

Abstract
Background
Complex regional pain syndrome (CRPS) is a painful, disabling and often chronic condition, where many patients transition from an acute phase with prominent peripheral neurogenic inflammation to a chronic phase with evident central nervous system (CNS) changes. Ketamine is a centrally-acting agent believed to work through blockade of N-methyl-D-aspartate (NMDA) receptors and is being increasingly used for the treatment of refractory CRPS, although the basis for the drug’s effects and efficacy at different stages of the syndrome remain unclear.
Methods
We used a mouse model of CRPS (n=8–12/group) involving tibia fracture/cast immobilization to test the efficacy of ketamine (2 mg/kg/day; 7 days) or vehicle infusion during acute (3weeks [3w] post-fracture) and chronic (7w post-fracture) stages.
Results
Acute phase fracture mice displayed elevated limb temperature, edema and nociceptive sensitization that were not reduced by ketamine. Fracture mice treated with ketamine during the chronic phase showed reduced nociceptive sensitization that persisted beyond completion of the infusion. During this chronic phase, ketamine also reduced latent nociceptive sensitization and improved motor function at 18 weeks post-fracture. No side effects of the infusions were identified. These behavioral changes were associated with altered spinal astrocyte activation and expression of pain-related proteins including NMDA receptor 2b (NR2b), Ca2+/calmodulin-dependent protein kinase ii (CaMK2), and brain-derived neurotrophic factor (BNDF).
Conclusions
Collectively, these results demonstrate that ketamine is efficacious in the chronic, but not acute stages of CRPS, suggesting that the centrally-acting drug is relatively ineffective in early CRPS when peripheral mechanisms are more critical for supporting nociceptive sensitization.
INTRODUCTION
CRPS is a painful, disabling and often chronic condition with an estimated 50,000 new cases in the United States each year.1 The syndrome encompasses a disparate collection of signs and symptoms involving the sensory, motor and autonomic nervous systems, bone demineralization, skin growth changes and vascular dysfunction all limited to a single extremity in most cases. Although CRPS is described as one entity, it is often characterized by two distinct phases: a “warm” acute and a “cold” chronic phase.2 In addition to changes in the clinical signs and symptoms, these two phases are accompanied by distinct biochemical changes both in human subjects3 and rodent models of CRPS.4 This transition from the acute to the chronic phase of CRPS suggests a shift in the underlying pain mechanisms, with peripheral mechanisms believed to underlie at least some of the acute manifestations and CNS changes evolving to support symptoms in the more chronic phases. Consequently, we might anticipate a shift in responses to therapeutic interventions depending on whether those interventions are peripherally or centrally targeted.
Ketamine is a centrally acting agent believed to work through blockade of NMDA receptors used for postoperative, cancer-related and neuropathic pain. This agent is being used increasingly for the treatment of refractory chronic CRPS where several day infusions are a common method of administration;5–8 however, neither the basis for the drug’s effects in CRPS or its efficacy at different stages of the syndrome are clearly defined at this point. Ketamine has been shown to have anti-depressive effects in rodents9 and is increasingly being used in patients with refractory depression and anxiety,10,11 symptoms commonly co-occurring with CRPS. Furthermore, in a clinical setting, chronic ketamine treatment in CRPS patients has been associated with improvement in motor function12 and the reversal of the “pain brain” state, compared to control subjects, suggesting central sites of action.13
In order to study the therapeutic effects of chronic ketamine on CRPS, we have utilized a previously characterized mouse model of distal tibia fracture/cast immobilization displaying the nociceptive, functional, vascular, trophic, inflammatory and immune aspects of this syndrome.4,14–17 Our results are consistent with the hypothesis that short-term systemic administration of low-dose ketamine is efficacious in reversing mechanical allodynia displayed by fracture/cast mice when administered in the chronic, but not acute, stage of the syndrome. Furthermore, this study provides some of the biochemical correlates of this behavioral outcome, including changes in neuroinflammation and signs of plasticity at the level of the spinal cord (SC).
MATERIALS AND METHODS
Animals
A total of 5 cohorts of mice were used. Male C57/B6J mice ages 12–14 weeks were purchased from a commercial supplier (Jackson Labs, Sacramento, CA, USA) and were allowed to habituate to the animal facility for a minimum of 10 days prior to the experiments. Mice were housed in groups of 4 on a 12-hr light/dark cycle and an ambient temperature of 22 ± 3°C, with food and water available ad libitum. All animal procedures and experimental designs were approved by the Veterans Affairs Palo Alto Health Care System Institutional Animal Care and Use Committee (Palo Alto, CA, USA) and followed the “animal subjects” guidelines of the International Association for the Study of Pain.
Limb fracture and cast immobilization
Following the random allocation to the control or the fracture/cast group, mice were anesthetized with 1.5% isoflurane and underwent a distal tibial fracture in the right leg. Briefly, a hemostat was used to make a closed fracture of the right tibia just distal to the middle of the tibia and the hindlimb was wrapped in casting tape (cat. # 82001, Scotchcast™ Plus, 3M, Leicestershire, UK), as previously described.18 After the procedure, the mice were given subcutaneous buprenorphine (0.05 milligrams per kilogram [mg/kg]) and enrofloxacin (5 mg/kg) for the next two days, as well as normal saline (1.5 ml once) for post-operative analgesia, prevention of infection and prevention of dehydration. Mice were inspected daily to ensure the cast was positioned properly through the 3-week period of cast immobilization. Mice were provided with chow pellets postoperatively ad libitum; dietary gels were also made available on the cage floor for mice having undergone surgery. Casts were removed 3 weeks after surgery under brief isoflurane anesthesia.
Naïve age- and sex-matched mice were used as control. We chose to use naïve mice instead of cast immobilization (sham) animals because cast immobilization results in an intermediate phenotype that shows signs of transient mechanical allodynia.14
Pharmacology
Fracture and control mice were randomly assigned to one of two treatment groups: ketamine or saline infusion. Ketamine (2 mg/kg/day) or sterile saline were administered for 7 days using subcutaneous osmotic mini-pumps (total volume=100 μl; rate of delivery= 0.5 μl /hour; Alzet model 1007D, Cupertino, CA, USA). Doses higher than 2 mg/kg/day were not used due to technical limitations pertaining to the commercially available concentration of the drug preparation, in addition to the volume of the pumps that can be used in mice.
At 3 or 7 weeks after fracture, animals were anesthetized with 1.5% isoflurane, and the hair was clipped in the dorsal cervical region. After disinfecting the skin, a 0.5 cm incision was made through the dermis (between the two shoulder blades) using a small pair of scissors, and a skin “pocket” was created using sterile forceps. The osmotic pump was then inserted and the wound was closed with staples. Antibiotic ointment was applied to prevent infection. All pumps were removed 8 days after insertion. For the mice that received ketamine 3 weeks after fracture, the pumps were inserted 2 days following cast removal.
A summary of the experimental time course is included in Fig. 1.
Fig. 1. Summary of experimental design.

Timeline of surgical/pharmacological interventions and behavioral/biochemical measurements when ketamine was administered at 3 weeks (1 cohort of mice, A) or 7 weeks (4 cohorts, B) post fracture. Fx=fracture; C=control; W=week; K=ketamine; S=saline.
