

Abstract
Huntington’s disease is a neurodegenerative disorder characterised primarily by motor abnormalities, and is caused by an expanded polyglutamine repeat in the huntingtin protein. Huntingtin dynamically shuttles between subcellular compartments, and the mutant huntingtin protein is mislocalised to cell nuclei, where it may interfere with nuclear functions, such as transcription. However, the mechanism by which mislocalisation of mutant huntingtin occurs is currently unknown. An immortalised embryonic striatal cell model of HD (StHdh Q111) was stimulated with epidermal growth factor in order to determine whether the subcellular localisation of huntingtin is dependent on kinase signalling pathway activation. Aberrant phosphorylation of AKT and MEK signalling pathways was identified in cells carrying mutant huntingtin. Activity within these pathways was found to contribute to the regulation of huntingtin and mutant huntingtin localisation, as well as to the expression of immediate-early genes. We propose that altered kinase signalling is a phenotype of Huntington’s disease that occurs prior to cell death; specifically, that altered kinase signalling may influence huntingtin localisation, which in turn may impact upon nuclear processes such as transcriptional regulation. Aiming to restore the balance of activity between kinase signalling networks may therefore prove to be an effective approach to delaying Huntington’s disease symptom development and progression.
Introduction
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder caused by a CAG expansion within the first exon of the HTT gene, which gives rise to an expanded polyglutamine tract in the huntingtin protein. HD is primarily characterised by progressive motor abnormalities that manifest in the third to fourth decades of life, but is also commonly associated with cognitive impairments and psychiatric disturbances [1–3]. The caudate and putamen exhibit the most prominent cell loss [4]; GABAergic medium spiny neurons (MSNs) are the first to be affected, and is later accompanied by widespread atrophy of cortical structures [5]. Neuronal dysfunction occurs prior to both striatal atrophy and overt motor symptom onset [6,7], and therefore cell death and degeneration in HD-affected neuronal cells are likely to develop following an initial period of dysregulation of numerous cellular processes [8].
Patients with HD have been reported as having a low incidence of tumour formation, as well as a lower glucose metabolism [9,10]. As the activity of kinase signalling cascades are well characterised in the development of tumorigenesis [11] and the regulation of glucose metabolism [12], these peripheral characteristics of HD could be a result of huntingtin-mediated alterations in growth factor-responsive kinase pathways. Anti-apoptotic kinase signalling pathways in particular have been found to be important for prolonging neuronal survival in HD models by inhibiting cellular changes elicited by mutant huntingtin [13].
The most thoroughly characterised survival pathway in HD is the protein kinase B (AKT) pathway; enhanced activation of AKT in models of HD has been attributed to the reduced expression of its inhibitor, PH domain leucine-rich repeat protein phosphatase 1 (PHLPP1), which is suppressed in HdhQ111, R6/1, R6/2 and HD94 striata, as well as in human putamen [14]. Aberrant interactions between mutant huntingtin and growth factor receptor bound protein 2 (GRB2) may also directly activate growth factor signalling cascades that are upstream of AKT activation, thus enhancing AKT phosphorylation [15]. Mitogen activated protein kinase kinase (MEK) has been less thoroughly investigated in the context of HD, and the neuroprotective effects of its activation remain under debate [16,17]. However, MEK1 activation has been found to enhance the phosphorylation of huntingtin [18], which can reduce mutant huntingtin toxicity [19–24].
Huntingtin has numerous potential cellular functions in both the cytoplasm and the nucleus; dynamic shuttling between these compartments is likely to be a mechanism by which a complex variety of huntingtin-associated activities can be regulated [25,26]. Within cell nuclei, huntingtin has been found to localise to nuclear structures such as promyelocytic leukaemia (PML) bodies and the nucleolus [27–29], associates with a variety of transcription factors, and is able to bind to DNA directly. These mechanisms can all be modified by the presence of an expanded polyglutamine repeat within mutant huntingtin [29–38]. These interactions implicate huntingtin as having a necessary role in transcription and RNA processing within the nucleus, as well as in the assembly of nuclear matrix bound protein complexes [29,39]. These observations support transcriptional dysregulation as an important mechanism contributing to HD pathogenesis. Subcellular mislocalisation of mutant huntingtin to cell nuclei may directly affect transcription: however, as AKT and MEK pathways are known to play a role in gene expression, either by direct regulation or through the phosphorylation of other proteins [16,40–43], disruption of these upstream kinase signalling pathways by mutant huntingtin may also be a mechanism by which transcriptional regulation is altered in HD.
By stimulating cells from the StHdh Q111 immortalised embryonic striatal cell model of HD, which carries 111 CAG repeats [44], with epidermal growth factor (EGF), we have created a dynamic model that demonstrates that both AKT and MEK signalling pathways may contribute to the regulation of huntingtin subcellular localisation, as well as to the control of gene expression. We find that both AKT and MEK pathways are aberrantly regulated in cells carrying an expanded polyglutamine repeat, and inhibition of these pathways corrected mutant huntingtin mislocalisation and gene expression to a phenotype more closely resembling that of wild type cells. We suggest that aberrant control of kinase signalling may be a mechanism influencing mutant huntingtin mislocalisation, which in turn may alter transcriptional dysregulation, thus contributing to HD pathogenesis.
Materials & Methods
Immortalised Cell culture
The StHdh Q111 immortalised embryonic striatal cell model used in this project was originally derived from primary striatal neurons cultured from the HdhQ111 knock-in mouse model of HD [44]; as such, wild type cells (StHdh Q7/7) carry two alleles each containing 7 CAG repeats, and heterozygote (StHdh Q7/111) and homozygote (StHdh Q111/111) cells carry either one or two mutant alleles carrying 111 CAG repeats, respectively. The StHdh Q7/7, StHdh Q7/111 and StHdh Q111/111 cell lines utilised here were a kind gift from Marcy MacDonald (Molecular Neurogenetics Unit, Massachusetts General Hospital, Massachusetts, USA). Cells were grown and maintained in high glucose Dulbecco’s Modified Eagle Medium (DMEM) (Life Technologies) containing 1% Penicillin-Streptomycin solution (Sigma Aldrich), 1% 40mg/ml Geneticin (Life Technologies) and 10% fetal bovine serum (FBS) (PAA) in a humid environment at 33°C with 5% C02.
Primary Cell Culture
HdhQ111 mice were bred in house from a colony established from a stock supplied by Jackson laboratories for the specific purpose of this experiment. All experiments were conducted under the UK Animals (Scientific procedures) Act of 2015 and approved by local ethical review by Cardiff University Biological Standards Committee. Staged pregnant mice were sacrificed by cervical dislocation at embryonic day 14 (E14) in the absence of analgesics; an approved Schedule 1 method under the UK Animals (Scientific Procedures) Act 2015 at Cardiff University, School of Biosciences, within the JBIOS unit. Embryos were removed, the brains removed, and microdissection of the ganglionic eminence was carried out as previously described [45]. The resulting dissections were digested in 0.1% Trypsin (Lorne Laboratories) and cultured in DMEM/F12 containing 1% antibiotic-antimycotic solution, 2% B-27 supplement (all Life Technologies) and 10% FBS (Sigma-Aldrich). Cells were grown in a humid environment at 37°C with 5% C02
Antibodies
Ab109115 (Abcam) was used at a dilution of 1:100, and Mab2166 (Merck Millipore) was used at 1:1000 for immunofluorescence. S421 was a kind gift from Dr. Sandrine Humbert, (Institut Curie, Paris, France), and was used at 1:1000 for immunofluorescence. Anti-EGFR (New England Biolabs) was used at 1:50 for immunofluorescence and 1:1000 for western blots. Calreticulin (Abcam) was used at 1:500 for immunofluorescence, and α-tubulin was used at 1:5000 for western blots. Anti-CTIP2 (Abcam) was used at 1:500, and Anti-DARPP32 (Santa Cruz Biotechnology Inc.) was used at 1:200; both for immunofluorescence. Secondary antibodies; Goat-anti-Rabbit AlexaFluor 568 and Donkey-anti-Mouse AlexaFluor 488 (Life Technologies) were both used at 1:100 for immunofluorescence. Goat-anti-Rabbit AlexaFluor 680 (Life Technologies) and Goat-anti-mouse DyLight 800 were used for Western blots at 1:10,000 and 1:20,000, respectively.
Immunofluorescence
Cells were fixed by incubation with 10% formalin solution (containing 4% paraformaldehyde; Sigma-Aldrich) for 15 min. at room temperature. Cells were permeabilised with 0.1% Triton x-100 (Life Technologies) and blocked with 1% (w/v) bovine serum albumin (BSA) (Life Technologies) before undergoing incubation with the appropriate antibodies. Cells were imaged using either a Leica SP5 confocal microscope, or a Leica DM6000B fluorescent microscope.
Western blotting
Proteins were denatured and resolved on a 4–12% Bis-Tris gel, then electroblotted onto a PVDF membrane. Blots were blocked in 5% milk powder in PBS with 0.1% Tween (PBS-T) before incubation with the appropriate antibodies. Protein bands were visualised and quantified on a Licor® Odyssey® quantitative fluorescent imaging system.
ELISA
Phospho-AKT1 (Ser473) and phospho-MEK1 (Ser217/221) PathScan® Sandwich ELISA kits (Cell Signalling Technology) were utilised to assay kinase activation and inhibition, and were carried out according to manufacturer’s instructions.
Kinase activation and inhibition and huntingtin localisation
EGF (Life Technologies) was reconstituted and diluted to a stock solution of 100μg/ml in PBS and stored at -20°C. Cells were serum starved for 24 hours prior to EGF treatment, then incubated with 100ng/ml EGF in fresh serum free media for the appropriate length of time.
AKT inhibitor VIII (Merck Millipore) was reconstituted in dimethyl sulfoxide (DMSO) (Sigma Aldrich) to a stock solution of 0.1mM, and MEK 1/2 inhibitor (Merck Millipore) was initially reconstituted in DMSO, before being diluted to a 0.05mM stock solution in serum free media and stored at -20°C. Cells were serum starved for 24 hours prior to treatment with either kinase inhibitor. Each inhibitor was diluted to the appropriate concentration in serum free media, and was incubated with the cells for 2h prior to EGF stimulation. Serum free media containing the equivalent volume of DMSO alone was used as a control. EGF was diluted to 100ng/ml in fresh serum free media that also contained the appropriate concentration of each kinase inhibitor or DMSO before addition to the cells.
RNA extraction
RNA was extracted from cells using phenol/chloroform, precipated in ethanol, and purified by silica membrane spin columns (RNeasy MinElute Cleanup kit (Qiagen)) according to manufacturer’s instructions. DNA contaminants were treated with TURBO™DNase (Life Technologies) according to manufacturer’s instructions.
Quantitative RTPCR
1μg RNA was reverse transcribed using the High Capacity RNA-to-cDNA™ kit (Life Technologies), and the resulting cDNA was amplified using Sybr®Green mastermix (Life Technologies) containing the relevant oligonucleotide primer pair. The oligonucleotides used were as follows; Egr1 forward: 5’- CCTATGAGCACCTGACCACA-3’ reverse: 5’- GGGATAACTCGTCTCCACCA-3’, Arc forward: 5’- GAGAGCTGAAAGGGTTGCAC-3’ reverse: 5’-ACGTAGCCGTCCAAGTTGTT-3’, Ngfib forward: 5’- CAATGCTTCGTGTCAGCACT-3’ reverse: 5’ TGGCGCTTTTCTGTACTGTG-3’. The thermal cycler protocol was as follows: Stage 1–95°C for 10 min. Stage 2 (x40 cycles) - 95°C for 15 s, 55°C for 1 s. Stage 3 (dissociation curve) - 95°C following a gradual ramp. All qRTPCR protocols were controlled and analysed by the Sequence Detection Systems (SDS) Version 2.3 programme by Life Technologies.
