step B) facilitates an additional turnover (step C),11 giving a theoretical maximum of 4 dCDP/α2 in the absence of TR/TRR/NADPH. In practice, only 3 dCDP/α2 are routinely measured with wt RNR due to partial oxidation of α2 (during purification and handling) and our inability to completely pre-reduce wt-α2 before an experiment.11 Our kinetic experiments with F3Y•-β2 contain 1:1 α2:F3Y•-β2 with only 40% of active F3Y•-β2 (Figure 2A). However, 3 dCDP/α2 are generated in the absence of a reducing system (Table 1) requiring that each F3Y• perform multiple turnovers (3.5 dCDP/F3Y•, Table 1) to service all α2s.
Figure 5.

Amount of dCDP generated in the absence of a reducing system. A theoretical maximum of 4 dCDP/α2 can be produced; however, only 3 dCDP/α2 are routinely measured. The reaction mixture contains only 40% active F3Y•-β2 (Figure 2A) supporting reorganization of active and inactive α2/F3Y•-β2 complexes to oxidize all α2s.
To assess if Y356• is on-pathway, the kinetics of dCDP formation were determined. Wt-α2, F3Y•-β2, [3H] CDP, and ATP were mixed (5 ms to 300 s) and quenched rapidly with 2% HClO4. CDP and dCDP were separated and analyzed by standard procedures,11,30 and the results are shown in Figure 3B. The data are best described by eq 2 with a fixed first exponential phase, a variable second exponential phase, and a very slow third linear phase. We initially attempted to fit the data with an exponential phase and a linear phase with poor results (Figure S2A). To obtain the fit shown in Figure 3B (black line), we fixed the amplitude and rate constant (k1) of the first phase at 0.5 dCDP/F3Y• and 30 s–1, respectively. Fixing this phase was required due to the scatter in the data at early time points. This scatter is a result of “two or none” and half-sites reactivity associated with RNR (Figure 2A,B). The [3H] dCDP measured between 5 and 100 ms is close to the background measured with [3H] CDP in the absence of α2. The range of choices considered for the amplitude and k1 of this phase were based on the amplitude and rate constant measured for Y356• formation by RFQ-EPR (Figure 3A) and the results obtained in the presence of the reducing system (presented in the next section). The detailed description of data fitting using different parameters for the first kinetic phase is shown in Figure S2A–D. An additional experiment to justify the fixed first phase is shown in Figure S3.
Once the first phase was fixed using eq 2, we obtained an amplitude and rate constant (k2) of 2.9 ± 0.1 dCDP/F3Y• and 0.5 ± 0.1 s–1, respectively, for the second phase and a rate constant (k3) of 0.012 ± 0.001 s–1 for the linear phase. This slow linear phase is associated with cytosine release and not dCDP formation. It occurs during the reaction of F3Y•-β2 with oxidized α2 as shown in Scheme S1.34 In a second manuscript, we show that the inability to monitor complete reverse RT at 25 °C (Figure 3A) is associated with reoxidation of Y356 (0.25 equiv) by F3Y•-β2/oxidized α2.
The fit shown in Figure 3B suggests that the pathway radical is kinetically and chemically competent for nucleotide reduction at 25 °C. These data require that Y356• accumulates during reverse RT. k2 for product formation correlates well with kapp for Y356• disappearance at this temperature (0.5 s–1 vs 0.4 s–1, Figure 3A).
Kinetics and dCDP Formation with F3Y•-β2 at 5 °C
The kinetics of dCDP formation were also measured at 5 °C, and the results are shown in Figure 4B. The data were fit to a biexponential equation providing amplitudes of 0.3 ± 0.1 dCDP/F3Y• and 2.9 ± 0.1 dCDP/F3Y• with k1 and k2 of 3 ± 1 s–1 and 0.08 ± 0.01 s–1, respectively (Table 1). A1 and k1 for dCDP formation are very similar to the amplitude and rate constant measured for Y356• formation by RFQ-EPR spectroscopy at the same temperature (Figure 4A). These data suggest that Y356• is kinetically and chemically competent for dCDP formation and accumulates during reverse RT. Similar to our observations at 25 °C, k2 of 0.08 s–1 for dCDP formation is similar to kapp of 0.06 s–1 for reoxidation of F3Y by Y356• (Figure 4A).