Physiological Measures
Hindpaw temperature
The temperature of the hindpaw was measured using a fine wire thermocouple (Omega, CT, USA) applied to the paw skin, as described previously.19 Three sites were tested over the dorsal aspect of the hindpaw: the space between the first and second metatarsals (medial), the second and third metatarsals (central), and the fourth and fifth metatarsals (lateral). The six measurements for each hindpaw were averaged for the mean temperature.
Hindpaw volume
A laser sensor device (measurement range of 200 millimeters [mm], 0.01 mm resolution; cat. #4381-Precicura, Limab, Göteborg, Sweden) was used to determine the dorsal–ventral thickness of the hindpaw, as described previously.19
Behavioral testing
The experimenter was blind to the identity and experimental condition of the animals throughout the behavioral experiments and data analysis. Mice were habituated to handling by the experimenter for a few minutes each day for 7 days before initiation of the behavioral tests.
Mechanical hypersensitivity
Calibrated monofilaments (Stoelting Co., IL, USA) were applied to the plantar surface of the hindpaw and the 50% threshold to withdraw (grams) was calculated as previously described.20 The stimulus intensity ranged from 0.004 to 1.7 grams, corresponding to filament numbers (1.65, 2.36, 2.44, 2.83, 3.22, 3.61, 3.84, 4.08, 4.17, and 4.31).
Rotarod
Locomotor capacity was measured by the use of an accelerating rotarod (cat. #47600, Ugo Basile, Comerio, Italy). The task includes a speed ramp from 0 to 30 rotations per minute over 60 seconds, followed by an additional 240 seconds at the maximal speed. The latency to fall was reported for the initial exposure to the rotarod, in addition to 5 consecutive trials (30-minute inter-trial interval).
Capsaicin-evoked behaviors
This assay was performed 18 weeks after fracture. This assay measures the duration of total behaviors (biting, scratching, licking, and shaking) during the 5 minutes after a local subcutaneous injection of capsaicin (2.5 μg in 5 μL, cat. # MT028, Sigma-Aldrich, MO, USA) or vehicle (0.25% Dimethyl sulfoxide, 0.25% ethanol, 0.125% Tween-80 in saline) in the plantar surface of the right hindpaw. Quantification was carried out in real time by a blind observer using a stopwatch.
Each animal was tested with both capsaicin and vehicle with a 5-day washout between treatments. To examine the long-term effects of capsaicin, mechanical hypersensitivity was measured on the plantar aspect of the right and left hindpaws 1 hour after injection.
Qualitative measures of sedation and motor impairment
Although the current study uses a very low dose of ketamine that is administered systemically, we nonetheless elected to study potential side effects of the drug in the home cage environment, particularly since they could present potential confounds in data collection and interpretation. Side effects were measured by scoring stereotypic behavior and activity levels, in addition to changes in body mass. Stereotypic behavior was scored on a 7-point scale as: −3: anesthesia, −2: sedation, −1: drowsiness, 0: normal, 1: moderately increased (increased explorative behavior), 2: increased (increased urge to move around the cage), 3: greatly increased (inability to hold still with weaving, shaking or twitching of the head and body). Activity level was defined as follows: −3: anesthesia, −2: sedation, −1: drowsiness, 0: normal, 1: moderately impaired (disturbances in paw support), 2: impaired (unable to maintain paw support with the ability to regain an upright position after falling over), 3: greatly impaired (inability to regain an upright position after falling over). Mice were observed for 10 minutes each between 9:00AM and 2:00PM.
Immunohistochemistry
All analysis was blinded to the identity and experimental condition of the animal/tissue. Mice were anesthetized by 300 ul of ketamine/xylazine cocktail, followed by trans-cardiac perfusion with 0.9% saline solution at 7 weeks after injury. Ipsilateral lumbar spinal cords were carefully removed, post-fixed in 4% paraformaldehyde for 2 days, and rinsed in phosphate-buffered saline for 3 days. Ipsilateral spinal cords were then embedded in agarose and cross sections were cut at room temperature at 40μm thickness on a vibratome (Leica vt 1200S). 10 sections/mouse were randomly chosen for staining. Free floating immunohistochemistry was done using tris-buffered saline + 0.1% tween-20 (TBST) as the wash buffer and 10% donkey normal serum (cat. #ab7475, Abcam, MA, USA) in phosphate-buffered saline as the blocking buffer. A permeabilization step was added (prior to blocking and staining) using 0.1% triton and 0.6% hydrogen peroxide in 1X TBST. The following primary antibodies were used (diluted in blocking buffer): mouse monoclonal anti-Glial Fibrillary Acidic Protein (GFAP, 1:5000, cat. #MAB3402, Millipore, CA, USA) and rabbit polyclonal anti-Ionized Calcium Binding Adapter Molecule 1 (Iba1, 1:800, cat. #019-19741, Wako, Osaka, Japan). The following secondary antibodies were used (diluted in 1X TBST): Donkey anti-mouse Immunoglobulin G (IgG, H+L) AlexaFluor 647 (AF647, 1:500, cat. # ab150107, Abcam, MA, USA) and donkey anti-rabbit IgG (H+L) AlexaFluor 488 (AF488, 1:500, cat. # 711-545-152, Jackson Immunoresearch, PA, USA).
Spinal cord sections were mounted on slides using fluoromount aqueous mounting medium (cat. # F4680, Sigma, MO, USA). Images were analyzed as a 3 μm-step z-stack of 10 slices (20× objective magnification) using fluorescent imaging (Keyence BZ-X700, IL, USA). 3 sections per mouse were randomly chosen for analysis. GFAP+ and Iba1+ cells were manually quantified in each z-stack using the “Mark and Count” tool, and tissue area was measured in image J (Bethesda, MD, USA). Data is presented as the number of cells/mm2.
Quantification of target proteins
All analysis was blinded to the identity and experimental condition of the animal/tissue. Mice were anesthetized (isoflurane) and sacrificed by decapitation at 9–10 weeks after injury. The ipsilateral lumbar spinal cord was carefully removed and stored at −80°C until use.
Extraction of total protein and synaptosome preparation
Tissues were homogenized using T-PER Protein Extraction Reagent (cat. #87793, Thermo scientific; PA, USA) in the presence of proteinase and phosphatase inhibitors (cat. #0490683700, Roche applied science 1, CA, USA) and centrifuged at 12,000 Xg for 5 min at 4°C. Supernatant fractions were then frozen at −80°C until use.
For the isolation of synaptosomes, tissue was homogenized in 1X Syn-PER reagent (cat. #87793, Thermo scientific; PA, USA) containing “Halt” protease inhibitor cocktail (cat. #87785, Thermo Scientific; PA, USA), centrifuged at 1000 Xg for 10 minutes to remove cell debris, and the supernatant was transferred to a new tube where it was centrifuged at 17,500 Xg for 20 minutes. The pellets, containing synaptosomes, were gently re-suspended in 40 ul of the reagent (Syn-PER + “Halt”).
