

Abstract
Background
N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) is a natural inhibitor of pluripotent hematopoietic stem cell proliferation. Ac-SDKP plasma concentration is increased 5-fold after angiotensin-converting enzyme inhibition. Here we studied the effect of Ac-SDKP on monocyte/macrophage infiltration, fibroblast proliferation, and collagen deposition in the rat heart in renovascular hypertension.
Methods and Results
We investigated whether long-term Ac-SDKP administration would prevent left ventricular (LV) hypertrophy and interstitial collagen deposition in rats with 2-kidney, 1-clip (2K-1C) hypertension. Ac-SDKP (400 μg · kg−1 · d−1) did not affect development of hypertension. Mean blood pressure was similar in rats with 2K-1C hypertension whether they were given vehicle or Ac-SDKP and was higher than in controls. Both LV weight and cardiomyocyte size were significantly increased in rats with 2K-1C hypertension compared with controls and were unaffected by Ac-SDKP. Proliferating cell nuclear antigen- and monocyte/macrophage-positive cells were increased in the LV of 2K-1C hypertensive rats; this increase was significantly blunted by Ac-SDKP (P<0.001). LV interstitial collagen fraction was also increased in 2K-1C hypertensive rats given vehicle (10.1±0.8%) compared with sham (5.3±0.1%, P<0.0001), and this increase was prevented by Ac-SDKP (5.4±0.4%, P<0.001).
Conclusions
Ac-SDKP inhibited monocyte/macrophage infiltration, cell proliferation, and collagen deposition in the LV of hypertensive rats without affecting blood pressure or cardiac hypertrophy, suggesting that it may be partly responsible for the cardioprotective effect of angiotensin-converting enzyme inhibitors.
Keywords: angiotensin; hypertension, renal; blood pressure; inhibitors; collagen; myocardium
N-acetyl-Ser-Asp-Lys-Pro (Ac-SDKP) is endogenously released from its protein precursor thymosin-β4 in bone marrow cells.1,2 Ac-SDKP naturally inhibits entry of pluripotent hematopoietic stem cells into the S phase, instead maintaining them in the G0/G1 phase and thus blocking DNA synthesis.3,4 This tetrapeptide is normally present in human plasma and circulating mononuclear cells5 and is widely distributed in vivo.6 Angiotensin-converting enzyme (ACE) has 2 homologous NH2- and COO-terminal catalytic domains. Ac-SDKP is found in tissues where ACE is present and is hydrolyzed almost exclusively by ACE,7 showing a 50-fold higher affinity for the N-domain than for the C-domain of recombinant mutant ACE.8 This peptide has a 4.5-minute half-life in the circulation and thus is probably released continuously.9 Long- or short-term administration of the ACE inhibitor (ACEI) captopril prevented degradation of endogenous Ac-SDKP, raised its circulating concentrations 5-fold,7,9 and inhibited in vitro plasma hydrolysis of [3H]Ac-SDKP by 90% to 99%.7
The antiproliferative effects of Ac-SDKP are not limited to the hematopoietic system. Indeed, after hepatectomy in rats, Ac-SDKP reduced hepatocyte proliferation by ≤50% as assessed by [3H]thymidine incorporation10 and thus may also help stabilize cell growth. In hypertension, cardiac fibroblasts are important target cells for overload pressure (mechanical forces) and hormones with trophic effects such as angiotensin II (Ang II), endothelin-1, and catecholamines.11,12 In response to these stimuli, cardiac fibroblasts proliferate and increase fibrillar collagen in the myocardium, including type I and III collagen.13 Moreover, inflammatory cells such as macrophages are significantly increased and co-localized with fibroblasts in the renovascular hypertensive rat heart.14 Inflammatory cells could also promote fibrosis by releasing mediators such as cytokines that act on fibroblasts.14 Disproportionate ventricular accumulation of fibrillar collagen has been observed in humans15 and animals with arterial hypertension,11,12 leading to myocardial fibrosis. Long-term ACE inhibition blunted cardiac hypertrophy and abnormally increased interstitial collagen deposition within the myocardium in hypertensive patients,16 spontaneously hypertensive rats,17 and Lewis rats with heart failure induced by myocardial infarction.18 Decreased Ang II generation associated with increased plasma and/or tissue levels of kinins could mediate the protective effect of ACEIs.18 However, Ac-SDKP might be another important mechanism for the therapeutic effect of ACEIs in hypertension and cardiac disease, because plasma Ac-SDKP levels are greatly increased after short- and long-term ACE inhibition7–9 and Ac-SDKP inhibits adult rat cardiac fibroblast proliferation and collagen synthesis in vitro.19 Therefore, the present study was designed to test the hypothesis that long-term Ac-SDKP administration prevents cardiac monocyte/macrophage infiltration, fibroblast proliferation, and collagen deposition in the left ventricle (LV) of rats with 2-kidney, 1-clip (2K-1C) hypertension.