The RCQ data were also analyzed relative to α2 to show that 2.7 ± 0.1 dCDPs/α2 are generated. DeoxyCDP formation was monitored for a total of 20 min, and in contrast to the 25 °C data, no third kinetic phase associated with cytosine was observed.
Kinetics in the Presence of a Reducing System
EPR Analysis of Y356• Concentration During Steady-State Turnover
The ability of F3Y•-β2 to perform multiple turnovers in the absence of a reducing system (3.5 dCDP/F3Y•, Table 1) and the observation of a plateau phase in the RFQ-EPR kinetic traces (Figures 3A and 4A) suggested that reverse RT is visualized subsequent to complete oxidation of α2. Thus, we predicted that the concentration of Y356• would vary minimally in the presence of the reducing system, TR/TRR/NADPH, as oxidized α2 is re-reduced. To test this prediction, F3Y•-β2, wt-α2, CDP, and ATP were combined in the presence of TR/TRR/NADPH, and samples were quenched by hand in liquid isopentane between 20 and 90 s. In accordance with our prediction, the amount of Y356• does not change: 0.26 to 0.28 equiv/F3Y• at 5 °C and 0.40 to 0.46 equiv/F3Y• at 25 °C (Table S2). No reverse RT was visualized during the time frame of the reaction.
Kinetics of dCDP Formation at 5 and 25 °C
The observation of Y356• accumulation during reverse RT (Figures 3 and 4) in the absence of a reducing system and the lack of variation in [Y356•] during steady-state turnover suggest that the rate-limiting step occurs subsequent to dCDP formation and Y356• reformation during reverse RT. This model predicts that RCQ experiments in the presence of the reducing system would show a burst of dCDP representing the first turnover followed by a linear phase for steady-state turnover. F3Y•-β2, wt-α2, [3H]CDP, and ATP were mixed in the presence of TR/TRR/NADPH, and the reaction was monitored from 5 ms to 100 s. The results of the F3Y•-β2 experiments, at 5 and 25 °C, are shown in Figure 6 and summarized in Table 1.
Figure 6.

Kinetics of product formation in F3Y•-β2 in the presence of TR/TRR/NADPH at (A) 25 °C and (B) 5 °C. The averages of 2–4 separate trials are shown. Data were fit to a two-phase model with the rate constants given in Table 1. The insets show dCDP formation during the first (A) 0.8 s and (B) 2.5 s. The burst phase represents the very first turnover by an α/β pair. Reverse PCET regenerating F3Y• is rate-limiting during steady-state turnover.
As predicted, a burst of dCDP formation is observed at both temperatures (0.26 ± 0.05 dCDP/F3Y• at 6 ± 3 s–1 at 5 °C and 0.40 ± 0.05 dCDP/F3Y• at 22 ± 9 s–1 at 25 °C) followed by a linear phase (0.20 ± 0.01 s–1 at 5 °C and 1.73 ± 0.04 s–1 at 25 °C). The large uncertainties observed in these parameters are associated with low amounts of dCDP arising from the “two or none” model (Figure 2A) and half-sites reactivity (Figure 2B). Unfortunately, we are unable to increase protein concentration in these experiments as studies with wt RNR have revealed kinetic complexities associated with the re-reduction process11 and potentially quaternary structure interconversions.35
The observed rate constant and amplitude for the burst phase are within error similar to the parameters observed for Y356• formation (Figures 3A and 4A) and support our conclusion that Y356• is on-pathway for dCDP formation. These data also correlate well with the kinetics of the first phase measured in the absence of TR/TRR/NADPH (0.3 dCDP/F3Y• at 3 s–1 at 5 °C and 0.5 dCDP/F3Y• at 30 s–1 at 25 °C) supporting that k1 in the absence of a reducing system and the burst phase in the presence of TR/TRR/NADPH report on the first turnover by one α/F3Y•-β pair in the α2/F3Y•-β2 complex (Figure 2B). Subsequent to dCDP formation and Y356• regeneration during reverse RT, reoxidation of F3Y and re-reduction of oxidized α2 facilitate further turnovers. We argue subsequently that the rate-limiting step in F3Y•-β2 is reoxidation of F3Y by Y356•.
