

Abstract
Objectives
Development and validation of a selective, robust high-performance liquid chromatography-tandem mass spectrometric (HPLC/MS-MS) method for the quantification of morphine, morphine-3-β-glucuronide, morphine-6-β-glucuronide, hydromorphone, and normorphine in human serum.
Design and methods
Drug-free human serum samples spiked with morphine, morphine-3-β-glucuronide, morphine-6-β-glucuronide, hydromorphone, and normorphine were prepared by protein precipitation using methanol containing the internal standards. Samples were injected onto a Thermo Scientific AccuCore PFP column for chromatographic separation. Detection was achieved using a Thermo Scientific TSQ Vantage mass spectrometer. Assay validation followed the new Clinical and Laboratory Standards Institute (CLSI) C62-A guidelines.
Results
The analytical measuring range for all analytes was determined to be 5 to 1,000 ng/mL. Intra- and inter-assay precision for three quality control levels were ≤ 7.0% and ≤ 13.5%, respectively. Carryover, stability, linearity, matrix effects, extraction and processing efficiency and method comparison characteristics were acceptable relative to the CLSI C62 guidelines.
Conclusion
The validation of this HPLC-MS/MS method demonstrated a robust and rapid assay for the quantification of morphine, morphine-3-β-glucuronide, morphine-6-β-glucuronide, hydromorphone, and normorphine.
Keywords: HPLC-MS/MS, neonatal abstinence syndrome, morphine, glucuronide, method validation
1. Introduction
Morphine, a United States Controlled Substance Act Schedule II medication because of its high potential for abuse which may lead to severe psychological or physical dependence, is a powerful opioid analgesic commonly used for management of severe pain. Morphine has a high potential for addiction as well for the development of tolerance to the medication, which requires increases in dosage and/or frequency of administration. Morphine is metabolized to form morphine-3-β-glucuronide, morphine-6-β-glucuronide via liver uridine diphosphate glucuronosyltransferase (UGT) enzymes 2B7 and 1A3, and in very small quantities normorphine, and hydromorphone [1]. Morphine-6-β-glucuronide, which comprises approximately 10% of the metabolites formed, is bioactive and has analgesic activity equal to or greater than morphine [1]. Morphine is also a metabolite of heroin, and is produced after initial metabolism to 6-acetylmorphine.
Opiate addiction and dependence are a growing public health concern, in large part because of the increasing non-medical abuse of prescription opiates [2]. The increased abuse of morphine and other prescription opiates, the regional prevalence of heroin abuse and treatment for opiate-dependent pregnant woman in methadone and buprenorphine maintenance programs have increased the rate of neonates that are born with opiate dependency and withdrawal symptoms in the United States [3]. In utero opiate exposure is associated with prematurity and long term alterations in neurodevelopment, as well as neonatal opiate withdrawal. The phenomenon of neonatal opiate withdrawal after in utero exposure is referred to as Neonatal Abstinence Syndrome (NAS). NAS is a constellation of clinical symptoms that include neurologic excitability, gastrointestinal dysfunction, failure to thrive, and autonomic dysregulation [4, 5]. Moderate to severely affected neonates require pharmacologic treatment, the first line of which is opiate replacement with morphine [6]. Due to the age and small size of the neonates with NAS, only small sample volumes can be obtained for testing. As such, a highly sensitive method for detection of these analytes is necessary.
In support of a human clinical pharmacokinetic study in infants with NAS, a serum/plasma based high-performance liquid chromatography (HPLC) tandem mass spectrometric (MS/MS) method was developed for the direct quantification of morphine and its metabolites, morphine-3-β-glucuronide, morphine-6-β-glucuronide, normorphine, and hydromorphone. Although other methods have been developed to analyze morphine and its two glucuronide metabolites, there are no serum methods that include normorphine and hydromorphone and small sample volumes [7-10]. Here we present a sensitive method to measure morphine and its metabolites using only 50 μL of serum.
2. Methods and Materials
2.1 Chemicals and reagents
Two independent lot numbers of certified reference standards of morphine, morphine-3-β-glucuronide, morphine-6-β-glucuronide, normorphine, hydromorphone as well as a single lot of the isotopically labeled internal standards (IS) morphine-d3 morphine-3-β-glucuronide-d3, morphine-6-β-glucuronide-d3, and hydromorphone-d6 were all purchased from Cerilliant Corporation (Round Rock, TX). Interference standards (see Supplemental Table 1) were obtained from Cerilliant Corporation. Drug-free serum was obtained from Bio-Rad Laboratories (Irvine, CA). High-performance liquid chromatography (HPLC) grade water and methanol were purchased from Fischer Scientific (Pittsburgh, PA) and formic acid was acquired from Sigma Aldrich (St. Louis, MO). For selectivity, matrix effects and endogenous studies, remnant human serum was acquired from vacutainer serum separator tubes (SST) (BD, Franklin Lakes, NJ) and plasma for the plasma to serum comparison from lithium heparin tubes (BD, Franklin Lakes, NJ) via an Institutional Review Board (IRB)-approved protocol through The Johns Hopkins University School of Medicine.
2.2 Preparation of standards and reagents
All certified reference standards previously mentioned were diluted in methanol (MeOH) to prepare separate working calibrator and quality control stock solutions using two different lot numbers of each compound to increase assay robustness. The calibrator working stock solutions were prepared at 10,000, 1,000 and 100 ng/mL while the QC stock solutions were prepared at 5,000 and 500 ng/mL. Calibration standards were prepared at 1,000, 700, 500, 250, 100, 50, 25, 10, and 5 ng/mL by spiking drug-free human serum with appropriate calibrator working solution. Quality controls were prepared at 850, 75, and 5.5 ng/mL by spiking drug-free human serum with appropriate QC working solution. Calibrators and QC samples were aliquoted (50 μL) into Eppendorf tubes and stored at −20 °C until used. A methanol protein precipitation solution containing 50 ng/mL of each IS was prepared and stored at −20 °C until used. Interference sample was prepared by spiking drug-free human serum with each of the interference standards at a concentration of 500 ng/mL. A separate set of interference samples were spiked with hydromorphone-3-β-glucuronide, morphine-3-β-glucuronide, and morphine-6-β-glucuronide in equimolar concentrations.
