

Abstract
Intestinal fibrosis is a common feature of Crohn's disease (CD) and may appear as a stricture, stenosis or intestinal obstruction. Fibrostenosing CD leads to a significantly impaired quality of life in affected patients and constitutes a challenging treatment situation. In the absence of specific medical anti-fibrotic treatment options endoscopic or surgical therapy approaches with their potential harmful side effects are frequently used. However, our understanding of mechanisms of fibrogenesis in general and specifically intestinal fibrosis has emerged. Progression of fibrosis in the liver, lung or skin can be halted or even reversed and possible treatment targets have been identified. In face of this observation and given the fact that fibrotic alterations in various organs of the human body share distinct core characteristics, this article aims to address, whether reversibility of intestinal fibrosis may be conceivable and to highlight promising research avenues and therapies.
Keywords: Inflammatory bowel disease, fibrosis, stricture, disease course, stenosis, reversibility
Introduction
Fibrosis is defined as the exaggerated accumulation of collagen-rich extracellular matrix (ECM) and expansion of mesenchymal cells and may affect various organs of the human body as a consequence of chronic inflammatory diseases.1, 2 With regard to Crohn's disease (CD) patients, epidemiological data indicate that more than 40% of CD patients with ileal disease manifestation will develop clinically apparent stricture formation3 and the vast majority of these patients will have to undergo surgery at least once during the course of their disease.4 While recent observations indicate that immunosuppressive therapy may lead to a delay in disease progression5 or reduction in the need for intestinal surgery in CD patients in the long-run,6 endoscopic balloon dilation as well as surgical approaches such as strictureplasty or bowel segment resection still remain major therapeutic approaches for CD patients with symptomatic strictures.7 Of note, a recent study from Sweden reports that 13% of CD patients presented with a stricturing CD phenotype already at diagnosis,8 highlighting that this complication is not exclusively a feature of the long-term disease course but may result in symptomatic strictures in the short-run that require early interventional therapy. This frequent clinical observation together with the chronic progressive nature of stricturing CD lead to the common belief that fibrosis is a one-way street from fibrosis to stricture formation with intestinal obstruction followed by the eventual need for surgical resection. This notion however has to be challenged. Recently, a large combined analysis including a total of 1.112 patients assessed the efficacy of strictureplasty in CD and found an overall symptomatic recurrence rate of 39% for jejunoileal strictures (161 out of 411 patients) and 36% for ileocolonic strictures (9 out of 25 patients). Strikingly, only 3% or 20%, respectively, of CD patients strictures were found at the previous site of strictureplasty.9 This finding indicates that surgical intervention has the potential to stop progressing or even to reverse intestinal fibrosis. Based on Cleveland Clinic data, following strictureplasty operations, patients have been encouraged to undergo follow-up small bowel series after an interval of at least six months. Of the 44 asymptomatic patients who complied, recurrence at the strictureplasty sites with narrowing of the caliber of the bowel was noted in only 11% of patients after a median interval of two years.10 Maconi et al. performed serial ultrasound examinations in patients after strictureplasty and found a reduced thickness of the intestinal wall, suggesting a possible mechanism and further fueling the promise of reversibility of intestinal strictures.11
The concept of reversibility of intestinal fibrosis is in concordance with various observations from other organs such as the improvement of skin scarring12 and reduced skin thickening in systemic sclerosis,13 decreased proteinuria in patients with renal interstitial fibrosis,14 the improvement of vital capacity in idiopathic pulmonary fibrosis15, 16 the successful therapeutic reduction of myocardial collagen content in hypertensive patients17 and reversibility of myocardial fibrosis after adrenalectomy.18 In addition, there is a growing body of evidence indicating that liver fibrosis is a potentially reversible and bidirectional process overcoming the former paradigm of liver cirrhosis being an irreversible process. Repetitive histological evaluation via liver biopsies could prove reduction of the fibrosis grade after removal of the liver injury-causing triggers in patients with hepatitis C,19 hepatitis B,20 non-alcoholic steatohepatitis (NASH)21 or autoimmune hepatitis.22
Although the gut comprises unique features compared to other organ fibroses, such as severity and chronicity of inflammation in the context of IBD, the quality and quantity of the commensal microbiota or environmental influences on the disease course, intestinal fibrosis shares essentially all core mechanistic features with fibrotic disease of the above-mentioned organs.1, 23-25 Therefore, it appears to be reasonable to consider these mechanisms and therapeutic approaches and apply them as promising approaches for the reversal of stricturing CD. In the following sections we will discuss putative mechanisms for the reversal of intestinal fibrosis as seen in the phenomenon of intestinal strictureplasty, such as suppression of inflammation, the two forms of cellular transformation – epi- and endothelial to mesenchymal transition, macrophage subtype switch, fibroblast deactivation, and fibroblast apoptosis.
