

Abstract
Cardiovirus Leader proteins (LX) inhibit cellular nucleocytoplasmic trafficking by directing host kinases to phosphorylate Phe/Gly-containing nuclear pore proteins (Nups). Resolution of the Mengovirus LM structure bound to Ran GTPase, suggested this complex would further recruit specific exportins (karyopherins), which in turn mediate kinase selection. Pull-down experiments and recombinant complex reconstitution now confirm that Crm1 and CAS exportins form stable dimeric complexes with encephalomyocarditis virus LE, and also larger complexes with LE:Ran. shRNA knockdown studies support this idea. Similar activities could be demonstrated for recombinant LS and LT from Theiloviruses. When mutations were introduced to alter the LE zinc finger domain, acidic domain, or dual phosphorylation sites, there was reduced exportin selection. These regions are not involved in Ran interactions, so the Ran and Crm1 binding sites on LE must be non-overlapping. The involvement of exportins in this mechanism is important to viral replication and the observation of trafficking inhibition by LE.
Keywords: nucleocytoplasmic trafficking inhibition, cardiovirus, Leader protein, Crm1
Introduction
Eukaryotic nuclei have double-layered membranes studded with an estimated 2800 channels called nuclear pore complexes (NPC). Each pore is comprised of about 30 nucleoporin proteins (Nups) arranged with octagonal symmetry (1). Many of these Nups, especially those lining the central channel, are hydrophobic, displaying characteristic Phe/Gly-repeat units which serve as a physical barrier to casual diffusion of molecules >40 kDa. Larger macromolecules cannot, by themselves, navigate the disordered, hydrophobic Nup tangle that constitutes the central channel. Instead, directionally targeted cargos display short nuclear localization signal motifs (NLS) or nuclear export signal motifs (NES) that are recognized and bound by karyopherin receptors (importins or exportins) traveling in the required direction. The cargo/receptor selection process is regulated by a Ran GTPase-moderated energy gradient, which determines the active, or inactive status of the various karyopherins (2).
There are multiple virus types which usurp these transport pathways to enhance their own replication or prevent activation of innate responses. Cardioviruses in the Picornaviridae family use the unique strategy of induced hyper-phosphorylation directed at multiple Phe/Gly Nups, to bring about a rapid, virtually complete inhibition of cellular import/export processes (3). The effect is toxic to cells, but allows these cytoplasmic viruses to replicate with apparent impunity. Encephalomyocarditis virus (EMCV), Mengovirus, Theiler’s murine encephalomyelitis virus (TMEV) and Saffold virus (Saf) are characteristic isolates which have been studied for these activities (4, 5).
Each Cardiovirus encodes a small, highly-charged Leader protein (LX, 67–76 amino acids) at the N-terminus of its polyprotein, which, when introduced into cells is sufficient to recapitulate the entire Nup hyper-phosphorylation phenomenon (3). Solution structures of the Mengo protein (LM) show a CHCC amino-proximal zinc finger domain as the dominant motif in an otherwise flexible conformation (6, 7). A central “hinge” region and C-proximal acidic region assume more defined induced-fit conformations only when LM interacts with Ran GTPase, a strong (3 nM) preferred binding target when tested with recombinant proteins, or as detected with native proteins within the infected cells’ nuclear rims (3, 6, 8). The complex locks Ran into an irreversible “active” structure, nearly identical (~1.6 Å RMSD) to nucleotide-bound RanGTP (6). In uninfected cells, this conformer of Ran would normally chaperone interactions between export karyopherins and their cargos, and then participate in shuttling these complexes through the NPC from nucleus to cytoplasm. But since Ran and LX are not kinases, it is clear that neither can be directly responsible for catalyzing infection-dependent Nup phosphorylation. Instead, it has been proposed that LM:Ran complexes nucleate subsequent (irreversible?) reactions with exportins, which in turn then carry the culpable activated kinase cargos directly to Nup targets (6). In recent support of this model, recombinant LE-GST (EMCV), when reacted with HeLa cytosol was shown to pulldown immunologically detectable amounts of Crm1 (chromosome region maintenance 1 protein, XPO1) and CAS (cellular apoptosis susceptibility protein, XPO2) exportins. The responsible kinases, as implicated with parallel inhibition studies, are known to include MAPK agents from the ERK1 and p38 pathways (9). Presumably, these enzymes, for which the activated forms have dominant nuclear localizations (10), are somehow preferentially selected for Nup phosphorylation activities by the LX:Ran:exportin complexes.
Other than the LX:Ran interactions, though, few of these steps have been broadly explored experimentally. Moreover, complicating the overall picture is an additional mechanistic requirement for dual LX phosphorylation. Ran interactions with LM do not require these modifications, but exportin extraction by recombinant LX (6), and ultimately Nup phosphorylation (5, 11), are sensitive to the motif status. Cardiovirus LX proteins vary somewhat in the sequence and lengths of their hinge and acidic regions, and it is within this variable context that the required phosphorylation residues occur. We now report new results showing direct, reciprocal binding interactions between native and recombinant LX proteins, with cellular and recombinant exportins, Crm1 and CAS. The data refine the existing mechanistic model and moreover suggest that these specific interactions may be mediated by the LX zinc finger, acidic domains and phosphorylation sites, which do not directly participate in Ran binding.