Discussion
The rate-limiting protein conformational change(s) that gate(s) E. coli class Ia RNR turnover has precluded insight into the 35 Å forward RT, nucleotide reduction, and reverse RT processes. Our current model for wt RNR based on studies similar to those described herein for F3Y•-β2 is shown in Scheme 2. Upon association of β2/α2/CDP/ATP, a conformational change(s) (5–10 s–1, Scheme 2, step A) triggers rapid RT into α2 and nucleotide reduction (>100 s–1, step B).12,36 DeoxyCDP formation is rate-limited by the conformational change(s) and occurs at 5–10 s–1 as measured by RCQ methods reported previously11 and reproduced here under the same conditions utilized for the F3Y•-β2 studies (Figure S4). Subsequent to dCDP formation, reverse RT to regenerate Y122• is required to be downhill12 and rapid (>103 s–1, step C) as modeled by Ge et al.11 to account for our inability to observe Y122• disappear and reappear during turnover (±TR/TRR/NADPH). The physical steps in wt RNR preclude detection of intermediates in these processes. Thus, studying the chemistry has required engineering specific perturbations to the system initially through site-directed mutagenesis37−40 and the use of mechanism-based inhibitors34,41,42 and, more recently, with site-specific incorporation of unnatural amino acids.12,13,23,24 While with many of these approaches we were able to monitor the disappearance of Y122• concomitant with formation of new radicals, in none of these cases was the catalytic cycle of RNR completed, and no insight was obtained into reverse RT.
Scheme 2. Kinetic Model for wt RNR in the Absence and Presence of a Reducing System.

The graphic shows only the key amino acids in the PCET pathway. Rate constants are shown for the reaction at 25 °C. Y356• cannot be visualized in wt RNR due to the rate-limiting conformational change(s). Figure adapted from ref (11).
Our recent engineering of an orthogonal tRNA-synthetase tRNA pair that can incorporate di- and trifluorotyrosines (FnY, n = 2, 3) with a range of reduction potentials and pKa’s in RNR19 allowed us to introduce a tunable thermodynamic perturbation of PCET kinetics with minimal steric perturbations. F3Y is predicted as ∼10 mV harder to oxidize than Y,17,43 assuming that the first step in forward RT involves PT from the water on the diferric cluster to F3Y• concomitant with ET from Y356 to F3Y• (Figure 1). F3Y•-β2 is capable of catalyzing multiple turnovers but allows detection of Y356• (±TR/TRR/NADPH) due to perturbed reverse RT kinetics. Our current model for F3Y•-β2 turnover is shown in Scheme 3. The ability to accumulate Y356• in F3Y•-β2 but not in wt RNR is directly related to the differences in the rate-limiting step in the two systems.
Scheme 3. Kinetic Model for F3Y•-β2 in the Absence and Presence of a Reducing System.

The figure represents a reductionist view of key amino acids in the PCET pathway. The rate constants for each step are shown in pink (25 °C), blue (5 °C), and black (both temperatures). It is not known if the first RT step involves PCET to generate the phenol (F3Y), as in wt RNR or ET to generate the phenolate (F3Y–), as in NO2Y•-β2.