Methods
2K-1C Hypertension
Male Sprague-Dawley rats (150 to 175 g; Charles River, Wilmington, DE) were anesthetized with pentobarbital sodium (50 mg/kg IP). The left renal artery was exposed by retroperitoneal flank incision and dissected free of the renal vein and connective tissue. A silver clip with a lumen of 0.22 mm was placed around the artery for partial occlusion; in sham operations, the artery was not clipped. After surgery, an osmotic minipump (Alzet 2 ML4, Alza Corp) was implanted subcutaneously in the neck to deliver either Ac-SDKP (Bachem) or saline plus 0.01N acetic acid,20 basing the route of administration and dosage on previous studies.21,22 Rats were subdivided into 4 groups: (1) sham, (2) 2K-1C plus vehicle (saline plus 0.01N acetic acid), (3) 2K-1C plus low-dose Ac-SDKP (400 μg · kg−1 · d−1), and (4) 2K-1C plus high-dose Ac-SDKP (800 μg · kg−1· d−1). This study was approved by the Henry Ford Hospital Care of Experimental Animals Committee.
Blood Pressure and Heart Weight
Systolic blood pressure was measured by the tail-cuff method twice a week for 8 weeks. At the end of the experiment, rats were anesthetized with sodium hexobarbital (50 mg/kg IP), and a catheter (PE-10 fused to PE-50, Clay Adams) filled with saline plus heparin (100 IU) was inserted into the abdominal aorta through the femoral artery; the distal end was tunneled under the skin and exteriorized on the neck. Twenty-four hours later, each conscious rat was placed in a restrainer, and mean blood pressure was recorded on a 4-channel recorder (Brush 220, Gould) once a minute for 60 minutes using a pressure transducer and processor (model P10EZ, Ohmeda). Animals were again anesthetized with 50 mg/kg pentobarbital sodium; the heart was stopped at diastole by intraventricular injection of 15% KCl, excised, cleaned of blood, and then blotted to dryness. The LV plus septum, right ventricle, and left and right atria were weighed, and the LV was sectioned transversely into 3 slices. The slices were rapidly frozen in isopentane precooled in liquid N2 and stored at −70°C. One LV sample was stored at −20°C until the hydroxyproline assay was performed. The LV plus septum, right ventricle, and left and right atria weights were normalized to 100 g of body weight.
Histological and Histochemical Analysis of Myocyte Cross-Sectional Area and Interstitial Collagen Fraction in the LV
Sections 10 μm thick from each frozen slice were stained separately with (1) fluorescein-labeled peanut agglutinin (Vector Laboratories) after pretreatment with 3.3 U/mL neuroaminidase type V (Sigma) to delineate myocyte cross-sectional area (an indicator of myocyte volume) and the interstitial space (consisting of collagen and capillaries) and (2) rhodamine-labeled Griffonia simplicifolia lectin I to show only the capillaries, because G simplicifolia lectin I selectively binds to capillaries.23,24 Three radially oriented microscopic fields were selected at random from each section and photographed. We used a fluorescence microscope (Nikon) equipped with a Spot digital camera (×100, Diagnostic Instruments).
Cross-Sectional Area of LV Myocytes
Stained sections were photographed on 35-mm film. Each field contained ≥100 myocytes. Images were projected with a photomagnifier, and myocyte cross-sectional area (radial fields) and length (longitudinal fields) were determined by computer-based planimetry (SigmaScan). An average cross-sectional area was calculated using data from all 3 slices.18
LV Interstitial Collagen Fraction
Total surface area (microscopic field), interstitial space (collagen plus capillaries), and area occupied by capillaries alone were measured by computer-assisted videodensitometry (JAVA, SPSS Inc). The interstitial collagen fraction was calculated as percent surface area occupied by the interstitial space minus the percent surface area occupied by capillaries. The interstitial collagen fraction was averaged using data from all 3 slices.18
LV Cell Proliferation (PCNA) and Inflammatory Cell Infiltration (ED1)
LV samples were isolated 2 weeks after the 2K-1C procedure,14,25 fixed in 2.5% paraformaldehyde, and mounted in a paraffin block. Four-micron-thick sections were deparaffinized, rehydrated, boiled in 0.2% citric acid (pH 6.0) for 10 minutes for antigen retrieval, washed 3 times in phosphate-buffered saline for 5 minutes each, preincubated with blocking serum (1% normal serum) for 30 minutes, and finally incubated with a mouse monoclonal antibody against either proliferating cell nuclear antigen (PCNA, 1:250 dilution) or rat monocytes and macrophages (ED1, 1:1000 dilution; Chemicon) at room temperature for 30 minutes. Each section was washed 3 times in phosphate-buffered saline, and PCNA or ED1 was assayed (Vectastain ABC kit, Vector Laboratories). Sections were developed with diaminobenzidine substrate (Vector) and counter-stained with hematoxylin. We used a Nikon microscope equipped with a charge-coupled device video camera (Optronics). The microscopic image was imported to a computer fitted with a Bioquant NOVA image analysis system (R&M Biometrics). Stained cells were counted at ×40, and because every selected image occupied the entire window, we measured window size with the same objective. The window area was fixed at 22 194 μm2 (0.022194 mm2). Cell density was calculated as the number of cells per window area (ie, number of PCNA- or ED1-positive cells per square millimeter). For each sample, 24 randomly selected fields were examined.
