

Abstract
Parasitic diseases caused by protozoan pathogens lead to hundreds of thousands of deaths per year in addition to substantial suffering and socioeconomic decline for millions of people worldwide. The lack of effective vaccines coupled with the widespread emergence of drug‐resistant parasites necessitates that the research community take an active role in understanding host–parasite infection biology in order to develop improved therapeutics. Recent advances in next‐generation sequencing and the rapid development of publicly accessible genomic databases for many human pathogens have facilitated the application of systems biology to the study of host–parasite interactions. Over the past decade, these technologies have led to the discovery of many important biological processes governing parasitic disease. The integration and interpretation of high‐throughput ‐omic data will undoubtedly generate extraordinary insight into host–parasite interaction networks essential to navigate the intricacies of these complex systems. As systems analysis continues to build the foundation for our understanding of host–parasite biology, this will provide the framework necessary to drive drug discovery research forward and accelerate the development of new antiparasitic therapies. WIREs Syst Biol Med 2015, 7:381–400. doi: 10.1002/wsbm.1311
For further resources related to this article, please visit the WIREs website.
INTRODUCTION
Protozoan parasites infect over a half billion people worldwide, and continue to play a significant role in shaping global mortality and morbidity rates despite decades of research.1 Some important human diseases caused by these pathogens include malaria, leishmaniasis, African sleeping sickness, toxoplasmosis, Chagas disease, and amoebiasis. The lack of government funding for many of these classically ‘neglected’ pathogens, the recent emergence of antiparasitic drug resistance, and the absence of licensed vaccines warrant global concern. In addition, host–parasite research is impeded by specific technical and resource limitations. New cost‐effective, high‐throughput strategies are therefore necessary to circumvent these obstacles and to develop novel therapeutics.
The postgenomic era has generated unparalleled opportunities for creating and integrating systems biology data (i.e., organism‐ or cellular‐scale data produced through a number of ‐omic, or system‐wide, technologies). This holistic approach is in direct contrast to conventional reductionist methods that ‘reduce’ systems into smaller, more tractable units. Systems‐based methods are particularly useful to study complex biological relationships that are: (1) open, with constant information exchange and a net flow of resources, and (2) stochastic, with spatial, temporal, and population heterogeneity.2 Host–parasite systems embody all of these defining characteristics. ‐Omic technologies are also much more efficient and economical when comparing the cumulative time, labor, and cost per gene to traditional reductionist strategies. Not surprisingly, these methods have been critical for improving our understanding of host–parasite relationships and accelerating antiparasitic drug discovery.3, 4 In this review, we discuss the current state of host–parasite systems biology research. This includes the various obstacles faced by parasite researchers, the advancements and feasibility of several genome‐wide technologies, and the key research areas benefiting from such approaches. We aim to emphasize major advances from the past few years as well as the specific hypotheses and gaps emerging from these studies.
UNIQUE CHALLENGES OF THE HOST–PARASITE INTERFACE
Complex Life Cycles Within Multiple Hosts
Parasites have evolved elegant strategies to survive and replicate within their hosts. One strategy includes constantly changing their cellular state in order to progress through their life cycle, while simultaneously evading recognition by the host immune system.1 The vast number of developmental stages, combined with distinct tissue tropisms, increases the complexity of host–parasite interactions (Table 1). In this review, we focus on the following species owing to their global impact on human health and influence in the research community: (1) the Apicomplexans Toxoplasma gondii, which causes toxoplasmosis, Plasmodium spp., which cause malaria, and Cryptosporidium spp., which cause the diarrheal disease cryptosporidosis; (2) the Kinetoplastids Trypanosoma brucei, which causes African sleeping sickness, Trypanosoma cruzi, which causes Chagas disease, and the Leishmania parasites, which cause both cutaneous and visceral leishmaniasis; (3) the Diplomonad Giardia lamblia, which causes the intestinal disease giardiasis; and (4) the Amoebozoa Entamoeba histolytica, which causes amoebic dysentery (Table 1). Many of these parasites, such as the Apicomplexans and the Kinetoplastids, are vector‐borne, intracellular pathogens that complete their life cycle within multiple hosts. One exception is T. brucei, which carries out its life cycle extracellularly. Others, such as Cryptosporidium, Entamoeba, and Giardia, can develop into infectious, resistant cysts that survive outside of their hosts and are generally spread via the fecal–oral route.
Table 1.
Protozoan Parasites That Cause Human Disease
| Species | Disease | Host(s) | Human Tissue Tropism | Parasite Developmental Stages | |
|---|---|---|---|---|---|
| Apicomplexans | Toxoplasma gondii | Toxoplasmosis | Domestic cats and humans | Intestine, muscle, neural tissue | Oocysts, tachyzoites, tissue cysts |
| Plasmodium spp. | Malaria | Infected female Anopheles mosquitos and humans | Hepatocytes, erythrocytes, central nervous system | Sporozoites, liver stages (trophozoites, shizonts, merozoites, hypnozoites in some species), blood stages (erythrocyte ring stages, mature trophozoites, shizonts, merozoites), gametocytes, mosquito stages (zygotes, ookinetes, oocysts) | |
| Cryptosporidium spp. | Cryptosporidiosis | Humans | Epithelial cells of gastrointestinal or respiratory tract | Oocysts, sporozoites, trophozoites, meronts, merozoites, gamonts, microgamonts and macrogamonts, zygotes | |
| Kinetoplastids | Trypanosoma brucei | African sleeping sickness | Tsetse fly and humans | Bloodstream, lymphatic system, central nervous system | Metacyclic trypomastigotes, bloodstream trypomastigotes, procyclic trypomastigotes, epimastigotes |
| Trypanosoma cruzi | Chagas disease | Triatomine bug and humans | A variety of cell types near the site(s) of infection, bloodstream | Metacyclic trypomastigotes, intracellular amastigotes, bloodstream trypomastigotes, epimastigotes | |
| Leishmania spp. | Leishmaniasis | Sandflies and humans | Mononuclear phagocytes in various tissues | Promastigotes, amastigotes | |
| Diplomonads | Giardia lamblia | Giardiasis | Humans | Small intestine, proximal small bowel, colon | Cysts, trophozoites |
| Amoebozoa | Entamoeba histolytica | Amoebic dysentery | Humans | Small intestine, large intestine, liver, brain, lungs | Cysts, trophozoites |
Information gathered from Centers for Disease Control (CDC), www.cdc.gov.
Because of the important differences in each life‐cycle stage, researchers must consider these unique developmental niches as separate systems when studying host–parasite interactions. This is especially important for systems‐based analysis, as parasites display periodic stage‐dependent gene expression.5 Accordingly, even slight asynchrony within parasite samples can result in inaccurate gene expression measurements, severely limiting statistical power. This is particularly challenging when analyzing clinical samples ex vivo, as parasite populations are rarely homogeneous. Therefore, researchers often utilize specialized techniques in order to synchronize parasites in culture, isolate specific cellular stages from mixed culture, or computationally remove stochastic noise.6 While this experimental isolation of developmental stages will aid in the understanding of stage‐specific host–parasite interactions, it will also be important for future systems‐based studies to integrate this knowledge into a multistage model more representative of physiological mixed parasite populations.
Challenging In Vitro Culture
While it is certainly possible to utilize systems‐based approaches for in vivo and ex vivo studies, the establishment of in vitro methods is particularly useful for many high‐throughput applications. The complex nature of each parasite's life cycle often requires multiple in vitro culture systems in order to study all of the developmental stages. While some parasite stages are easily propagated in culture, others are not.7, 8 For example, blood‐stage Plasmodium falciparum parasites can be maintained almost indefinitely in culture if supplied with fresh erythrocytes; however, sporozoites are generally freshly isolated from the salivary glands of infected mosquitos when studying liver‐stage infection. In order to study hypnozoites (the clinically dormant hepatic stage of Plasmodium vivax and Plasmodium ovale), researchers rely on technically challenging, time‐intensive assays only available in locations where the species are accessible.9 The absence of methods to isolate developmentally synchronized cysts presents a major hurdle for the study of encystation and excystation by enteric parasites such as E. histolytica 10 and Cryptosporidium parvum.11 Furthermore, low in vitro infection rates for many protozoan pathogens often lead to insufficient material for systems‐based analysis.
Large Uncharacterized Genomes
Not only are parasite genomes generally larger and more complex than their prokaryotic counterparts, but their functional characterization and annotation is severely limited by lack of both genetic tools and resources.3 Fully sequenced and annotated genomes greatly strengthen many areas of systems‐biology research; this includes determination of coding and noncoding reading frames, alternative splice variants, and the assignment of gene functions. Although many important protozoan parasite genomes have been sequenced, an overwhelming percentage of genes are still assigned ‘hypothetical’ functions, as illustrated in Figure 1. This lack of functional characterization is correlated with the relative magnitude of the research (Figure 1), as well as other factors such as the genetic intractability of certain species. The 24‐Mb P. falciparum genome, for example, is extremely AT‐rich (80.6%) and therefore traditional genetic approaches are particularly challenging.3 Not surprisingly, about 40% of the genome is still uncharacterized. For other parasites such as T. gondii and Leishmania major, this number is even higher, with more than half of annotated genes assigned ‘hypothetical’ functions (Figure 1).
Figure 1.
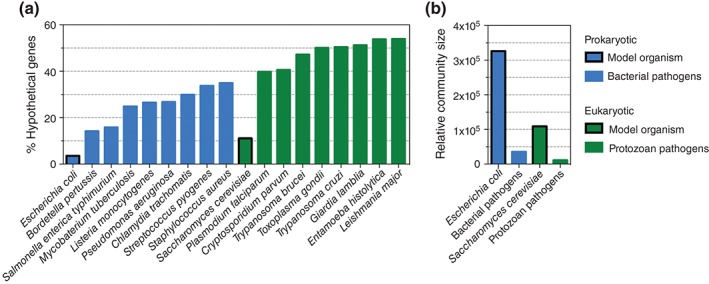
Percentage of ‘hypothetical’ genes and relative community size for important unicellular human pathogens and their model organisms. (a) The percentage of ‘hypothetical’ genes for selected prokaryotic and eukaryotic pathogens compared to their relevant model organism, Escherichia coli and Saccharomyces cerevisiae, respectively. Percentages for each species were calculated from the number of genes including ‘hypothetical,’ ‘unknown,’ or ‘uncharacterized’ in the gene description compared to the total number of pathogen genes from the NCBI database for model organisms and bacterial pathogens, and from the corresponding EuPathDB databases for protozoan pathogens. (b) The relative community size for model organisms, and the mean relative community size for the bacterial and protozoan pathogens listed in A, based on the number of results generated from a Pubmed (http://www.ncbi.nlm.nih.gov/pubmed) search of the species name.