In our model, the F3Y•-β2/wt-α2/CDP/ATP complex undergoes a conformational change prior to generation of Y356• in one α/β pair (Scheme 3, step A). The RFQ-EPR data reported in Figures 3A and 4A provide the rate constants for this step and suggest that F3Y•-β2 perturbs the conformational gate relative to the wt enzyme (20–30 vs 5–10 s–1). We expect that forward RT into α and dCDP production (step B in Schemes 2 and 3) occur with similar rate constants to wt RNR (>100 s–1).12,36 DeoxyCDP formation is rate-limited by the slow, conformationally gated generation of Y356• during forward RT as measured by the first phase in the absence of a reducing system (Figures 3B and 4B) or the burst phase in the presence of TR/TRR/NADPH (Figure 6) in the RCQ studies. Subsequent to product formation, we propose that reverse RT to regenerate Y356• is fast12 as modeled in wt RNR where reverse RT to regenerate Y122• is 103 s–1. However, unlike in wt RNR, slow reoxidation of F3Y (step C) rate-limits subsequent turnovers.
In the absence of a reducing system, the RFQ-EPR data (Figures 3A and 4A) provide the rate constants for step C. Upon regeneration of F3Y•, β2 rapidly dissociates from a partially oxidized α2, associates with a second reduced α2 and cycles through steps A–C until all α2s are completely oxidized.44 Y356• concentration does not vary significantly during this time as visualized by the plateaus in the RFQ-EPR kinetic traces (Figures 3A and 4A). The second phase of the RCQ studies described in Figures 3B and 4B provides the rate constants for turnover in the absence of TR/TRR/NADPH (Scheme 3, branch I). Altered reverse RT kinetics in F3Y•-β2 allow us to observe for the first time the disappearance and reappearance of the radical at position 122 subsequent to complete oxidation of α2. The molecular bases for our ability to observe reverse RT are not well-understood but are likely related to the initiating step in the PCET process. In addition to perturbing the driving force for RT, the fluoro substitutions could alter the distance between the phenolic oxygen and the water on the diferric cluster, thus affecting PT between the two (Figure 1). F3Y also perturbs the pKa at position 122 compared to Y (solution pKa 6.4 vs 10).43 Depending on the pKa of the water on the diferric cluster, the phenolate F3Y– could be generated instead of the anticipated phenol F3Y.
Additional insight into the differences between wt and F3Y•-β2 catalysis is obtained from the amplitudes for dCDP formation in the absence of a reducing system (Table 1 and Figure S4). In the F3Y•-β2 system, the first phase (0.5 dCDP/F3Y• at 25 °C) reports on the very first turnover by an α/β pair (Figure 2B), while the second phase (2.9 dCDP/F3Y•) reports on consumption of all remaining reduced α2s, rate-limited by reverse RT. This result is distinct from our previous11 and current observations for wt RNR (Figure S4) where two phases are also measured for dCDP formation. The first phase is presumed to report on the conformationally gated generation of 2 dCDPs by all α2s (the experimental observation is 1.3 ± 0.2 dCDP/α2, 6 ± 1 s–1), and the second phase is interpreted to report on the generation of 2 additional dCDPs subsequent to re-reduction of the active site disulfide (the experimental observation is 1.6 ± 0.2 dCDP/α2, 0.5 ± 0.1 s–1). The variation in the amplitudes of the two phases between wt and F3Y•-β2 is consistent with different rate-limiting steps in the two systems. However, in both cases the total number of dCDPs generated is the same: 3 dCDP/α2 (Schemes 2 and 3, branch I).
The EPR and RCQ data collected in the presence of a reducing system also lend support to Scheme 3. In F3Y•-β2, a burst of dCDP formation prior to steady-state turnover is observed. The amplitude of this phase (∼0.5 dCDP/F3Y• at 25 °C) again reflects that turnover occurs only on one α/β pair prior to the rate-limiting step and is consistent with slow reverse RT. Upon regeneration of F3Y• after one turnover (Scheme 3, step C), re-reduction of oxidized α2 by TR/TRR/NADPH resets the system for additional turnovers (branch II). Y356• under these conditions behaves in a similar fashion to Y122• in wt RNR; i.e., no changes in its concentration are detected during steady-state conditions (Table S2). Slow reoxidation of F3Y followed by rapid re-reduction, forward RT, nucleotide reduction, and regeneration of Y356• (Scheme 3, steps A–C) precludes observation of its disappearance and reappearance. We interpret the linear phases in Figure 6A,B as representative of the rate constants for reverse RT in the presence of a reducing system (Scheme 3, step C) and the rate constant limits for re-reduction of oxidized α2. Although we set a lower limit for re-reduction from our experiments, we note that the rate constant for this step must be relatively fast, >5–10 s–1, to account for our inability to monitor changes in Y356• concentration.