Statistical analysis
Microscopy images were analysed blind to genotype, EGF stimulation and kinase inhibition by renaming image files to randomised numbers by individuals who were not involved in the project. Mean pixel intensities from nuclear, cytoplasmic and perinuclear regions of interest were measured from grayscale images using the GNU Image Manipulation Program 2.6.11, copyright 1995–2008 (S1 Fig). Every cell within each image was analysed; higher antibody binding elicited a higher signal. Nuclear/cytoplasmic (N/C) and nuclear/perinuclear (N/P) ratios were calculated for each cell individually in order to account for variation in background noise and to determine the presence of any alterations in huntingtin localisation between subcellular regions. All data was subject to analysis by the appropriate ANOVA, followed by post-hoc Tukey HSD tests.
Results
Epidermal growth factor receptor localisation in StHdh Q111 cell lines
To affirm the ability of the StHdh Q111 cell lines to respond to EGF stimulation, the presence and localisation of the EGF receptor (EGFR) was investigated by immunofluorescence and western blotting (Fig 1). The EGFR was present in all three cell lines, and exhibited no significant differences in its expression or subcellular localisation between genotypes, which is consistent with previous observations in the same model [46].
Fig 1. A. The EGFR is present in StHdh Q7/7, StHdh Q7/111 and StHdh Q111/111, cells and has a similar subcellular localisation Scale bar = 20μm. B. Western Blot indicating similar expression of the EGFR protein in all three cell lines, α-tubulin was used as a loading control. C. Quantification of fluorescence intensity of protein bands in B normalised to loading control and expressed as a proportion of StHdh Q7/7 (FI/StHdh Q7/7); there are no significant differences in the levels of EGFR between StHdh Q7/7, StHdh Q7/111 and StHdh Q111/111 cells.
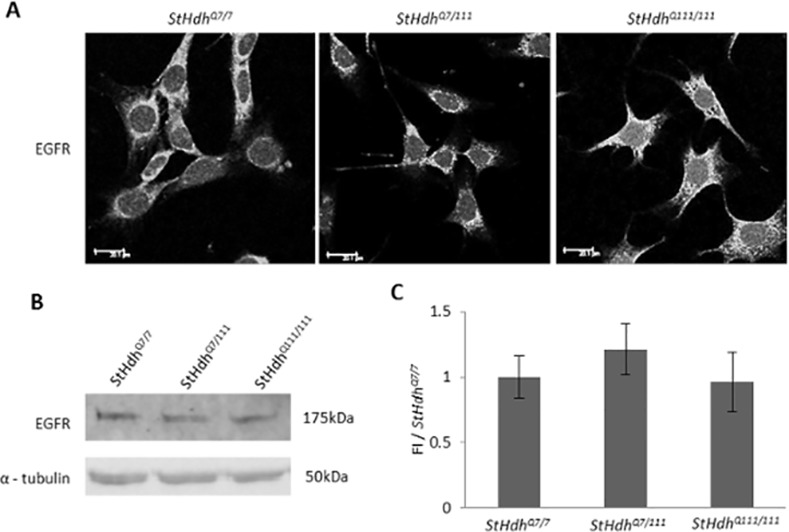
Error bars = ± SEM. Images are representative of multiple experiments. N = 3 replications.
Differential kinase pathway activation in StHdh Q111/111 cells
To determine whether kinase signalling pathways downstream of EGF stimulation show differential effects in the StHdh Q111 cell model, we examined the phosphorylation of kinases AKT and MEK in StHdh Q7/7 and StHdh Q111/111 cells in response to EGF stimulation by ELISA assay (Fig 2).
Fig 2. Mean absorbance (Abs) read at 450nm following a sandwich ELISA protocol for the detection of phosphorylated A. AKT1 and B. MEK1 in StHdh Q7/7 and StHdh Q111/111 cells at 0 mins and 10 mins of 100ng/ml EGF stimulation, either with or without a prior 2 hour incubation with 500nM AKT inhibitor VIII or 1μM MEK 1/2 inhibitor.
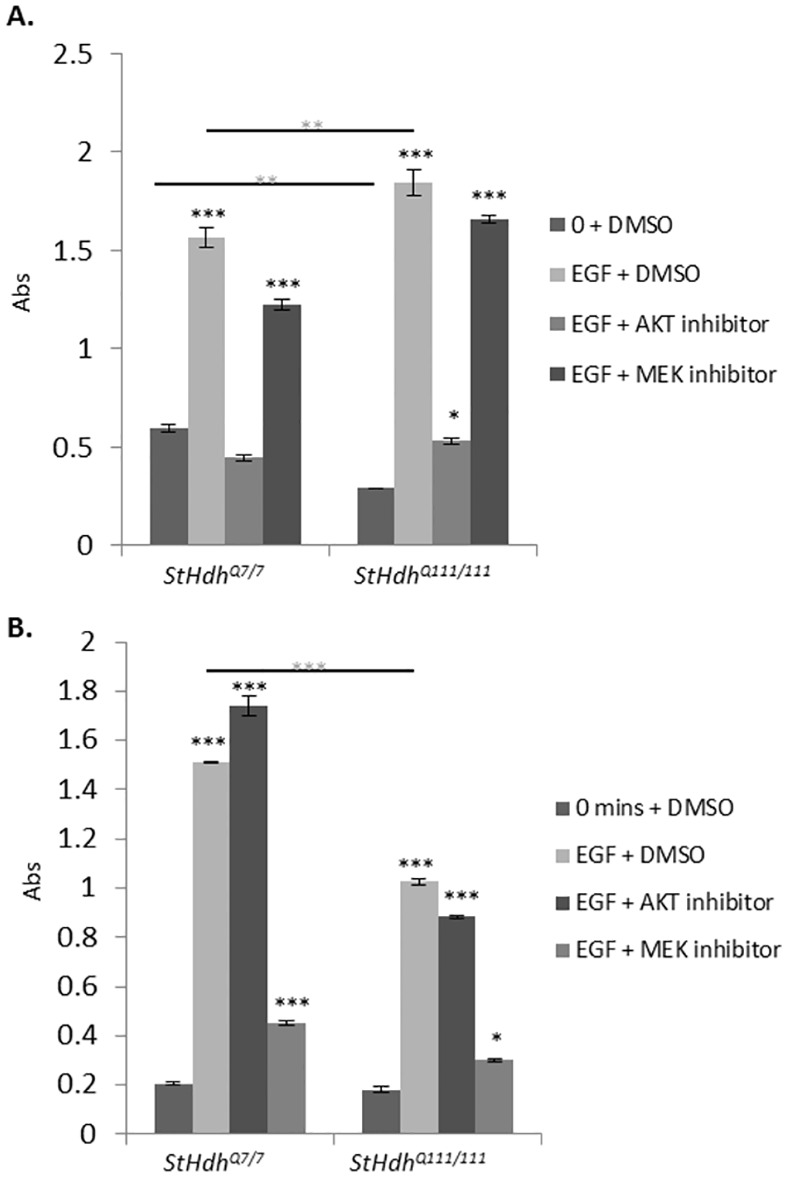
In cases where inhibitors were not used, cells were incubated with the equivalent volume of DMSO for the same amount of time prior to treatment and processing. Error bars = ±SEM. Black asterisks denote a significant difference from 0 mins + DMSO. Grey asterisks indicate genotypic differences. N = 3 replications *p<0.05, ** p<0.01, *** p<0.001.
There was a significant effect of both genotype (F1, 8 = 1044, p<0.001) and EGF stimulation (F3, 8 = 2504.74, p<0.001) on the level of AKT1 phosphorylated on serine 473 (S473) and MEK1 phosphorylated on serines 217/221 (S217/221). Post-hoc Tukey HSD analyses indicated that although StHdh Q111/111 cells exhibited suppressed AKT1 phosphorylation compared to StHdh Q7/7 cells at baseline (p<0.01), AKT1 phosphorylation was significantly increased in both genotypes following EGF stimulation (both p<0.001). However, when comparing the magnitude of phosphorylation following EGF stimulation, StHdh Q111/111 cells showed an augmented response (p<0.05), thus resulting in a significantly increased level of phosphorylated AKT1 in StHdh Q111/111 in comparison to StHdh Q7/7 cells (p<0.01) (Fig 2).
While there was no difference in the level of phosphorylated MEK1 between genotypes at baseline, there was a strong phosphorylation response in both StHdh Q7/7 (p<0.001) and StHdh Q111/111 cells (p<0.001) following EGF stimulation. However, a suppressed response in StHdh Q111/111 cells resulted in a significantly lower level of phosphorylated MEK1 in StHdh Q111/111 cells following EGF stimulation than in StHdh Q7/7 cells (p<0.001) (Fig 2).
Inhibition of AKT and MEK phosphorylation
The efficacy and specificity of selective kinase inhibitors against AKT1 and MEK1 phosphorylation were tested in both StHdh Q7/7 and StHdh Q111/111 cells by ELISA assay (Fig 2). Treatment with AKT inhibitor VIII suppressed EGF-stimulated AKT1 phosphorylation in both cell lines, while having no significant effect on the phosphorylation of MEK1. Following AKT1 inhibition, there was no longer a significant difference in AKT1 phosphorylation between genotypes in response to EGF stimulation.
Treatment with MEK 1/2 inhibitor prior to EGF stimulation significantly suppressed the MEK1 phosphorylation response in both genotypes, but did not alter AKT1 phosphorylation. Despite significant inhibition of phosphorylation compared to EGF stimulation alone, MEK1 phosphorylation remained significantly higher than at baseline for both StHdh Q7/7 cells (p<0.001) and StHdh Q111/111 cells (p<0.05), and remained higher in StHdh Q7/7 cells compared with StHdh Q111/111 cells (p<0.01) (Fig 2).
Alterations in huntingtin subcellular localisation in response to EGF stimulation
Amino-terminal huntingtin co-localises with endoplasmic reticulum
As kinase activation occurs upstream of huntingtin phosphorylation [47] and has been identified as a modulator of huntingtin nuclear localisation [21,22], we investigated whether EGF stimulation would alter the subcellular localisation of non-phosphorylated amino-terminal huntingtin epitopes. Prior experience with amino-terminal huntingtin antibodies within this lab has identified that they detect an isoform of huntingtin that localises in a region surrounding the nucleus. In order to better define this region for accurate quantification of immunofluorescence images, we looked at the co-localisation of amino-terminal huntingtin with the ER marker, calreticulin (S2 Fig). Calreticulin is present in all three cell lines in a region immediately surrounding the nucleus, and is not altered by the presence of mutant huntingtin. Furthermore, it co-localises with amino-terminal huntingtin and mutant huntingtin detected by Mab2166 (S2 Fig). We therefore defined the area in which HTT co-localises with calreticulin as the ‘perinuclear’ region for forthcoming experiments and image analyses (for further details of the analyses, refer to Methods).