Moreover, the lack of functional characterization of parasite genomes makes the interpretation of large datasets difficult. This is especially true when building system‐wide networks based on gene ontology, as differentially expressed genes with unknown function are often excluded, which may lead to an inaccurate representation of the system. Because approximately half of parasite genes fall into this ‘hypothetical’ category, caution must be taken to express the degree of uncertainty when clustering datasets into biological processes. Accordingly, a current focus of parasite biology is to assign global gene function, and thus genome‐wide technologies rooted in systems biology are essential.
RECENT ADVANCES IN SYSTEMS‐BASED APPROACHES TO HOST–PARASITE RESEARCH
Application of ‐Omic Technologies
Systems biology utilizes multiple platforms in order to survey global cellular processes. These include the classic ‐omic technologies, namely transcriptomics, proteomics, and metabolomics. The recent whole‐genome sequencing of many important human parasite genomes has led to significant progress in the development of these approaches to the study of parasitic disease. In this section, we will summarize these strategies and their application to host–parasite interactions.
Transcriptomics
Transcriptomics has been fundamental in shaping our current understanding of parasite infection biology. Probe‐dependent cDNA microarrays have been a historically useful tool for gene validation and discovery, as well as determining differential transcript expression in parasites.12, 13 In T. gondii, for example, microarray analysis led to the identification of developmentally regulated genes that clustered into distinct processes such as immune avoidance and sugar metabolism.14 Microarray data have also revealed that P. falciparum genes in specific pathways are co‐regulated,15 and that many genes that are co‐transcribed share common regulatory elements.16 cDNA microarray chips for a number of protozoan pathogens are now commercially available, and recently, the first C. parvum‐specific microarray17 was developed and made accessible to the research community. Microarrays have also provided global insights into the host response to parasitic infections.18, 19 Cross‐hybridization severely limits the scope of probe‐dependent techniques, however, as high background reduces the dynamic range of the assay and parasite and host transcriptomes must be analyzed separately.20 Probe‐independent, tag‐based methods such as serial or cap analysis of gene expression (SAGE or CAGE, respectively) can provide a more quantitative picture of the transcriptome, and have been particularly useful for gene expression analysis in eukaryotic pathogens, as reviewed elsewhere.21
All of the aforementioned techniques are restricted by the inability to detect specific mRNA isoforms, unannotated noncoding RNAs, and precise splice junctions. Recent advances in next‐generation sequencing platforms have allowed for deep sequencing of RNA, known as RNA‐Seq.22 This approach provides quantitative full‐genome coverage, is extremely sensitive, and can identify RNA species and alternative splicing events that are undetectable by microarray or tag‐based analyses. Recent advances in strand‐specific RNA‐Seq23, 24, 25 have also revealed widespread transcription of natural antisense transcripts (NATs) in many eukaryotic parasites. So far, evidence for NATs has been found in species such as P. falciparum, T. gondii, T. brucei, Leishmania spp., and G. lamblia.26 RNA‐Seq also enables the simultaneous sequencing of both host and parasite transcriptomes,20 allowing for unprecedented insights into host–pathogen interactions. In a study by Pittman et al.,27 in vivo dual RNA‐Seq analysis of T. gondii and its murine host revealed significant influence of both the host environment on parasite gene expression as well as parasite development on host transcription. Simultaneous sequencing of both human and P. falciparum RNA isolated from peripheral blood from 116 malaria patients28 also provided important insights into host–parasite interactions, including the identification of host and pathogen genes that correlate with clinical disease severity. While this paired analysis provides a much more powerful approach than single RNA‐Seq, the vast majority of ‐omic datasets currently survey either the host or the pathogen during infection. As dual RNA‐Seq increases in both resolution and cost‐effectiveness, it will no doubt continue to provide novel molecular insights into host–parasite transcriptomics.
Proteomics
Because of innovations in existing technologies and the development of new methodologies, the field of proteomics has made significant progress in surveying the complex repertoire of proteins that define host–parasite systems. Mass spectroscopy (MS),29 which measures the mass‐to‐charge ratio and abundance of ions, has been by far the most widely used method for proteomic analysis. Prior to the advent of genome sequencing, intact proteins had to be directly analyzed by MS through technically challenging and low‐throughput ‘top‐down’ procedures. The postgenomic era has significantly benefited from the implementation of ‘bottom‐up’ approaches that instead utilize enzymatic or chemical fragmentation of proteins.30 The protein sequence is then inferred by mapping of the MS fragmentation spectra to databases built from annotated genomic information. MS‐based approaches have been fundamental in the assembly of the whole‐cell proteomes of protozoan parasites during multiple life‐cycle stages, as reviewed elsewhere.30 Despite the increasing number of proteomic datasets publicly available for these organisms, there remains a massive deficit in experimentally validated proteome coverage (i.e., the percentage of protein‐coding genes that have evidence for protein expression) (Table 2). Moreover, this insufficiency is highly variable among parasites. While more well‐studied protozoan parasites such as P. falciparum, T. gondii, and T. brucei have more than 50% proteomic coverage, others, such as Giardia intestinalis and E. histolytica, have less than 30% coverage, and for L. major, less than 5% of the predicted proteome is experimentally validated (Table 2). MS‐based methods have also been instrumental in profiling the host proteome in response to parasitic infections. For example, Nelson et al.31 utilized 2D electrophoresis, difference gel electrophoresis, and MS to profile the host proteomic response to infection with T. gondii, and analysis of the resulting dataset suggested extensive global reprogramming of host metabolic pathways. In a truly integrative study32 of both parasite and host cell transcriptomic and proteomic data during the intraerythrocytic developmental cycle of P. falciparum, 24 human proteins were identified in significant quantities within the parasite. Interestingly, these host proteins, like many parasite proteins found in the same study, displayed distinct abundance profiles throughout parasite development.
Table 2.
Experimental Proteome Coverage for Protozoan Parasites
| Species | Strain | Total Genes | Protein‐Coding Genes | Proteomic Expression | Proteome Coverage (%) |
|---|---|---|---|---|---|
| Toxoplasma gondii | GT1 | 8637 | 8460 | 4488 | 53 |
| Plasmodium falciparum | 3D7 | 5777 | 5542 | 4104 | 74 |
| Cryptosporidium parvum | Iowa II | 3886 | 3805 | 1320 | 35 |
| Trypanosoma brucei | TREU927 | 12,094 | 11,567 | 6632 | 57 |
| Trypanosoma cruzi | CL‐Brener Esmeraldo‐like | 10,597 | 10,339 | 3674 | 36 |
| Leishmania major | Friedlin | 9378 | 8400 | 329 | 4 |
| Giardia lamblia | Assemblage A Isolate WB | 9747 | 9667 | 2166 | 22 |
| Entamoeba histolytica | HM‐1:IMSS | 8333 | 8306 | 2443 | 29 |
Information gathered from EuPathDB databases, http://eupathdb.org.
The current field of parasite proteomics is moving toward more sensitive and specific methods. While it is critical that we continue to map and annotate both host and parasite proteomes during infection, it is also important that we directly measure differential protein expression in order to better understand host–parasite biology. This general approach, commonly referred to as ‘quantitative proteomics,’33 includes relative quantification methods such as isobaric tagging for relative and absolute quantification (iTRAQ) and stable isotope labeling by amino acids in cell culture (SILAC), and label‐free methods such as spectral counting. Quantitative proteomics has been particularly useful in mapping the phosphoproteomes of many parasites,34, 35, 36 as well as phosphorylated host proteins in response to parasitic infection.37 These studies have revealed that reversible protein phosphorylation, mediated by protein kinases and phosphatases, is an important regulator of many aspects of host–parasite biology. In addition to quantitative proteomics, there has been an increasing interest in mapping the proteomes of subcellular organelles, called ‘organellar proteomics.’38 Organelle isolation prior to proteomic analysis is commonly achieved by cellular fractionation or specific labeling and purification methods. This type of proteomic analysis has enhanced our understanding of subcellular protein localization for many protozoan parasites, such as the nuclear proteome for P. falciparum 39 and the mitochondrial outer membrane proteome for T. brucei.40
Metabolomics
The systems‐based application of metabolomics, or the global survey of small molecules (<1 kDa), has provided significant insight into the metabolic processes governing host–parasite infection biology over the last decade, and has been expertly reviewed elsewhere.41 Because the majority of antiparasitic drugs target enzymes involved in parasite metabolism, mapping host–parasite metabolomes will be critical for the development of novel therapeutics. The study of parasite metabolism has historically relied on the use of low‐throughput radioactive labeling or enzymatic‐based assays. Today, high‐throughput MS‐based technologies as well as nuclear magnetic resonance spectroscopy are the major tools used by researchers investigating metabolomes.42, 43 Pioneering studies have utilized these technologies to survey the metabolomes for many parasite life‐cycle processes, including Entamoeba cyst formation,44 Leishmania promastigote development,45 Toxoplasma tachyzoite replication,46 and Plasmodium intraerythrocytic progression.47 Metabolic labeling coupled with MS is an effective strategy for measuring metabolic pathway flux. For example, Ke et al.48 utilized 13C labeling of P. falciparum genetic knockout lines that have deletions in mitochondrial tricarboxylic acid (TCA) cycle enzymes to show that mitochondrial metabolism is surprisingly flexible throughout the parasite life cycle. Additionally, profiling the host metabolome49, 50 has generated valuable information as to how parasites scavenge host resources and how the host alters its own metabolism to fight infection. Host metabolomic studies also have clinical importance, as this information has been utilized in the identification of diagnostic biomarkers of protozoan infections.51, 52, 53 Albeit the application of metabolomics to the study of host–parasite interactions is relatively recent, significant progress has already been made toward understanding the dynamic metabolic networks that regulate parasitic infections.