A comparison of the burst phase for F3Y•-β2 and that for wt RNR in the presence of TR/TRR/NADPH reveals distinct behavior in the two systems (Figure 6 and Figure S5).11 Although the conformational change is rate-limiting for dCDP formation in wt RNR, we have previously noted that the rate-limiting step in the presence of a reducing system can switch to re-reduction of oxidized α2 at the high protein concentrations required for RCQ studies (10 μM).11 In wt RNR, this results in a conformationally gated burst of 2 dCDPs by all α2s (the experimental observation is 1.9 ± 0.1, 9 ± 2 s–1) prior to steady-state turnover (Figure S5).11 The burst phase reflects oxidation of all α2 active sites despite the presence of only 60% active wt-β2 (1.2 Y•/β2, Figure 2A) and does not represent a single turnover. In contrast to the wt system, we have isolated the very first turnover by an α2β2 complex with F3Y•-β2 due to rate-limiting reverse RT.
As a final point, the rate constant measured for dCDP formation in the presence of a reducing system is 3-fold faster that that measured in its absence (Scheme 3, branch I vs I). A similar variation has been previously noted for dCDP formation in the wt system.11 It is possible that re-reduction of the active site disulfide by the C-terminal tail (Figure 5, step B) is reversible and only driven to completion when the TR/TRR/NADPH system is included in the assays. However, we currently cannot rule out other modes by which TR accelerates re-reduction of the α2 active site.
Conclusions
Radical initiation in the class I RNRs is proposed to involve long-range PCET through three pathway tyrosines.9,10,17 Using an engineered RNR system, we have observed one of the proposed intermediates and demonstrate for the very first time chemically competent reverse RT that completes the RNR catalytic cycle. We additionally obtained insight into radical stoichiometry within β2, half-sites reactivity, and the ability of β2 to act catalytically during turnover. This work highlights the utility of unnatural amino acids in engineering specific perturbations for the study of redox active tyrosine residues in proteins; F3Y could facilitate understanding of a number of additional tyrosyl radical mediated metabolic processes.45−48
Acknowledgments
This work was supported by NIH Grant GM29595 to J.S.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b09189.
Detailed experimental procedures, tables, figures, and scheme of cytosine release from CDP (PDF)
Author Present Address
∥ Merck Research Laboratories, 33 Avenue Louis Pasteur, Boston, MA 02115, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- Stubbe J.; van der Donk W. A. Chem. Rev. 1998, 98, 705–62 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- Jordan A.; Reichard P. Annu. Rev. Biochem. 1998, 67, 71–98 10.1146/annurev.biochem.67.1.71. [DOI] [PubMed] [Google Scholar]
- Brown N. C.; Canellakis Z. N.; Lundin B.; Reichard P.; Thelander L. Eur. J. Biochem. 1969, 9, 561–73 10.1111/j.1432-1033.1969.tb00646.x. [DOI] [PubMed] [Google Scholar]
- Licht S.; Gerfen G. J.; Stubbe J. Science 1996, 271, 477–81 10.1126/science.271.5248.477. [DOI] [PubMed] [Google Scholar]