Hydroxyproline Assay
Myocardial collagen was measured by hydroxyproline assay.26 LV samples from 2K-1C rats (180 to 200 mg) were freeze-dried, weighed, and pulverized. Each sample was homogenized in 0.1 mol/L NaCl and 5 mmol/L NaHCO3, washed with the same solution 5 times, and hydrolyzed in 1 mL of 6N HCl for 16 hours at 110°C. Samples were filtered, vacuum-dried, and then dissolved in distilled water. Hydroxyproline was measured using a colorimetric assay and a standard curve of 0 to 5 μg hydroxyproline. Data were expressed as micrograms of collagen per milligram dry weight, assuming that collagen contains an average of 13.5% hydroxyproline.27
Ac-SDKP Plasma Levels
After mean blood pressure was measured, arterial blood from 4 rats per group was collected in a heparinized tube containing captopril (final concentration, 10 μmol/L) and centrifuged at 2000g for 15 minutes at 4°C. Recovered plasma was stored at −70°C until the Ac-SDKP assay was performed. Plasma Ac-SDKP was quantified with a competitive enzyme immunoassay5 and expressed as nanomoles per liter.
Statistical Analysis
The mean response in the 2K-1C/vehicle group was compared separately with each Ac-SDKP group (400 or 800 μg · kg−1 · d−1) and controls. Significance was evaluated with Student’s t test and Holm’s procedure, which adjusts individual rejection criteria to ensure a family-wise P value of 0.05. Procedural assumptions were verified, and data are expressed as mean±SEM.
Results
Effect of Long-Term Ac-SDKP Infusion on Blood Pressure and Heart Rate
Ac-SDKP at 400 or 800 μg · kg−1 · d−1 for 8 weeks had no effect on mean blood pressure or the development of hypertension in 2K-1C rats (Figure 1). At the end of the experiment, mean blood pressure was significantly higher in rats with 2K-1C/vehicle (161±5 mm Hg, n=10) than in sham (118±2 mm Hg, n=14; P<0.001). However, heart rate was unchanged (Figure 2).
Figure 1.
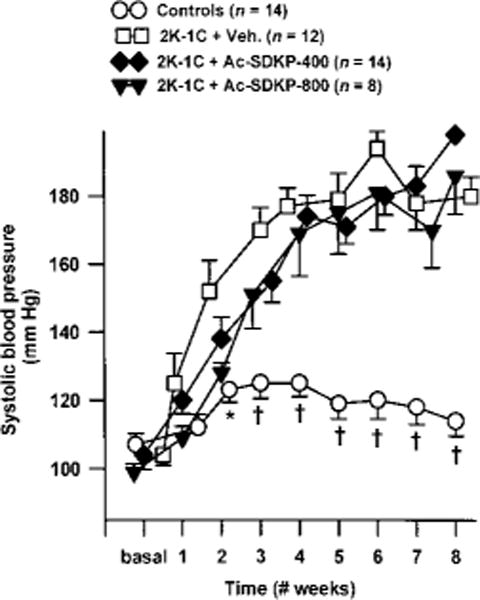
Changes in systolic blood pressure in sham group and rats with 2K-1C hypertension given vehicle (Veh.) or Ac-SDKP for 8 weeks. Ac-SDKP was started a few minutes after the left renal artery was clipped but did not affect development of 2K-1C hypertension. Each point represents mean±SEM, n=5 to 14 for each group. *P<0.05 and †P<0.001 vs 2K-1C/vehicle.
Figure 2.
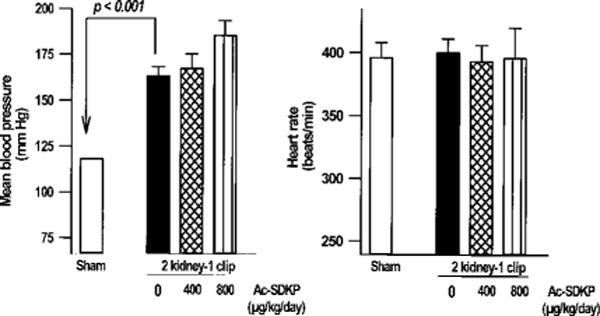
Mean blood pressure (left) and heart rate (right) in restrained conscious controls and rats with 2K-1C hypertension given vehicle or Ac-SDKP for 8 weeks. Ac-SDKP had no effect on blood pressure or heart rate. Each bar represents mean±SEM, n=5 to 14 for each group.
Effect of Long-Term Ac-SDKP Infusion on LV Hypertrophy and Cardiomyocyte Cross-Sectional Area
LV hypertrophy was significantly greater in 2K-1C rats than in sham (P<0.05). Ac-SDKP did not reduce LV hypertrophy in hypertensive rats (Figure 3). Myocyte cross-sectional area was also significantly increased in 2K-1C/vehicle rats versus sham (P<0.05) and was similar in Ac-SDKP–treated hypertensive rats.
Figure 3.