EGF stimulation alters the localisation of amino-terminal huntingtin epitopes
Statistical analyses were focussed on the nuclear/cytoplasmic (N/C) and nuclear/perinuclear (N/P) ratios (refer to Methods for details), as these proved to be most informative and representative of the immunofluorescence images. There was a significant effect of genotype on the localisation of huntingtin as measured by the N/C ratio, when visualised using both Mab2166 (F2, 847 = 46.93, p<0.001) and Ab109115 (F2, 856 = 132.21, p<0.001; (Fig 3, S3 Fig), which was a result of an increase in the detection of nuclear huntingtin epitopes in StHdh Q7/111 and StHdh Q111/111 cells compared to StHdh Q7/7 cells. A significant effect of genotype was also present in the analysis of the N/P ratio for both antibodies (Mab2166: F2, 847 = 134.48, p<0.001; Ab109115: F2, 856 = 228.6, p<0.001). Additionally, there was also a significant effect of EGF stimulation in both N/C and N/P ratios when using Mab2166 (N/C: F3, 847 = 26.43, p<0.001, N/P: F3, 847 = 49.2, p<0.001) and Ab109115 (N/C: F3, 856 = 6.08, p<0.001, N/P: F3, 856 = 18.64, P<0.001; Fig 3, S3 Fig). Post-hoc analyses indicate that this is due to a modest reduction in nuclear huntingtin in the N/P ratio for StHdh Q7/7 cells following 15 mins of EGF stimulation (p<0.05) when detected by Ab109115, and after 5 mins when detected by Mab2166 in both N/C and N/P ratios (both p<0.05). Similarly, StHdh Q7/111 cells exhibited a similar pattern, with a reduction in nuclear detection as measured by the N/P ratio from 15 mins of EGF stimulation when using Ab109115 (p<0.05) and in both the N/C and N/P ratios when using Mab2166 at 5 mins (both p<0.001). The reduced nuclear localisation following EGF stimulation when visualised using Ab109115 was abrogated in StHdh Q111/111 cells, suggesting an impaired cellular response to EGF stimulation in the presence of mutant huntingtin. In contrast, there was a reduction in the N/P ratio alone at 5 mins in these cells when visualised using Mab2166 (p<0.001), indicating that the impairment may be huntingtin epitope-specific (Fig 3, S3 Fig).
Fig 3. A. Subcellular localisation of an N-terminal epitope of huntingtin and mutant huntingtin in StHdh Q7/7, StHdh Q7/111 and StHdh Q111/111 cell lines. Cells were fixed following 0, 5, 15 and 30 min. of stimulation with 100ng/ml EGF, labelled with Mab2166 against amino acids 181–810, then analysed by confocal microscopy. Scale bar = 20μm. B. Quantitative analysis of immunofluorescence images in A. Nuclear/Cytoplasmic (N/C) and Nuclear/Perinuclear (N/P) mean pixel intensity ratios (MPI) for StHdh Q7/7, StHdh Q7/111 and StHdh Q111/111 cells following 0, 5, 15 and 30 min. of stimulation with 100ng/ml EGF.
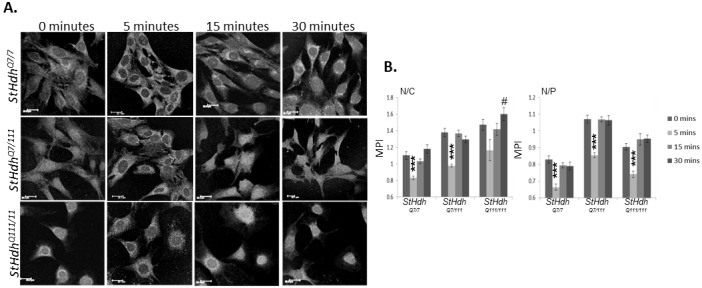
Mean pixel intensities were calculated from confocal microscopy images using GNU Image Manipulator. All images were randomised and analysed blind to genotype and length of time stimulated with EGF. Each condition consisted of 9 confocal microscopy images taken from 3 separate coverslips. n = 66–91. Error bars = ± SEM. Data representative of 3 experiments; * Denotes a significant difference from 0min.; # Denotes a significant difference from 5mins; */# p<0.05, **/## p<0.01, ***/### p<0.001.
Alterations in the subcellular localisation of amino-terminal huntingtin in response to EGF is independent of phosphorylation on serine 421
Although the phosphorylation of huntingtin on S421 has been shown to occur following AKT activation [48], which results in a neuroprotective reduction of nuclear huntingtin [22,47], we did not recognise any change in the subcellular localisation of huntingtin phosphorylated on S421 in these cells in response to EGF stimulation in any genotype (S4 Fig).
Replicating the effect of EGF stimulation on huntingtin localisation in primary HdhQ111 striatal cells
Primary cells were cultured from microdissected E14 HdhQ111 mouse model striatum, from which the StHdh Q111 cell model was originally derived [44]. In order to confirm the presence of striatal cells in primary cell cultures, a typical selection of cells was immunostained with antibodies against striatal cell markers dopamine- and cAMP-regulated neuronal phosphoprotein (DARPP-32) and CTIP2 (S5 Fig), both of which have been identified as being enriched within medium spiny neurons [49–51]. The majority of cells present in dissections were labelled with striatal markers, and therefore were considered to reflect the characterisation of an embryonic striatal cell population.
Confirming the presence of the EGFR in HdhQ111 cells
Immunofluorescence analysis revealed that the EGFR was present and had the same subcellular localisation in primary striatal cells from all three genotypes; HdhQ7/7, HdhQ7/111 and HdhQ111/111 (Fig 4). There was a trend towards reduced levels of the EGFR in HdhQ7/111 and HdhQ111/111 cells as indicated by western blot, although this difference does not reach significance.
Fig 4. A. The EGFR is present in HdhQ7/7, HdhQ7/111 and HdhQ111/111, cells and has a similar subcellular localisation. Scale bar = 20μm B. Western Blot indicating similar expression of the EGFR protein in all three genotypes, α-tubulin was used as a loading control. C. Quantification of fluorescence intensity of protein bands in B normalised to loading control and expressed as a proportion of HdhQ7/7 (FI/HdhQ7/7).
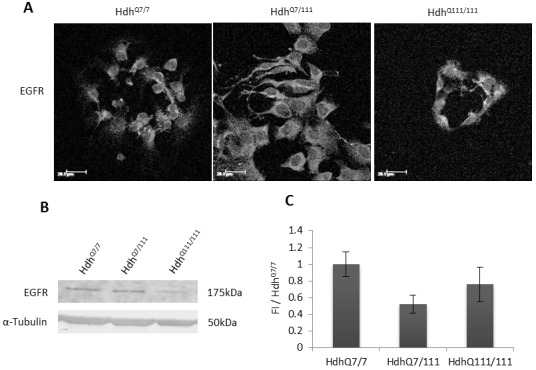
Error bars = ± SEM. Images are representative of multiple experiments. N = 3 replications.
Kinase pathway activation in HdhQ111 primary cells
The phosphorylation of AKT1 and MEK1 following EGF stimulation was assayed in HdhQ111 primary striatal cell cultures (Fig 5). Treatment of primary cultures with EGF significantly increased AKT1 and MEK1 phosphorylation in both HdhQ7/7 and HdhQ111/111 cells (all p<0.001), although in contrast to StHdh Q111 cell lines, there was no observable difference between genotypes.
Fig 5. Mean absorbance (Abs) read at 450nm following a sandwich ELISA protocol for the detection of phosphorylated A. AKT1 and B. MEK1 in HdhQ7/7 and HdhQ111/111 primary cells at 0 mins and 10 mins of 100ng/ml EGF stimulation.
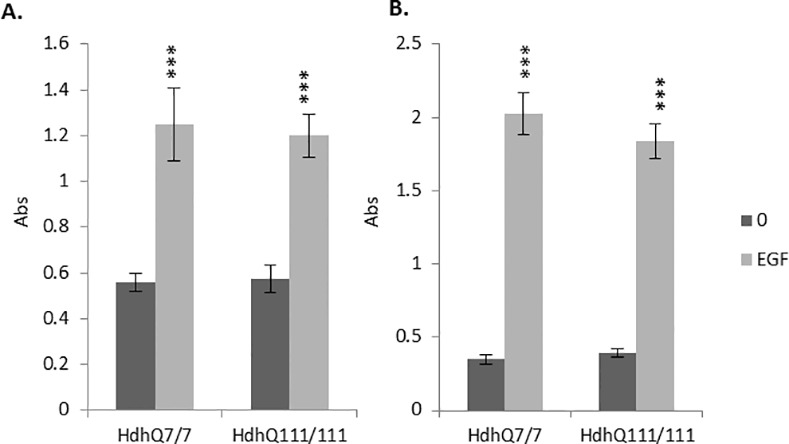
Error bars = ±SEM. Black asterisks denote a significant difference from 0 mins + DMSO. N = 5 replications. *** p<0.001.
EGF stimulation alters the localisation of amino-terminal huntingtin epitopes in HdhQ111 primary cells
The localisation of the huntingtin epitope detected by Mab2166 was primarily perinuclear in HdhQ7/7, HdhQ7/111 and HdhQ111/111 cells, as observed in immortalised striatal cell lines (Fig 6). Analysis of mean pixel intensity ratios indicated a significant reduction in the N/C ratio in HdhQ7/7 cells after 5 mins of EGF stimulation (p<0.05), and in the N/P ratio after both 5 and 15 mins (both p<0.01) of EGF stimulation. In contrast, there was no significant effect of EGF stimulation for either mutant huntingtin-carrying genotypes at any time point. There was a similar pattern of effect when cells were immunostained with Ab109115, although this was only mildly significant in the N/P ratio for HdhQ7/7 cells (p<0.05; S6 Fig).
Fig 6. Subcellular localisation of an N-terminal epitope of huntingtin and mutant huntingtin in HdhQ7/7, HdhQ7/111 and HdhQ111/111 primary cell lines.
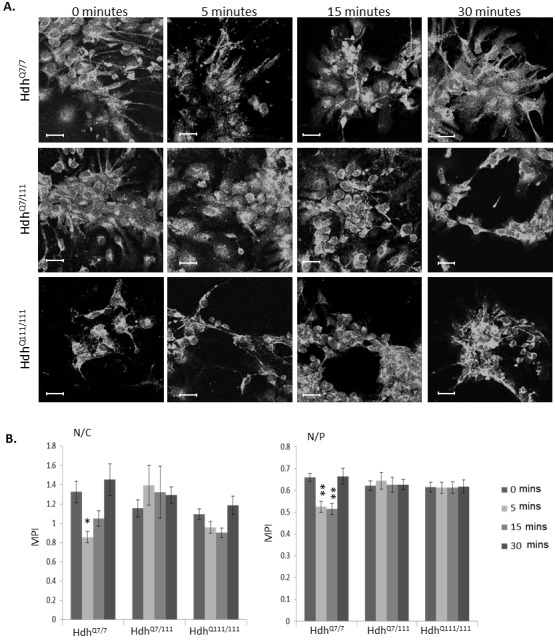
Cells were fixed following 0, 5, 15 and 30 min. of stimulation with 100ng/ml EGF, labelled with Mab2166, then analysed by confocal microscopy. Scale bar = 20μm. B. Quantitative analysis of immunofluorescence images in A. Nuclear/Cytoplasmic (N/C) and Nuclear/Perinuclear (N/P) mean pixel intensity ratios (MPI) for HdhQ7/7, HdhQ7/111 and HdhQ111/111 primary cells following 0, 5, 15 and 30 min. of stimulation with 100ng/ml EGF. Mean pixel intensities were calculated from confocal microscopy images using GNU Image Manipulator. All images were randomised and analysed blind to genotype and length of time stimulated with EGF. Each condition consisted of 9 confocal microscopy images taken from 3 separate coverslips. n = 49–70. Error bars = ± SEM. Data representative of 3 experiments; * Denotes a significant difference from 0min.; *p<0.05, ** p<0.01.
Inhibition of AKT1 phosphorylation alters EGF-regulated huntingtin localisation in StHdh Q111 cell lines
As EGF stimulation elicited AKT1 phosphorylation and altered the subcellular localisation of amino-terminal huntingtin in both StHdh Q111 and primary HdhQ111 cell lines, we sought to investigate whether the phosphorylation of AKT1 may be a mechanism that regulates huntingtin subcellular localisation. The subcellular localisation experiment was repeated, with the addition of a 2 hour incubation period with AKT VIII inhibitor, or the equivalent volume of DMSO, prior to stimulation with EGF (Fig 7, S7 Fig). Mab2166 and Ab109115 were again used to detect the localisation of amino-terminal huntingtin, and images were analysed in the same manner. There was a significant interaction between genotype and treatment condition for both N/C and N/P ratios (N/C F18, 2785 = 10.34, p<0.001; N/P F18, 2785 = 17.24, p<0.001), therefore suggesting a differential effect of AKT inhibition between genotypes (Fig 7, S7 Fig).
