

Abstract
One of the key mechanisms involved in renal Na+ retention in chronic heart failure (CHF) is activation of epithelial Na+ channels (ENaC) in collecting tubules. Proteolytic cleavage has an important role in activating ENaC. We hypothesized that enhanced levels of proteases in renal tubular fluid activate ENaC resulting in renal Na+ retention in rats with CHF. CHF was produced by left coronary artery ligation in rats. By immunoblotting, we found that several urinary serine proteases were significantly increased in CHF rats compared to sham rats (fold increases: furin 6.7, prostasin 23.6, plasminogen 2.06 and plasmin 3.57 vs. sham). Similar increases were observed in urinary samples from patients with CHF. Whole-cell patch-clamp was conducted in cultured renal collecting duct M-1 cells to record Na+ currents. Protease-rich urine (from rats and patients with CHF) significantly increased the Na+ inward current in M-1 cells. Two weeks of protease inhibitor treatment significantly abrogated the enhanced diuretic and natriuretic responses to ENaC inhibitor benzamil in rats with CHF. Increased podocyte lesions were observed in the kidneys of rats with CHF by transmission electron microscopy. Consistent with these results, podocyte damage markers desmin and podocin expressions were also increased in rats with CHF (increased ~2 folds). These findings suggest that podocyte damage may lead to increased proteases in the tubular fluid which in turn contributes to the enhanced renal ENaC activity, providing a novel mechanistic insight for Na+ retention commonly observed in CHF.
Keywords: Na+ and water retention, renal function, protease, heart failure, podocyte damage
Introduction
An impaired ability to excrete Na+ load is commonly seen in patients with chronic heart failure (CHF). 1, 2 Chronic heart failure and chronic kidney disease often co-exist. 3, 4 This coexistence of CHF and chronic kidney disease is commonly referred to the cardio-renal syndrome. However, elucidating the contribution of specific molecular mechanism/s for the inappropriate Na+ and water retention commonly seen in CHF remains largely undefined.
Renal epithelial Na+ channels (ENaC) represent the “fine tuning”, rate-limiting step in Na+ reabsorption in the distal nephron. ENaC are composed of three subunits: α, β and γ-ENaC localized in the distal convoluted tubules, cortical collecting ducts (CCD), and medullary collecting ducts of the distal nephron. 5, 6 The activity of ENaC is regulated by angiotensin (ANG) II and aldosterone, which markedly increases the apical permeability of the collecting duct to Na+. 7-9 Interventional studies in patients have shown that systemic ENaC inhibition by ANG II receptor blockade or aldosterone mineralocorticoid receptor blockade is an important treatment option in arterial hypertension and CHF. 10 Our previous study has shown that increased renal ENaC subunits expression and activity may contribute to the renal Na+ and water retention observed in rats with CHF. 11
Recently, proteolytic cleavage of ENaC has been recognized as one of the important and major mechanisms related to ENaC activation 12, 13 and subsequent Na+ retention. These studies suggest that proteolytic activation of ENaC results from proteolytic release of inhibitory tracts within the α- and γ- subunits. Among the several proteases in urine, previous studies indicate a critical role for cleavage of ENaC subunits by prostasin, plasmin and furin for the activation of ENaC. 14, 15 Prostasin and plasmin are extracellular proteases whereas furin is intracellular protease. Furin is a member of a family of proprotein convertases that reside primarily in the trans-Golgi network. Prostasin was identified initially as a secreted prostate gland product with trypsin-like activity although it was eventually realized that it was the shed version of the glycophosphatidylinositol (GPI)-anchored protease. Plasmin is not present in the nephron lumen under normal conditions. Recent studies suggest that in the setting of glomerular diseases associated with podocyte injury, plasminogen is filtered by the glomerulus and is converted to plasmin by urokinase that is present within the tubular lumen. 16, 17 Under such circumstances protease inhibitors could represent potential antihypertensive agents with renoprotective effects. For example, camostat mesilate, an orally active synthetic serine protease inhibitor, has been shown to reduce blood pressure and renal injury in salt-sensitive hypertension. 18
The protease mechanism related to ANG II-aldosterone axis has also been shown to stimulate ENaC activity in the renal tubule. 19, 20 Thus, we hypothesized that tubular proteases contribute to the activating ENaC in rats with CHF. In the present study we attempted to address the following questions, 1) Are the levels of proteases increased in the urine of rats with CHF and patients with CHF? 2) Do urinary proteases increase ENaC activity of renal tubules and Na+ retention in rats with CHF? 3) Is there increased podocyte injury which would result in increased leakage of proteases into renal tubular fluid in rats with CHF?
Methods
Study approval
All the procedures on animals in this study were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee. The experiments were conducted according to the APS Guiding Principles for Research Involving Animals and Human Beings and the “NIH guide for the care and use of laboratory animals”.
The human study was approved by the local institutional review committee (“Ethik-Kommission”) of the Martin-Luther University Halle-Wittenberg (Study number 2011-64). All subjects gave informed consent prior to study participation.
Statistics
Data were subjected to a two-way ANOVA followed by a Multiple Range (for multiple comparisons) or Student-Newman Keuls test. P < 0.05 were considered to indicate statistical significance.