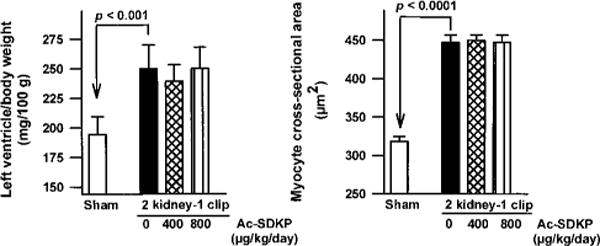
Ratio of LV weight to body weight (left) and myocyte cross-sectional area (right) in controls and rats with 2K-1C hypertension given vehicle or Ac-SDKP for 8 weeks. Hypertension significantly increased LV hypertrophy and myocyte cross-sectional area, which were not affected by long-term Ac-SDKP treatment. Each bar represents mean±SEM, n=5 to 14 for each group.
Effect of Long-Term Ac-SDKP Infusion on Cell Proliferation (PCNA) and Monocyte/Macrophage (ED1) Infiltration in the LV Interstitium
PCNA-positive cells were found only in the LV interstitial space in controls but were distributed diffusely throughout both interstitial and perivascular spaces in the 2K-1C/vehicle rats (Figure 4). PCNA-positive cells were significantly increased in the 2K-1C/vehicle group (P<0.0001). Ac-SDKP significantly reduced the number of PCNA-positive cells in the LV interstitial and perivascular spaces, resembling control levels (P<0.01). There were also significantly more ED1-positive cells in the LV in the 2K-1C/vehicle group than in the controls (Figure 4). Ac-SDKP kept monocytes/macrophages (ED1) at normal levels in 2K-1C rats.
Figure 4.
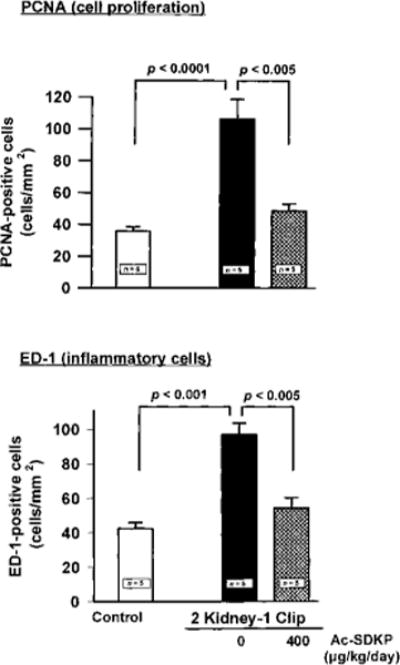
Effect of Ac-SDKP on number of PCNA- and ED1-positive cells in the LV interstitial space in 2K-1C hypertensive rats given vehicle or Ac-SDKP for 8 weeks. PCNA- and ED1-positive cells were significantly increased in the 2K-1C/vehicle group, but were lowered by Ac-SDKP. Each bar represents mean±SEM, n=5 for each group.
Effect of Long-Term Ac-SDKP Infusion on LV Interstitial Collagen Deposition
LV collagen estimated by hydroxyproline content was significantly increased in untreated 2K-1C rats versus controls (Figure 5). Hypertensive rats treated with Ac-SDKP (400 μg · kg−1 · d−1) had less LV collagen than did untreated rats (7.56±0.44 vs 11.85±1.05 μg/mg dry LV, P<0.005). Higher doses of Ac-SDKP did not appear to reduce LV collagen more than lower doses (8.82±1.3 μg/mg dry LV). The interstitial collagen fraction was significantly increased in untreated 2K-1C rats versus sham (10.1±0.8% vs 5.3±0.1%, P<0.0001) but normalized in 2K-1C rats given 400 μg · kg−1 · d−1 Ac-SDKP (5.4±0.4%) and was somewhat less reduced at higher doses (6.78±0.81%, Figure 5), confirming the hydroxyproline data. Myocyte cross-sectional area and interstitial collagen deposition are shown in Figure 6.
Figure 5.
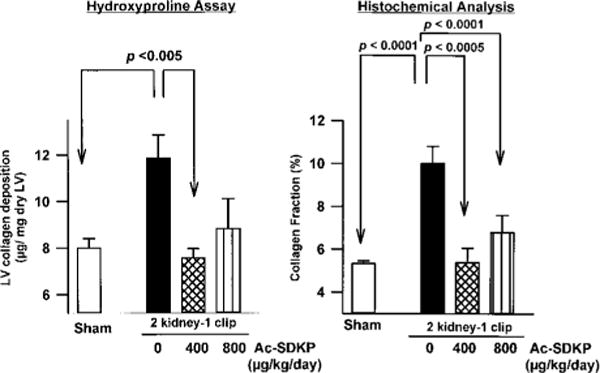
Interstitial collagen fraction (left) and LV collagen deposition measured by hydroxyproline assay (right) in controls and rats with 2K-1C hypertension given vehicle or Ac-SDKP for 8 weeks. Ac-SDKP significantly blunted collagen deposition in hypertensive rats. Each bar represents mean±SEM, n=5 to 14 for each group.
Figure 6.