Fig 7. A. StHdh Q7/7, B. StHdh Q7/111 and C. StHdh Q111/111 cells treated with either AKT inhibitor VIII, MEK 1/2 inhibitor, or the equivalent volume of DMSO for 2 hours prior to 0, 5, 15 and 30 mins stimulation with 100ng/ml EGF, then probed with amino-terminal huntingtin antibody Mab2166. Scale bar = 20μm. D-E. Quantification of mean pixel intensity (MPI) from images represented in A-C for the D. Nuclear/Cytoplasmic (N/C) ratio and E. Nuclear/Perinuclear (N/P) ratio.
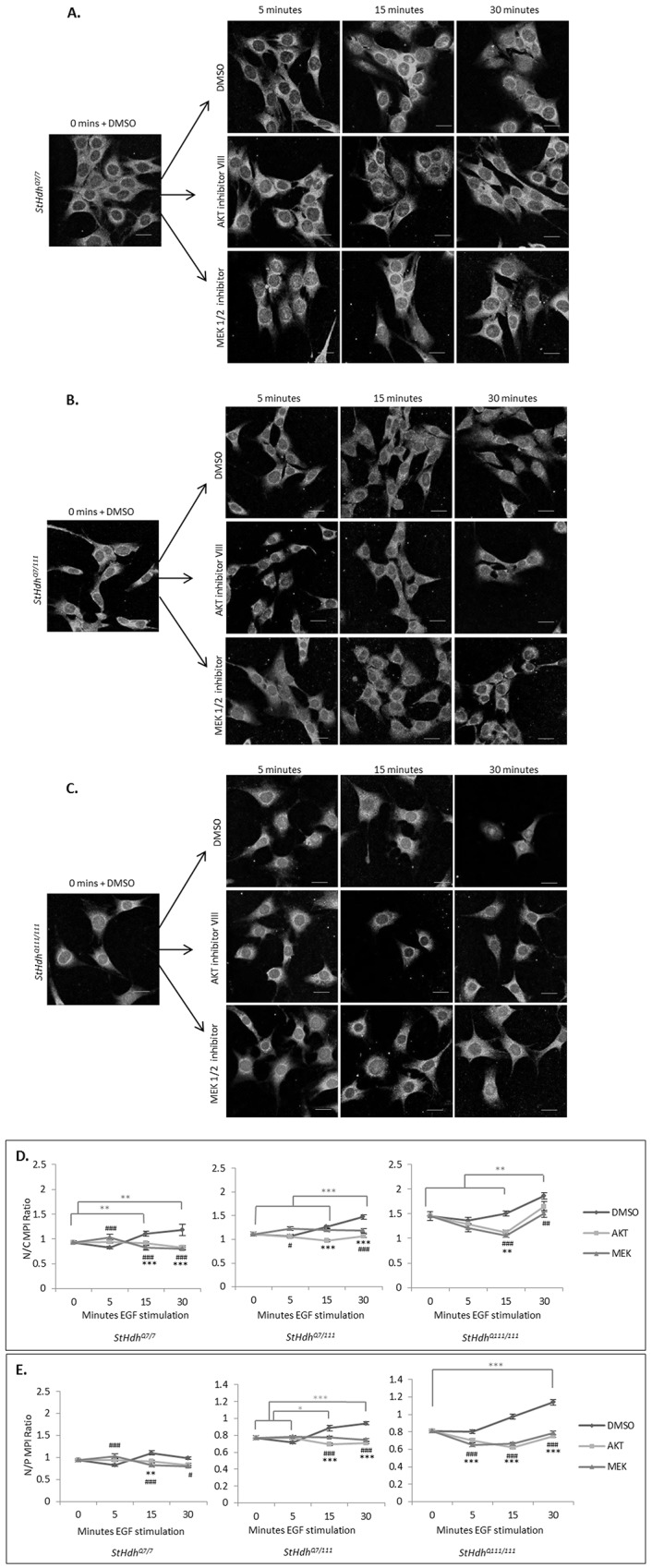
Error bars = SEM. Light grey bars and asterisks signify statistically significant differences between DMSO conditions. Black asterisks and hashes indicate statistically significant differences between DMSO vs AKT inhibitor conditions and DMSO vs MEK inhibitor conditions, respectively. Data representative of three experiments. n = 85–135. */# p<0.05, **/## p<0.01, ***/### p<0.001.
AKT1 inhibition alters the apparent localisation of huntingtin in StHdh Q7/7 cells in an epitope-dependent manner
A significant reduction in nuclear huntingtin was not replicated in StHdh Q7/7 cells following EGF stimulation when detected by Mab2166, which may be due to the presence of DMSO (Fig 7). However, there was still a trend towards reduced nuclear huntingtin detection at 5 mins of stimulation in both N/C and N/P ratios, and the original observation was replicated when Ab109115 was used for detection (S7 Fig). AKT1 inhibition had no significant effect at the 5 minute time point following EGF stimulation in either Ab109115 or Mab2166 immunostained cells. However, AKT1 inhibition did appear to prevent increased nuclear Mab2166 detection of huntingtin at 15 and 30 mins, which was likely elicited by DMSO (Fig 7, both p<0.01). In contrast, there was no effect of AKT1 inhibition on huntingtin localisation when detected by Ab109115.
AKT1 inhibition reduces the nuclear detection of huntingtin in StHdh Q7/111 and StHdh Q111/111 cells
Similar to the effect of EGF stimulation on the subcellular localisation of huntingtin in Sthdh Q7/7 cells in the presence of DMSO, a large reduction in nuclear huntingtin detection by Mab2166 at 5 mins of EGF stimulation was not fully replicated in either StHdh Q7/111 or StHdh Q111/111 cells when DMSO was present, although both lines exhibited a trend towards this effect. In both cell lines there was an increase in nuclear huntingtin with increased length of EGF stimulation, which is observable in both N/C and N/P ratios (Fig 7, S7 Fig). This effect could be visualised using either Mab2166 or Ab109115 antibodies, and may be a result of DMSO-induced toxicity. Inhibition of AKT1 counteracts this effect; in StHdh Q7/111 cells, there is a significant reduction in both N/C (p<0.001) and N/P (p<0.001) ratios from 15 mins of EGF stimulation when AKT1 is inhibited. In StHdh Q111/111 cells, the same nuclear reduction is apparent from 5 mins in the N/P ratio (p<0.001) and from 15 mins in the N/C ratio (p<0.01; Fig 7). A similar result is produced when visualised with Ab109115; nuclear detection of huntingtin is significantly reduced from 5 mins in both StHdh Q7/111 (N/C p<0.05) and StHdh Q111/111 (N/C p<0.05) cells, although this effect is considerably more marked in StHdh Q111/111 cells, and persists for the entirety of the experiment (S7 Fig).
MEK inhibition alters huntingtin localisation in a similar manner to AKT inhibition
Inhibition of MEK1 was carried out in parallel with the AKT1 inhibition experiment, and therefore shared the same DMSO controls. MEK1 inhibition had a strikingly similar effect on the nuclear presence of huntingtin in response to EGF stimulation as AKT1 inhibition did in all three genotypes (Fig 7, S7 Fig). StHdh Q7/7 cells exhibited increased nuclear huntingtin detection in the N/C (p<0.001) and N/P (p<0.001) ratios following 5 mins of EGF stimulation when MEK1 was inhibited, as detected by Mab2166 (Fig 7). This effect was also observed when detected by Ab109115 (S7 Fig).
Similar to the effect of AKT1 inhibition in StHdh Q7/111 cells, there was a modest effect of MEK1 inhibition on huntingtin localisation when detected by both Mab2166 and Ab109115; there was a reduction in the N/P ratio at 15 mins and 30 mins (both p<0.001) when visualised with Mab2166 (Fig 7), and a modest reduction in the N/C ratio at 15 mins (p<0.05) when visualised with Ab109115 (S7 Fig).
StHdh Q111/111 cells showed a similar pattern of huntingtin localisation change following MEK1 inhibition as following AKT1 inhibition (Fig 7, S7 Fig). There was a substantial reduction in the detection of nuclear huntingtin from 5 mins post- EGF stimulation following MEK1 inhibition, which was observed primarily in the N/P ratio (all time points p<0.001) when using Mab2166 (Fig 7). This effect was particularly strong when Ab109115 immunostaining was analysed, with both N/C and N/P ratios being significantly lower when MEK1 was inhibited at all time points (all p<0.001; S7 Fig).
Dysregulation of immediate early gene expression is downstream of AKT and MEK signalling
The regulation of huntingtin localisation has been implicated as a mechanism for transcriptional control [19,23,25,26,52]. We therefore compared the expression of three immediate early (IE) genes downstream of the EGF signalling pathway between StHdh Q7/7 and StHdh Q111/111 cells, as these exhibited the most disparate huntingtin localisation responses to EGF stimulation and kinase inhibition. The expression of Egr1, Arc and Ngfib were measured by qRTPCR in response to EGF stimulation alone, and following either AKT1 or MEK1 inhibition. The transcriptional responses of these genes were typically more reactive in StHdh Q111/111 cells, which elicited larger gene expression fold changes in response to EGF stimulation than in StHdh Q7/7 cells (Fig 8). EGF stimulation significantly increased the expression of Egr1 in both genotypes (F3, 24 = 17.95, p<0.001) although this effect was augmented in StHdh Q111/111 cells (fold change x5.5, p<0.001) compared to StHdh Q7/7 cells (fold change x2.24, p<0.05). This was an effect that was also observed in primary HdhQ7/7 and HdhQ111/111 cells (S8 Fig). AKT1 inhibition had no effect on Egr1 expression in StHdh Q7/7 cells, and although there was a trend towards a reduction in Egr1 expression in StHdh Q111/111 cells, this did not reach significance. In contrast, MEK1 inhibition suppressed the effect of EGF stimulation on Egr1 expression in both genotypes (StHdh Q7/7 p<0.05, StHdh Q111/111 p<0.001; Fig 8).
Fig 8. Relative quantitation (RQ) values representing gene expression fold change of Egr1, Arc and Ngfib in StHdh Q7/7 and StHdh Q111/111 cells following inhibition with either 500nM AKT inhibitor VIII, 1μM MEK 1/2 inhibitor or the equivalent volume of DMSO, followed by 0 or 2 hours of 100ng/ml EGF stimulation.
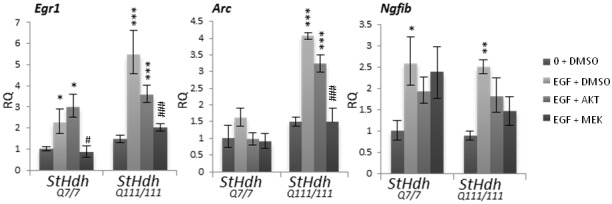
Statistical analysis was conducted on ΔCt values. Error bars = ± SEM. Asterisks denote a significant difference from 0 + DMSO, hashes indicate a significant difference from EGF + DMSO. Data representative of two experiments. N = 6.*/# p<0.05, **/## p<0.01, ***/### p<0.001.
The expression of Arc exhibited a similar pattern; while there was a larger effect of EGF in StHdh Q111/111 cells (fold change x4, p<0.001) compared to StHdh Q7/7 cells (fold change x1.6, n.s.), AKT1 inhibition had no effect on Arc expression in either genotype. By contrast, MEK1 inhibition prevented the large increase in Arc expression observed in StHdh Q111/111 cells following EGF stimulation (p<0.001). The expression of Ngfib increased to a similar extent in both genotypes following EGF stimulation (F3, 24 = 8.07, p<0.001. StHdh Q111/111 fold change x2.5, StHdh Q7/7 fold change x2.59, both p<0.05); this effect was partially suppressed by both AKT1 and MEK1 inhibition for both StHdh Q7/7 and StHdh Q111/111 cells, but did not reach significance (Fig 8).