2.3 Sample preparation
Protein precipitation was accomplished by adding 500 μL of the methanol containing IS to 50 μL of each standard, control or sample. The resulting solution was vortexed for 30 seconds and centrifuged at 12,000 × g for 5 minutes. The supernatant was transferred to a 96-well plate, evaporated to dryness using a 60 °C air stream and reconstituted in 500 μL water containing 0.1% formic acid (mobile phase A). An aliquot of 15 μL was injected for high-performance liquid chromatographic-tandem mass spectrometric analysis.
2.4 HPLC conditions
A Thermo Scientific (San Jose, CA) Prelude system comprised of an Aria TLX1 system equipped with 1250 Transcend pumps and a CTC PAL autosampler with a sample stack maintained at 10 °C was used. Compounds were chromatographically separated using a Thermo Scientific AccuCore PFP (50 × 2.1 mm, 2.6 μm particle size) column that was maintained at 27 °C. Mobile phases consisted of water containing 0.1% formic acid (v/v) (mobile phase A) and methanol containing 0.1% formic acid (v/v) (mobile phase B). The analytes and their respective internal standards were eluted using a solvent gradient that started at 5% mobile phase B with a 40 second hold, then ramped to 75% mobile phase B over 150 seconds. The column was re-equilibrated at 5% mobile phase B for 60 seconds before the next injection. The total analytical run time for each injection was 4.2 minutes.
2.5 Mass spectrometric parameters
Monitoring of the analytes and their respective internal standards was achieved using a TSQ Vantage tandem mass spectrometer (Thermo Scientific, San Jose, CA) equipped with a heated electrospray ionization source (HESI). Mass spectrometric conditions were optimized by direct infusion of each of the five analytes at a flow rate of 10 μL/min and a mobile phase conditions of 50%:50% A:B into the mass spectrometer. The instrument was operated in selected reaction monitoring (SRM) and positive ionization mode. The following mass analyzer settings were manually optimized for the analytes of interest: vaporizer temperature (450 °C), spray voltage (4500 V), capillary temperature (275 °C), sheath gas pressure (60), ion sweep gas pressure (0), and auxiliary gas pressure (15).
Using the optimized conditions, the [M + H]+ products of all target analytes and their respective internal standards were observed as both parent and collision-induced product ions. Instrument-specific collision energies and product ions for each analyte are listed in Table 1. Product ions were selected based on both ion abundance and consistency in fragment ion formation over multiple infusions. The most highly abundant and consistent fragment ion was used for drug measurement. These ions matched other previously published articles [9, 11]. Quantification of drug concentrations was based on the specific analyte/IS peak area ratio. The only exception was normorphine in which morphine-6-β-glucuronide-d3 was used as the IS as discussed below.
Table 1.
Instrument specific parameters for the analysis of morphine, morphine-3-β-glucuronide, morphine-6-β-glucuronide, hydromorphone and normorphine.
| Analyte | Retention Time (min) | Parent mass (m/z) | Product mass (m/z) | Collision Enery (V) | S-Lens (V) |
|---|---|---|---|---|---|
| Morphine | 1.37 | 286.1 | 165.1 | 40 | 125 |
| 185.0 | 30 | 125 | |||
| Morphine-3-β-glucuronide | 0.56 | 462.2 | 165.1 | 52 | 171 |
| 286.1 | 31 | 171 | |||
| Morphine-6-β-glucuronide | 1.14 | 462.2 | 165.1 | 52 | 171 |
| 286.1 | 31 | 171 | |||
| Hydromorphone | 1.93 | 286.1 | 165.1 | 40 | 125 |
| 185.0 | 30 | 125 | |||
| Normorphine | 0.87 | 272.1 | 152.1 | 38 | 110 |
| 209.0 | 24 | 110 | |||
| Morphine-D3 | 1.37 | 289.2 | 152.1 | 60 | 126 |
| Morphine-3-β-glucuronide-D3 | 0.56 | 465.2 | 289.1 | 30 | 167 |
| Morphine-6-β-glucuronide-D3 | 1.14 | 465.2 | 289.1 | 30 | 167 |
| Hydromrphone-D6 | 1.93 | 292.1 | 185.1 | 29 | 110 |
3. Method Validation
The HPLC-MS/MS method was validated based on the recommendations published in the Clinical and Laboratory Standards Institute (CLSI) C62-A: Liquid Chromatography-Mass Spectrometry Methods [12]. Evaluated metrics are described below.
3.1 Pre-validation steps
3.1a Simple imprecision
Simple imprecision (intra- or within-day) was assessed by analyzing twenty individually prepared samples at the low, mid, and high QC levels for each analyte. The means, standard deviations (SD), and coefficients of variation (%CV) were determined. Accuracy or trueness was determined by calculating the percent bias (%BIAS) of the mean measured values from the theoretical value of the QC concentrations.
3.1b Selectivity
Selectivity was assessed by evaluating for any potential isobaric interference from endogenous substances at the expected retention times for all five analytes and the four internal standards. Remnant serum samples (n=3) without spiked analyte or internal standards and the interference samples without internal standards (n=3) were analyzed to evaluate the presence of any isobaric interferences.