Two-color fluorescent western blot analysis
To assess the protein levels of the N-methyl-D-aspartate receptor 2b (NR2b) and Ca2+/calmodulin-dependent protein kinase ii (CaMK2), Western blot analysis was performed according to standard procedures. Briefly, after sodium dodecyl sulfate - polyacrylamide gel electrophoresis and blotting, proteins on the membranes were detected by overnight incubation at 4°C with the primary antibody (rabbit polyclonal anti NR2b, 1:500, cat. #ab65783, Abcam, MA, USA; mouse monoclonal anti-CaMK2, 1:2000, cat. #MA1-048, Thermo Scientific, PA, USA) followed by incubation with an infrared dye (IRDye) 800CW goat anti-rabbit IgG (H+L) (1:20,000; cat. #925-32211, LI-COR biosciences; NA, USA) or goat anti-mouse IgG (H+L) (1:20,000; cat.. #926-32210, LI-COR biosciences; NA, USA). β-actin was used as an internal control and was detected with the mouse monoclonal anti-β-actin antibody (1:5000; cat. # ab6276, Abcam, MA, USA) followed by incubation with an IRDye 680CW goat anti-mouse IgG (H+L) (1:20,000; cat. #926-32220, LI-COR biosciences, NA, USA). The signals were detected using Odyssey (LI-COR biosciences, NA, USA) and quantified using the Image J software (MD, USA).
Enzyme-linked immunosorbent assay (ELISA)
The mouse Brain-Derived Neurotrophic Factor (BDNF) ELISA kit (total protein; cat. #EK0309, Boster Immunoleader, CA, USA) and the mouse synaptophysin (SYP) ELISA kit (synaptosomal preparation; cat. #SEA425Mu, Cloud Clone Corp, TX, USA) were used as per the manufacturers’ instructions. Measures of the target proteins were normalized with respect to total protein as quantified by the Bradford assay (cat. # 500-0001, Bio-Rad, CA, USA).
Statistical Analysis
All data are expressed as mean ± standard error of the mean (SEM). Statistical analysis between experimental groups was carried out using a two-way analysis of variance (2-way ANOVA) followed by the Holm-Sidak method to correct for multiple comparisons (Fig. 2, 3, 4, 5, 6, Fig. 7, 8). Repeated measures 2-way ANOVA was used for figures 3, 4D–Fs, and 5. Significance was set at p < 0.05. (Prism 5; GraphPad Software, CA, USA). Grubb’s outlier test was used for the detection and subsequent exclusion of outlier data points. Analyses conducted on n ≤ 4 are presented as scatter plots (instead of bar graphs) to place the reader closer to the actual data (Fig. 7, 8). Sample size determination was guided by prior experience with the reported assays and is indicated in the figure legends.
Fig. 2. Physiological and behavioral changes in complex regional pain syndrome (CRPS) mice.

CRPS mice display increased edema (A) and temperature (B) on the affected hindpaw at 3, but not 7, weeks post-fracture. In addition, they show signs of mechanical allodynia in the ipsilateral (Ipsi, 3- and 7-week timepoints, C) and contralateral (Contra, 3-week timepoint, D) hindpaw. *p<0.05, *** p<0.001. n=16 mice for each of the 4 groups.
Fig. 3. Ketamine is efficacious when administered at the late, but not early, phase of complex regional pain syndrome.

Ketamine shows no efficacy in reversing mechanical allodynia when administered 3 weeks after fracture (A, B). In contrast, ketamine reverses mechanical allodynia on the ipsilateral (Ipsi) hindpaw when administered at the 7-week timepoint (C, D). No changes were observed in the contralateral (Contra) hindpaw. *p<0.05, ** p<0.005, *** p<0.001 compared to the Fracture+Saline group. #p<0.05, ## p<0.005, ### p<0.001 compared to the Control+Saline group. Panels A and B: n= 6 mice for Control+Saline, n= 6 mice for Contol+Ketamine, n= 8 mice for Fracture+Saline, and n= 11mice for Fracture+Ketamine. Panels C and D: n= 8 mice for Control+Saline, n= 8 mice for Contol+Ketamine, n= 6 mice for Fracture+Saline, and n= 9 mice for Fracture+Ketamine. The red lines indicate the duration of ketamine administration.
Fig. 4. Ketamine exposure improves rotarod performance 18 weeks after fracture.

3 different cohorts were used to measure initial rotarod performance at 3 different timepoints. Fracture animals show no obvious impairment in the rotarod assay at the 7- (A) and 8- (B) week timepoints, however, they do show impairment at the 18-week timepoint, which can be ameliorated by ketamine administration (C). Subsequent training in fracture/cast mice showed no statistically-significant differences between the ketamine- and saline-treated groups (D–F). *p<0.05, *** p<0.001. Panels A and D: n= 10 mice for Control+Saline, n= 8 mice for Contol+Ketamine, n= 8 mice for Fracture+Saline, and n= 8mice for Fracture+Ketamine. Panels B and E: n= 6 mice for each of the 4 groups. Panels C and F: n= 8 mice for Control+Saline, n= 8 mice for Contol+Ketamine, n= 6 mice for Fracture+Saline, and n= 12 mice for Fracture+Ketamine.
Fig. 5. Ketamine exposure improves capsaicin-induced hypersensitivity 18 weeks after fracture.

18 weeks after injury, fracture mice spend more time engaging in capsaicin-evoked behaviors (licking, biting, scratching, and shaking) compared to control mice (A, B, red arrows, p<0.001). This is indicative of persistent hindpaw sensitization in the absence of notable differences in mechanical thresholds. Furthermore, the ketamine-treated group displayed less capsaicin-evoked behaviors when compared to the saline-treated group (B). No changes were observed in mechanical thresholds one hour following capsaicin or vehicle treatments in the ipsilateral (Ipsi, C, D) and contralateral (Contra, E, F) hindpaws. *** p<0.001. n= 8 mice for Control+Saline, n= 8 mice for Contol+Ketamine, n= 6 mice for Fracture+Saline, and n= 8 mice for Fracture+Ketamine.
Fig. 6. No side-effects were observed during chronic ketamine administration.

No changes in body weight (A), stereotypic behaviors (B) and activity levels in the home cage (C) were observed between ketamine- and saline-treated groups 5 days after ketamine exposure (at 7 weeks post-fracture). n= 7 mice for Control+Saline, n= 8 mice for Contol+Ketamine, n= 6 mice for Fracture+Saline, and n= 12 mice for Fracture+Ketamine.
Fig. 7. Ketamine exposure ameliorates complex regional pain syndrome-related upregulation of spinal astrocytes.

Immunohistochemical staining for GFAP shows that, in comparison with control animals, fracture mice exhibit increased numbers of astrocytes in the dorsal horn of the ipsilateral spinal cord 10 weeks after fracture. This increase is absent in the ketamine treated group (A). In contrast, no changes were seen in the number of Iba1+ cells at this timepoint (B). Examples of GFAP- and Iba1-stained sections are included in panels C–F. The areas enclosed by the dotted squares are further enlarged in the central panels. Scale bar = 100 μm *p<0.05. Sample sizes are shown in the scatter graph. GFAP= Glial fibrillary acidic protein, Iba1= Ionized calcium binding adaptor molecule 1, Ctl/Sal=Control + Saline, Ctl/Ket= Control + Ketamine, Fx/Sal= Fracture + Saline, Fx/Ket= Fracture + Ketamine.
Fig. 8. Ketamine exposure is accompanied by biochemical changes in the lumbar spinal cord.