Detailed description of procedures is available in Methods in the online-only Data Supplement
Results
Basal hemodynamic characteristics
The myocardial infarct model in the rat mimics the most common cause of CHF in humans and is employed in numerous laboratories. 11, 21-24 Table 1 summarizes the salient characteristics of sham and CHF rats utilized in the present study. Heart weight, body weight and wet lung weight were significantly higher in CHF rats compared to sham rats (P < 0.05). The CHF group displayed an average myocardial infarct of greater than 30% of the left ventricle. In contrast, sham rats had no observable damage to the myocardium. Left ventricular end-diastolic pressure (LVEDP) was significantly elevated in CHF rats compared to sham rats (P < 0.05). +dP/dtmax was significantly decreased in CHF rats indicating decreased contractility of the left ventricles. This contractile dysfunction was likely the cause for the increase in LVEDP. -dP/dtmax had a similar trend in rats with CHF. These data suggest that rats in the CHF group had decreased cardiac contractility and were experiencing diastolic dysfunction. Overall, these characteristics indicate that CHF rats were in heart failure and possibly retaining fluid.
Table 1.
Basal hemodynamic characteristics of sham-operated and heart failure rats
| Measures | Sham (n = 12) | CHF (n = 12) |
|---|---|---|
| Body weight (g) | 395 ± 21 | 444 ± 22* |
| Heart weight (g) | 1.2 ± 0.2 | 2.1 ± 0.4* |
| Lung wet weight (g) | 1.7 ± 0.2 | 2.3 ± 0.1* |
| Infarct size (% of epicardial LV) | 0 | 37 ± 6* |
| LVEDP (mmHg) | 1 ± 1 | 24 ± 6* |
| + dP/dtmax (mmHg/sec) | 6945 ± 299 | 5428 ± 154* |
| − dP/dtmax (mmHg/sec) | −5365 ± 320 | −3765 ± 293* |
P < 0.05 vs. respective sham-operated rats. LV: left ventricle; LVEDP: left ventricular end diastolic pressure.
Table 2 summarizes the renal and hemodynamic characteristics of patients with CHF and control subjects recruited in the present study. Patients with CHF (NYHA III-IV) had significantly increased levels of plasma brain natriuretic peptide (BNP) and creatinine compared to the control subjects without CHF. Further, left ventricular ejection fraction (LVEF) was significantly decreased (32 ± 6 % vs 68 ± 1 %, P < 0.05) in the patients with CHF compared to the control subjects.
Table 2.
Basal renal and hemodynamic characteristics of control and heart failure patients
| Groups | Age | BNP (pg/ml) | Creatinine (μmol/l) | Estimated GFR (ml/min) | LVEF (%) |
|---|---|---|---|---|---|
| Control (n = 3) | 66 ± 5 | 51 ± 5 | 86 ± 9 | > 60 | 68 ± 1 |
| CHF (n = 6) | 68 ± 6 | 1039 ± 246* | 112 ± 12* | > 60 | 32 ± 6* |
P < 0.05 vs. respective control patients. BNP: brain natriuretic peptide; GFR: glomerular filtration rate; LVEF: left ventricular ejection fraction.
Urinary serine proteases were increased in CHF
We examined several urinary serine proteases in sham and CHF rats (Figure 1A). Four weeks after coronary ligation or sham surgery, 24 hrs urine samples were collected from metabolic cages and processed for protease measurement by immunoblotting. We found that several urinary serine proteases were significantly increased in CHF compared to the sham rats (fold increases: furin 6.7, prostasin 23.6, plasminogen 2.06 and plasmin 3.57 vs. sham, P < 0.05). Urinary protease activity measured using zymogram also demonstrated increases in CHF rats (~ 5 folds vs sham, Figure 1B). Similarly, we found that these three proteases in the urine were also increased in the patients with CHF (fold increases: furin 7.0, prostasin 7.7, plasminogen 5.4 and plasmin 3.9 vs. control, P < 0.05) (Figure 2). This is the first evidence showing increased urinary serine proteases in the CHF condition to our knowledge.
Figure 1.

A. Protein expression of urinary proteases (furin, prostacin, plasminogen and plasmin) in sham and CHF rats measured by immunoblotting. B. Zymogram of urine showing increased gelatinase activity in urine from CHF rats compared to sham rats. *P < 0.05 different from respective sham group.
Figure 2.
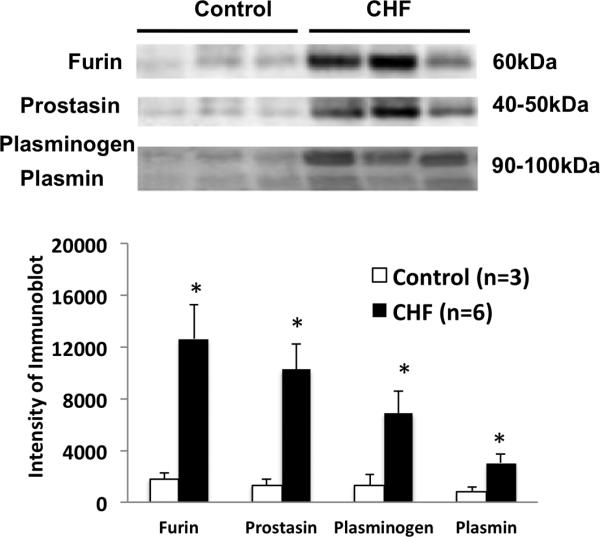
Protein expression of urinary proteases (furin, prostacin, plasminogen and plasmin) in the control and CHF patients measured by immunoblotting. *P < 0.05 different from respective control group.