Review criteria
A comprehensive literature search was performed to assess all relevant citations found in Embase, Medline (service of the US National Library of Medicine (NLM) and the National Institutes of Health (NIH)) and the Cochrane Library for the following key words: (‘Crohn's disease (CD’) OR ‘Crohn's’ AND (‘stricture’ OR ‘fibrosis’), (‘kidney’ OR ‘liver’ OR ‘skin’ OR ‘lung’ OR ‘systemic nephrogenic’ AND ‘fibrosis’ OR ‘anti-fibrotic therapy’). Additionally, references of cited original articles and reviews were further assessed for relevant work. The search included studies between 1960 and 2014. These data together with the authors' personal experience in the field represent the basis of this review.
Mechanisms orchestrating reversibility of fibrosis
Multiple mechanisms that have been proven to be crucially involved in the reversal of fibrosis in organs other than the gut are candidates in the orchestration of resolution of intestinal fibrosis and could represent promising targets for anti-fibrotic therapy (Figure 1). To date only few of those mechanisms have been tested in the intestine.
Figure 1. Mechanisms contributing to reversibility of intestinal fibrosis.
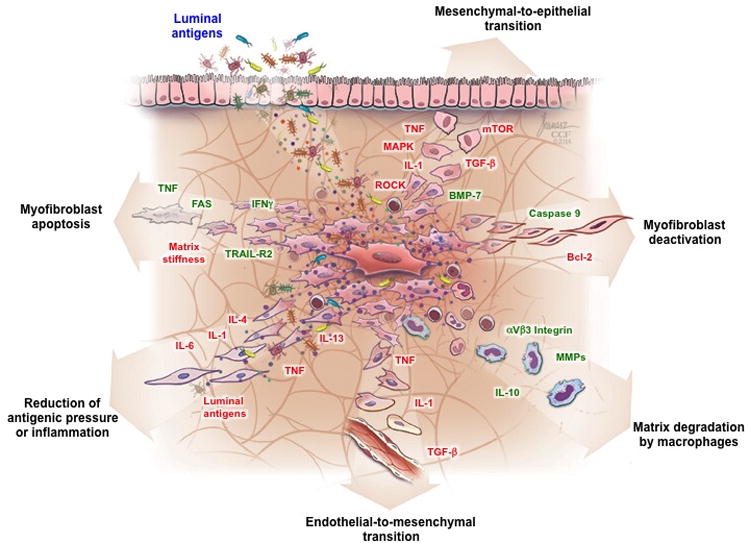
Depicted are the mechanisms and mediators that drive (green color) or inhibit (red color) reversibility of strictures in Crohn's disease. Abbreviations: Bcl-2: B-cell lymphoma-2; BMP-7: Bone morphogenetic protein; IL, Interleukin; IFN, Interferon; MAPK, MAP kinase; MMP, Matrix metalloproteinase; mTOR, mammalian target of rapamycin; ROCK, Rho-associated protein kinase; TGF, transforming growth factor; TNF, Tumor necrosis factor; TRAIL-R2: TNF-related apoptosis-induced ligand receptor 2
Suppression of inflammation
Emerging data suggests a diminished degree of inflammation at the site of strictureplasty after surgical intervention.26-28 This observation could be confirmed by histological and molecular assessment and determination of pro-inflammatory cytokine production of the ileal mucosa.29 While at the time of strictureplasty mucosal expression of Interleukin(IL)-1 β, IL-6, IL-8 or tumor necrosis factor (TNF)- α was markedly increased compared to expression levels in the non-inflamed mucosa, analysis 12 month after surgery revealed no difference in expression of these pro-inflammatory cytokines in the prior inflamed and non-inflamed areas.29 It was hypothesized that the decrease in inflammation may be a result of the reduced mechanical and antigenic pressure after successful increase in luminal diameter leading to decreased microbial and food allergen contact with inflamed and permeable mucosal areas of the intestine.29 As a consequence, at least in strictureplasties, a reduced inflammatory burden may be associated with a decrease in pro-fibrotic activity or even reversal due to a reduced fibrogenic pressure.
Over the recent years, the intestinal microbiome has been recognized to potentially modulate colitis and intestinal fibrogenesis due to its pleiotropic actions.30 For example, isolated human colonic fibroblasts from CD patients that were stimulated via bacterial components and toll-like receptors (TLR) mediated pathways revealed a significantly enhanced proinflammatory and profibrotic cytokine production.31 Additionally, in experimental models of colitis, the absence of the microbiota protects animals from intestinal inflammation hinting at the regulatory role of the microbiota.32 In line with these findings, human IBD patients presenting with alterations in genes coding bacterial recognition and processing were reported and antibodies against microbial components were shown to be associated with a severe disease phenotype and stricturing course of disease.33-35 In face of these observations, aside reduced inflammatory activity, an ameliorated antigenic pressure after stricture dilation may be one explanation for fibrosis reversal.