Results
Screen for LE binding partners
Ran GTPase, as a binding partner for GST-LE, was previously identified by reciprocal pull-down activities and confirmed with Western analyses using anticipated reactive antibodies (3, 6). Related experiments suggested the phosphorylation status of LE was a key determinant for additional relevant interactions, particularly with exportins (6). As a broader screen for dominant cytosolic partners, HeLa lysates were pre-cleared with glutathione beads and then reacted with GST-LE in the presence of added ATP. These conditions allow efficient dual phosphorylation of LE (11) while reducing background binding by non-specific proteins. After SDS-PAGE fractionation, 7 prominent band regions (Fig. 1) were excised, pooled, digested with trypsin and submitted for orbi-trap mass spectrometry. Table 1 summarizes the strongest sequence hits as defined by positive identification of at least 30 unique peptides each. The list includes exportin 1 (Crm1) and exportin 2 (CAS), the nuclear transport-associated karyopherins which had been modeled previously with probable LE:Ran interactions (6). The sizes of these proteins, 123 kDa and 110 kDa, respectively, are consistent with the principal band 2 included in pool A. Therefore, LE, as it is phosphorylated in this cytoplasmic context, does indeed react strongly with exportins. Interestingly, 6 of the 9 strongest hits (Cover %) covering both pools identified several other proteins with structurally analogous, HEAT repeat motifs, similar to those characteristic of the exportins, perhaps indicating a topological class of preferred contact interactions with LE.
Figure 1.
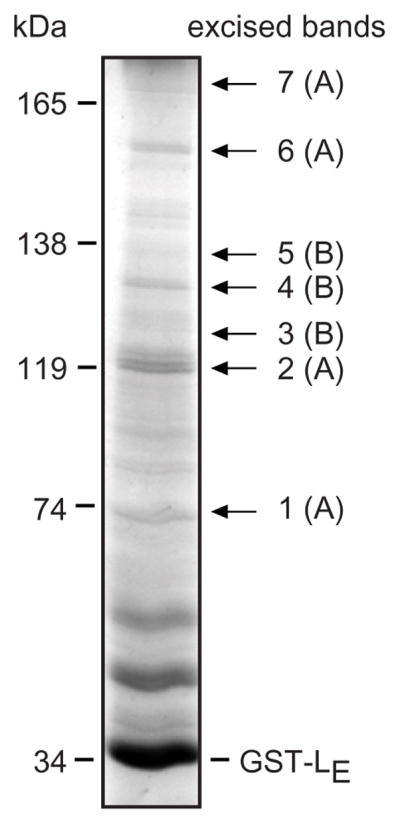
Cytosol selection by GST-LE. HeLa cytosol proteins (prey) reactive with GST-LE (bait) were collected, gel fractionated and visualized with silver stain. Bands of interest (1–7) were excised, pooled (A or B), digested with trypsin and analyzed by orbi-trap mass spectroscopy for protein identity. kDa indicates the MW of proteins in a parallel marker lane.
Table 1.
Proteins identified by mass spectroscopy
| Pool A | Protein Name | B Heat repeat | GenBank (ID) | C Size (kDa) | D Frags (#) | E Cover (%) |
|---|---|---|---|---|---|---|
| A | DNA-dependent protein kinase catalytic subunit | Yes | P78527 | 460 | 127 | 39 |
| A | E3 ubiquitin-protein ligase UBR5 | No | O95071 | 309 | 35 | 20 |
| A | Translational activator GCN1 | Yes | Q92616 | 234 | 85 | 41 |
| A | CAD protein | No | P27708 | 242 | 52 | 34 |
| B | Proteasome-associated protein ECM29 homolog | Yes | Q5VYK3 | 204 | 53 | 38 |
| B | Ras GTPase-activating-like protein IQGAP1 | No | P46940 | 189 | 30 | 26 |
| B | Bifunctional aminoacyl-tRNA synthetase | No | P07814 | 171 | 33 | 31 |
| B | Mitocondrial leucine-rich PPR motif-containing protein | No | P42704 | 158 | 48 | 48 |
| B | Condensin complex subunit 1 | Yes | Q15021 | 157 | 39 | 40 |
| B | Isoleucyl-tRNA synthetase, cytoplasmic | No | P41252 | 144 | 30 | 31 |
| B | DNA-directed RNA polymerase II subunit RPB2 | No | P30876 | 134 | 32 | 40 |
| B | DNA polymerase delta catalytic subunit | No | P28340 | 124 | 33 | 42 |
| A | Exportin-1 (Crm1) | Yes | O14980 | 123 | 37 | 50 |
| A | Exportin-2 (CAS) | Yes | P55060 | 110 | 43 | 57 |
| B | Protein KIAA1967 | No | Q8N163 | 103 | 33 | 50 |
| A | Alpha-actinin-4 | No | O43707 | 105 | 44 | 61 |
| A | Heat shock cognate 71 kDa protein | No | P11142 | 71 | 31 | 55 |
Figure 1 gel bands 1,2,6,7 were pool A, bands 3,4,5 were pool B
protein has HEAT repeats (Huntington elongation factor 3, regulatory subunit A of protein phosphatase A, and TOR1), consisting of pairs of anti-parallel helices arranged in a consecutive parallel stack (22).