3.1c Matrix effects, extraction and processing efficiency
Matrix effects on analyte ionization were quantitatively assessed as described by Matuszewski et al. [13]. Three sets of samples were prepared by spiking low (50 ng/mL), mid (500 ng/mL), and high (1000 ng/mL) concentrations of all five analytes into five different pooled remnant serum samples. Another non-matrix ‘un-extracted’ sample set was prepared in mobile phase A at the same concentrations. The post-extraction pooled remnant serum samples were prepared at the same concentrations after following the previously described extraction procedure but prior to drying the samples down. Raw peak areas for all five analytes and their internal standards were analyzed to assess matrix effects, extraction efficiency, and processing efficiency. Matrix effects (ME) were assessed by obtaining the ratio of mean raw peak areas of the post-extracted samples to un-extracted samples (ME = (mean post-extracted peak area/mean un-extracted peak area) × 100). Extraction or recovery efficiency (RE) was calculated as the ratio of mean peak areas of pre-extracted samples to post-extracted samples (RE = (mean pre-extracted peak area/mean post-extracted peak area) × 100). Finally, the overall processing efficiency (PE) was evaluated by obtaining the ratio of mean peak areas of pre-extracted samples to un-extracted samples (PE = (mean Pre-extracted peak area/mean un-extracted peak area) × 100).
3.1d Carryover
Carryover was assessed by measuring high (H = 3000 ng/mL) and low (5.5 ng/mL) samples individually prepared and injected in the following order: L1, L2, L3, H1, H2, L4, H3, H4, L5, L6, L7, L8, H5, H6, L9, H7, H8, L10, H9, H10, and L11. The results were analyzed using EP Evaluator Release 8 (Data Innovations, South Burlington, VT). Acceptability criteria for the carryover were based on guidelines described in the CLSI protocol EP10-A3-AMD [14].
3.2 Assay validation
3.2a Calibration curve analysis and limit of quantitation
Calibrators were analyzed at the beginning and end of each run. Standard curves spanning the analytical measuring range of the assay (5-1000 ng/mL for each analyte) were constructed using a 1/X weighted least squares-fitted linear regression for all analytes. The lower limit of quantitation (LLOQ) for this assay was defined as the lowest concentration that could be detected with acceptable precision (%CV ≤ 15%) and accuracy (%BIAS ≤ ±15%).
3.2b Linearity and dilution
Linearity was evaluated by spiking drug-free serum at a nominal concentration of 1,000 ng/mL of each analyte, and serially diluting seven consecutive times with drug-free serum. This provided a total of 8 specimen levels that spanned the calibration curve, and two individually prepared samples at each concentration were analyzed. The mean calculated concentration at each level was plotted using least squares linear regression.
Dilution studies included 1) within measuring range; 2) outside measuring range; and, 3) a post-sample preparation dilution study. All dilutions for the within and outside studies were performed with drug-free serum. For the within measuring range or low sample volume study, a mid-QC sample was diluted 2-, 3-, 4-, and 5-fold. For the outside of the measuring range study, three samples were prepared at 1,500, 2,000 and 3,000 ng/mL in drug-free serum. These samples were then diluted 2-, 3-, and 4-fold, respectively. A post-sample preparation dilution study was performed on a freshly prepared sample spiked at the mid-QC level. Four samples, additionally labeled 1 through 4, were prepared and reconstituted as described above. These samples were then used to prepare individual diluted samples(2-, 3-, 4-, and 5-fold with Mobile phase A) in triplicate in a fashion that each diluted sample came from a uniquely prepared source (e.g., for the 2-fold dilution, tubes 1 through 3 were used, for the 3-fold dilution, tubes 4, 1, and 2 where used, etc.). The remaining aliquot of each prepared sample was analyzed in triplicate (n=12) thereby providing a mean concentration to be used for calculation of percent difference (%DIFF).
3.3 Imprecision
Imprecision (inter- or between-day) was assessed by analyzing twenty individually prepared samples at the low, mid, and high QC levels for each analyte that were analyzed across multiple batches and days. The means, standard deviations (SD), and coefficients of variation (%CV) were determined. Accuracy was determined by calculating the percent bias (%BIAS) of the mean measured values from the theoretical value of the QC concentrations.
3.4 Endogenous Assay interference
For the human clinical pharmacokinetic (PK) study for which this method was developed, the method of collection (heel-stick) had the potential to cause increased levels of hemolysis in samples. Two additional drug free remnant specimens that were visually hemolyzed were simultaneously spiked at the low and high QC level along with drug-free serum. Quantitative hemoglobin measurements of the remnant specimens were obtained on a Sysmex XN-9000 (Sysmex Corporation, Kobe, Japan). Samples from the remnant serum and drug-free serum were individually prepared and analyzed in triplicate.
3.5 Stability
Stability was evaluated using high and low QC samples. In the first experiment, 50 μL aliquots (n=3) were allowed to sit at room temperature for 24 hrs. In the second experiment, 50 μL aliquots (n=3) were frozen at −20 °C and were analyzed after three freeze-thaw cycles. In the third experiment, previously analyzed samples were stored in the auto-sampler at 10 °C and analyzed at 8- and 24-hours. Long term stability of the QC samples stored at −20 °C was measured at 6 months (n=3) and compared to freshly prepared QC samples.
3.6 Method Comparison
Ten specimens of drug-free serum were spiked with four analytes (excluding normorphine) at various concentrations that span the analytical measurement range. The specimens were aliquoted into two sets containing 3 mL each. One set was analyzed by the current method, and the second set was analyzed by NMS Laboratories (Willow Grove, PA). NMS Labs sample preparation includes solid phase extraction prior to sample analysis by Ultra Performance Liquid Chromatography (UPLC) tandem mass spectrometer (MS/MS). Due to differences in linear range, NMS Labs was required to dilute specimens (Upper Limit of Quantification: morphine 500 ng/mL and hydromorphone 200 ng/mL) and was unable to quantify all samples due to lower limit of quantification (LLOQ) (morphine-3-β-glucuronide 300 ng/mL and morphine-6-β-glucuronide 50 ng/mL). Results obtained from the external laboratory were compared to those obtained by this method by Deming Regression analysis via EP Evaluator Release 8.