Macrophage subtypes
Besides antigenic pressure and cellular transition, activation of inflammatory cells such as monocytes and macrophages is a crucial step in fibrogenesis. Interestingly, these cells may facilitate pleiotropic and divergent actions depending on the stage of fibrosis development. In the early stages of liver fibrosis, macrophages may enable pro-fibrotic actions through production of transforming growth factor-beta (TGF- β) and platelet-derived-growth-factor (PDGF)-mediated hepatic stellate cell (HSC) activation. Thus, targeted macrophage depletion during the injury phase of experimental liver fibrosis resulted in decreased scaring and reduced numbers of myofibroblasts.36 In contrast, their depletion during the recovery phase of experimental liver fibrosis culminated in impaired scar resolution36 indicating that this cell type has not only a role in initiation, but also in resolution of fibrosis depending on the timing of action. These ambiguous properties could be explained by: (1) macrophages are a source of matrix metalloproteinases (MMPs). More specifically, MMP-2, -9 and 13 digest ECM compounds and therefore contribute to ECM degradation;36, 37 (2) macrophages mediate elimination of apoptotic cells resulting in clearance of pro-fibrotic triggers;38, 39 (3) anti-inflammatory IL-10 is secreted by macrophages and can limit TGF-β-induced collagen production in fibroblast;40 (4) macrophages can express HSC-specific death ligands.41, 42 In particular, M2 macrophages, a specific subset of macrophages, that is activated by IL-4 or IL-13, are crucially involved in fibrogenesis.43 The phenotype of these cells is defined by distinct products such as the mannose receptor, chitinase-3-like-protein-3 and the enzyme arginase-1 (Arg1), which regulates l-proline production, a prerequisite for collagen production 2, 44. Additionally, it is known that M2 cells compete with T-helper 2 cells and fibroblasts for l-arginine, which is required for l-proline synthesis.45 M2 cells link fibrosis and inflammation due to their capability to induce T reg cells.46 In vivo, M2 macrophages were shown to decelerate fibrosis development upon helminth infection 47. A more detailed description of the role of various macrophage subtypes as well as other cell types relevant for fibrogenesis has been published elsewhere48-52.
In addition to macrophages, natural killer (NK) cells have the ability to promote fibrosis resolution. In murine models of liver fibrosis, increased NKG2D receptor expression on NK cells resulted in enhanced HSC deletion through interaction with its ligand retinoic acid early inducible-1 on HSC and facilitated amelioration of experimental fibrosis.53 Interestingly, NK cells could be activated by the pro-inflammatory, but anti-fibrotic cytokine interferon-gamma (IFN-γ).53
The above-mentioned findings emphasize the potential pro- as well as anti-fibrotic actions of inflammatory cells, depending on the quality, quantity and timing of their response. Accordingly, targeted intervention during fibrosis development by the use of anti-inflammatory agents or cells may imply a high therapeutic potential. With regard to intestinal stenosis, it is well likely that a change in macrophage polarization or activation of NK cells contribute to the reversal of strictureplasty associated fibrosis in IBD.
Epi- and endothelial to mesenchymal transition
Another mechanism crucially involved in fibrosis establishment is transition of epithelial and endothelial cells into mesenchymal cells and hence their contribution to the pool of fibroblasts in fibrogenesis. Evidence exists about epi- (EMT) and endothelial to mesenchymal (EndoMT) transition occurring in intestinal inflammation.54, 55 Of note, this process is believed to be reversible. Although there is no study available demonstrating reversibility of EMT or EndoMT within in the intestine, there is a growing body of evidence in vitro and in vivo suggesting a presence of this mechanism in organs other than the gut. This is promising since this represents a means to reduce the excessive number of fibrotic effector cells in strictured areas in CD. A wide variety of distinct pathways have been identified to reverse EMT. In vitro examples include activation of protein phosphatase 2a in prostate cancer cell lines56, inhibition mTOR signaling pathway via rapamycin in gallbladder cancer cells.57, combined treatment with a TGF-β receptor I (TβRI) inhibitor and rho-associated protein kinase (ROCK) inhibitor in murine renal tubular epithelial cells58 or fibroblast growth factor (FGF)-1 and heparin treatment in TGF-β1-stimulated murine and alveolar epithelial-like cells.59 In vivo observations fuel the hope for induction of MET as a possible treatment approach to inhibit and reverse fibrotic diseases. In a murine model of renal fibrosis, MET resulted in accurate repair of injured epithelium.60 Moreover, anti-fibrotic effects via MET were reported in experimental models of pulmonary fibrosis by FGF-1 through MAPK/ERK kinase signaling, ERK-1 phosphorylation and Smad2 dephosphorylation.61 Zeisberg and coworkers suggested that bone morphogenic protein (BMP)-7, a member of the TGF-β superfamily, may be a promising target to facilitate the reversal of TGF-β-induced EMT. More specifically, BMP-7 administration resulted in reversal of chronic kidney disease in experimental models62 and mediated anti-fibrotic effects through the inhibition of HSC activation leading to amelioration of CCL4-induced liver fibrosis.63 While less information is available comparable factors have been identified promoting mesenchymal-to-endothelial transition (MEndoT), including rapamycin in pulmonary arterial hypertension,64 or BMP-7 in cardiac fibrosis65. Nevertheless, the ultimate proof of MET would include reversibility of EMT in primary cells from human fibrotic organs. Inflammatory pressure appears to be the main driver of cellular transformation and hence the reduced degree of inflammation after strictureplasty could inhibit or even reverse this process.