As per genecard.org
Number of peptide fragments by mass spec, identifying this protein
Sum of peptide fragment coverage of full protein sequence
LE association with exportins
Mutational mapping (12) and determination of the solution structure of LM bound to Ran GTPase (6) explored sequence contributions from the central hinge region (aa 35–40), the zinc finger (aa 14–20), the acidic domain (aa 37–59) and phosphorylation sites (Tyr41, Thr47) as important to the observation of LX-dependent Nup phosphorylation. However, Cys19Ala (zinc finger), 4D4A (acidic domain) and Tyr41Ala/Thr47Ala phosphorylation changes did not significantly affect LE:Ran interactions with endogenous or recombinant proteins. Those contacts are mediated primarily by the LX hinge region, including W40 (12). With regard to exportins though, a subset panel GST- tagged LE proteins with same sequence changes (3D3A not 4D4A) were all less efficient than unmodified protein (wt) at extracting endogenous Crm1 and CAS from HeLa cell lysates (Fig 2A). Relative to the acidic domain deletion (ΔA) a negative control, which misfolds the remainder of the protein (12), the 4 tested substitutions in the other crucial Nup phosphorylation areas reduced exportin extraction by 50–90%. Exportin extraction was not unique to the LE sequences. Equivalent experiments with LS (Saf) and LT (TMEV) recombinant proteins showed these materials too, reacted readily (Fig 2B). In a reverse challenge, antibodies to Crm1, when attached to beads, were similarly able to find both intended target (native Crm1) and GST-LE spiked into the lysates (Fig 2C). Again, the most efficient conditions for these pulldowns required the full LE sequence. Mutational mapping and structure determinations place the W40 site primarily within the GST- LM:Ran interface, although NMR suggests there is considerable conformational motion (6, 12). The inefficiency with which Crm1 (bait) is extracted by αCrm1 (compare Fig 2C mock to wt) and the stoichiometric excess (>100x) of GST-LE make this particular experiment somewhat less sensitive to changes in binding efficiencies, relative to Fig 2A, particularly with regard to this specific mutation.
Figure 2.

GST-tagged LE mutations. Bait proteins, GST-LE and related mutant derivatives (A) as well as LE-GST, LS-GST and LT-GST (B) were incubated with HeLa cytosol. After glutathione sepharose bead extractions, Western analyses determined the amount of bound Crm1 and CAS, relative to the wt samples (% intensity). (C) GST-LE and mutant derivatives (prey) were mixed with HeLa cytosol (+ 0.5 μM ATP) then incubated with protein-G sepharose beads linked to αCrm1 (bait). After boiling and protein fractionation, the Crm1 and GST signals were detected by Western analyses. In these blots, the VH and VL coupling antibody proteins were also reactive with the secondary antibodies. (*) indicates LE-GST mutant derivatives.
Direct interaction with Crm1
HeLa lysates contain endogenous Ran GTPase. Moreover, these lysate-based systems will efficiently phosphorylate the first site and partially phosphorylate the second site of any added cardiovirus Leader (LX) proteins displaying intact sequences (5). But with cell-free GST-LE systems, the phosphorylation status can be precisely controlled by sequential addition of the relevant kinases. Syk activity at Tyr41 requires pre-phosphorylation with AmpK or CK2 at Thr47 (5, 11). GST-LE (singly phosphorylated) was able to extract recombinant Crm1 (rCrm1) from buffers that contained 300 or 500 mM salt, indicating rather tight complexes (Fig 3A). Surprisingly, the presence or absence of recombinant Ran (rRan) did not seem to have an effect on rCrm1 pulldown (in vitro), and rRan itself was also extracted whenever present. These experiments were carried out at stoichiometry, so it isn’t clear whether the GST-LE was partitioning between its potential partners or facilitating an inclusive complex. In either case, as previously reported with native Crm1/CAS from cell lysates (6), the relative extraction efficiency was sensitive to the phosphorylation status of LE (Fig 3B). With or without rRan, 1 or preferably 2 pre-phosphorylation GST-LE events, reproducibly (n=4) gave 50–70% stronger rCrm1 signals per unit of GST(-LE).
Figure 3.

Recombinant selection by GST-LE. (A) GST-LE reacted with CK2 (adds 1 phosphate) was incubated with stoichiometric amounts of rCrm1 and rRan (+RCC1) at 300 mM or 500 mM NaCl. After glutathione bead extraction, bound proteins were visualized by Western analyses. (B) Similar to A, with no added NaCl, GST-LE without no phosphates (0), 1 added phosphate (1, CK2) or 2 added phosphates (2, CK2 plus Syk) was mixed with combinations of rCrm1, rRan and RCC1. Glutathione bead-extracted proteins were visualized by Western analyses. The bands were quantified by densitometry. For each lane, the ratio of Crm1 to GST signal was normalized to the 0 phosphate, no rRan sample (100%). This full experiment was carried out 4 times. The averaged values per sample type are indicated (SD ave 28)
Complex formation
When the Fig 3A experiment was repeated with stepwise addition of the components, again, GST-LE (1 added phosphate) extracted rCrm1 regardless of whether rRan (+RCC1) was added before (“a”), or after (“b”) as the tertiary component. As a preliminary estimate of whether these 3 known proteins could collectively interact in vitro, dual phosphorylated GST-LE, rRan and rCrm1 were mixed, incubated and then fractionated over a Superdex 200 column. The absorbance values (Fig 4B) and parallel silver-stained gels (Fig 4C) placed about 20% of the signals from all 3 proteins within early eluates, near the trailing edge of the void volume (fractions 35–40). The column sensitivity in this region is too low to assign specific stoichiometries, but trimeric associations (~190 kDa) and/or larger combinations of GST-LE:rCrm1:rRan would be expected to elute here. There were additional strong peaks consistent with GST-LE:rCrm1 complexes (fractions 67–75) and also monomers of rRan (fractions 85–90). It can’t be excluded that some material at the front edge of the late-eluting rRan peak also contains Leader material (GST-LE:rRan?) that was carried below the limits detectable by silver-stain. In any case, the central fractions show conclusively that rCrm1 (120 kDa) and phosphorylated GST-LE (35 kDa) do readily interact in apparent 1:1 stoichiometry. Similar to the pulldown experiments (Fig 3B), parallel chromatographs confirmed that the intensity of this peak and the observation of complexes near the void volume, were dependent on the phosphorylation status GST-LE (not shown).