Discussion
We demonstrate that EGF stimulation elicits differential effects on the activation of the MEK1 and AKT1 kinase signalling pathways downstream of the EGF receptor in StHdh Q7/7 and StHdh Q111/111 cell lines, despite similar expression levels and localisation of the EGFR. We also demonstrate that dysregulation of these pathways may alter the subcellular localisation of huntingtin in both StHdh Q111 and HdhQ111 cell lines, which may impact upon downstream transcriptional control (Fig 9).
Fig 9. Simplified cartoon of the hypothesised relationship between EGF-stimulated kinase activation, huntingtin localisation and transcriptional regulation.

Upon binding to the EGFR, EGF elicits the phosphorylation of MEK and AKT. In the presence of mutant huntingtin, aberrant interactions with GRB2 and suppressed PHLPP1 expression serve to alter the phosphorylation response. In turn, active MEK and AKT regulate the subcellular localisation of huntingtin, possibly by post-translational modification; however this regulation is impaired due to mutant huntingtin-associated aberrant kinase phosphorylation. Regulation of huntingtin nuclear localisation may then influence transcriptional control via multiple mechanisms. As such, altered nuclear localisation of mutant huntingtin could disrupt the control of these mechanisms and would result in altered transcriptional regulation.
Baseline levels of AKT are relatively low in adult brain [53], however its activation rises following cellular stress and injury, and it invokes a protective response [54]. AKT exerts its protective effects by phosphorylating and inactivating components of the cell death machinery, such as B-cell lymphoma-2 associated death promoter (BAD), caspase-9, forkhead box proteins (FOXOs) and inhibitor of kappa-β kinase (IKK) [48,55]. Enhanced AKT activity has previously been identified in the StHdh Q111/111 cell line [13,23,42], as well as in a neural stem cell model of HD [56], which may reflect a neuroprotective compensatory response in order to counteract the excitotoxic effects of mutant huntingtin [13,54]. For example, increasing AKT activity in a Drosophila model of HD reduced neurotoxicity [57], and reduced the number of intranuclear inclusions formed in a striatal cell model expressing mutant huntingtin [48].
Less information is currently available regarding the role of MEK1 signalling in models of HD, although its activation has previously been found to be reduced in StHdh Q111 cells [46]. Consistent with Ginés et al. [13], we have identified suppressed MEK1 phosphorylation in StHdh Q111/111 cells following EGF stimulation. There is evidence that huntingtin may be phosphorylated by MEK1 activation [18], although the implications of this mechanism are yet to be elucidated. A constitutively active form of MEK1 was found to be protective against cell death in a truncated-mutant huntingtin expressing PC12 model of HD [16], although by contrast, the induction of MEK1 activity provided no protection against polyglutamine induced lethality in a polyglutamine-overexpressing Drosophila model [17]. ERK1/2 activity has been found to be neuroprotective in models of HD [16,58] and as it lies downstream of MEK1 activation, it may be inferred that its action is representative of MEK1 activity.
However, we did not observe differential phosphorylation responses to EGF stimulation in primary HdhQ111 cell cultures. This could be because mutant huntingtin requires a longer period of time to accumulate the underlying level of dysregulation responsible for these differential effects to be observed. Alternatively, the differential phosphorylation response may be model specific; multiple opposing effects of mutant huntingtin presence on kinase signalling pathways have previously been reported that vary depending on the model being investigated [59]. Regardless of the lack of a differential genotype effect on AKT1 and MEK1 phosphorylation in primary cells, EGF stimulation still altered huntingtin localisation and gene expression in HdhQ111 cells in a manner similar to that observed in StHdh Q111 cell lines, which could suggest that the extent of kinase phosphorylation may not be the most instructive indicator of kinase pathway integrity.
As kinase activation and inhibition has previously been associated with the regulation of the subcellular localisation of huntingtin [21–23], we investigated whether the differential responses in AKT1 and MEK1 phosphorylation following EGF stimulation could be reflected in the subcellular localisation of huntingtin. We demonstrate that EGF stimulation elicits differential apparent subcellular localisation of huntingtin and mutant huntingtin amino-terminal epitopes in both StHdh Q111 and primary HdhQ111 cells, and suggest that endogenous kinase signalling pathways may contribute to the regulation of either huntingtin localisation or conformation (Fig 9).
The neuroprotective effects of AKT activity have been proposed to be exerted through its phosphorylation of huntingtin on S421 [48,55], which has been found to reduce the toxicity of mutant huntingtin and the nuclear accumulation of toxic mutant huntingtin fragments, as well as restoring axonal transport [22,57,60–62]. However, we did not identify any changes in the localisation of huntingtin phosphorylated on S421 in response to EGF stimulation, which may be a result of the experimental design; other studies have elicited huntingtin S421 phosphorylation by the overexpression of constitutively active AKT or by insulin-like growth factor (IGF-1) stimulation [48,60,63], or have investigated the effects of phospho-mimetic mutations of huntingtin S421 [22], whereas the phosphorylation of endogenous huntingtin by EGF stimulation has not previously been assessed.
While other post-translational modifications may be contributing to the regulation of huntingtin subcellular localisation, which were not assayed here [21,23,24,64–68], it has not been established whether an alteration in the absolute localisation of huntingtin is observed following EGF stimulation, or whether there has been a change in conformation that would result in the exposure of alternative epitopes undetectable by the antibodies used in this study. While our results are consistent between both Mab2166 and Ab109115 antibodies, some variation in huntingtin detection was observed; this may be due to differences in binding affinity to specific huntingtin conformations. However, using both Mab2166 and Ab109115 we observe that the presence of nuclear huntingtin is increased in the context of the expanded polyglutamine; a phenotype previously described in this cell model [23,25,44,64] and in other models of HD [27,29,69–74].
Stimulation with EGF reduced the nuclear pool of huntingtin in StHdh Q7/7 and HdhQ7/7 cells, although this effect was apparent primarily between the nucleus and the region of the cell we defined as ‘perinuclear,’ which is consistent with the observation that huntingtin accumulates in the perinuclear region prior to nuclear translocation [75]. Huntingtin is also known to localise to the Golgi body and late endosomes [76,77], so increased localisation in the perinuclear region following stimulation may be representative of a role for huntingtin in the recycling and degradation of the EGFR, as has been established in human fibroblasts [78].
We also demonstrate that the effect of EGF stimulation on the localisation of mutant huntingtin is suppressed in StHdh Q111/111 and HdhQ111/111 cells; this suppression is consistent with the inhibited kinase phosphorylation and gene expression responses to EGF stimulation in other cell and Drosophila models of HD [79,80], as well as with the delayed EGFR degradation and altered trafficking that has been observed in human HD fibroblasts [78]. However, as we observe a trend towards a reduction of the EGFR in HdhQ7/111 and HdhQ111/111 cells, it is possible that this may also be contributing to the suppressed response in these cells.
The nuclear translocation of huntingtin has been previously proposed as a mechanism for regulating its function [25,26]. Multiple mechanisms may explain the ability of huntingtin to shuttle between the nucleus and cytoplasm[24,26,64,66,81,82]. The association of huntingtin with exogenous chaperones to traverse the nuclear pore, and the availability of nuclear export/nuclear localisation signals (NES/NLS) on huntingtin itself may change with alterations in its conformation [26,83–85], and are therefore potential mechanisms by which EGF stimulation could be altering the localisation of huntingtin. However, the process that may be regulating huntingtin localisation in our current model requires clarification.
In order to determine whether the dysregulation of AKT1 and MEK1 phosphorylation observed in StHdh Q111/111 cells could be responsible for altered mutant subcellular localisation, we repeated the experiment whilst inhibiting either AKT1 or MEK1 activation in response to EGF stimulation. Inhibition of either kinase elicited an increase in nuclear huntingtin in StHdh Q7/7 cells, indicating that the subcellular localisation of huntingtin can be regulated by AKT1 and MEK1 activity. The increase in nuclear huntingtin is not as marked when visualised with Mab2166 compared to Ab109115, which may be due to detection of an alternate epitope of huntingtin, or variation in antibody specificity. Alternatively, differences in subcellular localisation following EGF stimulation may be due to their ability to detect different huntingtin cleavage products. While both antibodies detect amino-terminal huntingtin, multiple aspartyl protease cleavage sites lie between the huntingtin binding sites for each antibody [86]; the resulting huntingtin fragments would therefore only be detectable by Ab109115, and would be able to more readily shuttle between cellular compartments by passive diffusion [19]. With continued EGF stimulation, this initial nuclear accumulation is followed by a slight reduction in nuclear huntingtin detection with both Ab109115 and Mab2166. Both AKT1 and MEK1 inhibition elicited similar effects on huntingtin localisation, and are therefore likely to be affecting huntingtin in a similar manner. However, whether this may be due to a shared downstream kinase or to similar, but distinct, mechanisms of action remains unclear and requires additional investigation.
Compared to the effect observed in StHdh Q111/111 cells, the result of kinase inhibition in StHdh Q7/7 cells was very modest. AKT1 and MEK1 inhibition in StHdh Q111/111 cells elicited a dramatic reduction in nuclear mutant huntingtin following treatment with EGF. The expanded polyglutamine may therefore be modifying the normal interactions between huntingtin and elements downstream of kinase activation, which consequently has an effect on mutant huntingtin subcellular localisation. A reduction in nuclear huntingtin was observed in StHdh Q7/7 cells following EGF stimulation alone, which may suggest that when aberrant activation of the MEK1 and AKT1 pathways is prevented, the StHdh Q111/111 cellular phenotype becomes more similar to that of StHdh Q7/7 cells. Increased nuclear localisation of huntingtin is typically considered to be pathogenic, and is associated with transcriptional dysregulation [87,88]; therefore reducing the presence of nuclear mutant huntingtin by inhibiting aberrant kinase signalling may prove to be beneficial for the appropriate regulation of cellular processes. To our knowledge, the manipulation of specific kinase signalling pathways has not yet been carried out in murine models of HD in vivo in order to determine any potential effects of kinase phosphorylation on phenotype and survival.
The shuttling of huntingtin between cellular compartments is associated with the regulation of its function [25,26], including the control of gene expression [19,23,37], so alterations in nuclear huntingtin localisation could be a regulatory mechanism for the aberrant transcriptional control that has been identified in StHdh Q111/111 cells and other models of HD [89–91]. We therefore decided to investigate whether EGF stimulation and kinase inhibition altered the expression of a panel of IE genes downstream of the EGF pathway [40,41]. The extent of IE gene expression changes following EGF stimulation was typically larger in StHdh Q111/111 cells, which is consistent with the AKT hyperactivity also observed in StHdh Q111/111 cells. However, the integrity of the MEK1 signalling pathway appears to be more relevant for the control of the expression of Egr1, Arc and Ngfib, as its inhibition had a much more substantial effect on IE gene expression than the inhibition of AKT1.
We propose that kinase signalling is capable of modulating huntingtin localisation in both immortalised StHdh Q111 and primary HdhQ111 cell models of HD. In particular, MEK1 and AKT1 signalling are able to regulate huntingtin localisation and transcriptional control in the StHdh Q111 cell model of HD, although the mechanism by which these processes may be associated remains unknown (Fig 9). MEK1 activity could be more relevant for transcriptional control in the StHdh Q111 cell model of HD than AKT1 due to the greater impact of its inhibition on IE expression. However, as MEK1 phosphorylation was suppressed in StHdh Q111/111 cells, and both AKT1 and MEK1 inhibition are able to alter huntingtin localisation in a similar manner, it is possible that a synergistic mechanism between multiple dysregulated pathways contributes to aberrant transcriptional control and huntingtin mislocalisation in models of HD. Alternatively, as similar huntingtin localisation and Egr1 expression was observed in HdhQ111 primary cells in the absence of differential MEK1 phosphorylation, it may be possible the extent of MEK1 phosphorylation does not accurately reflect downstream activity that is more directly impacting huntingtin localisation and transcriptional regulation. It also remains unclear from our model whether nuclear mutant huntingtin is having a direct effect upon transcriptional regulation, or whether another mechanism downstream of AKT1 and MEK1 signalling pathways is impacting upon transcriptional regulation in parallel with the regulation of huntingtin localisation.