Fibroblast deactivation
Given the fact that myofibroblasts are a major player in intestinal fibrosis development due to massive production and secretion of multiple collagen types66, 67 and ECM compounds,25, 68 therapeutic manipulation of these cells appears promising to stop or reverse fibrosis. Similarly, in liver fibrogenesis, HSCs, which are considered to be the main precursor of liver myofibroblasts, are a main driver in fibrogenesis. Upon chronic injury, HSC not only get activated and change their phenotype adopting myofibroblast features, but also increase ECM deposition.69 In experimental models, upon removal of the fibrosis-inducing agent, activated liver myofibroblasts were shown to reduce their numbers. This effect is mediated by senescence or apoptosis, but activated hepatic myofibroblasts can also revert back to a quiescent phenotype, characterized by decreased expression of fibrogenic molecules, such as collagen I and α–smooth muscle actin (SMA).70, 71 Fibroblast deactivation and apoptosis appear to share common pathways including activation of cell-death mediated pathways and caspase activation,72 increase of pro-apoptotic stimuli (for example p53, caspase 9) and reduction of anti-apoptotic genes like Bcl-2 and Hspa1a/b.71, 72 as shown in hepatic fibroblast deactivation. In consequence, reversal of fibrotic scars in liver fibrosis was observed. Troeger et al. used a model of transgenic mice to study activated HSC in CCl4-induced liver fibrosis. They found that activated HSCs persisted in the liver up to 45 days after CCl4 challenge. Subsequently, the reversal of HSC activation resulted in inhibition of fibrogenesis during fibrosis resolution.73
Fibroblast apoptosis
Aside deactivation or senescence, apoptosis could account for the reduced numbers of myofibroblasts in reversal of fibrosis. Evidence can be derived from multiple organs outside of the intestine.69, 74, 75 For example, TNF-related apoptosis-induced ligand receptor (TRAIL-R)2 is expressed on activated HSC76 and was found to initiate apoptosis after its activation, which appears to be its critical function. Recently, a subset of CCR6 expressing γδ T cells were shown to accumulate in fibrotic livers of mice and to ameliorate fibrosis development through Fas-mediated induction of apoptosis in HSC.77 Additionally, immune cells, including natural killer cells and γδ T cells, stimulated by the pro-inflammatory cytokine IFN-γ were shown to possess a high degree of cytotoxicity towards HSC, contributing to resolution of fibrosis.53 The principle of apoptosis of pro-fibrotic cells during the recovery phase is observed in other organs as well. For example, apoptosis of renal mesangial cells occurs during mesangial proliferative nephritis.24 Similarly, the apoptosis of dermal myofibroblasts is crucial in the context of cutaneous wound healing to facilitate remodeling of the collagen-rich scar tissue.78 Fibroblast apoptosis has been described in the intestine of IBD patients as well. Proliferation rates of human intestinal fibroblasts isolated from ileal specimen of CD patients, which were ex vivo treated with TNF-α or IFN-γ alone were not significantly decreased as compared to untreated controls.79 However, combined stimulation with TNF-α and INF-γ lead to a 4-fold increased apoptosis rate of intestinal fibroblasts.79 Data supporting this mechanism in vivo are missing.
Furthermore, the matrix microenvironment also modulates myofibroblast viability. Enhanced ECM stiffness, which is present in scar tissue and fibrosis results in increased activation and survival of myofibroblasts through cell to matrix interactions.80 For example, spatial changes in hepatic matrix mechanics may lead to the development of fibrous septa and reorganize the liver architecture.81 Additionally, stage and progression of liver fibrosis are associated with septal width since massive septae are characteristic for advanced stage liver cirrhosis. Mechanistic studies identified liver stiffness as a core feature of the ECM for the development of liver fibrosis. More specifically, it was demonstrated in vitro, that HSC differentiation into myofibroblasts requires a mechanically stiff substrate, with adhesion to matrix proteins and the generation of mechanical tension. In vitro, activation of rat HSC was found to be significantly influenced by the stiffness of coating matrices. While HSC exposed to intermediate stiffness adopted stable intermediate phenotypes, cells challenged with stiff support differentiated into myofibroblasts.82
Conceptually, inflammation is undoubtedly an initiator and likely driver of intestinal fibrosis, but it becomes apparent that the quality, quantity and the timing of the inflammatory infiltrate are critical in determining its pro- or anti-fibrotic properties. This fact is underlined by the observation that effective anti-inflammatory intervention does not necessarily result in reduced frequency of fibrostenotic complications in CD patients and fibrotic alterations in animal models 5, 83. Understanding the core mechanisms of fibroblast apoptosis could serve as a tool for novel anti-fibrotic therapies by limiting a main source of collagen-producing cells.