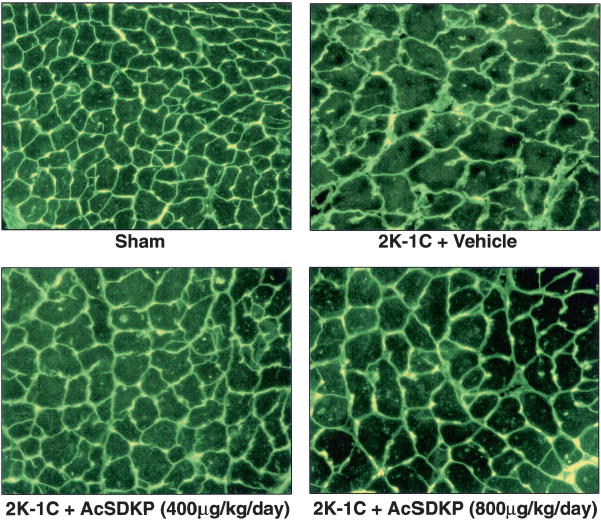
Histological sections illustrating myocyte cross-sectional area, interstitial collagen deposition (green), and capillaries (yellow; because images were exposed under fluorescent light, red color given by rhodamine is not apparent) from sham group and 2K-1C rats treated with vehicle, Ac-SDKP at 400 μg · kg−1 · d−1, or Ac-SDKP at 800 μg · kg−1 · d−1.
Ac-SDKP Plasma Concentration
Ac-SDKP plasma concentration was the same in 2K-1C/vehicle and sham groups (Figure 7) but nearly doubled in 2K-1C rats treated with Ac-SDKP versus vehicle, although this difference was significant only at the 800 μg · kg−1 · d−1 dose (P<0.02).
Figure 7.
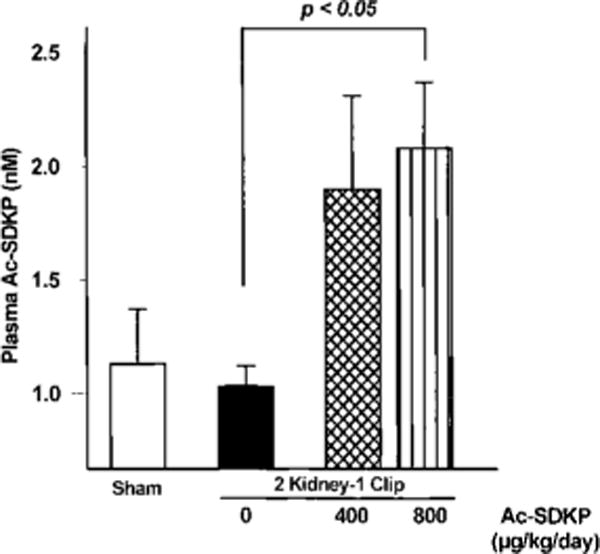
Plasma Ac-SDKP in sham group (open bar) and 2K-1C rats given vehicle (0), low-dose Ac-SDKP (400 μg · kg−1 · d−1), or high-dose Ac-SDKP (800 μg · kg−1 · −1). Ac-SDKP was increased in treated rats with 2K-1C hypertension compared with vehicle. Each bar represents mean±SEM, n=4 for each group.
Discussion
We found that Ac-SDKP inhibits collagen deposition, cell proliferation (cardiac fibroblasts), and infiltration by inflammatory cells (monocytes and macrophages) within the LV interstitial space of rats with 2K-1C hypertension without affecting blood pressure, cardiac hypertrophy, or myocyte cross-sectional area (an indicator of myocyte volume). At the end of the experiment, the plasma Ac-SDKP level was doubled in rats given Ac-SDKP compared with untreated hypertensive 2K-1C rats, values similar to those found in patients on long-term enalapril (an ACEI) for 25 days (3.97±0.75 nmol/L),28 normal rats given captopril for 28 days (3.51±0.87 nmol/L),29 or irradiated mice given lisinopril (another ACEI) for 72 hours.30 Higher doses (800 μg · kg−1 · d−1) were slightly less effective than lower doses, exhibiting almost the same pharmacological profile as our in vitro studies19 as well as others.31,32
The antifibrotic effect of Ac-SDKP in vivo could result from inhibition of fibroblast proliferation and collagen synthesis, as shown in our in vitro study.19 The present results may indirectly indicate that the beneficial effect of ACEIs in hypertension could be partially mediated by Ac-SDKP. In fact, long-term ACE inhibition has been associated with (1) a significant reduction in cardiac collagen deposition in spontaneously hypertensive rats,17 in rats with renovascular33 or aldosterone-salt–induced hypertension,34 or in hypertensive patients35 and (2) a significant increase in plasma Ac-SDKP in patients, normal rats, or irradiated mice.9,28,30 Long-term ACE inhibition in hypertension is associated with decreased collagen deposition, blood pressure, total peripheral resistance, and cardiac hypertrophy and improved cardiovascular function. However, these ACEI effects do not necessarily indicate whether reduced LV collagen deposition is due to hemodynamic changes, inhibition of conversion of Ang I to Ang II, increased tissue and/or circulating kinins, and/or increased Ac-SDKP plasma levels per se. Nicoletti et al14 likewise reported that myocardial macrophages (ED1-positive cells) were significantly increased in rats with 2K-1C hypertension and co-localized with collagen-synthesizing fibroblasts. Inflammatory cells could promote fibrosis by releasing growth factors or cytokines, which act on fibroblasts. Therefore, Ac-SDKP may prevent LV collagen deposition in hypertensive rats through inhibition of fibroblast proliferation and collagen synthesis and/or increased metalloproteinase activity, either directly or indirectly by inhibiting myocardial inflammatory cells. Both antihypertensive and non-antihypertensive doses of ACEIs abolished myocardial collagen deposition in hypertension, and the fact that this antifibrotic effect was unaffected by icatibant, a potent B2 kinin receptor antagonist,36 suggests that kinins do not mediate the antifibrotic effect of ACEIs in hypertensive rats and that Ac-SDKP may be a good candidate for this therapeutic effect. This hypothesis is based on our results as well as those of Junot et al,37 who found that at 0.1 to 0.3 mg/kg, captopril inhibited Ac-SDKP hydrolysis without affecting the renin-angiotensin system or blood pressure. Such an association between the therapeutic effect of ACEIs and Ac-SDKP is not unique to the cardiovascular system, perhaps occurring also after radiotherapy and/or chemotherapy. Indeed, ACEI prevented entry of murine hematopoietic stem cells into the cell cycle after irradiation in vivo, and this inhibitory effect correlated positively with a 6-fold increase in plasma Ac-SDKP.30,38 Ac-SDKP also blunts the cytotoxic effect of 3′-azido-3′-deoxythymidine (AZT), commonly used to treat AIDS patients.39 Moreover, long-term ACEI treatment inhibits renal fibroblast proliferation and fibrosis and improves function in animal models of renal disease40; thus, one has to wonder whether this renal protective effect of ACEIs could be mediated by Ac-SDKP.