Figure 4.

Complex formation. (A) GST-LE pretreated with CK2 (adds 1 phosphate) was pre-incubated with stoichiometric amounts of rCrm1 and rRan (+RCC1) for lanes labeled with “a”. Then, rCrm1 and rRan (+RCC1) were added to previously deficient samples (“b”) before a second incubation period. Glutathione bead extraction and Western visualization was as in Fig 3. (B) Dual phosphorylated GST-LE (CK2 plus Syk) was incubated with rCrm1 and rRan (+RCC1). The sample was fractionated on a size exclusion column pre-calibrated with MW markers. Fraction absorbance aligned with (C) SDS-PAGE (silver stain) identified eluted complexes. Bands labeled “rCrm1*” are minor bacterial contaminants from the rCrm1 purification procedure.
shRNA knockdown of Crm1
Ultimately, Nup phosphorylation itself is the test for mechanistic involvement in LX-dependent activities. Crm1 is an important protein with multiple cellular roles (13), especially during mitosis. Its functions are not entirely compensated by alternative exportins like CAS (reviewed in (14)). Nonetheless, HeLa cells transduced with an shRNA against Crm1 remained viable, divided and survived without visible cytopathic effect for more than a week after doxycycline induction. Under such conditions, within the first 2 days, ~60% of the Crm1 protein was turned over (Fig 5A). By day 3, this value increased to ~80%. Induction alone did not allow Nup phosphorylation, but if the cells were also infected with vEC9 (attenuated, recombinant EMCV), the diagnostic upward smear of Nup62 was observed within 4 hr PI. If the infection were delayed until 2, 3 or 4 days post-knockdown (PK) there was correspondingly lower levels of Nup62 phosphorylation and the virus replicated more poorly. This was evidenced both by the lower levels (normalized to tubulin per sample) of viral polymerase proteins 3D and 3CD (Fig 5A), and by reductions in progeny titer, as determined by plaque assay (Fig 5C). Clearly, vEC9 became disadvantaged in cells with reduced levels of Crm1. This phenomenon is directly attributable to the LX mechanism reliance on Crm1, because when similar shRNA-induced cells were transfected (4 days PK) with cDNAs for the LE, LT or LS proteins, Nup62 phosphorylation was again reduced (Fig 5B).
Figure 5.

Crm1 shRNA knockdown. (A) Plated HeLa cells transduced with an shRNA against human Crm1 were treated with doxycycline. After 0–4 days post-knockdown (PK) the cells were infected with vEC9 (MOI of 10). Lysates harvested 4 hr later were fractionated for Western analyses. Band intensities for 4 separate experiments, normalized to day 0 values and to the tubulin loading controls, are indicated. (B) Similar cells with (+) and without (−) induction were transfected (4 days PK) with LX-GST cDNAs. Harvest was at 16 hr post-transfection. Nup62 phosphorylation was determined as in A (n=2). (C) Infected cell lysates from A, after 0 days (−) and 4 days (+) PK were titered on normal HeLa cells. Plaque formation was evaluated after 28 hr. Observed titers (PFU/ml) are averaged from (n=3) replicate plates (SD: 0.02–0.28).
Crm1 inhibition
Leptomycin B (LMB) is a well-studied fungal antibiotic (15) which alkylates most iterations of eukaryotic Crm1 proteins. It binds covalently to Cys528, effectively blocking the interaction with cargo nuclear export signals (NES) that would normally intercalate into a cleft between HEAT repeats H11 and H12 (16). Essentially, LMB is a diagnostic for Crm1 partners which use this canonical binding site. When added to the GST-LE pull-down assays (Fig 6A), the recombinant proteins (singly phosphorylated GST-LE) behaved the same as they did in Fig 3A. None of the rRan and rCrm1 pulldowns were obviously altered by the presence of this drug. Infected cells, monitored for Nup62 phosphorylation over a range of vEC9 MOI, gave similar results (Fig 6B) in that the presence of the drug was essentially ineffective. Such cells gave equivalent progeny titers regardless of whether LMB was added (Fig 6C). The GST-LE recognition of Crm1, then, must be outside of this binding site, or can be compensated for by CAS activities.
Figure 6.

Crm1 inhibition. (A) Leptomycin B (LMB, 4 nM) was pre-incubated with rCrm1 before the addition of bait proteins (singly phosphorylated GSL-LE or GST) and (putative) auxiliary prey proteins (rRan with RCC1). The complexes were extracted with glutathione-sepharose beads, fractionated on gels and detected by Western analyses. (B) Plated HeLa cells were pre-incubated with LMB or its carrier (MeOH) before infection with vEC9 at the indicated MOI. The cells were harvested at 4 hr PI. Nup62 phosphorylation levels (% intensity) were determined by Western analyses and densitometry relative to samples with no inhibitor or virus, and as normalized to each lane’s tubulin levels. (C) Plates of confluent HeLa cells were infected with vEC9 (MOI of 10) after pretreatment (or not) with LMB. After 6 hrs, recovered progeny virus was titered by plaque assay. Observed PFU/ml averaged from 3 independent experimental iterations are indicated (SD: 0.44–0.49).