Conclusion and outlook
The ultimate goal to reverse fibrosis and to induce physiological tissue regeneration is challenging and will not readily be realized in CD patients in the near future. However, necessary steps on the way towards at least partial amelioration of intestinal fibrotic complications are anticipated. The balance of synthesis and degradation of various matrix protein compounds crucially influences the excessive ECM accumulation in CD-associated strictures. The commonly believed notion that scar tissue is a hypometabolic or even ametabolic compartment needs to be revisited. This could be shown by measuring collagen turnover rates by administration of heavy water.84 In experimental pulmonary fibrosis, bleomycin-treatment resulted not only in increased matrix deposition, but also increased matrix turnover rates in the lung.85 Hence even the most severe fibrotic areas, such as established strictures in CD, could be amendable to anti-fibrotic therapy leading to its complete reversal, given a continuous matrix turnover.
Multiple distinct signaling cascades and mechanisms have been identified to contribute to fibrosis. Therefore, potential therapeutic approaches should not only address a single but multiple targets in form of a combined approach. The complexity of their interactions is high. For example, cytokines such as IFN- γ or TNF-α may represent anti-fibrotic, but pro-inflammatory mediators at the same time. They did not lead to fibroblast apoptosis on their own, but only together. It is obvious that especially in CD patients the quality, quantity and timing of profibrotic mediators deserves thoughtful evaluation.
Targeting cell types over mediators could be a promising approach: stimulation of subpopulations of macrophages to increase MMP-expression and to induce apoptosis of myofibroblasts may be suitable to emerge the anti-fibrotic therapeutic armamentarium.2 Another approach could be the stimulation of collagenase production or MMPs and has been successfully performed in animal studies.86 Finally, gastroenterologists should be stimulated by fibrosis research in other organs. An example could be a knowledge transfer to the intestine from scleroderma research, like the anti-fibrotic protein melanocortin 87, 88.
From the clinical point of view, early risk stratification of individual CD patients for stricturing disease courses is paramount and may be evaluated by appropriate biomarkers, which are currently not available. Data from the liver suggests that once established, the possibility to spontaneously reverse fibrotic complications seems to be dependent on the stage of fibrosis.89-91 While early stages may be reversed by the removal of the etiological factor, advanced stages may not regress on their own. As shown for liver fibrosis, identification of morphological indicators or biochemical ECM analyses of advanced fibrosis could define mechanisms for a point of no return.89 In CD the impact of anti-inflammatory treatment in patients with established fibrostenotic strictures needs to be further elucidated in clinical trials.92 Despite the required complex study designs including long follow up periods and large patient cohorts, there are currently first studies ongoing investigating the potential of anti-inflammatory drugs to modify the course of disease and to prevent fibrostenotic complications in CD patients (Clinical Trials.gov Identifier: NCT01698307).
We presented multiple mechanisms that could play a role in the reversal of intestinal fibrosis, a phenomenon that can be observed after strictureplasty surgery, such as suppression of inflammation, the two forms of cellular transformation – epi-and endothelial to mesenchymal transition, macrophage subtypes, fibroblast deactivation, and fibroblast apoptosis. Multiple compounds, whose mechanisms were discussed in this article, are already in clinical development or clinical practice in other organs than the intestine that could be employed in the therapy of Crohn's disease.1, 93 We propose strictureplasty as a novel and understudied human model for reversal of fibrosis and potential anti-fibrotic drug approaches should make use of this knowledge to explore novel therapies.
Acknowledgments
Source of funding: This work was supported by a research fellowship from the Faculty of Medicine, Westfälische Wilhelms-Universität Münster to D.B. and grants from the National Institutes of Health (T32DK083251 and P30DK097948) and the European Crohn's and Colitis Foundation to F.R.
Abbreviations
- BMP
bone morphogenic protein
- CD
Crohn's disease
- CCl4
carbon tetrachloride
- ECM
extracellular matrix
- EMT
epi to mesenchymal transition
- EndoMT
endothelial to mesenchymal transition
- FGF
fibroblast growth factor
- ERK
extracellular regulated kinase
- HCC
hepatocellular carcinoma
- HSC
hepatic stellate cell
- IL
interleukin
- MMP
matrix metalloproteinase
- NASH
non-alcoholic steatohepatitis
- TIMP
tissue inhibitor metalloproteinases
- MET
mesenchymal-to-epithelial transition
- NK cells
natural killer cells
- TNF- α
tumor necrosis factor alpha
- IFN- γ
interferon gamma
- SMA
smooth muscle actin
- ROCK
rho-associated protein kinase
- STAT
signal transducer and activator of transcription
- TRF
tocotrienol rich fraction
- TGF
transforming growth factor
- TLR
toll-like receptor
- TRAIL
TNF-related apoptosis-induced ligand receptor
Footnotes
Conflicts of interest: None
I declare the authors have no competing interests or other interests that might be perceived to influence the interpretation of the article.