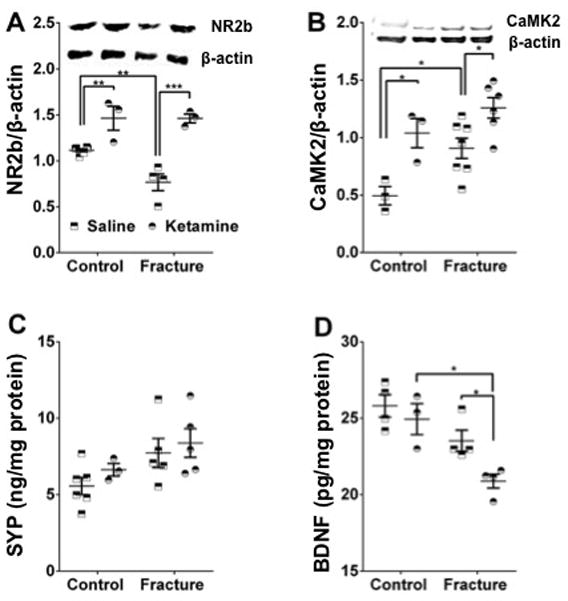
10 weeks following fracture, our data shows a decrease in the protein levels of NR2b in the fracture group, with ketamine exposure being associated with increased NR2b in both the fracture and control groups (A). Additionally, we show an increase in CAMK2 in the fracture group, with ketamine exposure linked to increased CAMK2 levels in both the fracture and control groups (B). No significant differences in synaptophysin levels were observed among the 4 groups (C). Finally, ketamine administration was accompanied by a decrease in BDNF levels in the fracture group only (D). *p<0.05, ** p<0.005, *** p<0.001. Sample sizes are shown in the scatter graph. NR2b= N-methyl-D-aspartate receptor 2b, CaMK2= Ca2+/calmodulin-dependent protein kinase ii, SYP= Synaptophysin, BDNF= Brain derived neurotrophic factor.
RESULTS
Fracture mice displayed a transient increase in edema and temperature in addition to persistent mechanical sensitivity in the affected hindpaw
Ipsilateral and contralateral measurements of hindpaw edema and temperature, measurements related to commonly observed signs in CRPS patients, were performed at 3 and 7 weeks post-fracture. Both temperature and edema demonstrated similar profiles, exhibiting transient increases identified at the 3-week (Temperature: 3w fracture versus [vs] control mean difference =0.59, 95% CI= 0.16 to 1.03, p<0.001; Edema: 3w fracture vs control mean difference =0.63, 95% CI= 0.41 to 0.84, p<0.001), but not 7-week timepoint (Fig. 2A, B, reported values reflect the difference between the values acquired from ipsilateral and contralateral hindpaw measurements). In contrast, mechanical sensitivity in the ipsilateral hindpaw, assessed using von Frey filaments, was evident in the in the 3- (fracture vs control mean difference =−1.25, 95% CI= −1.45 to −1.04, p<0.001), as well as 7-week timepoints (fracture vs control mean difference =−1.36, 95% CI= −1.56 to −1.15, p<0.001) (Fig. 2C). Furthermore, a transient decrease in mechanical thresholds was observed in the contralateral hindpaw of the fracture group at the 3-week timepoint only (Fig. 2D).
Chronic systemic ketamine infusion was anti-allodynic when administered at the late (7-weeks), but not early (3-weeks), timepoint after fracture
The infusion of ketamine for 1 week via subcutaneous osmotic pumps did not result in increased mechanical thresholds when administered 3 weeks after fracture (Fig. 3A, B). In contrast, when administered 7 weeks after fracture, ketamine had a significant anti-allodynic effect in fracture mice, starting at day 7 post-treatment. Furthermore, this anti-allodynic effect was maintained for 4 weeks after the termination of the ketamine infusion (2-way ANOVA surgery factor p-value<0.0001, time factor p-value <0.0001), until the resolution of mechanical hypersensitivity was observed in the saline treated fracture mice (at 12 weeks post-fracture). No significant ketamine effects were observed in control mice or in the ipsilateral hindpaw (Fig. 3C,D).
Due to the lack of efficacy at the acute (3-weeks) phase, all subsequent experiments were carried out in cohorts of mice treated at the chronic (7-weeks) phase.
Chronic systemic ketamine infusion had long-term effects on motor performance
Our findings regarding the anti-allodynic effects of ketamine prompted us to examine the short- and long-term functional outcomes of ketamine treatment administered at 7 weeks after fracture. We used 3 different cohorts since our aim was to examine rotarod performance following first-time exposure to the apparatus. At the 7-weeks timepoint, when ketamine was chronically administered via the osmotic pumps for 7 days, we observed no significant changes between the saline-treated control and fracture groups, and no ketamine efficacy in the fracture group (Fig. 4A, rotarod testing was performed on the 3rd day of chronic infusion, one animal was excluded from the fracture + saline group based on Grubb’s outlier test). Similarly, at the 8-week timepoint, which marks the removal of the osmotic pumps, there were no differences among the 4 experimental groups (Fig. 4B). These data suggest that fracture-induced functional deficits are not easily observed at these timepoints (although trends of diminished motor performance are seen), and that ketamine does not impair performance at the dose administered. It is noteworthy that the lack of significant fracture effect at this timepoint could be due to reduced motor performance in the control mice due to the surgical procedures used for implanting and removing the pumps. In contrast, when the mice were first introduced to the rotarod apparatus 18 weeks after fracture (long after mechanical thresholds were normalized), fracture mice showed significant impairment in the rotarod assay (ketamine group: fracture vs control mean difference=−61.03, 95% CI=−94.76 to −27.29, p<0.001; saline group: fracture vs control mean difference=−112.0, 95% CI=−155.1 to −68.97, p<0.001), which was reduced in the group of mice that was treated with chronic ketamine many weeks prior (Fig. 4C, fracture group: saline vs ketamine mean difference= −34.93, 95% CI=−66.73 to −3.140, p=0.033). These data suggest that sustained ketamine infusion can positively affect performance of a novel motor task many weeks after exposure to the drug.
Finally, training the fracture mice on the rotarod for 5 consecutive trials (inter-trial interval = 30 minutes) showed no difference between the treatment groups on the 3rd day of infusion (7-weeks), at the completion of infusion (8-weeks), and at 18 weeks after fracture (at this time-point, there was a significant effect of training, but post-hoc comparisons show no significant differences between the two groups at any of the trials) (Fig. 4D–F), suggesting that the observed results are unique to the initial exposure to a new motor task (one animal was excluded from the fracture + saline group at the 8w timepoint based on Grubb’s outlier test).
Fracture mice displayed hypersensitivity to subcutaneous capsaicin that was ameliorated by prior ketamine exposure
The rotarod data suggested that low-dose chronic ketamine administration (a 7 day infusion at 7 weeks post-fracture) had functional effects at 18 weeks post-fracture, long after the ketamine treatment ended. We then further explored the chronic effects of ketamine infusion on overall nociception. Since mechanical thresholds had already normalized by 18 weeks post-fracture, we challenged the 4 groups of mice with a subcutaneous injection of capsaicin. Capsaicin-evoked behaviors were interpreted as an indication of latent sensitization not detectable by measuring hindpaw mechanical sensitivity thresholds.