4. Results
Analytes demonstrated good peak shape and consistent chromatography for the selected SRM (Figure 1). Retention times (Table 1) remained consistent and system pressure increased only minimally throughout method validation. The analytical measuring range was determined to be 5 – 1000 ng/mL for each analyte. A plasma to serum comparison was performed and the results did not vary by more than 10% random variation for each analyte (data not shown).
Figure 1.
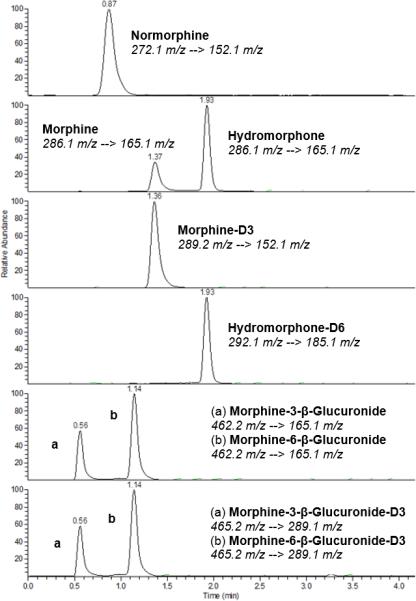
Chromatograms of drug-free human serum spiked with analytes and internal standards. Chromatograms represent a 100 ng/mL drug mixture. The selected transition is indicated for each chromatogram and elution times are indicated above each peak.
4.1 Imprecision and accuracy
Intra-day imprecision was assessed by preparing and analyzing 20 replicates at each quality control (QC) level. Inter-day imprecision was assessed by preparing and analyzing 2 replicates of each quality control (QC) level per batch for 10 batches (n=20). Accuracy was determined for both intra- and inter-day means by calculating the %BIAS from the theoretical concentration value. Imprecision mean, SD, %CV, and %BIAS are shown in Table 2.
Table 2.
Intra- and inter-assay imprecision and accuracy for morphine, morphine-3-β-glucuronide, morphine-6-β-glucuronide, hydromorphone and normorphine QC levels.
| QC Level | Intra-assay imprecision and accuracy (n=20) |
Inter-assay imprecision and accuracy (n=20) |
||||||
|---|---|---|---|---|---|---|---|---|
| Mean (ng/mL) | SD | %CV | %BIAS | Mean (ng/mL) | SD | %CV | %BIAS | |
| Morphine | ||||||||
| Low | 5.64 | 0.21 | 3.6 | 2.5 | 4.98 | 0.36 | 7.3 | −9.4 |
| Mid | 76.91 | 1.80 | 2.3 | 2.5 | 82.01 | 2.30 | 2.8 | 9.3 |
| High | 883.14 | 22.30 | 2.5 | 3.9 | 905.00 | 39.10 | 4.3 | 6.5 |
| Morphine-3-β-Glucuronide | ||||||||
| Low | 5.57 | 0.32 | 5.8 | 1.3 | 4.83 | 0.27 | 5.7 | −12.2 |
| Mid | 75.35 | 2.52 | 3.3 | 0.5 | 77.58 | 3.98 | 5.1 | 3.4 |
| High | 840.81 | 34.57 | 4.1 | −1.1 | 815.73 | 58.99 | 7.2 | −4.0 |
| Morphine-6-β-Glucuronide | ||||||||
| Low | 5.55 | 0.32 | 5.8 | 0.9 | 4.72 | 0.26 | 5.6 | −14.2 |
| Mid | 76.46 | 1.93 | 2.5 | 1.9 | 74.46 | 2.95 | 4.0 | −0.7 |
| High | 855.21 | 18.16 | 2.1 | 0.6 | 859.18 | 41.51 | 4.8 | 1.1 |
| Hydromorphone | ||||||||
| Low | 5.57 | 0.19 | 3.4 | 1.3 | 5.58 | 0.33 | 5.9 | 1.4 |
| Mid | 82.06 | 2.37 | 2.9 | 9.4 | 75.12 | 3.20 | 4.3 | 0.2 |
| High | 866.43 | 16.35 | 1.9 | 1.9 | 929.45 | 22.61 | 2.4 | 9.3 |
| Normorphine | ||||||||
| Low | 5.42 | 0.38 | 7.0 | −1.5 | 5.60 | 0.76 | 13.5 | 1.8 |
| Mid | 86.76 | 2.41 | 2.8 | 15.7 | 81.26 | 4.35 | 5.4 | 8.3 |
| High | 852.14 | 28.50 | 3.3 | 0.3 | 909.96 | 44.47 | 4.9 | 7.0 |
4.2 Selectivity
Selectivity was determined by measuring drug free human remnant serum and a 49-component mixture of common medications and illicit drugs, to include hydromorphone-3-β-glucuronide (see Table S1) for the presence of endogenous and isobaric compounds at the retention times corresponding to the analytes of interest. The chromatographic spectra did not generate any isobaric peaks at the selected retention time demonstrating that the method is selective for the analytes of interest (See Supplemental Figure 1). Hydromorphone-3-β-glucuronide peak apex was chromatographically resolved by about 0.1 minutes earlier than the morphine-6-β-glucuronide peak. However, when equimolar concentrations of both compounds are analyzed, hydromorphone-3-β-glucuronide contributed 67% to 80% (mean 75%) to the peak integration of morphine-6-β-glucuronide (See Supplemental Figure 2). However, in this study, the expectation was that hydromorphone and hydromorphone-3-β-glucuronide will be non-detectable (see Discussion Section).