Protease-rich urine activated ENaC in cultured renal cortical collecting duct M-1 cells
Since we found significantly increased urinary proteases in rats with CHF, this study was designed to investigate the functional effects of protease-rich urine (from rats and patients with CHF) on ENaC activity in cultured renal CCD M-1 cells. M-1 mouse CCD cell line (purchased from ATCC) was established from normal renal tissue. These cells retain many characteristics of CCD cells including the morphology and CCD antigens. Whole-cell patch-clamp was conducted on these M-1 cells to record Na+ currents before and after exposure to the urine (collected from sham and CHF rat, or control and CHF patient). The results show that protease-rich urine, from rats or patients with CHF, increases the Na+ inward current in M-1 cells compared to the sham rats or control patients urine exposure, respectively (Figure 3A and 3C). The stimulatory effect of urine on the Na+ inward current was reduced by ENaC inhibitor amiloride and pretreatment with protease inhibitor aprotinin in the urine (Figure 3A, 3B). On the contrary, the urine collected from sham rats or control patients did not increase the ENaC activity in M-1 cells. Addition of aprotinin to the urine collected from sham rats did not affect the inward Na+ current. This study supports the notion that proteases in the renal tubule have a potentially important role in activating ENaC of renal tubular cells in rats with CHF.
Figure 3.

Whole-cell patch-clamp was conducted in M-1 cells to record Na+ current before and after exposure to urine collected from; A) sham and CHF rats with or without amiloride, B) sham and CHF rats with or without aprotinin added, and C) control (Human) and CHF patients. n = 4/group * P < 0.05 vs. sham/control. # P < 0.05 vs. CHF.
Diuretic and natriuretic responses to ENaC inhibitor
After 2 weeks of protease inhibitor, aprotinin treatment, there were no significant differences in kidney weights between the groups of rats with or without aprotinin (Table 3). The basal urine flow before benzamil injection was not significantly different between the two sham groups. The basal Na+ excretion before benzamil injection was significantly increased in CHF group with aprotinin treatment.
Table 3.
Basal renal characteristics of sham-operated and heart failure rats
| Measures | Sham (n = 5) | CHF (n = 5) | Sham-aprotinin (n = 5) | CHF-aprotinin (n = 7) |
|---|---|---|---|---|
| Kidney weight (g) | 1.3 ± 0.2 | 1.9 ± 0.2* | 1.3 ± 0.1 | 1.9 ± 0.3* |
| Urine flow (μl/min/gkw) | 2.2± 0.3 | 2.3 ± 0.2 | 2.5 ± 0.4 | 2.7 ± 0.3 |
| Na+ excretion (μEq/min/gkw) | 0.14 ± 0.04 | 0.14 ± 0.01 | 0.17 ± 0.03 | 0.21 ± 0.03# |
P < 0.05 vs. respective sham-operated rats
P < 0.05 vs. respective group without aprotinin treatment
Benzamil injection produced diuresis and natriuresis in both groups of rats (Figure 4). Both diuresis and the natriuresis responses were significantly increased in CHF group compared to the corresponding sham rats after benzamil injection (P < 0.05), consistent with our previous observations. 11 Aprotinin significantly reduced the diuretic and natriuretic responses to benzamil in CHF (Figure 4A and 4B). Aprotinin did not significantly change the diuretic and natriuretic responses to benzamil in the sham group. This study provides additional evidence to support the idea that proteases in the renal tubule have important roles to activate ENaC. Further, protease inhibitors may reduce Na+ retention in CHF by attenuation of ENaC activity in the kidney.
Figure 4.

Urine flow (A) and Na+ excretion (B) in response to benzamil injection in sham and CHF rats with or without pretreatment with aprotinin for 2 weeks. (C) Whole-cell patch-clamp was conducted in M-1 cells to record inward current before and after exposure to urine collected from CHF rats with or without aprotinin treatment. * P < 0.05 vs. sham. # P < 0.05 vs. CHF.
In the patch clamp study, protease-rich urine from rats with CHF increased the Na+ inward current in M-1 cells compared to the sham urine exposure as shown above. However, the stimulatory effect of urine on the Na+ inward current was reduced by the urine from CHF rats with aprotinin treatment (Figure 4C) suggesting that long term protease treatment is effective in preventing the enhanced activation of ENaC observed in CHF.