Discussion
In cells or recombinant assays, cardiovirus Leader proteins react tightly with Ran GTPase, forcing this nuclear trafficking regulator into a conformation indistinguishable from the “active” GTP-bound format required for subsequent exportin binding (6). Formation of this complex is absolutely required as a preliminary step in the observation of virus-induced Nup hyper-phosphorylation, the ultimate cause of nucleocytoplasmic trafficking failure by these viruses (4). Resolution of the LM:Ran structure confirmed the central hinge region of LM (e.g. Trp40), tucked under the C-tail of Ran, forms the key interaction domain. The LM zinc finger (e.g. C19), phosphorylation sites (Y41, T47), and the majority of C-proximal acidic domain (e.g. Asp48, Asp51, Asp52), do not contribute to these contacts, even though each of these regions has proven essential to the observation of LX-dependent Nup phosphorylation (3, 12). The resolved structure predicts a model whereby these non-Ran binding segments might instead mediate subsequent interactions with the karyopherin class of exportins (e.g. Crm1, CAS), which require exactly this GTP-like Ran conformation for nuclear egress (6). Validation of this idea, that LX and exportins can directly interact, was the rationale for the experiments described here.
Native cardiovirus LX proteins are refractive to Western analyses and antibody induction because of their small size and high charge. Recombinant GST-tagged LX materials readily recapitulate most of the protein phenotypes in cells and extracts and are typically used to explore these mechanisms (4, 12, 17). With both native (HeLa cytosol) and recombinant (rCrm1) sources, reciprocal pulldowns indicated strong, salt-resistant direct interactions between 3 different cardiovirus (GST-)LX sequences and Crm1/CAS (Fig 2, Fig 3). Mass spec analyses showed these particular exportins, as well as other structurally analogous proteins with HEAT-repeat folds, were among the dominant GST-LE binding partners extracted from HeLa cytosol (Table 1). Ran would not have been expected as a partner here, because its efficient interaction with LE requires the catalytic presence of nuclear-bound guanine exchange factor, RCC1, to facilitate essential conformational morphs (8).
Once identified as the majority example exportin, Crm1 was then further examined both as native and recombinant protein as well as in combination with rRan, for GST-LE binding. Neither cellular protein was found to block the other’s extraction, nor was extraction of either or both apparently sensitive to the sequence of addition (Fig 3, Fig 4, Fig 6). Such results were not entirely surprising, because the structure model predicts that the LM zinc finger, acidic domain and phosphorylation sites, which do not make Ran contacts, are exactly the segments which should mediate putative exportin contacts (6). In other words, the binding domains are non-overlapping. In support of this idea, mutations in GST-LE affecting any of the 3 of the predicted contact regions significantly diminished the extraction of native and recombinant Crm1. The reduced affinity was particularly apparent when the LE phosphorylation sites (Tyr41, Thr47) were mutated, or when GST-LE in vitro, was restricted in its phosphorylation status. Dual phosphorylated GST-LE, reacted with rCrm1, fractionated by gel filtration at the size expected for a stoichiometric complex. An additional finding in favor of the current model is that LE interaction with Crm1 apparently did not involve the NES cargo recognition site (HEAT repeats H11 and H12) inhibited by LMB. Within fully assembled Crm1:LE:Ran complexes, the Nup phosphorylation kinases (activated ERK and p38 pathway kinases) would presumably need to bind here, or nearby, as preferred export cargos (6).
Clearly, exportins, particularly Crm1 and CAS, are strong viable partners for LX. An inducible Crm1 knockout cell line was created to link the requirements for Crm1 to EMCV replication. As was shown previously for the Ran mechanistic requirement (3), Nup62 phosphorylation and viral fecundity, as measured by 3D/3CD synthesis and by progeny titers, were strongly dependent upon the pool of native Crm1. This was also true when induced cells were transfected with cDNAs from any 3 cardiovirus LX proteins, indicating a common requirement for Crm1, if not other analogous exportins, in the collective Nup phosphorylation pathways.
The current mechanism for LX-dependent Nup hyper-phosphorylation expects that within the nuclear rim, LX:Ran complexes select exportins carrying activated kinases such as ERK1 and p38, and it is this multiplex which tethers within the nuclear pores and phosphorylates the Nups (6). Interestingly, we could not define a required sequence of addition for rRan and rCrm1 to a mutual GST-LE complex with the current reactions. Both proteins appeared to interact independently, especially if LE was properly phosphorylated. Near the void volume of the fractionation column, though, about 20% of the added mass eluted with signals from all 3 proteins. Whether these are the expected trimers or other high-mass complexes is not clear at present. Three additional anomalies from this experiment, the significant peak of free Ran, the dominance of GST-LE:rCrm1, and the lack of rRan:rCrm1 complexes, suggest that we don’t yet understand all the interplay among these proteins. In cells, especially within the context of nuclear pores, the cooperative, active formats of LE, Ran and Crm1 (or CAS) are sure to be influenced by the local environment, especially the GNP nucleotide gradient and perhaps other cellular (catalytic?) proteins.