References
- 1.Friedman SL, Sheppard D, Duffield JS, et al. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 2013;5:167sr161. doi: 10.1126/scitranslmed.3004700. [DOI] [PubMed] [Google Scholar]
- 2.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nature medicine. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cosnes J, Gower-Rousseau C, Seksik P, et al. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140:1785–1794. doi: 10.1053/j.gastro.2011.01.055. [DOI] [PubMed] [Google Scholar]
- 4.Cosnes J, Cattan S, Blain A, et al. Long-term evolution of disease behavior of Crohn's disease. Inflamm Bowel Dis. 2002;8:244–250. doi: 10.1097/00054725-200207000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Magro F, Rodrigues-Pinto E, Coelho R, et al. Is it Possible to Change Phenotype Progression in Crohn's Disease in the Era of Immunomodulators? Predictive Factors of Phenotype Progression. Am J Gastroenterol. 2014 doi: 10.1038/ajg.2014.97. [DOI] [PubMed] [Google Scholar]
- 6.Chatu S, Subramanian V, Saxena S, et al. The role of thiopurines in reducing the need for surgical resection in Crohn's disease: a systematic review and meta-analysis. Am J Gastroenterol. 2014;109:23–34. doi: 10.1038/ajg.2013.402. quiz 35. [DOI] [PubMed] [Google Scholar]
- 7.Rieder F, Zimmermann EM, Remzi FH, et al. Crohn's disease complicated by strictures: a systematic review. Gut. 2013;62:1072–1084. doi: 10.1136/gutjnl-2012-304353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sjoberg D, Holmstrom T, Larsson M, et al. Incidence and clinical course of Crohn's disease during the first year - results from the IBD Cohort of the Uppsala Region (ICURE) of Sweden 2005-2009. J Crohns Colitis. 2014;8:215–222. doi: 10.1016/j.crohns.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto T, Fazio VW, Tekkis PP. Safety and efficacy of strictureplasty for Crohn's disease: a systematic review and meta-analysis. Dis Colon Rectum. 2007;50:1968–1986. doi: 10.1007/s10350-007-0279-5. [DOI] [PubMed] [Google Scholar]
- 10.Fazio VW, Tjandra JJ, Lavery IC, et al. Long-term follow-up of strictureplasty in Crohn's disease. Dis Colon Rectum. 1993;36:355–361. doi: 10.1007/BF02053938. [DOI] [PubMed] [Google Scholar]
- 11.Maconi G, Sampietro GM, Cristaldi M, et al. Preoperative characteristics and postoperative behavior of bowel wall on risk of recurrence after conservative surgery in Crohn's disease: a prospective study. Ann Surg. 2001;233:345–352. doi: 10.1097/00000658-200103000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferguson MW, Duncan J, Bond J, et al. Prophylactic administration of avotermin for improvement of skin scarring: three double-blind, placebo-controlled, phase I/II studies. Lancet. 2009;373:1264–1274. doi: 10.1016/S0140-6736(09)60322-6. [DOI] [PubMed] [Google Scholar]
- 13.Kuhn A, Haust M, Ruland V, et al. Effect of bosentan on skin fibrosis in patients with systemic sclerosis: a prospective, open-label, non-comparative trial. Rheumatology (Oxford) 2010;49:1336–1345. doi: 10.1093/rheumatology/keq077. [DOI] [PubMed] [Google Scholar]
- 14.el-Agroudy AE, Hassan NA, Foda MA, et al. Effect of angiotensin II receptor blocker on plasma levels of TGF-beta 1 and interstitial fibrosis in hypertensive kidney transplant patients. Am J Nephrol. 2003;23:300–306. doi: 10.1159/000072820. 72820. [DOI] [PubMed] [Google Scholar]
- 15.King TE, Jr, Brown KK, Raghu G, et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184:92–99. doi: 10.1164/rccm.201011-1874OC. [DOI] [PubMed] [Google Scholar]
- 16.Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377:1760–1769. doi: 10.1016/S0140-6736(11)60405-4. [DOI] [PubMed] [Google Scholar]
- 17.Diez J, Querejeta R, Lopez B, et al. Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation. 2002;105:2512–2517. doi: 10.1161/01.cir.0000017264.66561.3d. [DOI] [PubMed] [Google Scholar]
- 18.Lin YH, Wu XM, Lee HH, et al. Adrenalectomy reverses myocardial fibrosis in patients with primary aldosteronism. J Hypertens. 2012;30:1606–1613. doi: 10.1097/HJH.0b013e3283550f93. [DOI] [PubMed] [Google Scholar]
- 19.Arthur MJ. Reversibility of liver fibrosis and cirrhosis following treatment for hepatitis C. Gastroenterology. 2002;122:1525–1528. doi: 10.1053/gast.2002.33367. [DOI] [PubMed] [Google Scholar]
- 20.Kweon YO, Goodman ZD, Dienstag JL, et al. Decreasing fibrogenesis: an immunohistochemical study of paired liver biopsies following lamivudine therapy for chronic hepatitis B. J Hepatol. 2001;35:749–755. doi: 10.1016/s0168-8278(01)00218-5. [DOI] [PubMed] [Google Scholar]