Podocyte integrity; desmin and podocin expression in rats with CHF
Podocyte lesions were found in the kidneys of rats with CHF. The number of podocytes and foot process width were detected by transmission electron microscopy (TEM, n = 3) (Figure 5A). Generally, there was a fairly large amount of damage in the kidneys of rats with CHF compared to sham rats. In particular, podocyte foot processes lost their normal shape, and displayed numerous areas of effacement. The glomerular basement membrane also displayed abnormal thickness in rats with CHF. Further, we found that desmin immunosignal, a marker for podocyte injury, 25 was enhanced in podocytes of CHF rats demonstrated by immunostaining (n = 6) (Figure 5B). Finally, both desmin and podocin protein expressions (another podocyte damage marker) were significantly increased in the cortex of rats with CHF (P < 0.05) (Figure 5C and 5D).
Figure 5.

A. Transmission electron microscopy showing glomerular podocyte in sham and CHF rats. Magnification; 59Kx. B Immunostaining for desmin (a marker of podocyte damage, red). Scale bar: 100 μm. C. Desmin protein expression in the cortex of the kidneys from sham and CHF rats. D. Podocin protein expression in the cortex of the kidneys from sham and CHF rats. * P < 0.05 vs. sham.
Discussion
The present study shows that several urinary serine proteases are significantly increased in patients as well as rats with CHF. This protease-rich urine (from rats with CHF vs. sham operated rats) significantly increased Na+ inward current in M-1 cells. Conversely, treatment with a protease inhibitor for 2 weeks significantly abrogated the enhanced diuretic and natriuretic responses to ENaC inhibitor, benzamil, in rats with CHF. At this phase of the disease we observed that there was dramatic damage to the podocytes in the kidneys from rats with CHF. Taken together, these data suggest a robust increase in ENaC activation due to tubular proteases, possibly owing to podocyte damage, which leads to the enhanced Na+ reabsorption in CHF.
Na+ and fluid retention are commonly seen in patients with CHF. Recently, the role of proteases in activating ENaC has been highlighted in a number of reviews. 26, 27 However, there is not much evidence for this in a disease state such as CHF, the focus of the current study. ENaC can be stimulated by soluble proteases such as trypsin, membrane-bound proteases such as plasmin and prostasin, and intracellular protease furin. Proteases have important roles in activation ENaC by cleaving parts of α- and γ-subunits and releasing intrinsic inhibitory tracts 27. Activation of ENaC may contribute to the Na+ retention and tissue congestion commonly observed in disease conditions such as CHF. In the present study, we found that several proteases prostasin, plasmin and furin were dramatically increased in the urine from both patients with CHF and rats with CHF. This is the first evidence showing increased urinary serine proteases in the CHF condition, to our knowledge.
To test the potency of these proteases to activate the ENaC in vitro, we used patch clamp measurement of Na+ current in cultured tubular M-1 cells exposed to urine collected from rats or patients with CHF and controls. The results demonstrate that urine collected from CHF rats or patients yield greater inward Na+ current compared to urine from controls. The increased inward Na+ current in CHF was normalized by protease inhibitor treatment. Our in-vivo and in-vitro studies provided complimentary evidence showing a direct effect of enhanced proteases on the ENaC activity and Na+ dysregulation in rats with CHF. All these results support the idea that increased proteases in tubular fluid increase ENaC activity and Na+ retention in rats with CHF.
The M-1 cell line is derived from microdissected CCD of transgenic mouse and displays the low-conductance, highly Na+-selective channel activity of the α, β and γ-ENaC subunits. It retains many antigenic and differentiated transport properties of the CCD. It has been used extensively to study amiloride-sensitive Na+ transport through ENaC. 17, 28, 29 In our patch clamp study, the amiloride-sensitive, protease-activated inward Na+ current identified here in M-1 cells is consistent with ENaC as the target but not definitive. The contribution from other amiloride-sensitive Na+ conducting channels such as ENaC like channels and a non-selective cation channels in M-1 cells cannot be conclusively excluded by these particular experiments.
Our previous work has shown increased ENaC subunit abundance and enhanced ENaC functional activity in rats with CHF. 11 This may also contribute to the Na+ retention observed in CHF. Interestingly, as mentioned above our data show that urinary serine proteases (furin, prostasin, plasminogen and plasmin) are all dramatically increased in CHF. Aprotinin, a broad spectrum serine protease inhibitor has displayed inhibitory effects on Na+-channel activity in the kidney. 30, 31 Based on these studies, we examined the contribution of proteases causing increased ENaC activity and Na+ retention in rats with CHF. In vitro studies demonstrated that addition of aprotinin to the urine of rats with CHF was able to abrogate the enhanced activation of ENaC suggesting that excess proteases in the urine of CHF rats was responsible for the enhanced functionality of the ENaC in the CHF condition. Further, our results showed that in rats with CHF, treatment with aprotinin to block endogenous proteases for two weeks, abrogated the enhanced ENaC activity responses to benzamil (increase urine volume and Na+ excretion). These results demonstrate that there is an enhanced functional activation of ENaC, which is dependent on tubular proteases in rats with CHF.
Aprotinin is an important member of a family of related protease inhibitors and has many clinically beneficial activities. 32 In the clinic, aprotinin has been widely used as pre- and intraoperative agents with antifibrinolytic activity. 33 It also has complex interactions with other drug therapies including angiotensin-converting enzyme inhibitors. 32 It may also affect hemodynamics in this regard. Our in-vivo and in-vitro studies provide evidence showing the direct effect of protease on the ENaC activity and Na+ dysregulation in CHF rats. A potential concern for this protocol is the efficacy of the protease inhibitors on renal ENaC activity. As a potential protease inhibitor, aprotinin has been shown to prolong the attenuation of ENaC function in the lung, airway and kidney. 28, 34, 35 Aprotinin has higher potency than other protease inhibitors such as soybean trypsin inhibitors to attenuate ENaC function and Na+ retention.