In cell-free reactions, a catalytic amount of RCC1 significantly accelerates binding saturation (8), locking LE and Ran into (virtually) in-dissociable complexes which mimic the topology of RanGTP even in the absence of a nucleotide (6). The same RanGTP format is required for Ran:Crm1 (or CAS) interactions (18, 19). From our experiments, it is now clear that GST-LE, preferably with at least 1 phosphate attached, can interact strongly and independently with both rRan and rCrm1 (or CAS). Void-volume complexes were observed with all three proteins, but we don’t yet know how to drive these assemblages to saturation. Possibly, the RCC1-dependent Ran morphing, an aid for good GST-LE interactions, works contrary to simultaneous rRan:rCrm1 binding by stripping endogenous nucleotides from the added Ran. This situation would explain the observed pool of free Ran, unable to dislodge GST-LE which is already tied up in rCrm1 complexes. It would also explain the lack of an observable rRan:rCrm1 peak, independent of the presence of GST-LE, as this should have been a strong native interaction, and indeed a driving force in normal nuclear trafficking. If true, then within the context of real nuclear pores, the innate nucleotide gradient must play a significant role in ternary complex assembly efficiency.
Materials and Methods
Recombinant proteins
Plasmids encoding GST- LE, GST-LEΔA, GST-LEW40A, and related derivatives are described (7, 8, 12), as are analogous LX-GST proteins from Saffold 2 and Theiler’s virus (BeAn), LS-GST, LT-GST, LE-GST and LE-GST Thr41Ala/Tyr47Ala (5, 20). LE-GST 3D3A was made similarly by introducing point mutations to alter the 3 codons for Asp48, Asp51 and Asp52 to Ala. For recombinant proteins, these cDNAs were transformed into BL21 E. coli. The cells were incubated in media (0.5% glucose, 20uM ZnCl2, 50 mg/mL ampicillin, 30°C, 3 hr) before induction with IPTG (1 mM) and harvest (6 hr). Cells were re-suspended in GST buffer (50 mM Tris pH 7.4, 125 mM NaCl, 20 uM ZnCl2, 1mM PMSF, 0.2 mM DTT, 0.5 mg/ml lysozyme), then sonicated. Debris was removed by centrifugation (20,000 x g, 45 min). The proteins were collected with GSTrap HP columns (GE Healthcare Bio-sciences, Pittsburgh PA) into modified GST buffer (10 mM glutathione disulfide, 50 mM Tris, pH 8.0). Protein fractions were dialyzed into buffer QA (10mM Bis-Tris Propane, 50mM NaCl, 2 mM DTT, pH 7.4), then applied to a Macro-Prep High Q anion exchange column (BioRad, Hercules, CA). The fractions were eluted (buffer QA, 1M NaCl) before dialysis (25 mM HEPES, 150 mM KCl, 2 mM DTT, pH 7.3), and assay for concentration and purity. A cDNA construct encoding amino-terminal, His-tagged murine Crm1 (rCrm1) was a kind gift from Dr. Dirk Görlich. Bacterial induction and protein isolation was as described (18). Recombinant His-tagged human Ran GTPase (rRan) and GST-RCC1 (human) were also prepared as described (8, 12). All recombinant proteins were snap frozen in liquid nitrogen and then stored at −80°C as single-experiment aliquots. Casein kinase 2 (CK2, phosphorylated, recombinant, New England Biolabs) and spleen tyrosine kinase (Syk, activated, recombinant, SignalChem) were used singly or doubly phosphorylate recombinant GST-LE at concentrations of 5 U/μg and 0.5 ng/μg, respectively (11).
Cell lysates
HeLa cells (ATCC CRL-1958) were grown in suspension (modified eagle’s medium, 10% calf serum, 5% CO2, 37°C). Lysates were prepared after collecting the cells with centrifugation (500 x g), washing the pellets (PBS), and resuspension (2x volume) in hypotonic buffer (0.75mM MgAc, 0.15 mM EGTA, 1mM PMSF, 0.01 mg/ml leupeptin, 20 μM pepstatin A, 3mM DTT). After a brief incubation (10 min, 4°C) the samples were homogenized (dounce) or sonicated (2 × 30 sec, 0°C) before clarification (10 min, 16,000 g). The soluble fraction was supplemented with 1/10th volume 10 x TB (200 mM HEPES pH 7.3, 10mM EGTA, 20 mM MgAc, 110 mM KAc), before aliquots were snap frozen and stored at −80°C. Before use in pull-down experiments, the lysates were pre-reacted with glutathione sepharose beads and then re-clarified (as below).
Protein extraction
GST-tagged LE and mutant derivatives (bait, generally 2 μg) was incubated (37°C, 45 min) with HeLa lysate (prey, 40 μl) before dilution (RIPA buffer, 50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% Na deoxycholate, 1% IGEPAL, pH 7.6), addition of glutathione sepharose beads (generally 10 μl per sample, GE Healthcare Bio-sciences) and agitation (3 hr, 4°C). When the pulldown prey was a recombinant protein, bait and prey were mixed stoichiometrically, diluted (to 100 μl, with TB, 200 mM HEPES pH 7.3, 1mM EGTA, 2mM MgAc, 110 mM KAc, 2 mM DTT), before bead addition as above. Both types of samples were then further diluted (2x) with binding buffer (50mM HEPES, pH 7.4, 150 mM NaCl, 0.5% IGEPAL), and mixed end-over-end (1 hr, 22°C). For immunopreciptation (IP), protein-G sepharose beads were conjugated to antibodies (bait, e.g. α-Crm1, 0.6 μl per 10μl beads, overnight, 4°C) before being rinsed, (1 x PBS, 0.02% Triton X-100), added to samples (prey, cytosol or recombinant protein, 10μl/sample) and incubated (with rocking, 3 hr, 4°C). After pulldown or IP, the beads were rinsed (6x, 500 μl PBS, triton X-100, then 2 × 120 mM Tris, pH 6.8, 10% glycerol w/v 2% SDS w/v, 200 mM DTT, 0.2% bromophenol blue) before the samples were denatured in a boiling water bath (10 min).