An abnormal increase in cardiac fibroblasts and extracellular matrix proteins during cardiac remodeling may be a major cause of increased myocardial stiffness and cardiac dysfunction.12,17,18 Collagen deposition in the myocardium of hypertensive rats begins within the adventitia of intramyocardial coronary arteries (perivascular fibrosis). As hypertension progresses, perivascular collagen radiates outward into neighboring intramuscular spaces, leading to established perivascular and interstitial fibrosis. ACEIs impair fibroblast proliferation, reverse excessive interstitial and perivascular fibrosis, and restore function.17 Because ACE activity has been found in various cells of the heart, including fibroblasts, and because Ac-SDKP is also found in the heart,6 a tissue ACE/Ac-SDKP system, acting as a paracrine hormone, might be involved in local regulation of myocardial Ac-SDKP levels, and thus regulation of fibroblast activity. The attractive feature of the antifibrotic effect of Ac-SDKP is that it occurs in the absence of decreased blood pressure or LV hypertrophy. The pharmacological effect of Ac-SDKP may be considered a novel mechanism whereby part of the cardioprotective effect of ACEIs in hypertension is achieved.
Acknowledgments
This study was supported by American Heart Association Grant-in-Aid 29GS978, American Heart Association Scientist Development Award 0130128N, and grant HL 2898217 from the National Institutes of Health, Bethesda, Md.
References
- 1.Grillon C, Reiger K, Bakala J, et al. Involvement of thymosin β4 and endoproteinase asp-N in the biosynthesis of the tetrapeptide Ac-Ser-Asp-Lys-Pro, a regulator of the haematopoietic system. FEBS Lett. 1990;274:30–34. doi: 10.1016/0014-5793(90)81322-f. [DOI] [PubMed] [Google Scholar]
- 2.Wdzieczak-Bakala J, Fache MP, Lenfant M, et al. AcSDKP, an inhibitor of CFU-S proliferation is synthesized in mice under steady-state conditions and secreted by bone marrow in long term culture. Leukemia. 1990;4:235–237. [PubMed] [Google Scholar]
- 3.Frindel E, Monpezat JP. The physiological role of the endogenous colony forming units-spleen (CFU-S) inhibitor acetyl-N-Ser-Asp-Lys-Pro (AcSDKP) Leukemia. 1989;3:753–754. [PubMed] [Google Scholar]
- 4.Monpezat JP, Frindel E. Further studies on the biological activities of the CFU-S inhibitory tetrapeptide AcSDKP, I: the precise point of the cell cycle sensitive to AcSDKP: studies on the effect of AcSDKP on GM-CFC and on the possible involvement of T-lymphocytes in AcSDKP response. Exp Hematol. 1989;17:1077–1080. [PubMed] [Google Scholar]
- 5.Pradelles P, Frobert Y, Créminon C, et al. Negative regulator of pluripotent hematopoietic stem cell proliferation in human white blood cells and plasma as analysed by enzyme immunoassay. Biochem Biophys Res Commun. 1990;170:986–993. doi: 10.1016/0006-291x(90)90489-a. [DOI] [PubMed] [Google Scholar]
- 6.Pradelles P, Frobert Y, Créminon C, et al. Distribution of a negative regulator of haematopoietic stem cell proliferation (AcSDKP) and thymosin β4 in mouse tissues. FEBS Lett. 1991;289:171–175. doi: 10.1016/0014-5793(91)81062-d. [DOI] [PubMed] [Google Scholar]
- 7.Azizi M, Rousseau A, Ezan E, et al. Acute angiotensin-converting enzyme inhibition increases the plasma level of the natural stem cell regulator N-acetyl-seryl- aspartyl-lysyl-proline. J Clin Invest. 1996;97:839–844. doi: 10.1172/JCI118484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rousseau A, Michaud A, Chauvet M-T, et al. The hemoregulatory peptide N-acetyl-Ser-Asp-Lys-Pro is a natural and specific substrate of the N-terminal active site of human angiotensin-converting enzyme. J Biol Chem. 1995;270:3656–3661. doi: 10.1074/jbc.270.8.3656. [DOI] [PubMed] [Google Scholar]
- 9.Azizi M, Ezan E, Nicolet L, et al. High plasma level of N-acetyl-seryl-aspartyl-lysyl-proline: a new marker of chronic angiotensin-converting enzyme inhibition. Hypertension. 1997;30:1015–1019. doi: 10.1161/01.hyp.30.5.1015. [DOI] [PubMed] [Google Scholar]