- Stubbe J. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 2723–4 10.1073/pnas.95.6.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht S.; Stubbe J. Compr. Nat. Prod. Chem. 1999, 5, 163–203 10.1016/B978-0-08-091283-7.00114-4. [DOI] [Google Scholar]
- Stubbe J.; Ackles D. J. Biol. Chem. 1980, 255, 8027–30. [PubMed] [Google Scholar]
- Nordlund P.; Sjöberg B. M.; Eklund H. Nature 1990, 345, 593–8 10.1038/345593a0. [DOI] [PubMed] [Google Scholar]
- Uhlin U.; Eklund H. Nature 1994, 370, 533–9 10.1038/370533a0. [DOI] [PubMed] [Google Scholar]
- Stubbe J.; Nocera D. G.; Yee C. S.; Chang M. C. Y. Chem. Rev. 2003, 103, 2167–201 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- Ge J.; Yu G.; Ator M. A.; Stubbe J. Biochemistry 2003, 42, 10071–83 10.1021/bi034374r. [DOI] [PubMed] [Google Scholar]
- Yokoyama K.; Uhlin U.; Stubbe J. J. Am. Chem. Soc. 2010, 132, 15368–79 10.1021/ja1069344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Stubbe J. J. Am. Chem. Soc. 2006, 128, 2522–3 10.1021/ja057776q. [DOI] [PubMed] [Google Scholar]
- Sjöberg B. M.; Gräslund A.; Eckstein F. J. Biol. Chem. 1983, 258, 8060–7. [PubMed] [Google Scholar]
- Salowe S.; Bollinger J. M. Jr.; Ator M.; Stubbe J.; McCracken J.; Peisach J.; Samano M. C.; Robins M. J. Biochemistry 1993, 32, 12749–12760 10.1021/bi00210a026. [DOI] [PubMed] [Google Scholar]
- Artin E.; Wang J.; Lohman G. J.; Yokoyama K.; Yu G.; Griffin R. G.; Bar G.; Stubbe J. Biochemistry 2009, 48, 11622–9 10.1021/bi901590q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnihan E. C.; Nocera D. G.; Stubbe J. Acc. Chem. Res. 2013, 46, 2524–35 10.1021/ar4000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wörsdorfer B.; Conner D. A.; Yokoyama K.; Livada J.; Seyedsayamdost M.; Jiang W.; Silakov A.; Stubbe J.; Bollinger J. M. Jr.; Krebs C. J. Am. Chem. Soc. 2013, 135, 8585–93 10.1021/ja401342s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnihan E. C.; Young D. D.; Schultz P. G.; Stubbe J. J. Am. Chem. Soc. 2011, 133, 15942–5 10.1021/ja207719f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama K.; Smith A. A.; Corzilius B.; Griffin R. G.; Stubbe J. J. Am. Chem. Soc. 2011, 133, 18420–32 10.1021/ja207455k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Chan C. T.; Mugnaini V.; Stubbe J.; Bennati M. J. Am. Chem. Soc. 2007, 129, 15748–9 10.1021/ja076459b. [DOI] [PubMed] [Google Scholar]
- Minnihan E. C. Ph.D. Dissertation, Massachusetts Institute of Technology, 2012. [Google Scholar]
- Minnihan E. C.; Seyedsayamdost M. R.; Uhlin U.; Stubbe J. J. Am. Chem. Soc. 2011, 133, 9430–40 10.1021/ja201640n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Xie J.; Chan C. T.; Schultz P. G.; Stubbe J. J. Am. Chem. Soc. 2007, 129, 15060–71 10.1021/ja076043y. [DOI] [PubMed] [Google Scholar]
- Chen H.; Gollnick P.; Phillips R. S. Eur. J. Biochem. 1995, 229, 540–9 10.1111/j.1432-1033.1995.tb20496.x. [DOI] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Yee C. S.; Stubbe J. Nat. Protoc. 2007, 2, 1225–35 10.1038/nprot.2007.159. [DOI] [PubMed] [Google Scholar]
- Chivers P. T.; Prehoda K. E.; Volkman B. F.; Kim B. M.; Markley J. L.; Raines R. T. Biochemistry 1997, 36, 14985–91 10.1021/bi970071j. [DOI] [PubMed] [Google Scholar]