As there is substantial overlap and shared targets between multiple kinase signalling pathways, it may be beneficial to target the restoration of the balance between pro- and anti-apoptotic mechanisms in HD rather than any one pathway in isolation. Despite being a neuroprotective anti-apoptotic pathway, the counter-intuitive inhibition of a hyper-activated AKT pathway may prove to be beneficial by restoring normal huntingtin localisation and IE gene expression by redressing the balance between pro- and anti-apoptotic pathways. However, suppressing AKT alone could ultimately enhance mutant huntingtin toxicity by failing to counter pro-apoptotic mechanisms. Therefore, a multi-target approach to restore the balance of compensatory and synergistic signal transduction mechanisms may be an efficacious therapeutic approach; this may be achieved by targeting upstream modulators of multiple kinase pathways, or by manipulating several targets simultaneously.
We demonstrate that the mislocalisation of mutant huntingtin is an early phenotype present in both an immortalised, and primary, embryonic cell model of HD, which can be modulated by the stimulation of kinase signalling pathways downstream of the EGFR. In particular, dysregulation of AKT1 and MEK1 kinase signalling pathways may contribute to aberrant huntingtin subcellular localisation. We hypothesise that the regulation of huntingtin localisation by kinase signalling could be a mechanism associated with transcriptional control, as the inhibition of both AKT1 and MEK1 activation was associated with alterations in both huntingtin localisation and IE gene expression. However, this study does not elucidate a direct functional relationship between huntingtin localisation and gene expression, and it is likely that mutant huntingtin impacts multiple mechanisms within cell nuclei. While we replicate the effect of EGF stimulation on the localisation of huntingtin in primary cells derived from the HdhQ111 mouse model of HD, our findings require additional validation in multiple models of HD before firm conclusions about the relationship between kinase signalling, huntingtin localisation and IE gene expression can be drawn. We have also not yet clarified the mechanism by which the alterations in huntingtin subcellular localisation have occurred. However, we suggest that the presence of an expanded huntingtin polyglutamine repeat may be responsible for alterations in kinase signalling that subsequently leads to the mislocalisation of mutant huntingtin, which may in turn contribute to the aberrant control of gene expression observed in HD. By using embryonic cell models of HD, we demonstrate that cellular dysfunction is likely to be present throughout development, which may have implications for the understanding of HD developmental pathogenesis. The inability of these mechanisms to maintain survival-associated functions with cumulative mutant huntingtin insult may then lead to the observable pathogenic disease cascade. Therefore the investigation of early signalling dysfunction in HD models may provide an efficacious approach to developing novel therapeutic targets to delay age of onset and slow symptom progression.
Supporting Information
For every cell, a circular region was individually selected within the nucleus (N), ‘perinuclear’ region (P) and cytoplasm (C), and a histogram displaying the mean pixel intensity for the selected region was generated. Within each cell, N/C and N/P pixel intensity ratios were calculated in order to compare the relative localisation of the huntingtin immunostaining, and to control for background variance between images and coverslips. Cell populations to be imaged were located by DAPI nuclear staining to avoid investigator bias. For each experiment, mean pixel intensities for N, P and C regions were measured blind to genotype and condition, and were taken from all cells present within 9 microscopy images derived from 3 individual coverslips. The presented data is a result of multiple experiments.
(TIF)
The amino-terminal epitope of huntingtin detected by Mab2166 colocalises with the ER marker calreticulin in the region immediately surrounding cell nuclei in StHdh Q7/7, StHdh Q7/111 and StHdh Q111/111 cells. This localisation pattern of huntingtin was then categorised as ‘perinuclear’ in following experiments. ‘Cytoplasmic’ localisation was considered as away from the more densely localised huntingtin in the ‘perinuclear’ area, which would not colocalise with calreticulin. B. 4x magnification of images in A. Magenta = huntingtin, green = calreticulin. Scale bar = 20μm.
(TIF)
Cells were fixed following 0, 5, 15 and 30 min. of stimulation with 100ng/ml EGF, labelled with Ab109115 against amino acids 1–100 of huntingtin, then analysed by confocal microscopy. Scale bar = 20μm. B. Quantitative analysis of immunofluorescence images in A. Nuclear/Cytoplasmic (N/C) and Nuclear/Perinuclear (N/P) mean pixel intensity ratios (MPI) for StHdh Q7/7, StHdh Q7/111 and StHdh Q111/111 cells following 0, 5, 15 and 30 min. of stimulation with 100ng/ml EGF. Mean pixel intensities were calculated from confocal microscopy images using GNU Image Manipulator. All images were randomised and analysed blind to genotype and length of time stimulated with EGF. Each condition consisted of 9 confocal microscopy images taken from 3 separate coverslips. Error bars = ± SEM. Data representative of 3 experiments. n = 60–87; * Denotes a significant difference from 0min.; # Denotes a significant difference from 5mins; */# p<0.05, **/## p<0.01, ***/### p<0.001.
(TIF)
Cells were fixed following 0, 5, 15 and 30 min. of stimulation with 100ng/ml EGF, labelled with anti-S421, then visualised by fluorescence microscopy. Each condition consisted of 9 confocal microscopy images taken from 3 separate coverslips. Scale bar = 20μm.
(TIF)
891 cells were assayed for these striatal cell markers, of which 93.83% were positively labelled.
(TIF)
Mean pixel intensities were calculated from confocal microscopy images using GNU Image Manipulator. All images were randomised and analysed blind to genotype and length of time stimulated with EGF. Each condition consisted of 9 confocal microscopy images taken from 3 separate coverslips. n = 49–70. Error bars = ± SEM. Data representative of 3 experiments; * Denotes a significant difference from 0min.; *p<0.05, ** p<0.01.
(TIF)
Error bars = SEM. Light grey bars and asterisks signify statistically significant differences between DMSO conditions. Black asterisks and hashes indicate statistically significant differences between DMSO vs AKT inhibitor conditions and DMSO vs MEK inhibitor conditions, respectively. Data representative of three experiments. n = 78–140. */# p<0.05, **/## p<0.01, ***/### p<0.001.
(TIF)
Statistical analysis was conducted on ΔCt values. Egr1 expression in both genotypes was significantly increased following EGF stimulation (both p<0.001), and the extent of this increase was significantly larger in HdhQ111/111 primary cells compared to HdhQ7/7 cells (p<0.05). Error bars = ± SEM. N = 5. * p<0.05, ** p<0.01, ***p<0.001.
(TIF)
Acknowledgments
We thank Professor Marcy MacDonald for the kind gift of the StHdh Q111 cells lines, and Dr. Sandrine Humbert for the generous gift of the S421 antibody. We also thank Yateen Patel for maintaining the HdhQ111 mouse colony and co-ordinating the staged pregnancies. KRB was supported by a Wellcome Trust studentship. The breeding colony is supported by CHDI.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This project was funded by a Wellcome Trust Scholarship awarded to KRB, grant number 089640/Z/09/Z (www.wellcome.ac.uk). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Buckley NJ, Johnson R, Zuccato C, Bithell A, Cattaneo E. The role of REST in transcriptional and epigenetic dysregulation in Huntington’s disease. Neurobiol Dis. Elsevier Inc.; 2010;39: 28–39. 10.1016/j.nbd.2010.02.003 [DOI] [PubMed] [Google Scholar]
- 2. Novak MJU, Tabrizi SJ. Huntington’s disease. BMJ. 2010;341: 34–40. 10.1136/bmj.c3109 [DOI] [Google Scholar]
- 3. Wexler NS. Huntington’s disease: advocacy driving science. Annu Rev Med. 2012;63: 1–22. 10.1146/annurev-med-050710-134457 [DOI] [PubMed] [Google Scholar]
- 4. Vonsattel JPG, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57: 369–384. [DOI] [PubMed] [Google Scholar]
- 5. Rosas HD, Salat DH, Lee SY, Zaleta AK, Pappu V, Fischl B, et al. Cerebral cortex and the clinical expression of Huntington’s disease: complexity and heterogeneity. Brain. 2008;131: 1057–1068. 10.1093/brain/awn025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aylward EH, Sparks BF, Field KM, Yallapragada V, Shpritz BD, Rosenblatt A, et al. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology. 2004;63: 66–72. [DOI] [PubMed] [Google Scholar]
- 7. Ciarmiello A, Cannella M, Lastoria S, Simonelli M, Frati L, Rubinsztein DC, et al. Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington’s disease. J Nucl Med. 2006;47: 215–222. [PubMed] [Google Scholar]
- 8. Levine MS, Cepeda C, Hickey MA, Fleming SM, Chesselet MF. Genetic mouse models of Huntington’s and Parkinson's diseases: illuminating but imperfect. Trends Neurosci. 2004;27: 691–697. 10.1016/j.tins.2004.08.008 [DOI] [PubMed] [Google Scholar]
- 9. Kuwert T, Lange HW, Langen KJ, Herzog H, Aulich A, Feinendegen LE. Cortical and subcortical glucose consumption measured by PET in patients with Huntingtons-disease. Brain. 1990;113: 1405–1423. 10.1093/brain/113.5.1405 [DOI] [PubMed] [Google Scholar]
- 10. Grafton ST, Mazziota JC, Pahl JJ, Stgeorgehyslop P, Haines JL, Gusella J, et al. Serial changes of cerebral glucose-metabolism and caudate size in persons at risk for Huntingtons-disease. Arch Neurol. 1992;49: 1161–1167. [DOI] [PubMed] [Google Scholar]
- 11. Brognard J, Hunter T. Protein kinase signalling networks in Cancer. Curr Opin Genet Dev. 2011;21: 4–11. 10.1016/j.gde.2010.10.012.Protein [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leng Y, Steiler TL, Zierath JR. Mitogen-Activated Protein Kinase Signaling in Skeletal Muscle From Lean and ob / ob Mice. Diabetes. 2004;53: 1436–1444. [DOI] [PubMed] [Google Scholar]
- 13. Gines S, Ivanova E, Seong I-S, Saura C a, MacDonald ME. Enhanced Akt signaling is an early pro-survival response that reflects N-methyl-D-aspartate receptor activation in Huntington’s disease knock-in striatal cells. J Biol Chem. 2003;278: 50514–22. 10.1074/jbc.M309348200 [DOI] [PubMed] [Google Scholar]
- 14. Saavedra A, García-Martínez JM, Xifró X, Giralt A, Torres-Peraza JF, Canals JM, et al. PH domain leucine-rich repeat protein phosphatase 1 contributes to maintain the activation of the PI3K/Akt pro-survival pathway in Huntington’s disease striatum. Cell Death Differ. 2010;17: 324–35. 10.1038/cdd.2009.127 [DOI] [PubMed] [Google Scholar]