Furin is an intracellular convertase-type protease that resides primarily in the trans-Golgi network. Urinary furin is more likely from epithelial cells of the kidney rather than from circulation or glomerular podocyte leakage. The source of urinary furin may be different from extracellular proteases plasmin and prostasin. Aprotinin can block the activity of proteases such as prostasin and plasmin, 36 however it has been reported that aprotinin at a dose of 0.1 mg/ml does not block furin. 37 Administration of the synthetic serine protease inhibitor camostat mesilate inhibits the protease activity of serine proteases such as prostasin and plasmin, but not furin. This protease inhibitor may inhibit the second cleavage of γ-ENaC and subsequently suppress ENaC activity on the plasma membrane. 38
The sympathetic nervous system and the renin-ANG II-aldosterone system have been suggested as possible cardio-renal mediators. 39 In CHF patients and rats with CHF, increased sympathetic nerve activity has been well documented. 40, 41 The plasma levels of norepinephrine, ANG II and aldosterone are all elevated in patients with CHF as well in rats with CHF. 42-44 ANG II and aldosterone are well recognized for their actions on ENaC in increasing Na+ reabsorption. 7-9 Interventional studies in patients have shown systemic ENaC inhibition by aldosterone mineralocorticoid receptor blocker may be an alternative to the treatment of hypertension and CHF. 10 Beyond this well-known action, a recent study suggests an additional mechanism by which ANG II and aldosterone could stimulate Na+ transport through ENaC by increasing the expression of proteases in the kidney. 45
Activated norepinephrine and ANG II-aldosterone systems are involved in several renal injury processes, including glomerular podocyte injury. 10, 46 Aldosterone is implicated in renal inflammatory and fibrotic processes, as well as in podocyte injury and mesangial cell proliferation. 10 Activation of the sympathetic nervous system augments kidney reninangiotensin system and oxidative stress. Intrarenal ANG II-induced increases in reactive oxygen species may contribute to the pathogenesis of glomerular podocyte injury and albuminuria. 46 Severe proteinuria and renal injury are also found in patients with CHF. 47, 48 The defective glomerular filtration barrier allows the filtration of ENaC-activating proteases, such as plasmin converted from plasminogen into the tubular fluid. In our study, we found that podocyte foot processes lost their normal shape, and displayed numerous areas of effacement in CHF rats. We also found that desmin and podocin protein expression, as markers for podocyte injury were enhanced in the podocytes of kidneys from rats with CHF. Thus, we propose that in CHF there is an increased damage to the podocytes that leads to leakage of proteases from the glomerulus into the tubular fluid which in turn causes an enhanced activation of ENaC leading to enhanced Na+ retention (Figure 6).
Figure 6.

Schematic of the excess protease activation of ENaC in the kidney (red arrows indicate the changes in CHF). ENaC activated by protease in the principle cell of renal tubule. ANG: angiotensin; ALDO: aldosterone; PN-1: protease nexin 1; uPA: urokinase-type plasminogen activator.
Cardio-renal syndrome describes the reciprocally detrimental interaction between chronic cardiac and renal dysfunction. The syndrome is highly prevalent and carries a high risk of mortality. The main therapy of cardio-renal syndrome is loop diuretics and angiotensin-converting enzyme inhibition. One emergent feature of this study is that CHF presents itself as exhibiting enhanced levels of urinary proteases and perhaps they represent urinary biomarkers of renal injury in patients with CHF. These data lead us to speculate that urinary proteases may perhaps be used as a biomarker of cardio-renal disease. Further studies need to evaluate a therapeutic and preventive strategy to delay the onset and progression of CHF and/or cardio-renal syndrome by using protease inhibitor administration.
In conclusion, these findings demonstrate that increased proteases in the tubular fluid contribute to the enhanced ENaC activity and thus Na+ retention commonly seen in patients with CHF. The significance of these studies is to provide insight into the molecular and cellular mechanisms that may contribute to activation of ENaC and consequently lead to dysfunction in Na+ balance, commonly observed in CHF.
Perspectives
The significance of these studies is to provide insight into the molecular and cellular mechanisms that contribute to ENaC activation associated with dysfunction in sodium balance in CHF. Additionally, the present studies provide novel information on the beneficial effects of protease inhibition on renal sodium regulation in CHF, which may allow us to develop new treatments and management programs for the complications and consequences of CHF. This is the first study that determines the role of proteases in affecting the activity of ENaC and its contribution to sodium balance dysfunction in CHF. These studies also examine the underlying possible mechanisms of enhanced protease activation of ENaC in CHF. The amalgamation of the observations in this fashion also represents a novel and an innovative approach to the study of renal handling of sodium and water in CHF.
Supplementary Material
Novelty and Significance.
What is New?