4.3 Matrix effects, extraction and processing efficiency
Calculated matrix effects, as well as extraction and processing efficiencies for the analytes and their internal standards are shown in Table 3. Mean matrix effects ranged from 88 to 114% and 90 to 121% for analyte and internal standard (IS) respectively. Recovery or extraction efficiency was calculated by comparing pre-extracted samples to post-extracted samples. The mean recovery efficiency for analyte and IS ranged from 72 to 84% and 73 to 85%, respectively. Processing efficiency (%PE) was determined by comparing post-extracted to un-extracted samples and the mean processing efficiency ranged from 64 to 87% and 67 to 91% for analyte and IS respectively.
Table 3.
Analysis of the mean matrix effects (%ME), mean recovery efficiency (%RE), and mean processing efficiency (%PE) for each analyte and corresponding internal standard (IS).
| %ME |
%RE |
%PE |
||||
|---|---|---|---|---|---|---|
| Analyte | IS | Analyte | IS | Analyte | IS | |
| Morphine | 104 | 102 | 84 | 85 | 87 | 87 |
| Morphine-3-β-Glucuronide | 97 | 90 | 72 | 73 | 70 | 67 |
| Morphine-6-β-Glucuronide | 114 | 121 | 74 | 75 | 85 | 91 |
| Hydromorphone | 101 | 101 | 81 | 84 | 82 | 84 |
| Normorphine | 88 | a | 73 | a | 64 | a |
Morphine-6-β-Glucuronide-D3 used as internal standard
4.4 Carryover
Carryover studies were performed by the previously described scheme. The high-low and low-low means as well as SD are shown in Table 4. These data demonstrate that a sample having a concentration as high as three times the upper limit of linearity (ULOL) does not cause substantial carryover into the next sample analyzed for the analytes in this method.
Table 4.
Carryover assessment from a highly concentrated sample and the low QC sample.
| Morphine | M3G | M6G | Hydromorphone | |
|---|---|---|---|---|
| Mean of low-low results (ng/mL) | 6.43 | 6.18 | 5.92 | 7.30 |
| Mean of high-low results (ng/mL) | 7.25 | 6.82 | 6.44 | 7.87 |
| SDlow-low results (ng/mL) | 0.43 | 0.43 | 0.31 | 0.34 |
| Carryovera (ng/mL | 0.82 | 0.64 | 0.52 | 0.56 |
Carryover = (mean of high-low results) - (mean of low-low results). Acceptable carryover is less than 3 × SDlow-low results.
4.5 Linearity and Dilution
Assay linearity was assessed by preparing a 1,000 ng/mL sample in drug-free serum, serially diluting eight times, and running in duplicate. The mean of the results obtained were plotted via linear regression analysis and yielded slopes of 1.014 to 1.049; y-intercepts between −5.4 to 2.1; and, Pearson coefficients >0.999 (Table 5).
Table 5.
Linearity results showing slope, y-intercept and Pearson's coefficient for each of the analytes.
| Slope | y-Intercept | Pearson's Coefficient | |
|---|---|---|---|
| Morphine | 1.049 | −4.3 | >0.999 |
| Morphine-3-β-Glucuronide | 1.043 | −5.4 | >0.999 |
| Morphine-6-β-Glucuronide | 1.014 | −4 | >0.999 |
| Hydromorphone | 1.029 | 1.3 | >0.999 |
| Normorphine | 1.037 | 2.1 | >0.999 |
Dilution studies were performed in the two traditional ways. Dilution of a low sample volume sample was assessed by diluting the mid-QC sample by 2-, 3-, 4-, and 5-fold (Table 6). This demonstrated that the method was capable of analyzing specimens that may not meet the required assay volume requirements. For samples that have concentrations outside of the linear range and require dilution into the linear range are shown in Table 7. The last dilution study, as indicated in the new CLSI C62-A guidelines, was performed as a post sample preparation study. In this study, even though the samples were diluted, no dilution factor was required to be applied since both the analyte and IS are diluted simultaneously. The diluted mean concentration for each analyte (n=3) was compared to the undiluted observed mean concentration for each analyte (n=12) to calculate the %DIFF. See Table 8. The importance of this study was to demonstrate that a sample that may exhibit unexpected matrix effects could be diluted to reduce the matrix effects and provide an accurate answer.
Table 6.
Low sample volume dilution study of a mid-QC sample that was diluted 2-, 3-, 4-, and 5-fold.
| Morphine | Morphine-3-β-Glucuronide | Morphine-6-β-Glucuronide | Hydromorphone | Normorphine | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2-fold | 3-fold 4-fold | 5-fold | 2-fold | 3-fold | 4-fold | 5-fold | 2-fold | 3-fold | 4-fold | 5-fold | 2-fold | 3-fold | 4-fold | 5-fold | 2-fold | 3-fold | 4-fold | 5-fold | ||
| Mean | 41.4 | 27.0 | 20.8 | 15.8 | 39.0 | 25.0 | 19.3 | 15.4 | 40.1 | 26.3 | 20.7 | 15.9 | 42.2 | 27.8 | 20.7 | 16.6 | 42.3 | 27.7 | 21.0 | 16.8 |
| SD | 1.7 | 1.3 | 0.3 | 1.2 | 2.0 | 0.9 | 0.2 | 1.1 | 1.4 | 0.7 | 0.6 | 1.4 | 0.3 | 1.1 | 0.5 | 0.4 | 0.5 | 0.9 | 0.1 | 0.4 |
| %CV | 4.0% | 4.8% | 1.4% | 7.5% | 5.2% | 3.7% | 0.8% | 7.4% | 3.4% | 2.5% | 2.9% | 9.0% | 0.7% | 4.1% | 2.4% | 2.5% | 1.3% | 3.1% | 0.5% | 2.3% |
| Final Conc. | 82.9 | 81.0 | 83.0 | 78.9 | 78.1 | 75.1 | 77.2 | 76.9 | 80.2 | 78.9 | 82.9 | 79.7 | 84.5 | 83.4 | 82.8 | 83.1 | 84.5 | 83.2 | 83.8 | 83.8 |
| %DEV | 10.5% | 8.0% | 10.7% | 5.2% | 4.1% | 0.2% | 2.9% | 2.5% | 6.9% | 5.2% | 10.5% | 6.3% | 12.6% | 11.2% | 10.4% | 10.8% | 12.7% | 11.0% | 11.8% | 11.8% |
Table 7.