Mass spectroscopy
Protein bands extracted by GST-LE pulldown, were fractionated by SDS-PAGE and visualized by Coomassie staining before excision and digestion with trypsin according to protocols recommended by the UW-Madison Biotechnology Center. Briefly, gel samples were treated with extraction buffer 1 (200 μL, mM NH4HCO3, 50% acetonitrile (ACN), 45 min), and then dehydrated (100% ACN, 5 min) under vacuum. The samples were resuspended (40 μl, 40 mM NH4CHO3, 10% ACN) in the presence of trypsin gold (Promega, 20 μg/ml, plus 0.025% Protease Max surfactant, 1 hr, 27°C). Subsequently, extraction buffer 2 was added (200 μl, 40mM NH4CHO3, 10% ACN) followed by incubation (overnight, 25°C) and re-extraction (2x, 60 min, 50 μl 50% ACN, 5% trifluoroacetic acid). Resultant peptides from select bands (see Table 1) were pooled, dehydrated (2 hr under vacuum) before purification and concentration via Ziptips (Millipore). The pool A and pool B samples were submitted for orbi-trap mass spectroscopy at the UW-Madison Biotechnology Center.
Western analyses
Prepared samples were resolved by SDS-PAGE. The proteins were electro-transferred to polyvinylidene difluoride membranes (Immobilon-P, Millipore). The membranes were blocked (1 hr, 10% NFD milk in TBST: 20 mM Tris pH 7.6, 150 mM NaCl, 0.5% Tween20) then incubated with a primary antibody (1% NFD milk in TBST, overnight, 4°C) before washing (3 x TBST) and reaction with an appropriate secondary antibody (1 hr, 20°C). Antibodies included: αNup (murine mAb414, IgG, Covance, 1:2000), αtubulin (murine Ab, IgG, Sigma, 1:10,000), αGST (murine mAb, IgG, Novagen, 1:10,000), αCrm1 (rabbit Ab, IgG, Abcam, 1:2000), αCAS (goat Ab, IgG, Santa Cruz Biotechnology; 1:1000), αRan (goat Ab, IgG, Santa Cruz Biotechnology; 1:1000), αmouse (secondary Ab, IgG, Sigma Aldrich, 1:8000), αrabbit (secondary Ab, IgG, Promega, 1:8000), αgoat (secondary Ab, IgG, Sigma, 1:8,000). For band visualization, the membranes were rinsed (3x, TBST), incubated (1 min) with enhanced chemiluminescence substrate (GE healthcare) and then exposed to film. Densitometry analyses were as previously described (11).
Gel filtration
A HiLoad Superdex 200 16/600 chromatography column (GE Lifesciences) was calibrated with a commercial kit (Sigma-Aldrich MWGF1000-1kt) according to manufacturer’s instructions. Recombinant protein combinations were incubated (37°C, 45 min), filtered (0.2 micron membrane) and volume adjusted (to 1 ml) before injection into the equilibrated column (50 mM HEPES, pH 7.4, 150 mM NaCl). After fractionation (0.2 ml/min, 4°C) the collected samples (1 ml) were concentrated by dehydration under vacuum (0.5 ml) and assayed by SDS-PAGE, with silver stain, or Western techniques.
Cell procedures
HeLa cells plated to confluence, were infected with vEC9 (21) at an MOI of 1–20. This strain is a recombinant, short poly(C) attenuated EMCV. At 3–6 hours post-infection (37°C, 5% CO2) the cells were rinsed (2x, PBS) and collected into SDS buffer for protein analyses, or alternatively, subject to freeze-thaw (3x), lysate clarification and assays for plaque-forming units (21). Transfection assays used GST-LE cDNA (2.6 μg per 106 cells in Lipofectamine 2000 (Life Technologies, Grand Island, NY)). After incubation (18 hr, 37°C, 5% CO2) the cells were rinsed (2x, PBS) and harvested in SDS buffer. Leptomycin B (LMB, Sigma Aldrich, St. Louis, MO), was added as required (to 4 nM), by pretreating cells or buffers for 1 hr. This concentration was then maintained throughout the experiment. A Crm1 knockout HeLa cell line was generated with the TRIPZ inducible lentiviral shRNA system (GE Dharmacon, shRNA V2THS-172053) according to manufacturer instructions. The line was maintained under selection with puromycin. Plated cells were induced with doxycycline (0.25 nM, Sigma Aldrich). The media was refreshed daily (1–4 days) as required by the experiment.