Integrating and Interpreting Large Datasets
Advances in systems‐based technologies have required the development of mathematical methods and computational tools to integrate and interpret multiple data types. This reliance will continue to grow as the size and number of datasets continue to exponentially increase. The computational approaches employed in systems biology span various mathematical disciplines. Here, we focus on a few examples where the analysis tools have proven useful. Efforts in data integration can be broadly grouped into the following approaches: (1) data organization and network construction, (2) network analyses, and (3) simulation and modeling.
Data Organization and Network Construction
Given the size and number of large ‐omic screens, organization of the resulting datasets into databases is critical to facilitate subsequent integration and analysis. Publicly available databases for eukaryotic parasites, as summarized in Table 3, provide the information required to populate the 1D annotation of the organisms, that is, the descriptive content summarizing each measured biological molecule. Examples of this include functional annotation or transcript levels for an individual gene. As exemplified in the summary plot of datasets uploaded to EuPathDB60 (Figure 2), there is generally much greater availability of transcriptomic data than proteomic data for protozoan pathogens. In turn, there is much greater availability of proteomic data than metabolomic data. This trend is in part due to technological advancements in the instrumentation and the degree of high‐throughput profiling that is feasible for each type of measurement. Public metabolomic databases are also relatively scarce. Metabolights68 is so far the only cross‐species, open‐access metabolomic database available, and there is currently no information submitted for protozoan parasites. However, as the value of metabolomic data is recognized,41 and the number of studies profiling host–parasite metabolomics increases, we anticipate an increase in open‐access metabolomic databases. In contrast to the 1D annotation of organisms, the 2D annotation includes defining interactions between biological molecules.69 For example, protein–protein interactions can be detailed as complexes or signaling pathways, and protein–metabolite interactions can be described as metabolic reactions occurring within the organism. The 2D annotation provides a platform in which different measurements are integrated and subsequently analyzed in order to gain meaningful insight into the capabilities and functions of biological organisms.69
Table 3.
Resources and Databases for Protozoan Parasites
| General or Parasite Species(s) | Database | Description | Web Address |
|---|---|---|---|
| General databases | PHI‐based: Pathogen‐Host Interactions48 | Expertly curated database of experimentally verified genes from pathogens | http://www.phi-base.org/ |
| Pathogen Portal | Integrative repository linking the NIAID Bioinformatics Resource Centers (BRCs) and providing ‐omics data for eukaryotic pathogens, all bacteria, and all viral families | http://www.pathogenportal.org/portal/portal/PathPort/Home | |
| ProtozoaDB54 | Gene‐based protozoan database with emphasis on distant similarities (HMM‐based) and phylogeny‐based annotations, including orthology analysis | http://protozoadb.biowebdb.org/ | |
| HPIDB: Host–Pathogen Interaction Database55 | Host–pathogen database integrating experimentally derived protein–protein interaction data from various public databases; BLASTP enabled | http://www.agbase.msstate.edu/hpi/main.html | |
| PRIDE Archive Proteomics Data Repository56 | European Bioinformatics Institute repository of mass spectrometry proteomics data | http://www.ebi.ac.uk/pride/archive/ | |
| EuPathDB (Eukaryotic Pathogen Database Resources)57 | Integrative database of eukaryotic pathogens housing sequencing data, microarray data, proteomics data, metabolic pathways, and phenotype information | http://eupathdb.org | |
| OMIC tools58 | Metadatabase providing a compendium of over 4400 web‐based tools for the analysis of genomic, transcriptomic, proteomic, and metabolomic data | http://omictools.com/ | |
| Cryptosporidium spp. | CryptoDB 59 | Part of the EuPathDB family of databases | http://cryptodb.org/cryptodb/ |
| Entamoeba histolytica | Entamoeba histolytica Assembly and Annotation60 | Assembly and annotation of E. histolytica with content imported from AmoebaDB | http://protists.ensembl.org/Entamoeba_histolytica/Info/Annotation/ |
| AmoebaDB 61 | Part of the EuPathDB family of databases | http://amoebadb.org/amoeba/ | |
| Giardia spp. | GiardiaDB 62 | Part of the EuPathDB family of databases | http://giardiadb.org/giardiadb/ |
| Leishmania spp. | Leishmania major—LeischCyc | Pathway/genome database for Leishmania major based on the BioCyc ontology | http://biocyc.org/LEISH/organism-summary?object=LEISH |
| TriTrypDB63 | Part of the EuPathDB family of databases, resource for Kinetoplastid species (including Leishmania spp.) | http://tritrypdb.org/tritrypdb/ | |
| Plasmodium spp. | PlasmoDB 64 | Part of the EuPathDB family of databases | http://plasmodb.org/plasmo/ |
| Full-Malaria | Full‐length cDNA database of Plasmodium species with web‐accessible analysis tool | http://fullmal.hgc.jp/index_ajax.html | |
| Toxoplasma gondii | ToxoDB 65 | Part of the EuPathDB family of databases | http://toxodb.org/toxo/ |
| Trypanosoma brucei on GeneDB 66 | Genomic and proteomic database resources for T. brucei that is part of the Sanger Institute Pathogen Program, GeneDB project | http://www.genedb.org/Homepage/Tbruceibrucei927 | |
| Trypanosoma spp. | TrypanoCyc67 | Pathway/genome database for T. brucei based on the BioCyc ontology | http://www.metexplore.fr/trypanocyc/ |
| TriTrypDB63 | Part of the EuPathDB family of databases, resource for Kinetoplastid species (including Trypanosoma spp.) | http://tritrypdb.org/tritrypdb/ |
Figure 2.
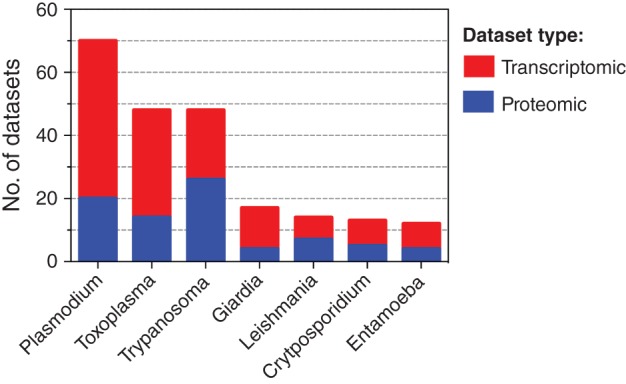
Distribution of transcriptomic and proteomic datasets uploaded to EuPathDB for selected Protozoan parasites. The number of transcriptomic and proteomic datasets submitted to the EuPathDB60 family of databases (see Table 3) for each Protozoan parasite genus. The total number of datasets is plotted for each parasite group, with the proportion of transcriptomic datasets (colored in red) and proteomic datasets (colored in blue) displayed within each bar graph.
Once the components of a system are defined, they need to be merged into a format amenable to the desired analysis style. The type of network that is constructed is dependent on the experimental data available. For example, protein–protein interaction networks can be constructed from MS spectra generated after yeast two‐hybrid screens or MS spectra generated after co‐immunoprecipitation.16 Metabolic network connectivity can be determined by individual metabolites that are linked together via enzymatic (and transport) reactions. Groups of reactions subsequently form pathways. Once such information is collected, the metabolic reactions can be linked together into pathways.70 Although advancements in network construction continue to be made with each new dataset, we must also be cognizant that we may never completely characterize all components of an organism nor comprehensively detail all interactions. Thus, analysis methods will need to be tolerant of the incomplete datasets.71
Network Analyses
Once data are organized and biological networks are constructed, many different methods can be deployed to leverage this information for valuable analyses. The connectivity of biological networks can be studied using methods from statistics and graph theory to systematically characterize relationships between different components (e.g., distance measures and connectivity) in a network and how the different elements within a network are organized. For example, 2846 protein–protein interactions were elucidated for 1312 proteins in P. falciparum using a high‐throughput yeast two‐hybrid assay.71 When the resulting network was analyzed using gene co‐expression data and ontology information, putative annotation for hypothetical proteins was feasible, and alternative biological functions were suggested for some annotated genes. Suthram et al.72 further analyzed this network using a network alignment approach called PathBLAST,73 an algorithm that identifies conserved pathways between organisms by identifying conserved proteins and then testing for conserved interactions. Through this, the authors found that few protein interactions were conserved between Plasmodium and several model organisms, thus demonstrating that the patterns of protein interaction in Plasmodium are quite distinct.
In Silico Modeling and Simulation
Beyond the analysis of network organization, various modeling approaches are being employed to simulate host–pathogen interaction pathways across spatial and temporal scales. Different types of calculations can be performed, depending on the modeling approach that is used. Cell‐scale modeling approaches are proving valuable for integrating and analyzing the increasingly large volumes of ‐omic data. One notable example is constraint‐based modeling,26 which uses metabolic network reconstructions.70 In this approach, all metabolic reactions in an organism are linked together and represented in a specific mathematical format that enables the calculation of network characteristics as well as simulation of different metabolic network flux states. As illustrated in Figure 3, every gene in the network reconstruction includes gene–protein‐reaction associations. These connections describe how transcripts are related to the proteins they encode, as well as their corresponding enzymatic reactions. Network reconstructions thus provide the foundation for the hierarchical data integration of biological models. Because the relationships between the components in these networks are defined by logical relationships, it is then relatively trivial to integrate multiple types of different datasets from the host and pathogen and analyze them simultaneously.
Figure 3.
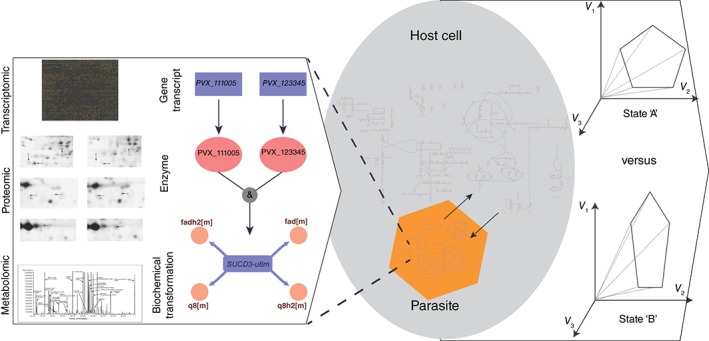
Organization, integration, and analysis of ‐omic datasets in metabolic network reconstructions used in constraint‐based modeling. Moving from left to right in the figure, various ‐omic data types (transcriptomic, proteomic, and metabolomic) are mapped onto the different components of the model. This includes the genes, enzymes, or small metabolites within a network for every reaction in the reconstruction (for which such data are available). Host–pathogen models can be constructed by connecting (or infecting) a host cell with the intracellular parasite. Subsequent simulations may characterize differences in the flux states in the noninfected versus infected state of the host cell. A gene–protein‐reaction relationship for Plasmodium succinate dehydrogenase is highlighted in the figure.