When capsaicin was injected into the hindpaw at the 18 weeks post-fracture time point, fracture mice spent more time engaging in capsaicin-evoked behaviors (licking, biting, scratching, and shaking the hindpaw) than control non-fractured mice (Fig 5A, B, red arrows). This observation could represent persistent hindpaw sensitization (due to peripheral or central mechanisms) in the absence of notable differences in mechanical thresholds,19 or alternatively, could be an indication of ongoing mechanical sensitivity that is not detected by reflexive measures. Furthermore, the group of mice that had been treated with a 7-day infusion of ketamine at 7 weeks post-fracture displayed less capsaicin-evoked behaviors when compared to the saline-treated fracture group (Fig 5B, Fracture group receiving capsaicin: saline vs ketamine mean difference=−49.92, 95% CI=−66.52 to −33.33, p<0.001). No changes were observed in mechanical thresholds one hour following capsaicin or vehicle treatments in the ipsilateral (Fig 5C, D) and contralateral hindpaws (Fig 5E, F).
It is important to note that this data doesn’t imply that the capsaicin hypersensitivity is necessarily due to central mechanisms, particularly since we limited our studies to the hindpaw corresponding to the fractured limb.
Low-dose systemic ketamine administration was not accompanied by observable side effects
Preclinical studies in animal models can be heavily confounded by motor ability and/or changes in overall activity levels. To test whether ketamine administration results in any visible side effects, changes in body weight (% change in body mass immediately before and after 1week of ketamine treatment), in addition to stereotypic behaviors and activity levels in the home cage were scored. While these measures might be better suited for a more global assessment of side effects and may not be able to detect side effects that are “mild”, our results show no differences in any of observed measures between ketamine- and saline-treated groups (Fig 6A–C). These observations are in agreement with our data from the rotarod (Fig 4A) assay, where ketamine treatment was not accompanied by motor dysfunction.
Fracture mice induced activation of spinal astrocytes was inhibited in mice previously treated with ketamine
In order to study the immunohistochemical correlates of the behavioral signs of anti-allodynia, we examined spinal levels of neuroinflammation markers at 10 weeks after fracture. This time point was chosen for the following reasons: first, the thresholds of mechanical sensitivity were different among the treatment groups at this timepoint, and second, any observed results would not be due to the acute effects of ketamine, since there would be a washout period of approximately 10 days before the biochemical measurements.
Immunohistochemical staining for GFAP showed that, in comparison with control animals, fracture mice exhibit increased numbers of immunostained astrocytes in the dorsal horn of the ipsilateral lumbar spinal cord at 10 weeks after fracture (saline group: fracture vs control mean difference=−141.4, 95% CI= −250.3 to −32.44, p=0.013). This increase was absent in the ketamine treated group (Fig. 7A, fracture group: saline vs ketamine mean difference= −135.6, 95% CI= −252.1 to −19.18, p=0.024). In contrast, no changes were seen in the number of Iba1+ immunostained spinal microglia at this timepoint (Fig. 7B).
Ketamine exposure was accompanied by biochemical changes in the lumbar spinal cord
In addition to our immunohistochemical studies, we studied biochemical changes in the lumbar spinal cord 10 weeks following fracture. NR2b was chosen based both on our preliminary studies and on a recent study showing NR2b-containing receptor/ion channels to play a prominent role in synaptic transmission in the superficial lamina of the rodent spinal cord.21 CaMK2 was included based on our ingenuity pathway analysis of our previously-published SC microarray data4 and its known binding to, and activation by, stimulated NR2bs; synaptophysin was chosen as a proxy indicator of synaptic abundance; and BDNF was chosen because of its known involvement in pain and central sensitization.
NR2b
There was a decrease in the protein levels of the glutamate receptor subunit NMDAR2b in spinal cord tissue from the fracture group at 10 weeks after fracture (saline group: fracture vs control mean difference=0.35, 95% CI= 0.12 to 0.57, p=0.004). Ketamine exposure was associated with increased NR2b in both the fracture (ketamine vs saline mean difference=0.70, 95% CI=0.43 to 0.96, p<0.001) and control (ketamine vs saline mean difference=0.35, 95% CI= 0.10 to 0.60, p=0.007) groups (Fig. 8A).
CaMK2
Protein levels of the serine/threonine-specific protein kinase CaMK2 were increased in the fracture group at 10 weeks after fracture, compared to non-fracture controls (saline group: fracture vs control mean difference=0.41, 95% CI=0.04 to 0.78, p=0.029), consistent with our previously published mRNA results from 3- and 7-weeks post-fracture mice.4 Furthermore, ketamine exposure was associated with further increases in CaMK2 levels in both the fracture (ketamine vs saline mean difference=−0.35, 95% CI=−0.65 to −0.05, p=0.021) and control (ketamine vs saline mean difference=−0.54, 95% CI=−0.98 to −0.10, p=0.015) groups (Fig. 8B).
Synaptophysin
No significant differences were observed in the protein levels of synaptophysin, a proxy marker for synaptic abundance caused by either fracture or ketamine treatment (Fig. 8C).
BDNF
There were no significant changes in spinal levels of BDNF 10weeks following fracture in comparison to control animals. However, ketamine exposure was associated with a decrease in BDNF in the fracture group only (Fig. 8D, ketamine vs saline mean difference=−2.66, 95% CI= −5.178 to −0.1352, p=0.039).
DISCUSSION
Many options for the treatment of CRPS have been described, but none are universally effective. Given its changing nature over time, there may be value in knowing when, over the course of the condition, specific therapies become most efficacious. One promising but controversial approach is the use of low-dose ketamine infusions,5,22 where periodic infusions are used repeatedly, with anti-hyperalgesic effects lasting for 6 months or more in some patients.23 Using the tibia fracture/cast model of CRPS in mice, we have shown that 1-week systemic infusions, at an allo-scaled dose (comparable to what is used in human subjects), is efficacious when administered in the late (7-weeks), but not early (3-weeks), stages of the syndrome. We believe that our preclinical data has significant clinical relevance since, when administered in the late phase, ketamine could ameliorate 3 key features relevant to the clinical management of this condition: 1) persistent allodynia: mechanical thresholds were reduced for up to 12 weeks after fracture and this allodynia completely resolved after ketamine treatment. 2) Latent sensitization: exaggerated capsaicin-induced nociceptive responses were observed in our model after reflexive measures of mechanical allodynia had subsided and were reversed or prevented by earlier ketamine treatment. 3) Motor dysfunction: similar to clinical observations,12 ketamine ameliorated motor dysfunction at a chronic timepoint long after the normalization of the mechanical sensitivity thresholds; and important finding, since pain relief and recovery of motor function go hand-in-hand when considering rehabilitation programs for CRPS patients.
Acute vs. Chronic Stages of CRPS
Similar to the two distinct phases (acute “warm” vs. chronic “cold”2) observed in patients, fracture/cast mice exhibit distinct physiological, behavioral, biochemical, and transcriptional changes in the early and late phases.4 We postulate that it is therefore likely that mechanism-based treatment outcomes would vary when administered at these different stages.