- 15. Liu YF, Deth RC, Devys D. SH3 Domain-dependent Association of Huntingtin with Epidermal Growth Factor Receptor Signaling Complexes. J Biol Chem. 1997;272: 8121–8124. [DOI] [PubMed] [Google Scholar]
- 16. Apostol BL, Illes K, Pallos J, Bodai L, Wu J, Strand A, et al. Mutant huntingtin alters MAPK signaling pathways in PC12 and striatal cells: ERK1/2 protects against mutant huntingtin-associated toxicity. Hum Mol Genet. 2006;15: 273–85. 10.1093/hmg/ddi443 [DOI] [PubMed] [Google Scholar]
- 17. Iijima-ando K, Wu P, Drier EA, Iijima K, Yin JCP. cAMP-response element-binding protein and heat-shock protein 70 additively suppress polyglutamine-mediated toxicity in Drosophila. Proc Natl Acad Sci U S A. 2005;102: 10261–10266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schilling B, Gafni J, Torcassi C, Cong X, Row RH, LaFevre-Bernt M a, et al. Huntingtin phosphorylation sites mapped by mass spectrometry. Modulation of cleavage and toxicity. J Biol Chem. 2006;281: 23686–97. 10.1074/jbc.M513507200 [DOI] [PubMed] [Google Scholar]
- 19. Bauer PO, Nukina N. The pathogenic mechanisms of polyglutamine diseases and current therapeutic strategies. J Neurochem. 2009;110: 1737–65. 10.1111/j.1471-4159.2009.06302.x [DOI] [PubMed] [Google Scholar]
- 20. Gu X, Greiner ER, Mishra R, Kodali R, Osmand A, Finkbeiner S, et al. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron. 2009;64: 828–40. 10.1016/j.neuron.2009.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thompson LM, Aiken CT, Kaltenbach LS, Agrawal N, Illes K, Khoshnan A, et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. 2009;187: 1083–99. 10.1083/jcb.200909067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Warby SC, Doty CN, Graham RK, Shively J, Singaraja RR, Hayden MR. Phosphorylation of huntingtin reduces the accumulation of its nuclear fragments. Mol Cell Neurosci. 2009;40: 121–7. 10.1016/j.mcn.2008.09.007 [DOI] [PubMed] [Google Scholar]
- 23. Atwal RS, Desmond CR, Caron N, Maiuri T, Xia J, Sipione S, et al. Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat Chem Biol. 2011;7: 453–60. 10.1038/nchembio.582 [DOI] [PubMed] [Google Scholar]
- 24. Maiuri T, Woloshansky T, Xia J, Truant R. The huntingtin N17 domain is a multifunctional CRM1 and Ran-dependent nuclear and cilial export signal. Hum Mol Genet. 2013;22: 1383–94. 10.1093/hmg/dds554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Truant R. Nucleocytoplasmic transport of huntingtin and Huntington’s disease. Clin Neurosci Res. 2003;3: 157–164. 10.1016/S1566-2772(03)00058-6 [DOI] [Google Scholar]
- 26. Truant R, Atwal RS, Burtnik A. Nucleocytoplasmic trafficking and transcription effects of huntingtin in Huntington’s disease. Prog Neurobiol. 2007;83: 211–27. 10.1016/j.pneurobio.2006.11.004 [DOI] [PubMed] [Google Scholar]
- 27. Hilditch-Maguire P, Trettel F, Passani L a, Auerbach a, Persichetti F, MacDonald ME. Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum Mol Genet. 2000;9: 2789–97. [DOI] [PubMed] [Google Scholar]
- 28. Tao T, Tartakoff a M. Nuclear relocation of normal huntingtin. Traffic. 2001;2: 385–94. [DOI] [PubMed] [Google Scholar]
- 29. Kegel KB, Meloni AR, Yi Y, Kim YJ, Doyle E, Cuiffo BG, et al. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J Biol Chem. 2002;277: 7466–76. 10.1074/jbc.M103946200 [DOI] [PubMed] [Google Scholar]
- 30. Boutell JM, Thomas P, Neal JW, Weston VJ, Duce J, Harper PS, et al. Aberrant interactions of transcriptional repressor proteins with the Huntington’s disease gene product, huntingtin. Hum Mol Genet. 1999;8: 1647–1655. 10.1093/hmg/8.9.1647 [DOI] [PubMed] [Google Scholar]
- 31. McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, et al. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet. 2000;9: 2197–2202. 10.1093/hmg/9.14.2197 [DOI] [PubMed] [Google Scholar]
- 32. Shimohata T, Nakajima T, Yamada M, Uchida C, Onodera O, Naruse S, et al. Expanded polyglutamine stretches interact with TAFII130, interfering with CREB-dependent transcription. Nat Genet. 2000;26: 29–36. 10.1038/79139 [DOI] [PubMed] [Google Scholar]
- 33. Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, et al. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97: 6763–6768. 10.1073/pnas.100110097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dunah AW, Jeong H, Griffin A, Kim Y-M, Standaert DG, Hersch SM, et al. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science. 2002;296: 2238–43. 10.1126/science.1072613 [DOI] [PubMed] [Google Scholar]
- 35. Li S, Cheng AL, Zhou H, Lam S, Rao M, Li H, et al. Interaction of Huntington Disease Protein with Transcriptional Activator Sp1. Mol Cell Biol. 2002;22: 1277–1287. 10.1128/MCB.22.5.1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bae B-I, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron. 2005;47: 29–41. 10.1016/j.neuron.2005.06.005 [DOI] [PubMed] [Google Scholar]
- 37. Benn CL, Sun T, Sadri-Vakili G, McFarland KN, DiRocco DP, Yohrling GJ, et al. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J Neurosci. 2008;28: 10720–33. 10.1523/JNEUROSCI.2126-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Johnson R, Zuccato C, Belyaev ND, Guest DJ, Cattaneo E, Buckley NJ. A microRNA-based gene dysregulation pathway in Huntington’s disease. Neurobiol Dis. 2008;29: 438–45. 10.1016/j.nbd.2007.11.001 [DOI] [PubMed] [Google Scholar]
- 39. Sun J, Xu H, Negi S, Subramony SH, Hebert MD. Differential effects of polyglutamine proteins on nuclear organization and artificial reporter splicing. J Neurosci Res. 2007;85: 2306–2317. 10.1002/jnr.21369 [DOI] [PubMed] [Google Scholar]
- 40. Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3: 802–8. 10.1038/ncb0901-802 [DOI] [PubMed] [Google Scholar]
- 41. Cabodi S, Morello V, Masi A, Cicchi R, Broggio C, Distefano P, et al. Convergence of integrins and EGF receptor signaling via PI3K/Akt/FoxO pathway in early gene Egr-1 expression. J Cell Physiol. 2009;218: 294–303. 10.1002/jcp.21603 [DOI] [PubMed] [Google Scholar]
- 42. Kong P-J, Kil M-O, Lee H, Kim S-S, Johnson GVW, Chun W. Increased expression of Bim contributes to the potentiation of serum deprivation-induced apoptotic cell death in Huntington’s disease knock-in striatal cell line. Neurol Res. 2009;31: 77–83. 10.1179/174313208X331572 [DOI] [PubMed] [Google Scholar]
- 43. Ro Y-T, Jang B-K, Shin CY, Park EU, Kim CG, Yang S-I. Akt regulates the expression of MafK, synaptotagmin I, and syntenin-1, which play roles in neuronal function. J Biomed Sci. 2010;17: 18 10.1186/1423-0127-17-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp a H, Persichetti F, et al. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9: 2799–809. [DOI] [PubMed] [Google Scholar]
- 45. Dunnett S, Bjorklund A. Dissecting embryonic neural tissues for transplantation In: Boulton A, Baker G, Dunnett S, editors. Neural Transplantation Methods. Totowa, New Jersey, USA: HUMANA PRESS INC; 1999. pp. 3–25. [Google Scholar]
- 46. Ginés S, Paoletti P, Alberch J. Impaired TrkB-mediated ERK1/2 activation in huntington disease knock-in striatal cells involves reduced p52/p46 Shc expression. J Biol Chem. 2010;285: 21537–48. 10.1074/jbc.M109.084202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Warby SC, Chan EY, Metzler M, Gan L, Singaraja RR, Crocker SF, et al. Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum Mol Genet. 2005;14: 1569–77. 10.1093/hmg/ddi165 [DOI] [PubMed] [Google Scholar]
- 48. Humbert S, Bryson EA, Cordelie FP, Connors NC, Datta SR, Finkbeiner S, et al. The IGF-1 / Akt Pathway is neuroprotective in Huntington’s disease and involves huntingtin phosphorylation by Akt. Dev Cell. 2002;2: 831–837. [DOI] [PubMed] [Google Scholar]
- 49. Slow EJ. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12: 1555–1567. 10.1093/hmg/ddg169 [DOI] [PubMed] [Google Scholar]
- 50. Carri AD, Onorati M, Lelos MJ, Castiglioni V, Faedo A, Menon R, et al. Developmentally coordinated extrinsic signals drive human pluripotent stem cell differentiation toward authentic DARPP-32+ medium-sized spiny neurons. Development. 2013;140: 301–12. 10.1242/dev.084608 [DOI] [PubMed] [Google Scholar]
- 51. Taylor DM, Moser R, Régulier E, Breuillaud L, Dixon M, Beesen AA, et al. MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in Huntington’s disease via additive effects of JNK and p38 inhibition. J Neurosci. 2013;33: 2313–25. 10.1523/JNEUROSCI.4965-11.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23: 217–247. 10.1146/annurev.neuro.23.1.217 [DOI] [PubMed] [Google Scholar]
- 53. Owada Y, Utsunomiya A, Yoshimoto T, Kondo H. Expression of mRNA for Akt, serine-threonine protein kinase, in the brain during development and its transient enhancement following axotomy of hypoglossal nerve. J Mol Neurosci. 1997;9: 27–33. 10.1007/BF02789392 [DOI] [PubMed] [Google Scholar]
- 54. Chong ZZ, Li F, Maiese K. Activating Akt and the brain’s resources to drive cellular survival and prevent inflammatory injury. Histol Histopathol. 2005;20: 299–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Humbert S. Huntingtin phosphorylation and signaling pathways that regulate toxicity in Huntington’s disease. Clin Neurosci Res. 2003;3: 149–155. 10.1016/S1566-2772(03)00057-4 [DOI] [Google Scholar]
- 56. Ritch JJ, Valencia A, Alexander J, Sapp E, Gatune L, Sangrey GR, et al. Multiple phenotypes in Huntington disease mouse neural stem cells. Mol Cell Neurosci. 2012; 10.1016/j.mcn.2012.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liévens J-C, Iché M, Laval M, Faivre-Sarrailh C, Birman S. AKT-sensitive or insensitive pathways of toxicity in glial cells and neurons in Drosophila models of Huntington’s disease. Hum Mol Genet. 2008;17: 882–94. 10.1093/hmg/ddm360 [DOI] [PubMed] [Google Scholar]
- 58. Saavedra A, Giralt A, Rué L, Xifró X, Xu J, Ortega Z, et al. Striatal-Enriched Protein Tyrosine Phosphatase Expression and Activity in Huntington’s Disease: A STEP in the Resistance to Excitotoxicity. J Neurosci. 2011;31: 8150–8162. 10.1523/JNEUROSCI.3446-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bowles KR, Jones L. Kinase Signalling in Huntington‘s Disease. 2014;3: 89–123. 10.3233/JHD-140106 [DOI] [PubMed] [Google Scholar]
- 60. Zala D, Colin E, Rangone H, Liot G, Humbert S, Saudou F. Phosphorylation of mutant huntingtin at S421 restores anterograde and retrograde transport in neurons. Hum Mol Genet. 2008;17: 3837–46. 10.1093/hmg/ddn281 [DOI] [PubMed] [Google Scholar]
- 61. Pineda JR, Pardo R, Zala D, Yu H, Humbert S, Saudou F. Genetic and pharmacological inhibition of calcineurin corrects the BDNF transport defect in Huntington’s disease. Mol Brain. 2009;2: 33 10.1186/1756-6606-2-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pennuto M, Palazzolo I, Poletti A. Post-translational modifications of expanded polyglutamine proteins: impact on neurotoxicity. Hum Mol Genet. 2009;18: R40–7. 10.1093/hmg/ddn412 [DOI] [PubMed] [Google Scholar]