Constraint‐based modeling has a growing number of methodologies74 enabling one to make diverse predictions including metabolic pathway usage, gene essentiality, and potential drug targets. Analysis of network reconstructions for L. major has demonstrated utility in predicting minimal requisite media conditions for growth75 and further for simulating lethality versus nonlethality responses to drug treatments. Network reconstructions contain the requisite pathways for biomass synthesis (i.e., growth); thus, in silico simulations can be carried out in a systematic fashion in which each gene is ‘deleted’ and the ability of the cell to grow can then be tested. Similarly, in silico experiments can be performed in which the response to a particular drug is tested. This is achieved by inhibiting or eliminating the activity of an enzymatic target of a drug within the network and then interrogating the capabilities of the in silico organism (e.g., testing the ability to generate biomass and carry flux through particular pathways).14, 68, 76 In a study by Chavali et al.,77 the authors elucidated potential combinatorial drug treatment regimens that would inhibit growth. Similar algorithms could be employed to study other parasites and complement drug‐screening efforts. Constraint‐based reconstructions and analyses have also provided insight into growth conditions and drug sensitivity for other important human parasites.78, 79, 80
In addition to the analysis of individual parasites, models are being developed that include host cell pathways. Specifically, based on host and parasite genome annotation, computational models can be reconstructed for both the host and parasite. Then computational simulations can elucidate how the pathways of the two different organisms influence each other. For example, the host–pathogen model analysis carried out by Bordbar et al.81 was the first integrated, simulation‐capable host–pathogen metabolic network reconstruction, in which two genome‐scale network reconstructions (a human host cell and a pathogen) were functionally integrated. Host–pathogen interactions in different infectious states were characterized through analysis of transcriptomic data, which revealed differences in flux states of the pathogen (Mycobacterium tuberculosis) in latent versus pulmonary versus meningeal tuberculosis. These differing metabolic states are the result of the different tissues as well as the different types of interactions between the pathogen and host cell. Further, such differences may suggest different treatment strategies, depending on the site of the infection. This methodology is likely to be critical in the analysis and interpretation of protozoan pathogen data. The development of host–parasite models represents a new avenue of application and much needed development. With the continued expansion and completion of 1D annotation of human parasites (Table 3), the concurrent and increasingly active 2D annotation of these pathogens will lead to an improved understanding of host–parasite interactions, and in the process, yield meaningful predictions and new hypotheses to test and validate in the wet lab.
Systems Biology to Help Mitigate the Challenges Associated with Host–Parasite Research
In addition to expanding the arsenal of tools available to researchers, advancements in systems‐biology‐based methods have helped address some of the most challenging obstacles associated with host–parasite research. As summarized earlier, some of these challenges include the complex, stochastic nature of parasitic life cycles, the lack of effective methods for culturing certain parasite developmental stages, and the overwhelming percentage of uncharacterized parasite genes. In this section, we will highlight some of the most recent systems‐biology‐based studies that have aimed to overcome these barriers.
Computational Methods to Deconvolute Complex Parasite Mixtures
Although a number of methods have been developed to synchronize or isolate specific developmental parasite stages in vitro, mixed parasite populations are often unavoidable in vivo, especially when analyzing patient samples. This is problematic when examining system‐wide expression data, as different stages have been shown to display distinct expression profiles. Clinical surveillance of patients harboring transmissible parasite stages, such as the Plasmodium sexual gametocyte stage, is critical for transmission reduction;82 however, this is difficult, as gametocytes comprise only a very small fraction of all blood‐stage Plasmodium parasites during infection. To address this issue for mixed populations of P. falciparum, researchers have developed a statistical method6 that estimates the relative contributions of cell cycle and developmental stage variation to the overall stochasticity of gene expression data. The method was based on both transcriptomic and microscopy analysis, and when applied to a published dataset of in vivo patient samples, they found that the previously reported variation in gene expression was directly correlated with a changing proportion of sexual‐stage parasites. In addition, a recent study83 utilized computational analysis of transcriptomic data in order to develop a novel qRT‐PCR‐based method that can estimate the amount of both asexual and sexual stages in patient samples. This strategy relied on the selection and validation of a small panel of developmentally associated transcriptional markers, a procedure deeply rooted in systems biology.
Systems Analysis to Understand and Improve In Vitro Parasite Culture
In vitro culture of parasites throughout their life cycle is a valuable technique to more easily study host–parasite biology. However, this is technically challenging for many pathogen developmental stages. A notable example is the stage conversion that occurs during E. histolytica encystation, or the development from pathogenic trophozoites into transmissible cysts, as this process cannot be currently reproduced in vitro. In Entamoeba invadens, a related Entamoeba species that infects reptiles, stage conversion can be induced in vitro. Several groups have recently mapped E. invadens encystation on a system‐wide scale in order to better understand the biological processes controlling cyst formation, and in the process they provide insight into the development of in vitro culture methods to induce E. histolytica encystation.10, 44 These studies led to the sequencing and assembly of the E. invadens genome and global characterization of both transcriptomic and metabolomic changes during encystation. Interestingly, RNA‐Seq analysis10 revealed that phospholipase D, an enzyme involved in lipid second messenger signaling, is required for efficient E. invadens stage conversion in vitro. In addition, MS‐based metabolomics44 revealed that despite an overall decrease in energy generation, there is an increase in the levels of certain biogenic amines as well as γ‐aminobutyric acid (GABA) during encystation. While it is still unclear how the biological processes revealed by these data specifically contribute to Entamoeba stage conversion, these studies provide important insight into pathways that may be targeted to induce E. histolytica in vitro. Additionally, the metabolic enzymes controlling these processes may be suitable targets for the development of transmission‐blocking drugs.
Genome‐Wide Strategies to Assign Gene Function to Hypothetical Genes
As previously emphasized, the majority of protozoan parasite genomes are only half annotated, with around 50% of genes assigned a hypothetical or unknown function (Figure 1). Because our understanding of host–parasite interactions requires knowledge of both host and parasite gene function, incomplete gene annotation significantly stifles progress in this field. This severely limits our basic understanding of parasite biology and stunts our progress toward improved antiparasitic therapies, as drug discovery research benefits from the functional annotation of parasite genes. There has been an increased effort in recent years to apply high‐throughput phenotypic screening and chemical genomics to identify novel parasite drug targets; however, often the genes targeted by promising compounds are uncharacterized.3 Accordingly, significant effort has been made to apply systems‐biology‐based methods in order to assign global gene function. Bioinformatic analysis using comparative genomics is a widely used strategy for predicting the function of uncharacterized proteins.84 This method relies on the evolutionary conservation of proteins with similar function. While comparative genomics has been important in the assignment of putative gene function for many parasite genes,85, 86 this analysis alone is not sufficient for the characterization of whole parasite genomes. There are notable examples where structurally similar proteins have divergent functions, and likewise, where proteins that have similar functions have divergent sequences.87 Additionally, there are a number of parasite proteins that do not have orthologs with known function, and therefore traditional comparative genomics would not be applicable. For example, C. parvum is particularly divergent, with only 4% of all the predicted open reading frames (ORFs) initially assigned putative functions based on sequence homology.88 Classical systems‐based profiling of parasite transcriptomes, proteomes, and metabolomes has helped to build biological context for a number of these uncharacterized proteins.54, 89, 90, 91 However, experimental evidence linking genotype to phenotype is still required in order to adequately characterize protein function.
In model systems, protein functional characterization has been largely achieved by phenotypic screens of genetically manipulated organisms. This ‐omics strategy, called functional genomics, includes forward genetic approaches, which identify the genetic basis for phenotype, and reverse genetic approaches, which identify the phenotypic consequence of genetic alteration. Unfortunately, these functional genetic strategies are challenging for many important human parasite systems, as genome manipulation is technically difficult. Despite the challenges, a number of recent genome‐wide screening strategies have been successfully executed. Transposon mutagenesis using the piggyBac transposable system has been particularly useful as a forward genetic strategy in Plasmodium species.92, 93 Additionally, improvements in forward genetic methods for chemical mutagenesis have facilitated functional genetics in organisms such as T. gondii.94 Reverse genetic strategies, including both gain‐of‐function and loss‐of‐function genetic screens, have also come a long way in recent years. Genome‐wide overexpression screens have been a valuable platform for characterizing protozoan parasite gene function. These studies have identified genes involved in phosphatidylinositol signaling as well as phagocytosis for the human protozoan pathogen E. histolytica.95, 96 While overexpression screens are useful because they can provide biological insight while avoiding the problem of genetic redundancy, loss‐of‐function screens can directly assess the phenotypic consequence of repressing endogenous gene expression, which in many cases is physiologically preferable. Moreover, the ease of gene knockdown technologies such as RNA interference (RNAi) has facilitated high‐throughput screening. While a number of protozoan pathogens such as P. falciparum lack the cellular machinery necessary for RNAi, others, like T. brucei, have a functional RNAi pathway amenable to reverse genetics.97 A number of recent genome‐wide RNAi screens have been carried out in T. brucei,98, 99, 100 and these studies have led to the identification of many parasite genes controlling important biological processes such as cell cycle progression, differentiation, and quorum sensing. Alternative methods to regulate gene expression in the genetically intractable Plasmodium parasite species are highly sought after. Significant progress has been made to this end, with the recent development of reverse genetic technologies including a tetracycline‐repressible transactivator system,101 a glmS ribozyme‐based post‐transcriptional knockdown system,102 and an inducible TetR‐aptamer system.103 Very recently, a genome‐scale library consisting of bar‐coded genetic modification vectors was developed as a reverse genetic screening resource for Plasmodium berghei.104 Application of the genome‐editing CRISPR‐Cas9 system to the study of malaria parasites has also been successful,55, 56, 105 and as this technology further develops, it will certainly improve our understanding of many hypothetical parasite proteins and thus host–parasite biology as a whole.