The differential efficacy of ketamine at the acute vs. chronic timepoints could be due to many factors. One possibility is that ketamine’s CNS mediated analgesic effects are obscured by peripheral pro-nociceptive mechanisms at the early stages of the syndrome. This hypothesis is supported by prior evidence where, during the acute phase, we have shown prominent effects of neuroinflammation and elevated levels of inflammatory mediators in the skin of the affected tissue including Interleukin-1beta, Tumor necrosis factor-alpha, Interleukin-6 and others, both in our rodent model15,19,24–26 and in human biopsy samples.27 Another potentially ketamine-resistant mechanism of nociceptive sensitization is the peripheral autoimmune response observed during the acute stage of the syndrome.28 A centrally-acting NMDA receptor antagonist may be ineffective in suppressing sensitization supported by these mechanisms. Consequently, once these peripheral inflammatory responses subside at 7 weeks post-fracture, as evidenced by the resolution of temperature elevation and edema, ketamine CNS analgesic action may be more effective. Alternatively, it is possible that ketamine acts on signaling pathways mediating central sensitization that are not present or fully developed at the acute timepoint. This hypothesis is supported by our behavioral evidence showing ketamine efficacy in providing persistent reversal of sensitization only during infusion in the chronic phase. Additionally, ketamine reduced latent sensitization to capsaicin injections performed after the resolution of allodynia (our experiments do not discern whether this latent sensitization is established entirely during the acute phase, the late chronic phase or requires both) and normalized motor performance on the rotarod; measures that are ostensibly, though not exclusively, sensitive to CNS changes. A summary of the peripheral and central changes at the acute and chronic stages of CRPS in the fracture/cast rodent model is shown in Fig. 9.
Fig. 9. Summary.
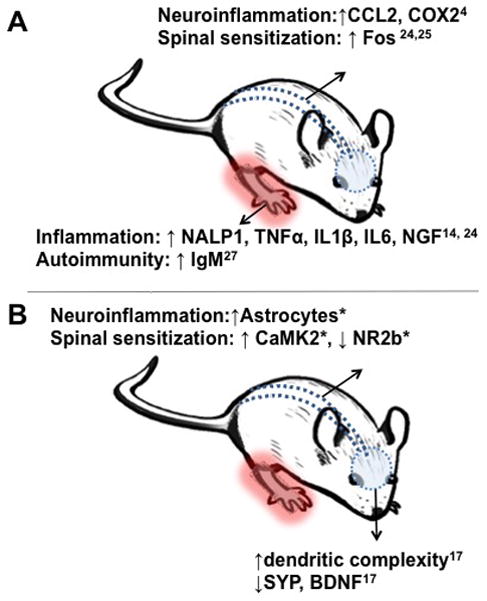
Illustration of potential nociceptive mechanisms observed in the acute (A) vs. chronic (B) stages of complex regional pain syndrome in the fracture/cast rodent model. * indicates data shown in the current manuscript. CCL2= chemokine (C-C motif) ligand 2, COX2= Cytochrome c oxidase subunit 2, NALP1= NAcht Leucine-rich repeat Protein 1, TNFα= Tumor necrosis factor alpha, IL1β= Interleukin 1 beta, IL6= Interleukin 6, NGF= Nerve growth factor, IgM= Immunoglobulin M, CaMK2= Ca2+/calmodulin-dependent protein kinase ii, NR2b= N-methyl-D-aspartate receptor 2b, SYP= Synaptophysin, BDNF= Brain derived neurotrophic factor.
Hypothesizing that ketamine’s effects might be most clearly demonstrated in the CNS, we performed immunohistochemical and molecular experiments to determine if CNS cells and proteins associated with persistent nociceptive sensitization were chronically inhibited after 7-days of ketamine infusion.
Spinal Mechanisms of Ketamine Activity
A 7-day infusion of ketamine chronically inhibited the post-fracture activation of SC dorsal horn astrocytes for several weeks after the pumps were removed. While initially considered as passive bystanders, it is now clear that glia constitute a dynamic part of the chronic pain equation. This is particularly true of the more abundant glial cells, astrocytes.29 At the chronic time point we studied, astrocytic but not microglial activation was evident. This is consistent with findings from preclinical models of pain where microglial upregulation has been shown during the acute phase30 while astrocytic activation is often prominently observed in the more persistent states.31 Data from pharmacological studies in preclinical pain models has shown that ketamine administration inhibits spinal nerve ligation-induced activation of astrocytes and the associated hyperalgesia.32 Additionally, our data is consistent with in vitro studies showing the inhibition of lipopolysaccharide-induced production of prostaglandin-E2 and tumor necrosis factor alpha in primary astrocytic cultures.33 Our data provides further evidence that glial inhibitory changes can be persistent following ketamine treatment.
The various immune mediators released in the SC after peripheral injury could result in central sensitization by direct modulation of excitatory synaptic transmission. Central sensitization is a mechanism common to many chronic pain syndromes including CRPS, where increased glutamate levels in the cerebrospinal fluid of patients are observed.34 Despite the fact that the exact role of NMDA receptors in this mechanism is unknown, it is generally felt that an important pathway through which peripheral noxious stimulation leads to persistent pain states is through central sensitization at the level of the SC, where, in concert with other systems, the co-release of peptides and glutamate from peripheral nerves induces the activation of NMDA receptors.35 For our biochemical studies, the NR2b subunit was chosen based both on our preliminary data and on a recent study showing NR2b-containing receptors to play a predominant role in synaptic transmission in the superficial lamina of the rodent SC;21 CaMK2 was included based on ingenuity pathway analysis of our previously-published SC microarray data4 and its known binding to, and activation by, stimulated NR2bs; synaptophysin was chosen as a proxy indicator of synaptic abundance; BDNF was chosen because of its established involvement in pain and central sensitization. We show that persistent sensitization in our model is associated with decreased NR2b and increased CaMK2, with no changes in the spinal levels of synaptophysin and BDNF. Furthermore, data from the ketamine-treated groups did not provide us with a clear biochemical mechanism that could account for the anti-allodynic effects of the drug. It is therefore possible that, at least at the 10-week timepoint, ketamine mechanisms are mainly glial and not neuronal, and might even be independent of NMDA receptors entirely. As such, future studies that fully evaluate the role of glial cells, particularly astrocytes, in this pain model are needed. We have observed in a rat model of CRPS that the intrathecal administration of the astroglial inhibitor L-alpha-aminoadipate reduces mechanical allodynia (data not shown). This concept is consistent with the lack of therapeutic efficacy of drugs that target aberrant neuronal activity alone36. It is also possible, however, that examining only a few biochemical targets did not provide an adequately comprehensive account of central sensitization in our model.