- Russel M.; Model P. J. Bacteriol. 1985, 163, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer G. Methods Enzymol. 1967, 10, 594–610 10.1016/0076-6879(67)10101-8. [DOI] [Google Scholar]
- Steeper J. R.; Steuart C. D. Anal. Biochem. 1970, 34, 123–30 10.1016/0003-2697(70)90092-8. [DOI] [PubMed] [Google Scholar]
- Tong W. H.; Chen S.; Lloyd S. G.; Edmondson D. E.; Huynh B. H.; Stubbe J. J. Am. Chem. Soc. 1996, 118, 2107–8 10.1021/ja952764y. [DOI] [Google Scholar]
- Bennati M.; Weber A.; Antonic J.; Perlstein D. L.; Robblee J. H.; Stubbe J. J. Am. Chem. Soc. 2003, 125, 14988–9 10.1021/ja0362095. [DOI] [PubMed] [Google Scholar]
- Bennati M.; Robblee J. H.; Mugnaini V.; Stubbe J.; Freed J. H.; Borbat P. J. Am. Chem. Soc. 2005, 127, 15014–5 10.1021/ja054991y. [DOI] [PubMed] [Google Scholar]
- Ator M. A.; Stubbe J. Biochemistry 1985, 24, 7214–21 10.1021/bi00346a029. [DOI] [PubMed] [Google Scholar]
- Ando N.; Brignole E. J.; Zimanyi C. M.; Funk M. A.; Yokoyama K.; Asturias F. J.; Stubbe J.; Drennan C. L. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 21046–51 10.1073/pnas.1112715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshansky L.; Pizano A. A.; Wei Y.; Stubbe J.; Nocera D. G. J. Am. Chem. Soc. 2014, 136, 16210–6 10.1021/ja507313w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence C. C.; Bennati M.; Obias H. V.; Bar G.; Griffin R. G.; Stubbe J. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 8979–84 10.1073/pnas.96.16.8979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao S. S.; Holler T. P.; Yu G. X.; Bollinger J. M. Jr.; Booker S.; Johnston M. I.; Stubbe J. Biochemistry 1992, 31, 9733–43 10.1021/bi00155a029. [DOI] [PubMed] [Google Scholar]
- Persson A. L.; Eriksson M.; Katterle B.; Pötsch S.; Sahlin M.; Sjöberg B. M. J. Biol. Chem. 1997, 272, 31533–41 10.1074/jbc.272.50.31533. [DOI] [PubMed] [Google Scholar]
- Kasrayan A.; Persson A. L.; Sahlin M.; Sjöberg B. M. J. Biol. Chem. 2002, 277, 5749–55 10.1074/jbc.M106538200. [DOI] [PubMed] [Google Scholar]
- van der Donk W. A.; Stubbe J.; Gerfen G. J.; Bellew B. F.; Griffin R. G. J. Am. Chem. Soc. 1995, 117, 8908–16 10.1021/ja00140a003. [DOI] [Google Scholar]
- van der Donk W. A.; Yu G. X.; Pérez L.; Sanchez R. J.; Stubbe J.; Samano V.; Robins M. J. Biochemistry 1998, 37, 6419–26 10.1021/bi9729357. [DOI] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Reece S. Y.; Nocera D. G.; Stubbe J. J. Am. Chem. Soc. 2006, 128, 1569–79 10.1021/ja055926r. [DOI] [PubMed] [Google Scholar]
- Olshansky L. Ph.D. Dissertation, Massachusetts Institute of Technology, 2015. [Google Scholar]
- Barry B. A.; El-Deeb M. K.; Sandusky P. O.; Babcock G. T. J. Biol. Chem. 1990, 265, 20139–43. [PubMed] [Google Scholar]
- Gupta A.; Mukherjee A.; Matsui K.; Roth J. P. J. Am. Chem. Soc. 2008, 130, 11274–5 10.1021/ja8042273. [DOI] [PubMed] [Google Scholar]
- Zhao X.; Suarez J.; Khajo A.; Yu S.; Metlitsky L.; Magliozzo R. S. J. Am. Chem. Soc. 2010, 132, 8268–9 10.1021/ja103311e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai A. L.; Kulmacz R. J. Arch. Biochem. Biophys. 2010, 493, 103–24 10.1016/j.abb.2009.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.