SYSTEMS ANALYSIS HAS ADVANCED OUR UNDERSTANDING OF KEY ASPECTS OF HOST–PARASITE BIOLOGY
An enormous amount of information has been produced from the generation of host–parasite systems‐biology datasets within the last decade. The proper integration and interpretation of this ‘big data’ is critical in order to link experimental findings to useful biological knowledge. Because of the system‐wide nature of these datasets, a vast number of important and interesting conclusions can usually be drawn from any given systems‐based study, although researchers often choose to pursue only a limited number of noteworthy findings. Interestingly, many systems‐based publications have followed up on data that enhance our understanding of specific subfields of host–parasite biology. Although this review will not attempt to encompass all of these findings, we will review some of the leading concepts arising from the analysis of recent genome‐wide datasets.
Regulation of Parasite Gene Expression
Understanding how parasites regulate gene expression throughout their life cycle within a host is necessary in order to fully appreciate the scope of host–parasite interactions. For example, parasites can actively interfere with host cell translation in order to hijack the cellular resources required for their own gene expression as well as suppress immune responses, as reviewed elsewhere.57, 58 The system‐wide investigation of parasite gene expression is also vital in understanding the coordinated set of events underlying important host–parasite interactions. The upregulation or downregulation of parasite proteins during specific developmental stages is dependent on the host cellular environment and needs to be carefully controlled to ensure parasite survival.
Genome‐wide approaches have been particularly important in the elucidation of the regulatory mechanisms governing parasite gene expression in recent years. Although transcriptomics has emerged as a powerful systems‐based approach, it must be emphasized that the quantitation of mRNA abundance is often an imperfect indicator of global gene expression. Indeed, for both prokaryotic and eukaryotic organisms, it has been demonstrated that mRNA levels correlate with protein expression for only 50–70% of genes,103 and for protozoan parasites, this number may be even lower.106 Systems‐based approaches have been especially useful for characterizing the dynamic control of parasite gene expression in recent years. These studies have revealed that precise control of gene expression is essential to drive the dramatic transformation that takes place as parasites cycle through developmental stages, and that the apparent lack of tight transcriptional regulation is remedied by extensive post‐transcriptional mechanisms.
In particular, translational delay, a process in which protein expression is actively suspended for expressed mRNA transcripts, is a common strategy employed by many protozoan parasites. Translational delay may be a particularly advantageous strategy for parasites, as they must quickly adapt to new environments and undergo developmental switching in order to survive; storing transcripts necessary for such adaptations allows for rapid changes in gene expression by circumventing the time needed for transcription. Genome‐wide next‐generation sequencing of both steady‐state mRNA as well as polysome‐associated transcripts during the asexual erythrocytic stage in P. falciparum 107 revealed widespread translational repression across the genome during different stages of the parasite life cycle. Surprisingly, more than 30% of parasite genes were found to be associated with translational delay. Many of the repressed genes appeared to be regulated by cell cycle stage and they clustered into discrete biological processes. For example, many genes associated with early‐stage processes, such as nutrient acquisition and erythrocyte remodeling, were transcribed during the trophozoite or schizont stages, and were only actively translated immediately following merozoite invasion. Another genome‐wide ribosomal profiling study of P. falciparum blood stages108 provides additional support for a model whereby transcription of important merozoite genes occurs during the previous stage and is translationally upregulated during invasion. Translational delay has also been demonstrated during the sexual gametocyte stage by temporary storage of specific transcripts in P‐bodies.109 Unlike the majority of other eukaryotic organisms, Trypanosomes transcribe almost all of their genes as large polycistronic clusters, and thus lack transcriptional control for most genes. Despite the absence of regulation at the level of transcription, transcriptomic surveys110 have revealed extensive variation in mRNA abundance across developmental stages, suggesting widespread post‐transcriptional control. Furthermore, the comparison of proteomic expression using SILAC and MS to transcriptomic datasets suggests that like Plasmodium, mRNA abundance does not predict protein expression for at least 30% of the T. brucei genome.111 The integration of data surveying global protein expression, polysome‐associated transcript abundance, and total mRNA during these stages revealed extensive translational repression during the time when T. brucei prepares for transmission.112
Parasite Utilization of Host Resources
While eukaryotic pathogens are often able to synthesize a number of nutrients required for growth de novo, it is often more advantageous to conserve the energy required for biosynthesis and to instead hijack host‐derived resources. This is especially true for the acquisition of host lipids, as protozoan parasites must quickly assemble a large amount of new membrane during replication within host cells. In Apicomplexan parasites such as P. falciparum and T. gondii, fatty acids are taken up from the host and converted into triacylglycerides, where they are then stored in lipid bodies.113 Recently, a system‐wide survey of the Plasmodium lipidome during liver‐stage infection49 revealed a significant enrichment in fatty acids important for membrane biogenesis, including phosphatidylcholine. Upon further investigation, it was found that the parasite actively acquires host‐derived phosphatidylcholine and that this process is essential for parasite survival within hepatocytes.49 It has also been shown that Leishmania parasites, while unable to synthesize sphingomyelin themselves, are able to hydrolyze host sphingomyelin in order to produce essential metabolites.114 A comparative genomics study114 identified a parasite enzyme, LaISCL, which is responsible for the degradation of host‐derived sphingomyelin, and showed that this process is necessary for the proliferation of L. major parasites within their mammalian hosts. More recently, the same group showed115 that this enzyme is also responsible for sphingomyelin turnover in Leishmania amazonensis, although in this species, the role of sphingomyelin degradation in promoting virulence is quite different.
Many protozoan parasites live an intracellular auxotrophic lifestyle, actively acquiring metabolites from their nutrient‐rich host in order to survive. For instance, blood‐stage Plasmodium parasites have lost the ability to biosynthesize purine rings or amino acids, and therefore scavenge host nucleotides to synthesize DNA and catabolize host hemoglobin to generate amino acids.47, 59 Recent system‐wide metabolomic studies have been instrumental in profiling the complex exchange of nutrients between parasites and their hosts. A comprehensive MS‐based approach47 revealed significant modulation of host metabolites during blood‐stage Plasmodium development. The authors found that host arginine depletion was particularly extensive, suggesting that this may contribute to human malarial hypoargininemia and progression to cerebral malaria. Another Apicomplexan parasite, T. gondii, relies on host nutrients, such as carbon, in order to proliferate within host cell vacuoles. In a combined metabolomic and stable isotope labeling approach, a recent study46 mapped the carbon metabolism pathway for T. gondii tachyzoites. This systems‐based analysis revealed that active catabolism of host glucose and glutamine through an oxidative TCA cycle is essential for parasite replication. Through these and similar systems‐biology‐based surveys, it is becoming clear that protozoan parasites have evolved complex strategies to both usurp and exploit host resources.
Host Immune Response to Parasitic Infection
In order to fully appreciate the complexity of host–parasite interactions, the host immune response must be considered. It is well established that while most protozoan infections are self‐limiting in immunocompetent hosts, however, immunocompromised individuals can develop severe and often life‐threatening disease, suggesting that an effective immune response is essential for regulating parasitic disease. Many ‐omic‐based strategies have contributed to our current knowledge of how the innate and adaptive immune systems resist parasitic infection, and in many cases, exacerbate disease. In particular, recent transcriptomic analyses of host–parasite systems have implicated the host innate Toll‐like receptor (TLR) and interferon (IFN)‐mediated proinflammatory pathways in the regulation of disease progression. Microarray analysis of malaria patient samples116 demonstrated an upregulation of TLR signaling genes that had sites for IFN‐inducible transcription factors. Upon subsequent analysis of Plasmodium‐infected rodents,116 it was revealed that TLR9 and MyD88 are critical to initiate the cytokine responses leading to acute malaria in vivo. Another transcriptomic analysis of patient responses117 further confirmed the enhancement of IFN‐stimulated genes (ISGs) upon infection with malaria parasites, and interestingly, the same study determined that TLR9‐independent sensing of AT‐rich Plasmodium DNA induces type I IFNs. In a dual RNA‐Seq approach,27 a recent report mapped host and pathogen transcriptomes during acute and chronic infection with T. gondii. Analysis of the differentially expressed transcripts revealed that many of the acute infection‐specific genes included ISGs such as guanylate‐binding proteins. Chronic infection‐specific transcripts were shown to comprise a unique set of immune genes, including those important for antigen recognition and presentation. Thus, these systems‐level analyses indicate that innate sensing of protozoan pathogens is important for the induction of proinflammatory responses aimed at controlling infection.
Parasitic disease is an evolutionary arms race; as our immune systems attempt to fight off infection, pathogens quickly respond by adapting to and subverting these attacks, often through elegant biological maneuvers. Multiple ‐omic‐based surveys have contributed to our knowledge of how protozoan parasites actively manipulate the host immune response in order to avoid detection. Over a decade of systems‐biology research has shown that T. gondii downregulates the innate immune response by multiple mechanisms. This includes preventing host nuclear translocation of proinflammatory transcription factors such as nuclear factor kappa β (NF‐κβ) and signal transducer and activator of transcription 1 (STAT1α), as well as upregulating anti‐inflammatory pathways such as those involving the suppressor of cytokine signaling (SOCS) proteins.118 A notable systems‐based study119 utilized transcriptomics and pathway analysis to show that Toxoplasma actively regulates host immune responses, and through forward genetics, discovered a parasite rhoptry kinase, ROP16, that is secreted into the host cytoplasm to interfere with STAT signaling. Additionally, Plasmodium parasites also secrete virulence factors that specifically block host innate immune signaling. During liver‐stage development, Plasmodium circumsporozoite protein (CSP) is exported and localized to the host cell nucleus where it interferes with the nuclear translocation of NF‐κβ, and microarray analysis confirms that at least 40 NF‐κβ‐responsive genes are downregulated with CSP expression.120 Likewise, in the blood stages of the parasite, a high‐throughput protein interaction screen121 found that Plasmodium merozoite surface protein 1 (MSP1) specifically binds to the human proinflammatory cytokine S100P, and that this interaction blocked activation of the host NF‐κβ‐mediated innate immune response. Through these and other genome‐wide investigations, it is clear that while the host innate immune system is essential in controlling parasitic infection, parasites have evolved complex strategies to effectively dampen these responses.