The present study shows that several urinary serine proteases are significantly increased in the renal tubular fluid during chronic heart failure. This protease-rich urine significantly increased the epithelial sodium channels (ENaC) mediated Na+ inward current.
Treatment with a protease inhibitor significantly abrogated the enhanced activation of ENaC in rats with chronic heart failure.
There was dramatic damage to the podocytes in the glomeruli from rats with chronic heart failure.
What is Relevant?
The clinical picture of advanced stages of chronic heart failure is often accompanied by the presence of edema and congestion, causing symptoms of dyspnea, fatigue, nausea and discomfort. Avid sodium retention has been suggested to contribute further to aggravate the progression of the disease.
The present studies provide significant new information and insight regarding the activation of ENaC channels influencing sodium and water balance, and the possibility of therapeutic intervention using protease inhibition on sodium retention which is endemic to chronic heart failure.
Summary
The data suggest a robust increase in ENaC activation due in part to proteases that potentially leak into the tubular fluid due to glomerular damage leading to the enhanced sodium retention in chronic heart failure.
Acknowledgments
Sources of Funding
This work was supported by NIH grants R56 HL124104 and P01 HL62222.
Footnotes
Disclosures
None.
References
- 1.Navas JP, Martinez-maldonado M. Pathophysiology of edema in congestive heart failure. Heart Dis Stroke. 1993;2:325–329. [PubMed] [Google Scholar]
- 2.Rasool A, Palevsky PM. Treatment of edematous disorders with diuretics. Am J Med Sci. 2000;319:25–37. doi: 10.1097/00000441-200001000-00003. [DOI] [PubMed] [Google Scholar]
- 3.Forman DE, Butler J, Wang Y, Abraham WT, O'Connor CM, Gottlieb SS, Loh E, Massie BM, Rich MW, Stevenson LW, Young JB, Krumholz HM. Incidence, predictors at admission, and impact of worsening renal function among patients hospitalized with heart failure. J Am Coll Cardiol. 2004;43:61–67. doi: 10.1016/j.jacc.2003.07.031. [DOI] [PubMed] [Google Scholar]
- 4.Heywood JT. The cardiorenal syndrome: Lessons from the adhere database and treatment options. Heart Fail Rev. 2004;9:195–201. doi: 10.1007/s10741-005-6129-4. [DOI] [PubMed] [Google Scholar]
- 5.Canessa CM, Shild L, Buell L, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Nature. 1994;Amiloride-sensitive epithelial na+ channel is made of three homologous subunits.367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 6.Rossier BC, Canessa CM, Schild L, Horisberger JD. Epithelial sodium channels. Curr Opin Nephrol Hypertens. 1994;3:487–496. doi: 10.1097/00041552-199409000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Beutler K, Masilamani S, Turban S, Nielsen J, Brooks HL, Ageloff S, Fenton RA, Packer RK, Knepper MA. Long-term regulation of enac expression in kidney by angiotensin ii. Hypertension. 2003;41:1143–1150. doi: 10.1161/01.HYP.0000066129.12106.E2. [DOI] [PubMed] [Google Scholar]
- 8.Ecelbarger CA, Kim GH, Terris J, Masilamani S, Mitchell C, Reyes I, Verbalis JG, Knepper MA. Vasopressin-mediated regulation of epithelial sodium channel abundance in rat kidney. Am J Physiol Renal Physiol. 2000;279:F46–53. doi: 10.1152/ajprenal.2000.279.1.F46. [DOI] [PubMed] [Google Scholar]
- 9.Blazer-Yost BL, Liu X, Helman SI. Hormonal regulation of enacs: Insulin and aldosterone. Am J Physiol. 1998;274:C1373–1379. doi: 10.1152/ajpcell.1998.274.5.C1373. [DOI] [PubMed] [Google Scholar]
- 10.Briet M, Schiffrin EL. Aldosterone: Effects on the kidney and cardiovascular system. Nat Rev Nephrol. 2010;6:261–273. doi: 10.1038/nrneph.2010.30. [DOI] [PubMed] [Google Scholar]
- 11.Zheng H, Liu X, Rao US, Patel KP. Increased renal enac subunits and sodium retention in rats with chronic heart failure. Am J Physiol Renal Physiol. 2011;300:F641–649. doi: 10.1152/ajprenal.00254.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughey RP, Mueller GM, Bruns JB, Kinlough CL, Poland PA, Harkleroad KL, Carattino MD, Kleyman TR. Maturation of the epithelial na+ channel involves proteolytic processing of the alpha- and gamma-subunits. J Biol Chem. 2003;278:37073–37082. doi: 10.1074/jbc.M307003200. [DOI] [PubMed] [Google Scholar]
- 13.Hughey RP, Carattino MD, Kleyman TR. Role of proteolysis in the activation of epithelial sodium channels. Curr Opin Nephrol Hypertens. 2007;16:444–450. doi: 10.1097/MNH.0b013e32821f6072. [DOI] [PubMed] [Google Scholar]