Dilution study of samples that have concentrations outside of the linear range of 1,500, 2,000, and 3,000 ng/mL and require dilution factors of 2-, 3-, and 4-fold respectively.
| 1500 ng/mL | 2000 ng/mL | 3000 ng/mL | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | %CV | %BIAS | Mean | SD | %CV | %BIAS | Mean | SD | %CV | %BIAS | |
| Morphine | 719.2 | 14.3 | 2.0 | −4.1 | 638.3 | 32.4 | 5.1 | 4.3 | 823.0 | 10.7 | 1.3 | 9.7 |
| Morphine-3-β-Glucuronide | 641.4 | 15.2 | 2.4 | −14.5 | 603.7 | 39.8 | 7.1 | −9.4 | 780.4 | 44.8 | 5.5 | −10.9 |
| Morphine-6-β-Glucuronide | 688.2 | 45.2 | 6.6 | −8.2 | 620.9 | 25.4 | 4.1 | −6.9 | 755.6 | 91.9 | 11.1 | 0.7 |
| Hydromorphone | 819.5 | 43.2 | 5.3 | 9.3 | 751.9 | 21.7 | 2.9 | 12.8 | 824.3 | 37.6 | 4.6 | 9.9 |
| Normorphine | 821.7 | 31.9 | 3.8 | 9.6 | 736.0 | 48.2 | 6.5 | −4.1 | 801.8 | 26.6 | 3.3 | 6.9 |
Table 8.
Post sample preparation dilution study at 2-, 3-, 4-, and 5-fold using Mobile phase A.
| Morphine (n=3) | Morphine-3-β-Glucuronide (n=3) | Morphine-6-β-Glucuronide (n=3) | Hydromorphone (n=3) | Normorphine (n=3) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2-fold | 3-fold | 4-fold | 5-fold | 2-fold | 3-fold | 4-fold | 5-fold | 2-fold | 3-fold | 4-fold | 5-fold | 2-fold | 3-fold | 4-fold | 5-fold | 2-fold | 3-fold | 4-fold | 5-fold | |
| Mean | 81.2 | 80.8 | 81.0 | 80.4 | 89.8 | 87.0 | 86.4 | 87.8 | 86.5 | 86.7 | 89.3 | 89.4 | 88.8 | 88.6 | 88.4 | 87.6 | 92.0 | 93.1 | 100.0 | 95.5 |
| SD | 3.2 | 3.9 | 2.9 | 1.1 | 1.5 | 1.6 | 2.7 | 4.1 | 5.0 | 1.5 | 2.6 | 1.6 | 2.4 | 2.3 | 2.7 | 2.1 | 10.6 | 1.1 | 7.1 | 5.1 |
| %CV | 3.9% | 4.8% | 3.6% | 1.4% | 1.6% | 1.9% | 3.2% | 4.7% | 5.8% | 1.7% | 2.9% | 1.8% | 2.7% | 2.6% | 3.0% | 2.4% | 11.6% | 1.2% | 7.1% | 5.3% |
| Undiluted Observed Mean (n=12) | 81.4 | 88.5 | 89.2 | 87.3 | 93.5 | |||||||||||||||
| %Diff | −0.2% | −0.8% | −0.5% | −1.2% | 1.5% | −1.8% | −2.4% | −0.8% | −3.1% | −2.8% | 0.1% | 0.2% | 1.7% | 1.4% | 1.2% | 0.3% | −1.6% | −0.4% | 7.0% | 2.1% |
4.6 Assay Interference
To ensure a reliable method for the PK study, we decided to perform an assay interference study in which samples containing high levels of hemoglobin were analyzed. We selected specimens by visual inspection for hemolysis, those specimens have a reddish hue after which quantitative hemoglobin levels were measured on a Sysmex XN9000 hematology analyzer. One specimen contained 1.9 g/dL of hemoglobin (grossly hemolyzed) and the second contained 0.2 g/dL hemoglobin (moderately hemolyzed). Before the samples were spiked with our analytes, each was analyzed to ensure they did not contain any of our panel compounds. The mean observed concentrations of the remnant samples was compared to the mean observed concentration of the drug-free serum that was spiked at the same time (Table 9). These values were used to calculate a %DIFF between the remnant samples to drug-free serum (n=3).
Table 9.
Percent difference from the mean observed drug-free serum to remnant specimens that contain hemoglobin (Hgb).
| High QC | Low QC | |||
|---|---|---|---|---|
| 1.9 g/dL Hgb | 0.2 g/dL Hgb | 1.9 g/dL Hgb | 0.2 g/dL Hgb | |
| Morphine | 6.63% | 9.74% | 4.69% | −0.58% |
| Morphine-3-β-Glucuronide | 9.61% | 8.25% | 6.52% | 6.72% |
| Morphine-6-β-Glucuronide | 8.09% | 9.97% | 6.72% | 3.31% |
| Hydromorphone | 7.56% | 6.72% | 7.07% | 5.91% |
| Normorphine | −2.85% | 5.48% | −2.13% | 2.33% |
4.7 Stability
Sample matrix stability was assessed by comparing QC samples stored at room temperature for 24-hours to QC samples removed from frozen storage. Normorphine had the greatest instability at −8% difference while all other analytes were <1% difference. Based on criteria of ≤ 15% difference, all analytes are stable at room temperature for up to 24-hours. Those specimens that went through three freeze-thaw cycles were also stable, according to this criteria. Again, normorphine demonstrated the greatest difference at −3% while all other analytes were ≤ 1%. Lastly, injection matrix stability was assessed by measuring samples that were prepared, analyzed and then re-analyzed at 8-hours and 24-hours. All analytes at the 8-hour mark were ≤ 1% different when compared to the original initial injection but at the 24-hour mark, morphine-6-β-glucuronide was −9%, morphine-3-β-glucuronide and normorphine were −7%, and morphine and hydromorphone were −5% lower than the original injection. All analytes in the QC samples stored at −20 °C were found to be stable for at least 6-months when compared to freshly prepared QC samples at all three levels. Normorphine demonstrated the largest difference at −11%, morphine-3-β-glucuronide and morphine-6-β-glucuronide were −7%, morphine was −5% and hydromorphone was −4% as compared to freshly prepared QC samples.