CONCLUSION
Parasitic disease research has significantly benefited from systems analyses. Host–parasite systems are complex, with stochasticity across and within developmental stages, are often technically challenging to model experimentally, and are built upon incompletely characterized genomic foundations. Despite the challenges, recent improvements in systems‐level technologies have facilitated the generation of ‘big data’ to model host–pathogen interactions. These analyses have improved our current knowledge of the basic biology driving parasitic infection, and have also yielded novel tools to facilitate further research. Many ‐omic surveys have been conducted, and global expression data for important human protozoan parasites are now publicly accessible through several pathogen databases. Furthermore, algorithms for the integration and interpretation of genomic, transcriptomic, and metabolomic data have elucidated novel insights and hypotheses into host–parasite interactions. In particular, systems biology approaches have shed light on how parasites utilize post‐transcriptional gene regulation to quickly adapt to changing host environments, hijack host‐derived resources to establish intracellular replication, and neutralize host immune responses to escape host proinflammatory attacks.
Each new ‐omics survey comes with the promise to ‘solve biology’ and serve as a singular framework for biological understanding. Inevitably, this fails, not because of overestimation of the utility of a particular measurement, but rather, the failure to recognize the need for multiple data types and for analysis to be carried out in an integrated, cohesive manner. Significant insights into host–parasite biology have been made with systems biology, but technical challenges still limit the application of systems approaches to parasite systems, leading to an uneven distribution of genome‐wide datasets across protozoan species and developmental stages. While a number of genomic and transcriptomic datasets have been generated for these pathogens, functional annotation is still absent for approximately half of all parasite genes, and proteomic coverage is severely lacking. Moreover, there is a complete absence of publicly accessible metabolomic databases for protozoan pathogens.
Moving forward, the field of host–parasite biology would greatly benefit from overcoming key deficiencies in systems biology research. Necessary advances include the optimization of parasite culturing methods, the development of functional genetic approaches (e.g., through the CRISPR‐Cas9 system), and computational models of host–parasite interactions. These tools will enable the generation of more genome‐wide datasets for functional characterization of parasite genes and provide tools for the analysis of these data. Thus, there are many opportunities for researchers to leverage systems biology to further a field that is far from saturated. There is no doubt that the increasing efficacy of systems‐based approaches will continue to improve our current understanding of host–parasite interactions, and accordingly, the treatment of parasitic disease.
ACKNOWLEDGMENTS
We would like to thank members of the Winzeler lab for critical review and discussion of the manuscript. This work was completed with generous support from the National Institutes for Health, the Bill and Melinda Gates Foundation, The Medicines for Malaria Venture, and the Novo Nordisk Foundation provided to the Center for Biosustainability at the Technical University of Denmark.
Conflict of interest: The authors have declared no conflicts of interest for this article.
REFERENCES
- 1. Sacks D, Sher A. Evasion of innate immunity by parasitic protozoa. Nat Immunol 2002, 3:1041–1047. [DOI] [PubMed] [Google Scholar]
- 2. Beiting DP, Roos DS. A systems biological view of intracellular pathogens. Immunol Rev 2011, 240:117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Winzeler EA. Applied systems biology and malaria. Nat Rev Microbiol 2006, 4:145–151. [DOI] [PubMed] [Google Scholar]
- 4. Sakata T, Winzeler EA. Genomics, systems biology and drug development for infectious diseases. Mol Biosyst 2007, 3:841–848. [DOI] [PubMed] [Google Scholar]
- 5. Hall N, Karras M, Raine JD, Carlton JM, Kooij TW, Berriman M, Florens L, Janssen CS, Pain A, Christophides GK, et al. A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science 2005, 307:82–86. [DOI] [PubMed] [Google Scholar]
- 6. Lemieux JE, Gomez-Escobar N, Feller A, Carret C, Amambua-Ngwa A, Pinches R, Day F, Kyes SA, Conway DJ, Holmes CC, et al. Statistical estimation of cell‐cycle progression and lineage commitment in Plasmodium falciparum reveals a homogeneous pattern of transcription in ex vivo culture. Proc Natl Acad Sci USA 2009, 106:7559–7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Visvesvara GS, Garcia LS. Culture of protozoan parasites. Clin Microbiol Rev 2002, 15:327–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Widmer G, Corey EA, Stein B, Griffiths JK, Tzipori S. Host cell apoptosis impairs Cryptosporidium parvum development in vitro. J Parasitol 2000, 86:922–928. [DOI] [PubMed] [Google Scholar]
- 9. Mueller I, Galinski MR, Baird JK, Carlton JM, Kochar DK, Alonso PL, del Portillo HA. Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet Infect Dis 2009, 9:555–566. [DOI] [PubMed] [Google Scholar]
- 10. Ehrenkaufer GM, Weedall GD, Williams D, Lorenzi HA, Caler E, Hall N, Singh U. The genome and transcriptome of the enteric parasite Entamoeba invadens, a model for encystation. Genome Biol 2013, 14:R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Karanis P, Aldeyarbi HM. Evolution of Cryptosporidium in vitro culture. Int J Parasitol 2011, 41:1231–1242. [DOI] [PubMed] [Google Scholar]
- 12. Gobert GN, Moertel LP, McManus DP. Microarrays: new tools to unravel parasite transcriptomes. Parasitology 2005, 131:439–448. [DOI] [PubMed] [Google Scholar]
- 13. Duncan R. DNA microarray analysis of protozoan parasite gene expression: outcomes correlate with mechanisms of regulation. Trends Parasitol 2004, 20:211–215. [DOI] [PubMed] [Google Scholar]
- 14. Cleary MD, Singh U, Blader IJ, Brewer JL, Boothroyd JC. Toxoplasma gondii asexual development: identification of developmentally regulated genes and distinct patterns of gene expression. Eukaryot Cell 2002, 1:329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Young JA, Fivelman QL, Blair PL, de la Vega P, Le Roch KG, Zhou Y, Carucci DJ, Baker DA, Winzeler EA. The Plasmodium falciparum sexual development transcriptome: a microarray analysis using ontology‐based pattern identification. Mol Biochem Parasitol 2005, 143:67–79. [DOI] [PubMed] [Google Scholar]
- 16. Young JA, Johnson JR, Benner C, Yan SF, Chen K, Le Roch KG, Zhou Y, Winzeler EA. In silico discovery of transcription regulatory elements in Plasmodium falciparum . BMC Genomics 2008, 9:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang H, Guo F, Zhou H, Zhu G. Transcriptome analysis reveals unique metabolic features in the Cryptosporidium parvum oocysts associated with environmental survival and stresses. BMC Genomics 2012, 13:647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Blader IJ, Manger ID, Boothroyd JC. Microarray analysis reveals previously unknown changes in Toxoplasma gondii‐infected human cells. J Biol Chem 2001, 276:24223–24231. [DOI] [PubMed] [Google Scholar]
- 19. Liehl P, Zuzarte-Luis V, Chan J, Zillinger T, Baptista F, Carapau D, Konert M, Hanson KK, Carret C, Lassnig C, et al. Host‐cell sensors for Plasmodium activate innate immunity against liver‐stage infection. Nat Med 2014, 20:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Westermann AJ, Gorski SA, Vogel J. Dual RNA‐seq of pathogen and host. Nat Rev Microbiol 2012, 10:618–630. [DOI] [PubMed] [Google Scholar]
- 21. Kronstad JW. Serial analysis of gene expression in eukaryotic pathogens. Infect Disord Drug Targets 2006, 6:281–297. [DOI] [PubMed] [Google Scholar]
- 22. Wang Z, Gerstein M, Snyder M. RNA‐Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009, 10:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, Gnirke A, Regev A. Comprehensive comparative analysis of strand‐specific RNA sequencing methods. Nat Methods 2010, 7:709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li S, Liberman LM, Mukherjee N, Benfey PN, Ohler U. Integrated detection of natural antisense transcripts using strand‐specific RNA sequencing data. Genome Res 2013, 23:1730–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Siegel TN, Hon CC, Zhang Q, Lopez‐Rubio JJ, Scheidig-Benatar C, Martins RM, Sismeiro O, Coppee JY, Scherf A. Strand‐specific RNA‐Seq reveals widespread and developmentally regulated transcription of natural antisense transcripts in Plasmodium falciparum . BMC Genomics 2014, 15:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Militello KT, Refour P, Comeaux CA, Duraisingh MT. Antisense RNA and RNAi in protozoan parasites: working hard or hardly working? Mol Biochem Parasitol 2008, 157:117–126. [DOI] [PubMed] [Google Scholar]
- 27. Pittman KJ, Aliota MT, Knoll LJ. Dual transcriptional profiling of mice and Toxoplasma gondii during acute and chronic infection. BMC Genomics 2014, 15:806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yamagishi J, Natori A, Tolba ME, Mongan AE, Sugimoto C, Katayama T, Kawashima S, Makalowski W, Maeda R, Eshita Y, et al. Interactive transcriptome analysis of malaria patients and infecting Plasmodium falciparum . Genome Res 2014, 24:1433–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yates JR, Ruse CI, Nakorchevsky A. Proteomics by mass spectrometry: approaches, advances, and applications. Annu Rev Biomed Eng 2009, 11:49–79. [DOI] [PubMed] [Google Scholar]
- 30. Wastling JM, Armstrong SD, Krishna R, Xia D. Parasites, proteomes and systems: has Descartes' clock run out of time? Parasitology 2012, 139:1103–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nelson MM, Jones AR, Carmen JC, Sinai AP, Burchmore R, Wastling JM. Modulation of the host cell proteome by the intracellular apicomplexan parasite Toxoplasma gondii . Infect Immun 2008, 76:828–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Foth BJ, Zhang N, Chaal BK, Sze SK, Preiser PR, Bozdech Z. Quantitative time‐course profiling of parasite and host cell proteins in the human malaria parasite Plasmodium falciparum . Mol Cell Proteomics 2011, 10:M110.006411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cox J, Mann M. Quantitative, high‐resolution proteomics for data‐driven systems biology. Annu Rev Biochem 2011, 80:273–299. [DOI] [PubMed] [Google Scholar]