- 14.Passero CJ, Carattino MD, Kashlan OB, Myerburg MM, Hughey RP, Kleyman TR. Defining an inhibitory domain in the gamma subunit of the epithelial sodium channel. Am J Physiol Renal Physiol. 2010;299:F854–861. doi: 10.1152/ajprenal.00316.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamm LL, Feng Z, Hering-Smith KS. Regulation of sodium transport by enac in the kidney. Curr Opin Nephrol Hypertens. 2010;19:98–105. doi: 10.1097/MNH.0b013e328332bda4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Passero CJ, Mueller GM, Rondon-Berrios H, Tofovic SP, Hughey RP, Kleyman TR. Plasmin activates epithelial na+ channels by cleaving the gamma subunit. J Biol Chem. 2008;283:36586–36591. doi: 10.1074/jbc.M805676200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Svenningsen P, Bistrup C, Friis UG, Bertog M, Haerteris S, Krueger B, Stubbe J, Jensen ON, Thiesson HC, Uhrenholt TR, Jespersen B, Jensen BL, Korbmacher C, Skott O. Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol. 2009;20:299–310. doi: 10.1681/ASN.2008040364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maekawa A, Kakizoe Y, Miyoshi T, Wakida N, Ko T, Shiraishi N, Adachi M, Tomita K, Kitamura K. Camostat mesilate inhibits prostasin activity and reduces blood pressure and renal injury in salt-sensitive hypertension. J Hypertens. 2009;27:181–189. doi: 10.1097/hjh.0b013e328317a762. [DOI] [PubMed] [Google Scholar]
- 19.Ruffieux-Daidie D, Staub O. Intracellular ubiquitylation of the epithelial na+ channel controls extracellular proteolytic channel activation via conformational change. J Biol Chem. 2011;286:2416–2424. doi: 10.1074/jbc.M110.176156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruffieux-Daidie D, Poirot O, Boulkroun S, Verrey F, Kellenberger S, Staub O. Deubiquitylation regulates activation and proteolytic cleavage of enac. J Am Soc Nephrol. 2008;19:2170–2180. doi: 10.1681/ASN.2007101130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfeffer MA, Pfeffer JM, Fishbein MC, Fletcher PJ, Spadaro J, Kloner RA, Braunwald E. Myocardial infarct size and ventricular function in rats. Circ Res. 1979;44:503–512. doi: 10.1161/01.res.44.4.503. [DOI] [PubMed] [Google Scholar]
- 22.Fishbein MC, Maclean D, Maroko PR. Experimental myocarial infarction in the rat. Qualitative and quantitative changes during pathologic evolution. Am J Pathol. 1978;90:57–70. [PMC free article] [PubMed] [Google Scholar]
- 23.Xu DL, Martin PY, Ohara M, St John J, Pattison T, Meng X, Morris K, Kim JK, Schrier RW. Upregulation of aquaporin-2 water channel expression in chronic heart failure rat. J Clin Invest. 1997;99:1500–1505. doi: 10.1172/JCI119312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lutken SC, Kim SW, Jonassen T, Marples D, Knepper MA, Kwon TH, Frokiaer J, Nielsen S. Changes of renal aqp2, enac, and nhe3 in experimentally induced heart failure: Response to angiotensin ii at1 receptor blockade. Am J Physiol Renal Physiol. 2009;297:F1678–1688. doi: 10.1152/ajprenal.00010.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yaoita E, Kawasaki K, Yamamoto T, Kihara I. Variable expression of desmin in rat glomerular epithelial cells. Am J Pathol. 1990;136:899–908. [PMC free article] [PubMed] [Google Scholar]
- 26.Rossier BC, Stutts MJ. Activation of the epithelial sodium channel (enac) by serine proteases. Annu Rev Physiol. 2009;71:361–379. doi: 10.1146/annurev.physiol.010908.163108. [DOI] [PubMed] [Google Scholar]
- 27.Kleyman TR, Carattino MD, Hughey RP. Enac at the cutting edge: Regulation of epithelial sodium channels by proteases. J Biol Chem. 2009;284:20447–20451. doi: 10.1074/jbc.R800083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li K, Guo D, Zhu H, Hering-Smith KS, Hamm LL, Ouyang J, Dong Y. Interleukin-6 stimulates epithelial sodium channels in mouse cortical collecting duct cells. Am J Physiol Regul Integr Comp Physiol. 2010;299:R590–595. doi: 10.1152/ajpregu.00207.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chalfant ML, Peterson-Yantorno K, O'Brien TG, Civan MM. Regulation of epithelial na+ channels from m-1 cortical collecting duct cells. Am J Physiol. 1996;271:F861–870. doi: 10.1152/ajprenal.1996.271.4.F861. [DOI] [PubMed] [Google Scholar]
- 30.Vallet V, Chraibi A, Gaeggeler HP, Horisberger JD, Rossier BC. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature. 1997;389:607–610. doi: 10.1038/39329. [DOI] [PubMed] [Google Scholar]