4.8 Method Comparison
The method described here was compared to an LC-MS/MS method used to quantify morphine, morphine-3-β-glucuronide, morphine-6-β-glucuronide, and hydromorphone as part of two different panels at NMS Labs. No reference lab could be found that offers normorphine analysis so it was excluded. 10 drug-free serum samples were spiked at various concentrations that span this method's linear range. Deming regression analysis showed strong correlation with a Pearson's correlation coefficient (R) of 0.990, 0.996, 0.971, and 0.993 for morphine, morphine-3-β-glucuronide, morphine-6-β-glucuronide, and hydromorphone respectively (Figure 2).
Figure 2.

Deming regression analysis of drug-free human serum spiked with morphine (A), morphine-6-β-glucuronide (B), morphine-3-β-glucuronide (C), and hydromorphone (D) comparing the described method (HPLC-MS/MS Vantage) to NMS Labs UPLC-MS/MS method. Slope, intercept, and Pearson's coefficient (R) are indicated for each analyte.
5. Discussion
The described method provides a rapid, simultaneous method to quantify morphine and its metabolites, morphine-3-β-glucuronide, morphine-6-β-glucuronide, hydromorphone and normorphine. The analytical measurement range was 5-1000 ng/mL for all analytes. The LLOQ in this method was appropriate for the pharmacokinetic study within the neonates. The difference between this method and that of NMS was purpose of use – pharmacokinetics (PK) versus forensic analysis. In the latter, concentrations would be expected to be higher than expected in the PK study that we are supporting. Newborns demonstrating NAS are typically small in size and require low doses of morphine for symptom control. Therefore, expected concentrations of morphine and its glucuronides would be around 50-100 ng/mL or lower. [17, 18] We decided to extend the ULOL to a reasonable level for future studies.
Method comparison studies demonstrated good correlation between our method and the method used at NMS Labs. For morphine and hydromorphone, agreement at the lower concentrations is to be expected since samples with morphine concentrations at or above 500 ng/mL and hydromorphone concentrations at or above 200 ng/mL required dilution to be analyzed at NMS Labs. The proportional error seen at the higher concentrations are most likely due to error associated with dilution. For the two glucuronides, agreement between the two methods is acceptable with morphine-6-β-glucuronide demonstrating the biggest scatter.
Normorphine demonstrated more consistent peak area ratio stability when morphine-6-β-glucuronide-d3 was used as the internal standard rather than morphine-d3. Previously reported methods have used deuterated morphine as the internal standard [15, 16]. In this method, the retention time difference between normorphine and morphine-d3 was 0.5 minutes while for morphine-6-β-glucuronide-d3 the retention time difference was 0.2 minutes. The proximal retention time was due to similar polar characteristics of the compounds with the described column and mobile phases used. Suitability of morphine-6-β-glucuronide-d3 as the IS for normorphine was compared with matrix and methanol with no effect on the calibration equation. Morphine-3-β-glucuronide-d3 was also considered as a possible internal standard for normorphine but was slightly less stable than morphine-d3.
Newborn specimens are routinely collected via a heel-stick, and the blood is pushed from the heel into the collection vial. This process has the potential to increase hemolysis in collected specimens. Bérubé et al. recently published about instability of phenolic compounds in the presence of methemoglobin at high pH during reconstitution steps [19]. With our study conditions, we did not see any degradation of the samples as Bérubé et al. had stated.
Several studies have shown that patients receiving high doses of morphine can cause detectable hydromorphone concentrations in urine (<<1% of the detected morphine concentration) [21-22]. In the study for which this method was developed, the neonates are receiving low doses of morphine. Therefore, the expectation to observe normorphine and hydromorphone in the neonate samples was very low. Furthermore, the contribution of hydromorphone-3- β-glucuronide isobar would be negligible or nonexistent. We ultimately decided to include hydromorphone and normorphine in the panel in case one of the neonates was a fast metabolizer which was not observed (manuscript in preparation).
6. Conclusion
We have developed and validated a robust HPLC-MS/MS method for the quantification of morphine and its metabolites using a low volume of serum for sample preparation. This assay has been successfully used to aid in a population pharmacokinetic study of enterally dosed morphine and its metabolites in infants with Neonatal Abstinence Syndrome. Our hope is that this very sensitive method will aid in future clinical trials where sampling volume is limited, dosing levels are variable, yet precise quantification is required.
Supplementary Material
Acknowledgements
This study was sponsored in part by an Educational Grant from Thermo Fisher Scientific. In addition, the Clinical Pharmacology NIH training grant 5T32GM066691 supported the second author during this work. Lastly, thank you to Tarrah Ezell, the research coordinator who coordinated sample collection and storage prior to drug quantification.