Acknowledgments
This work was supported by NIH grant RO1-AI017331 to ACP. The authors thank Dr. Nathan Sherer and Laraine Zimdars for assistance in creating the Crm1 shRNA cell line, and Dr. Dirk Görlich for his kind gift of the His-Crm1 plasmid.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Terry LJ, Wente SR. Flexible gates: dynamic topologies and functions for FG nucleoporins in nucleocytoplasmic transport. Eukaryotic Cell. 2009;8:1814–1827. doi: 10.1128/EC.00225-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakielny S, Dreyfuss G. Transport of proteins and RNAs in and out of nucleus. Cell. 1999;99:677–690. doi: 10.1016/s0092-8674(00)81666-9. [DOI] [PubMed] [Google Scholar]
- 3.Porter FW, Bochkov YA, Albee AJ, Wiese C, Palmenberg AC. A picornavirus protein interacts with Ran-GTPase and disrupts nucleocytoplasmic transport. Proc Natl Acad Sci USA. 2006;103:12417–12422. doi: 10.1073/pnas.0605375103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter FW, Palmenberg AC. Leader-induced phosphorylation of nucleoporins correlates with nuclear trafficking inhibition of cardioviruses. J Virol. 2009;83:1941–1951. doi: 10.1128/JVI.01752-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basta HA, Palmenberg AC. AMP-activated protein kinase phosphorylates EMCV, TMEV, and SafV Leader proteins at different sites. Virology. 2014;262:236–240. doi: 10.1016/j.virol.2014.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bacot-Davis VR, Ciomperlik JJ, Cornilescu CC, Basta HA, Palmenberg AC. Solution structures of Mengovirus leader protein, its phosphorylated derivatives, and in complex with Ran GTPase. Proc Natl Acad Sci USA. 2014;111:15792–15797. doi: 10.1073/pnas.1411098111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cornilescu CC, Porter FW, Zhao Q, Palmenberg AC, Markley JL. NMR structure of the Mengovirus leader protein zinc-finger domain. FEBS Letters. 2008;582:896–900. doi: 10.1016/j.febslet.2008.02.023. [DOI] [PubMed] [Google Scholar]
- 8.Petty RV, Palmenberg AC. Guanine-nucleotide exchange factor RCC1 facilitates a tight binding between EMCV Leader and cellular Ran GTPase. J Virol. 2013;87:6517–6520. doi: 10.1128/JVI.02493-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porter FW, Brown B, Palmenberg A. Nucleoporin phosphorylation triggered by the encephalomyocarditis virus leader protein is mediated by mitogen-activated protein kinases. J Virol. 2010;84:12538–12548. doi: 10.1128/JVI.01484-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basta HA, V, Bacot-Davis R, Ciomperlik JJ, Palmenberg AC. Encephalomyocarditis virus Leader is phosphorylated by CK2 and syk as a requirement for subsequent phosphorylation of cellular nucleoporins. J Virol. 2014;88:2219–2226. doi: 10.1128/JVI.03150-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bacot-Davis VR, Palmenberg AC. Encephalomyocarditis virus Leader protein hinge domain is responsible for interactions with Ran GTPase. Virology. 2013;443:177–185. doi: 10.1016/j.virol.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hutten S, Kehlenbach RH. Nup214 is required for CRM1-dependent nuclear protein export in vivo. Mol and Cell Biol. 2006;26:6772–6785. doi: 10.1128/MCB.00342-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu ZM, Jiang Q, Clarke PR, Zhang C. Phosphorylation of Crm1 by CDK1-cyclin-B promotes Ran-dependent mitotic spindle assembly. Journal of Cell Science. 2013;126:3417–3228. doi: 10.1242/jcs.126854. [DOI] [PubMed] [Google Scholar]
- 15.Hamamoto T, Seto H, Beppu T. Leptomycins A and B, new antifungal antibiotics. II. Structure and elucidation. Journal of Antibiotics. 1983;36:646–650. doi: 10.7164/antibiotics.36.646. [DOI] [PubMed] [Google Scholar]
- 16.Sun Q, Carrasco YP, Hu Y, Guo X, Mirzaei H, Macmillan J, Chook YM. Nuclear export inhibition through covalent conjugation and hydrolysis of Leptomycin B by CRM1. Proc Natl Acad Sci USA. 2012;110:1303–1308. doi: 10.1073/pnas.1217203110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petty RV, Basta HA, Bacot-Davis VR, Brown BA, Palmenberg AC. Binding interactions between the encephalomyocarditis virus leader and protein 2A. J Virol. 2014;88:13503–13509. doi: 10.1128/JVI.02148-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guttler T, Madl T, Neumann P, Deichsel D, Corsini L, Monecke T, Ficner R, Sattler M, Gorlich D. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat Struct Mol Biol. 2010;17:1367–1376. doi: 10.1038/nsmb.1931. [DOI] [PubMed] [Google Scholar]
- 19.Guttler T, Gorlich D. Ran-dependent nuclear export mediators: a structural perspective. EMBO J. 2011;30:3457–3474. doi: 10.1038/emboj.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciomperlik JJ, Basta HA, Palmenberg AC. Three cardiovascular leader proteins equivalently inhibit four different nucleocytoplasmic trafficking pathways. Virology. 2015;484:194–202. doi: 10.1016/j.virol.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dvorak CMT, Hall DJ, Hill M, Riddle M, Pranter A, Dillman J, Deibel M, Palmenberg AC. Leader protein of encephalomyocarditis virus binds zinc, is phosphorylated during viral infection and affects the efficiency of genome translation. Virology. 2001;290:261–271. doi: 10.1006/viro.2001.1193. [DOI] [PubMed] [Google Scholar]
- 22.Groves MR, Barford D. Topological characteristics of helical repeat proteins. Curr Opin Str Biol. 1999;9:383–389. doi: 10.1016/s0959-440x(99)80052-9. [DOI] [PubMed] [Google Scholar]