Non-spinal Mechanisms of Ketamine Activity
We examined the spinal effects of a 7–day systemic infusion of ketamine in a mouse model of CRPS. However, ketamine may have effects on both supraspinal structures and peripheral tissues as well. For example, human imaging shows that sub-anesthetic doses of ketamine reduce connectivity between the somatosensory network and areas involved in the affective processing of pain.37 In our data, a 7-day systemic ketamine infusion had no immediate effects of measures of anxiety in the fracture model (data not shown); however, it is nonetheless possible that some of the anti-hyperalgesic/anti-allodynic effects could be attributed to supraspinal mechanisms. Furthermore, sub-anesthetic doses of ketamine have been shown to have anti-depressant activity. Ketamine used in mouse models of depression and anxiety has been demonstrated to reverse deleterious neuroplastic changes, e.g. dendritic, synaptic protein and BDNF changes, in the prefrontal cortex and hippocampus.38 Similarly, improvements in neuropsychological testing along with normalization of resting state networks have been documented clinically in CRPS patients treated with ketamine.13
In addition to their role in the CNS, NMDA receptors are expressed at the peripheral processes of unmyelinated fibers, and their activation is linked to the development of hyperalgesia in neuropathic rats.39 Additionally, topical ketamine treatment has shown mixed effects in reducing mechanical allodynia in CRPS patients,7,40 where analgesic effects appear to be superior during the acute phase of the syndrome. These local mechanisms were not fully evaluated in our studies and that we cannot exclude peripheral in addition to central sites of action for ketamine in our model. Furthermore, ketamine has well known anti-inflammatory effects through interactions with toll-like receptors on immune cells, which could account for some of its analgesic actions.41 This is an important mechanism to consider in CRPS, since, in addition to peripheral inflammation that is associated with the syndrome, peripheral vascular changes could also be sensitive to ketamine treatment which is thought to be involved in improved blood flow and even direct relaxation of blood vessels.42
Limitations and Future Directions
Animal models have inherent limitations regarding the validity and translational value of the findings. Using a non-specific NMDA antagonist, while being clinically relevant, does leave the need to establish that the effects observed are, in fact, mediated by NMDA receptors, particularly since ketamine could act through NMDA-independent mechanisms of action, by binding to opioid receptors, monoamine transporters, muscarinic and nicotinic cholinergic receptors, serotonin receptors, etc.41 In the current study, ketamine was administered systemically. Despite the fact that ketamine is thought to cross the blood-SC and blood-brain barriers, a more thorough approach would have been to study, in parallel, the administration of ketamine to the periphery, SC, and various brain regions. Additionally, only a single dose of ketamine was used; it is possible that higher doses could have resulted in some analgesic action, though our methods were not suitable to address this question. Finally, the current manuscript focused on the long-term effects of ketamine administration; more granular timecourse studies might better elucidate the effects of ketamine infusion on CRPS-related behaviors and cellular and biochemical changes.
Acknowledgments
Financial support: This study was supported by National Institute of Health grant NS072168 to WSK and JDC (Palo Alto Institute of Research and Education, Palo Alto, CA, USA).
The authors would like to thank Dr. Tian-zhi Guo, PhD (Palo Alto Veterans Institute of Research and Education, Palo Alto, CA, USA), Dr. Wenwu Li, PhD (Palo Alto Veterans Institute of Research and Education, Palo Alto, CA, USA), and Ms. Hamda Khan (Palo Alto Veterans Institute of Research and Education, Palo Alto, CA, USA) for technical assistance.
Footnotes
Parts of this work have been presented at the American Pain Society Annual meeting (APR 30-May 3, 2014) in Tampa, Florida
Conflict of interest: The authors report no conflict of interest.
References
- 1.de Mos M, de Bruijn AG, Huygen FJ, Dieleman JP, Stricker BH, Sturkenboom MC. The incidence of complex regional pain syndrome: a population-based study. Pain. 2007;129:12–20. doi: 10.1016/j.pain.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Bruehl S. An update on the pathophysiology of complex regional pain syndrome. Anesthesiology. 2010;113:713–25. doi: 10.1097/ALN.0b013e3181e3db38. [DOI] [PubMed] [Google Scholar]
- 3.Parkitny L, McAuley JH, Di Pietro F, Stanton TR, O’Connell NE, Marinus J, van Hilten JJ, Moseley GL. Inflammation in complex regional pain syndrome: a systematic review and meta-analysis. Neurology. 2013;80:106–17. doi: 10.1212/WNL.0b013e31827b1aa1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gallagher JJ, Tajerian M, Guo T, Shi X, Li W, Zheng M, Peltz G, Kingery WS, Clark JD. Acute and chronic phases of complex regional pain syndrome in mice are accompanied by distinct transcriptional changes in the spinal cord. Mol Pain. 2013;9:40. doi: 10.1186/1744-8069-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwartzman RJ, Alexander GM, Grothusen JR, Paylor T, Reichenberger E, Perreault M. Outpatient intravenous ketamine for the treatment of complex regional pain syndrome: a double-blind placebo controlled study. Pain. 2009;147:107–15. doi: 10.1016/j.pain.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 6.Sigtermans MJ, van Hilten JJ, Bauer MC, Arbous MS, Marinus J, Sarton EY, Dahan A. Ketamine produces effective and long-term pain relief in patients with Complex Regional Pain Syndrome Type 1. Pain. 2009;145:304–11. doi: 10.1016/j.pain.2009.06.023. [DOI] [PubMed] [Google Scholar]
- 7.Finch PM, Knudsen L, Drummond PD. Reduction of allodynia in patients with complex regional pain syndrome: A double-blind placebo-controlled trial of topical ketamine. Pain. 2009;146:18–25. doi: 10.1016/j.pain.2009.05.017. [DOI] [PubMed] [Google Scholar]
- 8.Azari P, Lindsay DR, Briones D, Clarke C, Buchheit T, Pyati S. Efficacy and safety of ketamine in patients with complex regional pain syndrome: a systematic review. CNS Drugs. 2012;26:215–28. doi: 10.2165/11595200-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 9.Belujon P, Grace AA. Restoring mood balance in depression: ketamine reverses deficit in dopamine-dependent synaptic plasticity. Biol Psychiatry. 2014;76:927–36. doi: 10.1016/j.biopsych.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mathew SJ, Shah A, Lapidus K, Clark C, Jarun N, Ostermeyer B, Murrough JW. Ketamine for treatment-resistant unipolar depression: current evidence. CNS drugs. 2012;26:189–204. doi: 10.2165/11599770-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salvadore G, Singh JB. Ketamine as a fast acting antidepressant: current knowledge and open questions. CNS Neurosci Ther. 2013;19:428–36. doi: 10.1111/cns.12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schilder JC, Sigtermans MJ, Schouten AC, Putter H, Dahan A, Noldus LP, Marinus J, van Hilten JJ. Pain relief is associated with improvement in motor function in complex regional pain syndrome type 1: secondary analysis of a placebo-controlled study on the effects of ketamine. J Pain. 2013;14:1514–21. doi: 10.1016/j.jpain.2013.07.013. [DOI] [PubMed] [Google Scholar]