- 34. Urbaniak MD, Martin DM, Ferguson MA. Global quantitative SILAC phosphoproteomics reveals differential phosphorylation is widespread between the procyclic and bloodstream form lifecycle stages of Trypanosoma brucei . J Proteome Res 2013, 12:2233–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsigankov P, Gherardini PF, Helmer‐Citterich M, Spath GF, Zilberstein D. Phosphoproteomic analysis of differentiating Leishmania parasites reveals a unique stage‐specific phosphorylation motif. J Proteome Res 2013, 12:3405–3412. [DOI] [PubMed] [Google Scholar]
- 36. Treeck M, Sanders JL, Elias JE, Boothroyd JC. The phosphoproteomes of Plasmodium falciparum and Toxoplasma gondii reveal unusual adaptations within and beyond the parasites' boundaries. Cell Host Microbe 2011, 10:410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu Y, Nelson MM, Quaile A, Xia D, Wastling JM, Craig A. Identification of phosphorylated proteins in erythrocytes infected by the human malaria parasite Plasmodium falciparum . Malar J 2009, 8:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Andersen JS, Mann M. Organellar proteomics: turning inventories into insights. EMBO Rep 2006, 7:874–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oehring SC, Woodcroft BJ, Moes S, Wetzel J, Dietz O, Pulfer A, Dekiwadia C, Maeser P, Flueck C, Witmer K, et al. Organellar proteomics reveals hundreds of novel nuclear proteins in the malaria parasite Plasmodium falciparum . Genome Biol 2012, 13:R108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Niemann M, Wiese S, Mani J, Chanfon A, Jackson C, Meisinger C, Warscheid B, Schneider A. Mitochondrial outer membrane proteome of Trypanosoma brucei reveals novel factors required to maintain mitochondrial morphology. Mol Cell Proteomics 2013, 12:515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kafsack BF, Llinas M. Eating at the table of another: metabolomics of host‐parasite interactions. Cell Host Microbe 2010, 7:90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bollard ME, Stanley EG, Lindon JC, Nicholson JK, Holmes E. NMR‐based metabonomic approaches for evaluating physiological influences on biofluid composition. NMR Biomed 2005, 18:143–162. [DOI] [PubMed] [Google Scholar]
- 43. Want EJ, Nordstrom A, Morita H, Siuzdak G. From exogenous to endogenous: the inevitable imprint of mass spectrometry in metabolomics. J Proteome Res 2007, 6:459–468. [DOI] [PubMed] [Google Scholar]
- 44. Jeelani G, Sato D, Husain A, Escueta‐de Cadiz A, Sugimoto M, Soga T, Suematsu M, Nozaki T. Metabolic profiling of the protozoan parasite Entamoeba invadens revealed activation of unpredicted pathway during encystation. PLoS One 2012, 7:e37740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Silva AM, Cordeiro‐da‐Silva A, Coombs GH. Metabolic variation during development in culture of Leishmania donovani promastigotes. PLoS Negl Trop Dis 2011, 5:e1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. MacRae JI, Sheiner L, Nahid A, Tonkin C, Striepen B, McConville MJ. Mitochondrial metabolism of glucose and glutamine is required for intracellular growth of Toxoplasma gondii . Cell Host Microbe 2012, 12:682–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Olszewski KL, Morrisey JM, Wilinski D, Burns JM, Vaidya AB, Rabinowitz JD, Llinas M. Host‐parasite interactions revealed by Plasmodium falciparum metabolomics. Cell Host Microbe 2009, 5:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ke H, Lewis IA, Morrisey JM, McLean KJ, Ganesan SM, Painter HJ, Mather MW, Jacobs-Lorena M, Llinas M, Vaidya AB. Genetic investigation of tricarboxylic acid metabolism during the Plasmodium falciparum life cycle. Cell Rep 2015, 11:164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Itoe MA, Sampaio JL, Cabal GG, Real E, Zuzarte‐Luis V, March S, Bhatia SN, Frischknecht F, Thiele C, Shevchenko A, et al. Host cell phosphatidylcholine is a key mediator of malaria parasite survival during liver stage infection. Cell Host Microbe 2014, 16:778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Daubener W, Spors B, Hucke C, Adam R, Stins M, Kim KS, Schroten H. Restriction of Toxoplasma gondii growth in human brain microvascular endothelial cells by activation of indoleamine 2,3-dioxygenase. Infect Immun 2001, 69:6527–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li JV, Wang Y, Saric J, Nicholson JK, Dirnhofer S, Singer BH, Tanner M, Wittlin S, Holmes E, Utzinger J. Global metabolic responses of NMRI mice to an experimental Plasmodium berghei infection. J Proteome Res 2008, 7:3948–3956. [DOI] [PubMed] [Google Scholar]
- 52. Wang Y, Utzinger J, Saric J, Li JV, Burckhardt J, Dirnhofer S, Nicholson JK, Singer BH, Brun R, Holmes E. Global metabolic responses of mice to Trypanosoma brucei brucei infection. Proc Natl Acad Sci USA 2008, 105:6127–6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ng Hublin JS, Ryan U, Trengove R, Maker G. Metabolomic profiling of faecal extracts from Cryptosporidium parvum infection in experimental mouse models. PLoS One 2013, 8:e77803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bozdech Z, Llinas M, Pulliam BL, Wong ED, Zhu J, DeRisi JL. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum . PLoS Biol 2003, 1:E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wagner JC, Platt RJ, Goldfless SJ, Zhang F, Niles JC. Efficient CRISPR‐Cas9‐mediated genome editing in Plasmodium falciparum . Nat Methods 2014, 11:915–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang C, Xiao B, Jiang Y, Zhao Y, Li Z, Gao H, Ling Y, Wei J, Li S, Lu M, et al. Efficient editing of malaria parasite genome using the CRISPR/Cas9 system. MBio 2014, 5:e01414‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mohr I, Sonenberg N. Host translation at the nexus of infection and immunity. Cell Host Microbe 2012, 12:470–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jaramillo M, Gomez MA, Larsson O, Shio MT, Topisirovic I, Contreras I, Luxenburg R, Rosenfeld A, Colina R, McMaster RW, et al. Leishmania repression of host translation through mTOR cleavage is required for parasite survival and infection. Cell Host Microbe 2011, 9:331–341. [DOI] [PubMed] [Google Scholar]
- 59. Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, et al. Genome sequence of the human malaria parasite Plasmodium falciparum . Nature 2002, 419:498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Aurrecoechea C, Brestelli J, Brunk BP, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, Heiges M, et al. EuPathDB: a portal to eukaryotic pathogen databases. Nucleic Acids Res 2010, 38:D415–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Aurrecoechea C, Barreto A, Brestelli J, Brunk BP, Caler EV, Fischer S, Gajria B, Gao X, Gingle A, Grant G, et al. AmoebaDB and MicrosporidiaDB: functional genomic resources for Amoebozoa and Microsporidia species. Nucleic Acids Res 2011, 39:D612–D619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Aurrecoechea C, Brestelli J, Brunk BP, Carlton JM, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, et al. GiardiaDB and TrichDB: integrated genomic resources for the eukaryotic protist pathogens Giardia lamblia and Trichomonas vaginalis . Nucleic Acids Res 2009, 37:D526–D530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, Depledge DP, Fischer S, Gajria B, Gao X, et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res 2010, 38:D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, et al. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res 2009, 37:D539–D543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gajria B, Bahl A, Brestelli J, Dommer J, Fischer S, Gao X, Heiges M, Iodice J, Kissinger JC, Mackey AJ, et al. ToxoDB: an integrated Toxoplasma gondii database resource. Nucleic Acids Res 2008, 36:D553–D556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Logan‐Klumpler FJ, De Silva N, Boehme U, Rogers MB, Velarde G, McQuillan JA, Carver T, Aslett M, Olsen C, Subramanian S, et al. GeneDB—an annotation database for pathogens. Nucleic Acids Res 2012, 40:D98–D108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shameer S, Logan‐Klumpler FJ, Vinson F, Cottret L, Merlet B, Achcar F, Boshart M, Berriman M, Breitling R, Bringaud F, et al. TrypanoCyc: a community‐led biochemical pathways database for Trypanosoma brucei . Nucleic Acids Res 2015, 43:D637–D644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Haug K, Salek RM, Conesa P, Hastings J, de Matos P, Rijnbeek M, Mahendraker T, Williams M, Neumann S, Rocca‐Serra P, et al. MetaboLights—an open‐access general‐purpose repository for metabolomics studies and associated meta‐data. Nucleic Acids Res 2013, 41:D781–D786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Palsson B. Two‐dimensional annotation of genomes. Nat Biotechnol 2004, 22:1218–1219. [DOI] [PubMed] [Google Scholar]
- 70. Feist AM, Herrgard MJ, Thiele I, Reed JL, Palsson BO. Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol 2009, 7:129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Date SV, Stoeckert CJ Jr. Computational modeling of the Plasmodium falciparum interactome reveals protein function on a genome‐wide scale. Genome Res 2006, 16:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Suthram S, Sittler T, Ideker T. The Plasmodium protein network diverges from those of other eukaryotes. Nature 2005, 438:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kelley BP, Yuan B, Lewitter F, Sharan R, Stockwell BR, Ideker T. PathBLAST: a tool for alignment of protein interaction networks. Nucleic Acids Res 2004, 32:W83–W88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lewis NE, Nagarajan H, Palsson BO. Constraining the metabolic genotype‐phenotype relationship using a phylogeny of in silico methods. Nat Rev Microbiol 2012, 10:291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chavali AK, Whittemore JD, Eddy JA, Williams KT, Papin JA. Systems analysis of metabolism in the pathogenic trypanosomatid Leishmania major . Mol Syst Biol 2008, 4:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kim HU, Kim SY, Jeong H, Kim TY, Kim JJ, Choy HE, Yi KY, Rhee JH, Lee SY. Integrative genome‐scale metabolic analysis of Vibrio vulnificus for drug targeting and discovery. Mol Syst Biol 2011, 7:460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chavali AK, Blazier AS, Tlaxca JL, Jensen PA, Pearson RD, Papin JA. Metabolic network analysis predicts efficacy of FDA‐approved drugs targeting the causative agent of a neglected tropical disease. BMC Syst Biol 2012, 6:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Roberts SB, Robichaux JL, Chavali AK, Manque PA, Lee V, Lara AM, Papin JA, Buck GA. Proteomic and network analysis characterize stage‐specific metabolism in Trypanosoma cruzi . BMC Syst Biol 2009, 3:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Raghunathan A, Shin S, Daefler S. Systems approach to investigating host‐pathogen interactions in infections with the biothreat agent Francisella. Constraints‐based model of Francisella tularensis . BMC Syst Biol 2010, 4:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Plata G, Hsiao TL, Olszewski KL, Llinas M, Vitkup D. Reconstruction and flux‐balance analysis of the Plasmodium falciparum metabolic network. Mol Syst Biol 2010, 6:408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bordbar A, Lewis NE, Schellenberger J, Palsson BO, Jamshidi N. Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol Syst Biol 2010, 6:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Alonso PL, Brown G, Arevalo‐Herrera M, Binka F, Chitnis C, Collins F, Doumbo OK, Greenwood B, Hall BF, Levine MM, et al. A research agenda to underpin malaria eradication. PLoS Med 2011, 8:e1000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Joice R, Narasimhan V, Montgomery J, Sidhu AB, Oh K, Meyer E, Pierre-Louis W, Seydel K, Milner D, Williamson K, et al. Inferring developmental stage composition from gene expression in human malaria. PLoS Comput Biol 2013, 9:e1003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci USA 1999, 96:4285–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Silber AM, Pereira CA. Assignment of putative functions to membrane “hypothetical proteins” from the Trypanosoma cruzi genome. J Membr Biol 2012, 245:125–129. [DOI] [PubMed] [Google Scholar]
- 86. Rout S, Warhurst DC, Suar M, Mahapatra RK. In silico comparative genomics analysis of Plasmodium falciparum for the identification of putative essential genes and therapeutic candidates. J Microbiol Methods 2015, 109:1–8. [DOI] [PubMed] [Google Scholar]
- 87. Saghatelian A, Cravatt BF. Assignment of protein function in the postgenomic era. Nat Chem Biol 2005, 1:130–142. [DOI] [PubMed] [Google Scholar]
- 88. Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, Deng M, Liu C, Widmer G, Tzipori S, et al. Complete genome sequence of the apicomplexan, Cryptosporidium parvum . Science 2004, 304:441–445. [DOI] [PubMed] [Google Scholar]
- 89. Le Roch KG, Zhou Y, Blair PL, Grainger M, Moch JK, Haynes JD, De La Vega P, Holder AA, Batalov S, Carucci DJ, et al. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 2003, 301:1503–1508. [DOI] [PubMed] [Google Scholar]
- 90. Bringaud F, Biran M, Millerioux Y, Wargnies M, Allmann S, Mazet M. Combining reverse genetics and NMR‐based metabolomics unravels trypanosome‐specific metabolic pathways. Mol Microbiol 2015, 96:917–926. [DOI] [PubMed] [Google Scholar]
- 91. Butler CL, Lucas O, Wuchty S, Xue B, Uversky VN, White M. Identifying novel cell cycle proteins in Apicomplexa parasites through co‐expression decision analysis. PLoS One 2014, 9:e97625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ikadai H, Shaw Saliba K, Kanzok SM, McLean KJ, Tanaka TQ, Cao J, Williamson KC, Jacobs‐Lorena M. Transposon mutagenesis identifies genes essential for Plasmodium falciparum gametocytogenesis. Proc Natl Acad Sci USA 2013, 110:E1676–E1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Fonager J, Franke-Fayard BM, Adams JH, Ramesar J, Klop O, Khan SM, Janse CJ, Waters AP. Development of the piggyBac transposable system for Plasmodium berghei and its application for random mutagenesis in malaria parasites. BMC Genomics 2011, 12:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Farrell A, Coleman BI, Benenati B, Brown KM, Blader IJ, Marth GT, Gubbels MJ. Whole genome profiling of spontaneous and chemically induced mutations in Toxoplasma gondii . BMC Genomics 2014, 15:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Koushik AB, Welter BH, Rock ML, Temesvari LA. A genomewide overexpression screen identifies genes involved in the phosphatidylinositol 3‐kinase pathway in the human protozoan parasite Entamoeba histolytica . Eukaryot Cell 2014, 13:401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. King AV, Welter BH, Koushik AB, Gordon LN, Temesvari LA. A genome‐wide over‐expression screen identifies genes involved in phagocytosis in the human protozoan parasite, Entamoeba histolytica . PLoS One 2012, 7:e43025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kolev NG, Tschudi C, Ullu E. RNA interference in protozoan parasites: achievements and challenges. Eukaryot Cell 2011, 10:1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Monnerat S, Clucas C, Brown E, Mottram JC, Hammarton TC. Searching for novel cell cycle regulators in Trypanosoma brucei with an RNA interference screen. BMC Res Notes 2009, 2:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Jones NG, Thomas EB, Brown E, Dickens NJ, Hammarton TC, Mottram JC. Regulators of Trypanosoma brucei cell cycle progression and differentiation identified using a kinome‐wide RNAi screen. PLoS Pathog 2014, 10:e1003886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Mony BM, MacGregor P, Ivens A, Rojas F, Cowton A, Young J, Horn D, Matthews K. Genome‐wide dissection of the quorum sensing signalling pathway in Trypanosoma brucei . Nature 2014, 505:681–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Pino P, Sebastian S, Kim EA, Bush E, Brochet M, Volkmann K, Kozlowski E, Llinas M, Billker O, Soldati-Favre D. A tetracycline‐repressible transactivator system to study essential genes in malaria parasites. Cell Host Microbe 2012, 12:824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Prommana P, Uthaipibull C, Wongsombat C, Kamchonwongpaisan S, Yuthavong Y, Knuepfer E, Holder AA, Shaw PJ. Inducible knockdown of Plasmodium gene expression using the glmS ribozyme. PLoS One 2013, 8:e73783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Goldfless SJ, Wagner JC, Niles JC. Versatile control of Plasmodium falciparum gene expression with an inducible protein‐RNA interaction. Nat Commun 2014, 5:5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Gomes AR, Bushell E, Schwach F, Girling G, Anar B, Quail MA, Herd C, Pfander C, Modrzynska K, Rayner JC, et al. A genome‐scale vector resource enables high‐throughput reverse genetic screening in a malaria parasite. Cell Host Microbe 2015, 17:404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ghorbal M, Gorman M, Macpherson CR, Martins RM, Scherf A, Lopez‐Rubio JJ. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR‐Cas9 system. Nat Biotechnol 2014, 32:819–821. [DOI] [PubMed] [Google Scholar]
- 106. Lahav T, Sivam D, Volpin H, Ronen M, Tsigankov P, Green A, Holland N, Kuzyk M, Borchers C, Zilberstein D, et al. Multiple levels of gene regulation mediate differentiation of the intracellular pathogen Leishmania. FASEB J 2011, 25:515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Bunnik EM, Chung DW, Hamilton M, Ponts N, Saraf A, Prudhomme J, Florens L, Le Roch KG. Polysome profiling reveals translational control of gene expression in the human malaria parasite Plasmodium falciparum . Genome Biol 2013, 14:R128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Caro F, Ahyong V, Betegon M, DeRisi JL. Genome‐wide regulatory dynamics of translation in the asexual blood stages. Elife 2014, 3:e04106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Mair GR, Braks JA, Garver LS, Wiegant JC, Hall N, Dirks RW, Khan SM, Dimopoulos G, Janse CJ, Waters AP. Regulation of sexual development of Plasmodium by translational repression. Science 2006, 313:667–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Jensen BC, Sivam D, Kifer CT, Myler PJ, Parsons M. Widespread variation in transcript abundance within and across developmental stages of Trypanosoma brucei . BMC Genomics 2009, 10:482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Gunasekera K, Wuthrich D, Braga‐Lagache S, Heller M, Ochsenreiter T. Proteome remodelling during development from blood to insect‐form Trypanosoma brucei quantified by SILAC and mass spectrometry. BMC Genomics 2012, 13:556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Capewell P, Monk S, Ivens A, MacGregor P, Fenn K, Walrad P, Bringaud F, Smith TK, Matthews KR. Regulation of Trypanosoma brucei total and polysomal mRNA during development within its mammalian host. PLoS One 2013, 8:e67069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Mazumdar J, Striepen B. Make it or take it: fatty acid metabolism of apicomplexan parasites. Eukaryot Cell 2007, 6:1727–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Zhang O, Wilson MC, Xu W, Hsu FF, Turk J, Kuhlmann FM, Wang Y, Soong L, Key P, Beverley SM, et al. Degradation of host sphingomyelin is essential for Leishmania virulence. PLoS Pathog 2009, 5:e1000692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Pillai AB, Xu W, Zhang O, Zhang K. Sphingolipid degradation in Leishmania (Leishmania) amazonensis . PLoS Negl Trop Dis 2012, 6:e1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Franklin BS, Parroche P, Ataide MA, Lauw F, Ropert C, de Oliveira RB, Pereira D, Tada MS, Nogueira P, da Silva LH, et al. Malaria primes the innate immune response due to interferon‐γ induced enhancement of toll‐like receptor expression and function. Proc Natl Acad Sci USA 2009, 106:5789–5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Sharma S, DeOliveira RB, Kalantari P, Parroche P, Goutagny N, Jiang Z, Chan J, Bartholomeu DC, Lauw F, Hall JP, et al. Innate immune recognition of an AT‐rich stem‐loop DNA motif in the Plasmodium falciparum genome. Immunity 2011, 35:194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Hunter CA, Sibley LD. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat Rev Microbiol 2012, 10:766–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Saeij JPJ, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC. Toxoplasma co‐opts host gene expression by injection of a polymorphic kinase homologue. Nature 2007, 445:324–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Singh AP, Buscaglia CA, Wang Q, Levay A, Nussenzweig DR, Walker JR, Winzeler EA, Fujii H, Fontoura BMA, Nussenzweig V. Plasmodium circumsporozoite protein promotes the development of the liver stages of the parasite. Cell 2007, 131:492–504. [DOI] [PubMed] [Google Scholar]
- 121. Waisberg M, Cerqueira GC, Yager SB, Francischetti IMB, Lu JH, Gera N, Srinivasan P, Miura K, Rada B, Lukszo J, et al. Plasmodium falciparum merozoite surface protein 1 blocks the proinflammatory protein S100P. Proc Natl Acad Sci USA 2012, 109:5429–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]