- 21.Dixon JB, Bhathal PS, Hughes NR, et al. Nonalcoholic fatty liver disease: Improvement in liver histological analysis with weight loss. Hepatology. 2004;39:1647–1654. doi: 10.1002/hep.20251. [DOI] [PubMed] [Google Scholar]
- 22.Czaja AJ, Carpenter HA. Decreased fibrosis during corticosteroid therapy of autoimmune hepatitis. J Hepatol. 2004;40:646–652. doi: 10.1016/j.jhep.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 23.Rieder F, Fiocchi C. Intestinal fibrosis in IBD--a dynamic, multifactorial process. Nat Rev Gastroenterol Hepatol. 2009;6:228–235. doi: 10.1038/nrgastro.2009.31. [DOI] [PubMed] [Google Scholar]
- 24.Baker AJ, Mooney A, Hughes J, et al. Mesangial cell apoptosis: the major mechanism for resolution of glomerular hypercellularity in experimental mesangial proliferative nephritis. J Clin Invest. 1994;94:2105–2116. doi: 10.1172/JCI117565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gelbmann CM, Mestermann S, Gross V, et al. Strictures in Crohn's disease are characterised by an accumulation of mast cells colocalised with laminin but not with fibronectin or vitronectin. Gut. 1999;45:210–217. doi: 10.1136/gut.45.2.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tonelli F, Fedi M, Paroli GM, et al. Indications and results of side-to-side isoperistaltic strictureplasty in Crohn's disease. Dis Colon Rectum. 2004;47:494–501. doi: 10.1007/s10350-003-0084-8. [DOI] [PubMed] [Google Scholar]
- 27.Farmer RG, Hawk WA, Turnbull RB., Jr Indications for surgery in Crohn's disease: analysis of 500 cases. Gastroenterology. 1976;71:245–250. [PubMed] [Google Scholar]
- 28.Post S, Betzler M, von Ditfurth B, et al. Risks of intestinal anastomoses in Crohn's disease. Ann Surg. 1991;213:37–42. doi: 10.1097/00000658-199101000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto T, Umegae S, Kitagawa T, et al. Postoperative change of mucosal inflammation at strictureplasty segment in Crohn's disease: cytokine production and endoscopic and histologic findings. Dis Colon Rectum. 2005;48:749–757. doi: 10.1007/s10350-004-0826-2. [DOI] [PubMed] [Google Scholar]
- 30.Honda K, Takeda K. Regulatory mechanisms of immune responses to intestinal bacteria. Mucosal Immunol. 2009;2:187–196. doi: 10.1038/mi.2009.8. [DOI] [PubMed] [Google Scholar]
- 31.Otte JM, Rosenberg IM, Podolsky DK. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology. 2003;124:1866–1878. doi: 10.1016/s0016-5085(03)00403-7. [DOI] [PubMed] [Google Scholar]
- 32.Eckmann L. Animal models of inflammatory bowel disease: lessons from enteric infections. Ann N Y Acad Sci. 2006;1072:28–38. doi: 10.1196/annals.1326.008. [DOI] [PubMed] [Google Scholar]
- 33.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rieder F, Schleder S, Wolf A, et al. Association of the novel serologic anti-glycan antibodies anti-laminarin and anti-chitin with complicated Crohn's disease behavior. Inflamm Bowel Dis. 2010;16:263–274. doi: 10.1002/ibd.21046. [DOI] [PubMed] [Google Scholar]
- 35.Kaul A, Hutfless S, Liu L, et al. Serum anti-glycan antibody biomarkers for inflammatory bowel disease diagnosis and progression: a systematic review and meta-analysis. Inflamm Bowel Dis. 2012;18:1872–1884. doi: 10.1002/ibd.22862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duffield JS, Forbes SJ, Constandinou CM, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henderson NC, Iredale JP. Liver fibrosis: cellular mechanisms of progression and resolution. Clin Sci (Lond) 2007;112:265–280. doi: 10.1042/CS20060242. [DOI] [PubMed] [Google Scholar]
- 38.Popov Y, Sverdlov DY, Bhaskar KR, et al. Macrophage-mediated phagocytosis of apoptotic cholangiocytes contributes to reversal of experimental biliary fibrosis. Am J Physiol Gastrointest Liver Physiol. 2010;298:G323–334. doi: 10.1152/ajpgi.00394.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Douglass A, Wallace K, Parr R, et al. Antibody-targeted myofibroblast apoptosis reduces fibrosis during sustained liver injury. J Hepatol. 2008;49:88–98. doi: 10.1016/j.jhep.2008.01.032. [DOI] [PubMed] [Google Scholar]
- 40.Wilson MS, Elnekave E, Mentink-Kane MM, et al. IL-13Ralpha2 and IL-10 coordinately suppress airway inflammation, airway-hyperreactivity, and fibrosis in mice. J Clin Invest. 2007;117:2941–2951. doi: 10.1172/JCI31546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou X, Murphy FR, Gehdu N, et al. Engagement of alphavbeta3 integrin regulates proliferation and apoptosis of hepatic stellate cells. J Biol Chem. 2004;279:23996–24006. doi: 10.1074/jbc.M311668200. [DOI] [PubMed] [Google Scholar]