References
- 1.Skarke C, Lötsch J. Morphine Metabolites: Clinical Implications. Seminars in Anesthesia, Perioperative Medicine and Pain. 2002;21:258–264. [Google Scholar]
- 2.Results from the 2006 National Survey on Drug Use and Health: National Findings. Studies SOoA; Rockville, MD: 2007. DHHS Publication No. SMA 07-4293. Series H-32. [Google Scholar]
- 3.Patrick SW, Schumacher RE, Benneyworth BD, Krans EE, McAllister JM, Davis MM. Neonatal abstinence syndrome and associated health care expenditures: United States, 2000-2009. JAMA. 2012;307(18):1934–1940. doi: 10.1001/jama.2012.3951. [DOI] [PubMed] [Google Scholar]
- 4.Wiles JR, Isemann B, Ward LR. Current Management of Neonatal Abstinence Syndrome Secondary to Intrauterine Opioid Exposure. J. Ped. 2014;165:440–446. doi: 10.1016/j.jpeds.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sutter MB, Leeman L, Hsi A. Neonatal Opioid Withdrawal Syndrome. ObstetGynecolClin N Am. 2014;41:317–334. doi: 10.1016/j.ogc.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 6.Patrick SW, Kaplan HC, Passarella M, Davis MM, Lorch SA. Variation in treatment of neonatal abstinence syndrome in US Children's Hospitals, 2004-2011. J Perinatol. 2014 doi: 10.1038/jp.2014.114. [DOI] [PubMed] [Google Scholar]
- 7.Treyfors N, Hyllbrant B, Ekman L, et al. Determination of morphine, morphine-3-glucuronide and morphine-6-glucuronide in human serum by solid-phase extraction and liquid chromatography-mass spectrometry with electrospray ionization. J. Chromatogr. A. 1996;729:279–285. doi: 10.1016/0021-9673(95)01090-4. [DOI] [PubMed] [Google Scholar]
- 8.Fountain KJ, Yin Z, Diehl DM. Simultaneous analysis of morphine-related compounds in plasma using mixed-mode solid phase extraction and ultra performance liquid chromatography-mass spectrometry. J. Sep. Sci. 2009;32:2319–2326. doi: 10.1002/jssc.200900117. [DOI] [PubMed] [Google Scholar]
- 9.Clavijo CF, Hoffman KL, Thomas JJ, et al. A sensitive assay for the quantification of morphine and its active metabolites in human plasma and dried blood spots using high-performance liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem. 2011;400:715–728. doi: 10.1007/s00216-011-4775-z. [DOI] [PubMed] [Google Scholar]
- 10.Langman LJ, Korman E, Stauble ME, et al. Therapeutic monitoring of opioids: A sensitive LC-MS/MS method for quantitation of several opioids including hydrocodone and its metabolites. Ther Drug Monit. 2013;35:352–359. doi: 10.1097/FTD.0b013e318283e29a. [DOI] [PubMed] [Google Scholar]
- 11.Dickerson JA, Laha TJ, Pagano MB, et al. Improved detection of opioid use in chronic pain patients through monitoring of opioid glucuronides in urine. J. Anal. Tox. 2012;36:541–547. doi: 10.1093/jat/bks063. [DOI] [PubMed] [Google Scholar]
- 12.CLSI . CLSI document C62-A. Clinical and Laboratory Standards Institute; Wayne, PA: 2014. Liquid Chromatography-Mass Spectrometry Methods; Approved Guidelines. [Google Scholar]
- 13.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003;75:3019–3030. doi: 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- 14.CLSI . CLSI document EP10-A3-AMD. Clinical and Laboratory Standards Institute; Wayne, PA: 2014. Preliminary Evaluation of Quantitative Clinical Laboratory Methods; Approved Guideline – Third Edition. [Google Scholar]
- 15.Al-Asmari AI, Anderson RA. Method for quantification of opioids and their metabolites in autopsy blood by liquid chromatography-tandem mass spectrometry. J. Anal. Tox. 2007;31:394–408. doi: 10.1093/jat/31.7.394. [DOI] [PubMed] [Google Scholar]
- 16.Taylor K, Elliott S. A validated hybrid quadrupole linear ion-trap LC-MS method for the analysis of morphine and morphine glucuronides applied to opiate deaths. For. Sci. Intern. 2009;187:34–41. doi: 10.1016/j.forsciint.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 17.Bouwmeester NJ, Anderson BJ, Tibboel D, Holford NHG. Developmental pharmacokinetics of morphine and its metabolites in neonates, infants and young children. Brit. J Anaes. 2004;92:208–17. doi: 10.1093/bja/aeh042. [DOI] [PubMed] [Google Scholar]
- 18.Mashayekhi SO, Ghandforoush-Sattari M, Routledge PA, Hain RDW. Pharmacokinetic and Pharmacodynamic Study of Morphine and Morphine 6-Glucuronide after Oral and Intravenous Administration of Morphine in Children with Cancer. Biopharm. Drug Dispos. 2009;30:99–106. doi: 10.1002/bdd.649. [DOI] [PubMed] [Google Scholar]
- 19.Bérubé ER, Lacasse MC, Furtado M, Garofolo F. Severe impact of hemolysis on stability of phenolic compounds. Bioanalysis. 2013;5:1491–1499. doi: 10.4155/bio.13.109. [DOI] [PubMed] [Google Scholar]
- 20.Cone EJ, Heit HA, Caplan YH, Gourlay D. Evidence of morphine metabolism to hydromorphone in pain patients chronically treated with morphine. J. Anal. Toxicol. 2006;30:1–5. doi: 10.1093/jat/30.1.1. [DOI] [PubMed] [Google Scholar]
- 21.Wasan AD, Michna E, Janfaza D. Interpreting urine drug tests: prevalence of morphine metabolism to hydromorphone in chronic pain patients treated with morphine. Pain Medicine. 2008;9:918–923. doi: 10.1111/j.1526-4637.2007.00354.x. [DOI] [PubMed] [Google Scholar]
- 22.McDonough PC, Levine B, Vorce S, et al. The detection of hydromorphone in urine specimens with high morphine concentrations. J. Forensic Sci. 2008;53:752–754. doi: 10.1111/j.1556-4029.2008.00730.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.