- 10.Lombard M-N, Sotty D, Wdzieczak-Bakala J, et al. In vivo effect of the tetrapeptide, N-acetyl-Ser-Asp-Lys-Pro, on the G1-S transition of rat hepatocytes. Cell Tissue Kinet. 1990;23:99–103. doi: 10.1111/j.1365-2184.1990.tb01336.x. [DOI] [PubMed] [Google Scholar]
- 11.Weber KT, Janicki JS, Pick R, et al. Myocardial fibrosis and pathologic hypertrophy in the rat with renovascular hypertension. Am J Cardiol. 1990;65:1G–7G. doi: 10.1016/0002-9149(90)90952-w. [DOI] [PubMed] [Google Scholar]
- 12.Jalil JE, Doering CW, Janicki JS, et al. Fibrillar collagen and myocardial stiffness in the intact hypertrophied rat left ventricle. Circ Res. 1989;64:1041–1050. doi: 10.1161/01.res.64.6.1041. [DOI] [PubMed] [Google Scholar]
- 13.Weber KT, Sun Y, Tyagi SC, et al. Collagen network of the myocardium: function, structural remodeling and regulatory mechanisms. J Mol Cell Cardiol. 1994;26:279–292. doi: 10.1006/jmcc.1994.1036. [DOI] [PubMed] [Google Scholar]
- 14.Nicoletti A, Heudes D, Mandet C, et al. Inflammatory cells and myocardial fibrosis: spatial and temporal distribution in renovascular hypertensive rats. Cardiovasc Res. 1996;32:1096–1107. doi: 10.1016/s0008-6363(96)00158-7. [DOI] [PubMed] [Google Scholar]
- 15.Pardo Mindan FJ, Panizo A. Alterations in the extracellular matrix of the myocardium in essential hypertension. Eur Heart J. 1993;14(suppl J):12–14. [PubMed] [Google Scholar]
- 16.Díez J, Laviades C, Monreal I, et al. Toward the biochemical assessment of myocardial fibrosis in hypertensive patients. Am J Cardiol. 1995;76:14D–17D. doi: 10.1016/s0002-9149(99)80486-x. [DOI] [PubMed] [Google Scholar]
- 17.Brooks WW, Bing OH, Robinson KG, et al. Effect of angiotensin-converting enzyme inhibition on myocardial fibrosis and function in hypertrophied and failing myocardium from the spontaneously hypertensive rat. Circulation. 1997;96:4002–4010. doi: 10.1161/01.cir.96.11.4002. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y-H, Yang X-P, Sharov VG, et al. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure: role of kinins and angiotensin II type 2 receptors. J Clin Invest. 1997;99:1926–1935. doi: 10.1172/JCI119360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rhaleb N-E, Peng H, Harding P, et al. Effect of N-acetyl-seryl-aspartyl-lysyl-proline on DNA and collagen synthesis in rat cardiac fibroblasts. Hypertension. 2001;37:827–832. doi: 10.1161/01.hyp.37.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li XM, Soulard C, Filipski E, et al. Circadian-based effects of AcSDKP, with or without rhG-CSF on hematologic toxicity of chemotherapy in mice. Eur J Haematol. 1998;60:181–188. doi: 10.1111/j.1600-0609.1998.tb01020.x. [DOI] [PubMed] [Google Scholar]
- 21.Cappelaere P, Hecquet B, Rolland F, et al. Randomized placebo trial of myeloprotection with goralatide in patients with squamous cell carcinoma of the upper respiratory and digestive tracts or esophagus, treated with a carboplatin-fluorouracil combination. Bull Cancer. 1995;82:732–737. [PubMed] [Google Scholar]
- 22.Watanabe T, Kelsey LS, Yan Y, et al. In vivo haemoprotective activity of tetrapeptide AcSDKP combined with granulocyte-colony stimulating factor following sublethal irradiation. Br J Haematol. 1996;94:619–627. doi: 10.1046/j.1365-2141.1996.d01-1853.x. [DOI] [PubMed] [Google Scholar]
- 23.Hansen-Smith FM, Watson L, Lu DY, et al. Griffonia simplicifolia I: fluorescent tracer for microcirculatory vessels in nonperfused thin muscles and sectioned muscle. Microvasc Res. 1988;36:199–215. doi: 10.1016/0026-2862(88)90022-2. [DOI] [PubMed] [Google Scholar]
- 24.Laitinen L. Griffonia simplicifolia lectins bind specifically to endothelial cells and some epithelial cells in mouse tissues. Histochem J. 1987;19:225–234. doi: 10.1007/BF01680633. [DOI] [PubMed] [Google Scholar]