- 13.Becerra L, Schwartzman RJ, Kiefer RT, Rohr P, Moulton EA, Wallin D, Pendse G, Morris S, Borsook D. CNS Measures of Pain Responses Pre- and Post-Anesthetic Ketamine in a Patient with Complex Regional Pain Syndrome. Pain Med. 2009 doi: 10.1111/pme.12939. [DOI] [PubMed] [Google Scholar]
- 14.Guo TZ, Offley SC, Boyd EA, Jacobs CR, Kingery WS. Substance P signaling contributes to the vascular and nociceptive abnormalities observed in a tibial fracture rat model of complex regional pain syndrome type I. Pain. 2004;108:95–107. doi: 10.1016/j.pain.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 15.Li WW, Guo TZ, Li XQ, Kingery WS, Clark JD. Fracture induces keratinocyte activation, proliferation, and expression of pro-nociceptive inflammatory mediators. Pain. 2010;151:843–52. doi: 10.1016/j.pain.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei T, Sabsovich I, Guo TZ, Shi X, Zhao R, Li W, Geis C, Sommer C, Kingery WS, Clark DJ. Pentoxifylline attenuates nociceptive sensitization and cytokine expression in a tibia fracture rat model of complex regional pain syndrome. Eur J Pain. 2009;13:253–62. doi: 10.1016/j.ejpain.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tajerian M, Leu D, Zou Y, Sahbaie P, Li W, Khan H, Hsu V, Kingery W, Huang TT, Becerra L, Clark JD. Brain neuroplastic changes accompany anxiety and memory deficits in a model of complex regional pain syndrome. Anesthesiology. 2014;121:852–65. doi: 10.1097/ALN.0000000000000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo TZ, Wei T, Shi X, Li WW, Hou S, Wang L, Tsujikawa K, Rice KC, Cheng K, Clark DJ, Kingery WS. Neuropeptide deficient mice have attenuated nociceptive, vascular, and inflammatory changes in a tibia fracture model of complex regional pain syndrome. Mol Pain. 2012;8:85. doi: 10.1186/1744-8069-8-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reichling DB, Levine JD. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009;32:611–8. doi: 10.1016/j.tins.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 21.Hildebrand ME, Pitcher GM, Harding EK, Li H, Beggs S, Salter MW. GluN2B and GluN2D NMDARs dominate synaptic responses in the adult spinal cord. Sci Rep. 2014;4:4094. doi: 10.1038/srep04094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Connolly SB, Prager JP, Harden RN. A systematic review of ketamine for complex regional pain syndrome. Pain Med. 2015;16:943–69. doi: 10.1111/pme.12675. [DOI] [PubMed] [Google Scholar]
- 23.Kiefer RT, Rohr P, Ploppa A, Dieterich HJ, Grothusen J, Koffler S, Altemeyer KH, Unertl K, Schwartzman RJ. Efficacy of ketamine in anesthetic dosage for the treatment of refractory complex regional pain syndrome: an open-label phase II study. Pain Med. 2008;9:1173–201. doi: 10.1111/j.1526-4637.2007.00402.x. [DOI] [PubMed] [Google Scholar]
- 24.Li WW, Guo TZ, Liang D, Shi X, Wei T, Kingery WS, Clark JD. The NALP1 inflammasome controls cytokine production and nociception in a rat fracture model of complex regional pain syndrome. Pain. 2009;147:277–86. doi: 10.1016/j.pain.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sabsovich I, Wei T, Guo TZ, Zhao R, Shi X, Li X, Yeomans DC, Klyukinov M, Kingery WS, Clark JD. Effect of anti-NGF antibodies in a rat tibia fracture model of complex regional pain syndrome type I. Pain. 2008;138:47–60. doi: 10.1016/j.pain.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sabsovich I, Guo TZ, Wei T, Zhao R, Li X, Clark DJ, Geis C, Sommer C, Kingery WS. TNF signaling contributes to the development of nociceptive sensitization in a tibia fracture model of complex regional pain syndrome type I. Pain. 2008;137:507–19. doi: 10.1016/j.pain.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Birklein F, Drummond PD, Li W, Schlereth T, Albrecht N, Finch PM, Dawson LF, Clark JD, Kingery WS. Activation of cutaneous immune responses in complex regional pain syndrome. J Pain. 2014;15:485–95. doi: 10.1016/j.jpain.2014.01.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li WW, Guo TZ, Shi X, Czirr E, Stan T, Sahbaie P, Wyss-Coray T, Kingery WS, Clark JD. Autoimmunity contributes to nociceptive sensitization in a mouse model of complex regional pain syndrome. Pain. 2014;155:2377–89. doi: 10.1016/j.pain.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao YJ, Ji RR. Targeting astrocyte signaling for chronic pain. Neurotherapeutics. 2010;7:482–93. doi: 10.1016/j.nurt.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Echeverry S, Shi XQ, Zhang J. Characterization of cell proliferation in rat spinal cord following peripheral nerve injury and the relationship with neuropathic pain. Pain. 2008;135:37–47. doi: 10.1016/j.pain.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 31.Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem. 2006;97:772–83. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- 32.Mei X, Wang W, Wang W, Li Y, Zhang H, Wu S, Li Y, Xu L. Inhibiting astrocytic activation: a novel analgesic mechanism of ketamine at the spinal level? J Neurochem. 2009;109:1691–700. doi: 10.1111/j.1471-4159.2009.06087.x. [DOI] [PubMed] [Google Scholar]
- 33.Shibakawa YS, Sasaki Y, Goshima Y, Echigo N, Kamiya Y, Kurahashi K, Yamada Y, Andoh T. Effects of ketamine and propofol on inflammatory responses of primary glial cell cultures stimulated with lipopolysaccharide. Br J Anaesth. 2005;95:803–10. doi: 10.1093/bja/aei256. [DOI] [PubMed] [Google Scholar]
- 34.Alexander GM, Perreault MJ, Reichenberger ER, Schwartzman RJ. Changes in immune and glial markers in the CSF of patients with Complex Regional Pain Syndrome. Brain Behav Immun. 2007;21:668–76. doi: 10.1016/j.bbi.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 35.Baron R, Hans G, Dickenson AH. Peripheral input and its importance for central sensitization. Ann Neurol. 2013;74:630–6. doi: 10.1002/ana.24017. [DOI] [PubMed] [Google Scholar]
- 36.Varrassi G. Management of chronic pain. Foreword Clin Drug Investig. 2010;30(Suppl 2):1. doi: 10.2165/1158415-S0-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 37.Niesters M, Khalili-Mahani N, Martini C, Aarts L, van Gerven J, van Buchem MA, Dahan A, Rombouts S. Effect of subanesthetic ketamine on intrinsic functional brain connectivity: a placebo-controlled functional magnetic resonance imaging study in healthy male volunteers. Anesthesiology. 2012;117:868–77. doi: 10.1097/ALN.0b013e31826a0db3. [DOI] [PubMed] [Google Scholar]
- 38.Duman RS, Li N, Liu RJ, Duric V, Aghajanian G. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. 2012;62:35–41. doi: 10.1016/j.neuropharm.2011.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jang JH, Kim DW, Sang Nam T, Se Paik K, Leem JW. Peripheral glutamate receptors contribute to mechanical hyperalgesia in a neuropathic pain model of the rat. Neuroscience. 2004;128:169–76. doi: 10.1016/j.neuroscience.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 40.Ushida T, Tani T, Kanbara T, Zinchuk VS, Kawasaki M, Yamamoto H. Analgesic effects of ketamine ointment in patients with complex regional pain syndrome type 1. Reg Anesth Pain Med. 2002;27:524–8. doi: 10.1053/rapm.2002.35517. [DOI] [PubMed] [Google Scholar]
- 41.Sawynok J. Topical and peripheral ketamine as an analgesic. Anesth Analg. 2014;119:170–8. doi: 10.1213/ANE.0000000000000246. [DOI] [PubMed] [Google Scholar]
- 42.Akata T, Izumi K, Nakashima M. Mechanisms of direct inhibitory action of ketamine on vascular smooth muscle in mesenteric resistance arteries. Anesthesiology. 2001;95:452–62. doi: 10.1097/00000542-200108000-00030. [DOI] [PubMed] [Google Scholar]