- 31.Passero CJ, Hughey RP, Kleyman TR. New role for plasmin in sodium homeostasis. Curr Opin Nephrol Hypertens. 2010;19:13–19. doi: 10.1097/MNH.0b013e3283330fb2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waxler B, Rabito SF. Aprotinin: A serine protease inhibitor with therapeutic actions: Its interaction with ace inhibitors. Curr Pharm Des. 2003;9:777–787. doi: 10.2174/1381612033455468. [DOI] [PubMed] [Google Scholar]
- 33.Smith MS, Muir H, Hall R. Perioperative management of drug therapy, clinical considerations. Drugs. 1996;51:238–259. doi: 10.2165/00003495-199651020-00005. [DOI] [PubMed] [Google Scholar]
- 34.Ble FX, Cannet C, Collingwood S, Danahay H, Beckmann N. Enac-mediated effects assessed by mri in a rat model of hypertonic saline-induced lung hydration. Br J Pharmacol. 2010;160:1008–1015. doi: 10.1111/j.1476-5381.2010.00747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan CD, Selvanathar IA, Baines DL. Cleavage of endogenous gammaenac and elevated abundance of alphaenac are associated with increased na(+) transport in response to apical fluid volume expansion in human h441 airway epithelial cells. Pflugers Arch. 2011;462:431–441. doi: 10.1007/s00424-011-0982-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galizia L, Ojea A, Kotsias BA. [amiloride sensitive sodium channels (enac) and their regulation by proteases]. Medicina. 2011;71:179–182. [PubMed] [Google Scholar]
- 37.Molloy SS, Bresnahan PA, Leppla SH, Klimpel KR, Thomas G. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence arg-x-x-arg and efficiently cleaves anthrax toxin protective antigen. J Biol Chem. 1992;267:16396–16402. [PubMed] [Google Scholar]
- 38.Uchimura K, Kakizoe Y, Onoue T, Hayata M, Morinaga J, Yamazoe R, Ueda M, Mizumoto T, Adachi M, Miyoshi T, Shiraishi N, Sakai Y, Tomita K, Kitamura K. In vivo contribution of serine proteases to the proteolytic activation of gammaenac in aldosterone-infused rats. Am J Physiol Renal Physiol. 2012;303:F939–943. doi: 10.1152/ajprenal.00705.2011. [DOI] [PubMed] [Google Scholar]
- 39.Bongartz LG, Cramer MJ, Doevendans PA, Joles JA, Braam B. The severe cardiorenal syndrome: ‘Guyton revisited’. Eur Heart J. 2005;26:11–17. doi: 10.1093/eurheartj/ehi020. [DOI] [PubMed] [Google Scholar]
- 40.Packer M. Neurohormonal interactions and adaptations in congestive heart failure. Circulation. 1988;77:721–730. doi: 10.1161/01.cir.77.4.721. [DOI] [PubMed] [Google Scholar]
- 41.Patel KP, Zhang K, Carmines PK. Norepinephrine turnover in peripheral tissues of rats with heart failure. Am J Physiol Regul Integr Comp Physiol. 2000;278:R556–R562. doi: 10.1152/ajpregu.2000.278.3.R556. [DOI] [PubMed] [Google Scholar]
- 42.Packer M, Lee WH, Kessler PD, Gottlieb SS, Bernstein JL, Kukin ML. Role of neurohormonal mechanisms in determining survival in patients with severe chronic heart failure. Circulation. 1987;75:IV80–92. [PubMed] [Google Scholar]
- 43.Pedersen EB, Danielsen H, Jensen T, Madsen M, Sorensen SS, Thomsen OO. Angiotensin ii, aldosterone and arginine vasopressin in plasma in congestive heart failure. Eur J Clin Invest. 1986;16:56–60. doi: 10.1111/j.1365-2362.1986.tb01308.x. [DOI] [PubMed] [Google Scholar]
- 44.Kleiber AC, Zheng H, Schultz HD, Peuler JD, Patel KP. Exercise training normalizes enhanced glutamate-mediated sympathetic activation from the pvn in heart failure. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1863–1872. doi: 10.1152/ajpregu.00757.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Narikiyo T, Kitamura K, Adachi M, Miyoshi T, Iwashita K, Shiraishi N, Nonoguchi H, Chen LM, Chai KX, Chao J, Tomita K. Regulation of prostasin by aldosterone in the kidney. J Clin Invest. 2002;109:401–408. doi: 10.1172/JCI13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rafiq K, Noma T, Fujisawa Y, Ishihara Y, Arai Y, Nabi AN, Suzuki F, Nagai Y, Nakano D, Hitomi H, Kitada K, Urushihara M, Kobori H, Kohno M, Nishiyama A. Renal sympathetic denervation suppresses de novo podocyte injury and albuminuria in rats with aortic regurgitation. Circulation. 2012;125:1402–1413. doi: 10.1161/CIRCULATIONAHA.111.064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ellekilde G, Holm J, Hemmingsen L, Koczulab B, von Eyben FE. Impact of inhibition of angiotensin-converting enzyme on urinary excretion of proteins in chronic heart failure. Clin Chem. 1992;38:1377–1378. [PubMed] [Google Scholar]
- 48.Hillege HL, Nitsch D, Pfeffer MA, Swedberg K, McMurray JJ, Yusuf S, Granger CB, Michelson EL, Ostergren J, Cornel JH, de Zeeuw D, Pocock S, van Veldhuisen DJ. Renal function as a predictor of outcome in a broad spectrum of patients with heart failure. Circulation. 2006;113:671–678. doi: 10.1161/CIRCULATIONAHA.105.580506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.