- 25.Campbell SE, Janicki JS, Weber KT. Temporal differences in fibroblast proliferation and phenotype expression in response to chronic adminis tration of angiotensin II or aldosterone. J Mol Cell Cardiol. 1995;27:1545–1560. doi: 10.1016/s0022-2828(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 26.Cleutjens JP, Verluyten MJ, Smiths JF, et al. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–338. [PMC free article] [PubMed] [Google Scholar]
- 27.Chiariello M, Ambrosio G, Cappelli-Bigazzi M, et al. A biochemical method for the quantitation of myocardial scarring after experimental coronary artery occlusion. J Mol Cell Cardiol. 1986;18:283–290. doi: 10.1016/s0022-2828(86)80410-2. [DOI] [PubMed] [Google Scholar]
- 28.Comte L, Lorgeot V, Volkov L, et al. Effects of the angiotensin-converting enzyme inhibitor enalapril on blood haematopoietic progenitors and acetyl-N-Ser-Asp-Lys-Pro concentrations. Eur J Clin Invest. 1997;27:788–790. doi: 10.1046/j.1365-2362.1997.1980737.x. [DOI] [PubMed] [Google Scholar]
- 29.Junot C, Nicolet L, Ezan E, et al. Effects of angiotensin-converting enzyme inhibition on plasma, urine, and tissue concentrations of hemoregulatory peptide Acetyl-Ser-Asp-Lys-Pro in rats. J Cardiovasc Pharmacol. 1999;291:982–987. [PubMed] [Google Scholar]
- 30.Rousseau-Plasse A, Wdzieczak-Bakala J, Lenfant M, et al. Lisinopril, an angiotensin I-converting enzyme inhibitor, prevents entry of murine hematopoietic stem cells into the cell cycle after irradiation in vivo. Exp Hematol. 1998;26:1074–1079. [PubMed] [Google Scholar]
- 31.Bonnet D, Lemoine FM, Pontvert-Delucq S, et al. Direct and reversible inhibitory effect of the tetrapeptide acetyl-N-Ser-Asp-Lys-Pro (Seraspenide) on the growth of human CD34+ subpopulations in response to growth factors. Blood. 1993;82:3307–3314. [PubMed] [Google Scholar]
- 32.Volkov L, Quere P, Coudert F, et al. The tetrapeptide AcSDKP, a physiological inhibitor of normal cell proliferation, reduces the S phase entry of continuous cell lines. Exp Cell Res. 1996;223:112–116. doi: 10.1006/excr.1996.0063. [DOI] [PubMed] [Google Scholar]
- 33.Jalil JE, Janicki JS, Pick R, et al. Coronary vascular remodeling and myocardial fibrosis in the rat with renovascular hypertension: response to captopril. Am J Hypertens. 1991;4:51–55. doi: 10.1093/ajh/4.1.51. [DOI] [PubMed] [Google Scholar]
- 34.Sun Y, Ratajska A, Zhou G, et al. Angiotensin-converting enzyme and myocardial fibrosis in the rat receiving angiotensin II or aldosterone. J Lab Clin Med. 1993;122:395–403. [PubMed] [Google Scholar]
- 35.Laviades C, Varo N, Fernández J, et al. Abnormalities of the extracellular degradation of collagen type I in essential hypertension. Circulation. 1998;98:535–540. doi: 10.1161/01.cir.98.6.535. [DOI] [PubMed] [Google Scholar]
- 36.Linz W, Wiemer G, Schaper J, et al. Angiotensin converting enzyme inhibitors, left ventricular hypertrophy and fibrosis. Mol Cell Biochem. 1995;147:89–97. doi: 10.1007/BF00944788. [DOI] [PubMed] [Google Scholar]
- 37.Junot C, Menard J, Gonzales M-F, et al. In vivo assessment of captopril selectivity of angiotensin I-converting enzyme inhibition: differential inhibition of acetyl-Ser-Asp-Lys-Pro and angiotensin I hydrolysis. J Pharmacol Exp Ther. 1999;289:1257–1261. [PubMed] [Google Scholar]
- 38.Suzuki A, Aizawa S, Araki S, et al. Enhanced engraftment of intravenously transplanted hematopoietic stem cells into bone marrow of irradiated mice treated with AcSDKP. Exp Hematol. 1998;26:79–83. [PubMed] [Google Scholar]
- 39.Grillon C, Bonnet D, Mary J-Y, et al. The tetrapeptide AcSerAspLysPro (seraspenide), a hematopoietic inhibitor, may reduce the in vitro toxicity of 3′-azido-3′-deoxythymidine to human hematopoietic progenitors. Stem Cells. 1993;11:455–464. doi: 10.1002/stem.5530110513. [DOI] [PubMed] [Google Scholar]
- 40.Weir MR. The effects of enalapril on blood pressure, renal hemodynam-ics, and renal function. Clin Ther. 1989;11:685–700. [PubMed] [Google Scholar]