- 63. Rangone Á, Poizat G, Troncoso J, Ross CA, Macdonald ME, Humbert S. The serum- and glucocorticoid-induced kinase SGK inhibits mutant huntingtin-induced toxicity by phosphorylating serine 421 of huntingtin. Eur J Neurosci. 2004;19: 273–279. 10.1111/j.1460-9568.2003.03131.x [DOI] [PubMed] [Google Scholar]
- 64. Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, Truant R. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum Mol Genet. 2007;16: 2600–15. 10.1093/hmg/ddm217 [DOI] [PubMed] [Google Scholar]
- 65. Havel LS, Wang C-E, Wade B, Huang B, Li S, Li X-J. Preferential accumulation of N-terminal mutant huntingtin in the nuclei of striatal neurons is regulated by phosphorylation. Hum Mol Genet. 2011; ddr023–. 10.1093/hmg/ddr023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Desmond CR, Maiuri T, Truant R. A multifunctional, multi-pathway intracellular localization signal in Huntingtin. Commun Integr Biol. 2013;6: e23318 10.4161/cib.23318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Aiken CT, Steffan JS, Guerrero CM, Khashwji H, Lukacsovich T, Simmons D, et al. Phosphorylation of threonine 3: implications for Huntingtin aggregation and neurotoxicity. J Biol Chem. 2009;284: 29427–36. 10.1074/jbc.M109.013193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304: 100–4. 10.1126/science.1092194 [DOI] [PubMed] [Google Scholar]
- 69. Hoogeveen a T, Willemsen R, Meyer N, de Rooij KE, Roos R a, van Ommen GJ, et al. Characterization and localization of the Huntington disease gene product. Hum Mol Genet. 1993;2: 2069–73. A. [DOI] [PubMed] [Google Scholar]
- 70. Dorsman JC, Smoor M a, Maat-Schieman ML, Bout M, Siesling S, van Duinen SG, et al. Analysis of the subcellular localization of huntingtin with a set of rabbit polyclonal antibodies in cultured mammalian cells of neuronal origin: comparison with the distribution of huntingtin in Huntington’s disease autopsy brain. Philos Trans R Soc Lond B Biol Sci. 1999;354: 1061–7. 10.1098/rstb.1999.0459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wheeler VC. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet. 2000;9: 503–513. 10.1093/hmg/9.4.503 [DOI] [PubMed] [Google Scholar]
- 72. Wheeler VC, Gutekunst C-A, Vrbanac V, Lebel L-A, Schilling G, Hersch S, et al. Early phenotypes that presage late-onset neurodegenerative disease allow testing of modifiers in Hdh CAG knock-in mice. Hum Mol Genet. 2002;11: 633–40. [DOI] [PubMed] [Google Scholar]
- 73. Rockabrand E, Slepko N, Pantalone A, Nukala VN, Kazantsev A, Marsh JL, et al. The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum Mol Genet. 2007;16: 61–77. 10.1093/hmg/ddl440 [DOI] [PubMed] [Google Scholar]
- 74. Seong IS, Woda JM, Song J-J, Lloret A, Abeyrathne PD, Woo CJ, et al. Huntingtin facilitates polycomb repressive complex 2. Hum Mol Genet. 2010;19: 573–83. 10.1093/hmg/ddp524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sawa A, Nagata E, Sutcliffe S, Dulloor P, Cascio MB, Ozeki Y, et al. Huntingtin is cleaved by caspases in the cytoplasm and translocated to the nucleus via perinuclear sites in Huntington’s disease patient lymphoblasts. Neurobiol Dis. 2005;20: 267–74. 10.1016/j.nbd.2005.02.013 [DOI] [PubMed] [Google Scholar]
- 76. Pal A, Severin F, Lommer B, Shevchenko A, Zerial M. Huntingtin-HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington’s disease. J Cell Biol. 2006;172: 605–618. 10.1083/jcb.200509091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Del Toro D, Alberch J, Lazaro-Dieguez F, Martin-Ibanez R, Xifro X, Egea G, et al. Mutant Huntingtin Impairs Post-Golgi Trafficking to Lysosomes by Delocalizing Optineurin/Rab8 Complex from the Golgi Apparatus. Mol Biol Cell. 2009;20: 1478–1492. 10.1091/mbc.E08-07-0726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Melone M a B, Calarco A, Petillo O, Margarucci S, Colucci-D’Amato L, Galderisi U, et al. Mutant huntingtin regulates EGF receptor fate in non-neuronal cells lacking wild-type protein. Biochim Biophys Acta. 2013;1832: 105–13. 10.1016/j.bbadis.2012.09.001 [DOI] [PubMed] [Google Scholar]
- 79. Song C, Perides G, Liu YF. Expression of full-length polyglutamine-expanded Huntingtin disrupts growth factor receptor signaling in rat pheochromocytoma (PC12) cells. J Biol Chem. 2002;277: 6703–7. 10.1074/jbc.M110338200 [DOI] [PubMed] [Google Scholar]
- 80. Liévens J-C, Rival T, Iché M, Chneiweiss H, Birman S. Expanded polyglutamine peptides disrupt EGF receptor signaling and glutamate transporter expression in Drosophila. Hum Mol Genet. 2005;14: 713–24. 10.1093/hmg/ddi067 [DOI] [PubMed] [Google Scholar]
- 81. Xia J. Huntingtin contains a highly conserved nuclear export signal. Hum Mol Genet. 2003;12: 1393–1403. 10.1093/hmg/ddg156 [DOI] [PubMed] [Google Scholar]
- 82. Desmond CR, Atwal RS, Xia J, Truant R. Identification of a karyopherin β1/β2 proline-tyrosine nuclear localization signal in huntingtin protein. J Biol Chem. 2012;287: 39626–33. 10.1074/jbc.M112.412379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Li X-J, Li S-H, Sharp AH, Nucifora FC Jr, Schilling G, Lanahan A, et al. A huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995;378: 398–402. [DOI] [PubMed] [Google Scholar]
- 84. Faber PW, Barnes GT, Srinidhi J, Chen J, Gusella JF, MacDonald ME. Huntingtin interacts with a family of WW domain proteins. Hum Mol Genet. 1998;7: 1463–74. A. [DOI] [PubMed] [Google Scholar]
- 85. Ferrigno P, Silver P a. Polyglutamine expansions: proteolysis, chaperones, and the dangers of promiscuity. Neuron. 2000;26: 9–12. [DOI] [PubMed] [Google Scholar]
- 86. Kim YJ, Sapp E, Cuiffo BG, Sobin L, Yoder J, Kegel KB, et al. Lysosomal proteases are involved in generation of N-terminal huntingtin fragments. Neurobiol Dis. 2006;22: 346–356. 10.1016/j.nbd.2005.11.012 [DOI] [PubMed] [Google Scholar]
- 87. Reddy PH, Williams M, Tagle DA. Recent advances in understanding the pathogenesis of Huntington’s disease. Trends Neurosci. 1999;22: 248–255. [DOI] [PubMed] [Google Scholar]
- 88. Crook ZR, Housman D. Huntington’s Disease: Can Mice Lead the Way to Treatment? Neuron. 2011;69: 423–435. 10.1016/j.neuron.2010.12.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lee J-M, Ivanova E V, Seong IS, Cashorali T, Kohane I, Gusella JF, et al. Unbiased gene expression analysis implicates the huntingtin polyglutamine tract in extra-mitochondrial energy metabolism. PLoS Genet. 2007;3: e135 10.1371/journal.pgen.0030135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Fossale E, Wheeler VC, Vrbanac V, Lebel L- A, Teed A, Mysore JS, et al. Identification of a presymptomatic molecular phenotype in Hdh CAG knock-in mice. Hum Mol Genet. 2002;11: 2233–41. [DOI] [PubMed] [Google Scholar]
- 91. Carnemolla A, Fossale E, Agostoni E, Michelazzi S, Calligaris R, De Maso L, et al. Rrs1 is involved in endoplasmic reticulum stress response in Huntington disease. J Biol Chem. 2009;284: 18167–73. 10.1074/jbc.M109.018325 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
For every cell, a circular region was individually selected within the nucleus (N), ‘perinuclear’ region (P) and cytoplasm (C), and a histogram displaying the mean pixel intensity for the selected region was generated. Within each cell, N/C and N/P pixel intensity ratios were calculated in order to compare the relative localisation of the huntingtin immunostaining, and to control for background variance between images and coverslips. Cell populations to be imaged were located by DAPI nuclear staining to avoid investigator bias. For each experiment, mean pixel intensities for N, P and C regions were measured blind to genotype and condition, and were taken from all cells present within 9 microscopy images derived from 3 individual coverslips. The presented data is a result of multiple experiments.
(TIF)
The amino-terminal epitope of huntingtin detected by Mab2166 colocalises with the ER marker calreticulin in the region immediately surrounding cell nuclei in StHdh Q7/7, StHdh Q7/111 and StHdh Q111/111 cells. This localisation pattern of huntingtin was then categorised as ‘perinuclear’ in following experiments. ‘Cytoplasmic’ localisation was considered as away from the more densely localised huntingtin in the ‘perinuclear’ area, which would not colocalise with calreticulin. B. 4x magnification of images in A. Magenta = huntingtin, green = calreticulin. Scale bar = 20μm.
(TIF)
Cells were fixed following 0, 5, 15 and 30 min. of stimulation with 100ng/ml EGF, labelled with Ab109115 against amino acids 1–100 of huntingtin, then analysed by confocal microscopy. Scale bar = 20μm. B. Quantitative analysis of immunofluorescence images in A. Nuclear/Cytoplasmic (N/C) and Nuclear/Perinuclear (N/P) mean pixel intensity ratios (MPI) for StHdh Q7/7, StHdh Q7/111 and StHdh Q111/111 cells following 0, 5, 15 and 30 min. of stimulation with 100ng/ml EGF. Mean pixel intensities were calculated from confocal microscopy images using GNU Image Manipulator. All images were randomised and analysed blind to genotype and length of time stimulated with EGF. Each condition consisted of 9 confocal microscopy images taken from 3 separate coverslips. Error bars = ± SEM. Data representative of 3 experiments. n = 60–87; * Denotes a significant difference from 0min.; # Denotes a significant difference from 5mins; */# p<0.05, **/## p<0.01, ***/### p<0.001.
(TIF)
Cells were fixed following 0, 5, 15 and 30 min. of stimulation with 100ng/ml EGF, labelled with anti-S421, then visualised by fluorescence microscopy. Each condition consisted of 9 confocal microscopy images taken from 3 separate coverslips. Scale bar = 20μm.
(TIF)
891 cells were assayed for these striatal cell markers, of which 93.83% were positively labelled.
(TIF)
Mean pixel intensities were calculated from confocal microscopy images using GNU Image Manipulator. All images were randomised and analysed blind to genotype and length of time stimulated with EGF. Each condition consisted of 9 confocal microscopy images taken from 3 separate coverslips. n = 49–70. Error bars = ± SEM. Data representative of 3 experiments; * Denotes a significant difference from 0min.; *p<0.05, ** p<0.01.
(TIF)
Error bars = SEM. Light grey bars and asterisks signify statistically significant differences between DMSO conditions. Black asterisks and hashes indicate statistically significant differences between DMSO vs AKT inhibitor conditions and DMSO vs MEK inhibitor conditions, respectively. Data representative of three experiments. n = 78–140. */# p<0.05, **/## p<0.01, ***/### p<0.001.
(TIF)
Statistical analysis was conducted on ΔCt values. Egr1 expression in both genotypes was significantly increased following EGF stimulation (both p<0.001), and the extent of this increase was significantly larger in HdhQ111/111 primary cells compared to HdhQ7/7 cells (p<0.05). Error bars = ± SEM. N = 5. * p<0.05, ** p<0.01, ***p<0.001.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.