- 42.Friedman SL. Mac the knife? Macrophages- the double-edged sword of hepatic fibrosis. J Clin Invest. 2005;115:29–32. doi: 10.1172/JCI23928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 44.Hesse M, Modolell M, La Flamme AC, et al. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of L-arginine metabolism. J Immunol. 2001;167:6533–6544. doi: 10.4049/jimmunol.167.11.6533. [DOI] [PubMed] [Google Scholar]
- 45.Pesce JT, Ramalingam TR, Mentink-Kane MM, et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 2009;5:e1000371. doi: 10.1371/journal.ppat.1000371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herbert DR, Holscher C, Mohrs M, et al. Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity. 2004;20:623–635. doi: 10.1016/s1074-7613(04)00107-4. [DOI] [PubMed] [Google Scholar]
- 48.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Akuthota P, Weller PF. Eosinophils and disease pathogenesis. Seminars in hematology. 2012;49:113–119. doi: 10.1053/j.seminhematol.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ray S, De Salvo C, Pizarro TT. Central role of IL-17/Th17 immune responses and the gut microbiota in the pathogenesis of intestinal fibrosis. Current opinion in gastroenterology. 2014;30:531–538. doi: 10.1097/MOG.0000000000000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hammerich L, Tacke F. Role of gamma-delta T cells in liver inflammation and fibrosis. World journal of gastrointestinal pathophysiology. 2014;5:107–113. doi: 10.4291/wjgp.v5.i2.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61:1066–1079. doi: 10.1002/hep.27332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Radaeva S, Sun R, Jaruga B, et al. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130:435–452. doi: 10.1053/j.gastro.2005.10.055. [DOI] [PubMed] [Google Scholar]
- 54.Rieder F, Kessler SP, West GA, et al. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol. 2011;179:2660–2673. doi: 10.1016/j.ajpath.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Flier SN, Tanjore H, Kokkotou EG, et al. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J Biol Chem. 2010;285:20202–20212. doi: 10.1074/jbc.M110.102012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bhardwaj A, Singh S, Srivastava SK, et al. Restoration of PPP2CA expression reverses epithelial-to-mesenchymal transition and suppresses prostate tumour growth and metastasis in an orthotopic mouse model. Br J Cancer. 2014;110:2000–2010. doi: 10.1038/bjc.2014.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zong H, Yin B, Zhou H, et al. Inhibition of mTOR pathway attenuates migration and invasion of gallbladder cancer via EMT inhibition. Mol Biol Rep. 2014 doi: 10.1007/s11033-014-3321-4. [DOI] [PubMed] [Google Scholar]
- 58.Das S, Becker BN, Hoffmann FM, et al. Complete reversal of epithelial to mesenchymal transition requires inhibition of both ZEB expression and the Rho pathway. BMC Cell Biol. 2009;10:94. doi: 10.1186/1471-2121--10-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramos C, Becerril C, Montano M, et al. FGF-1 reverts epithelial-mesenchymal transition induced by TGF-{beta}1 through MAPK/ERK kinase pathway. Am J Physiol Lung Cell Mol Physiol. 2010;299:L222–231. doi: 10.1152/ajplung.00070.2010. [DOI] [PubMed] [Google Scholar]
- 60.Zeisberg M, Shah AA, Kalluri R. Bone morphogenic protein-7 induces mesenchymal to epithelial transition in adult renal fibroblasts and facilitates regeneration of injured kidney. J Biol Chem. 2005;280:8094–8100. doi: 10.1074/jbc.M413102200. [DOI] [PubMed] [Google Scholar]
- 61.Lamouille S, Subramanyam D, Blelloch R, et al. Regulation of epithelial-mesenchymal and mesenchymal-epithelial transitions by microRNAs. Curr Opin Cell Biol. 2013;25:200–207. doi: 10.1016/j.ceb.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zeisberg M, Hanai J, Sugimoto H, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nature medicine. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 63.Wang LP, Dong JZ, Xiong LJ, et al. BMP-7 attenuates liver fibrosis via regulation of epidermal growth factor receptor. Int J Clin Exp Pathol. 2014;7:3537–3547. [PMC free article] [PubMed] [Google Scholar]
- 64.Ranchoux B, Antigny F, Rucker-Martin C, et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation. 2015;131:1006–1018. doi: 10.1161/CIRCULATIONAHA.114.008750. [DOI] [PubMed] [Google Scholar]
- 65.Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nature medicine. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 66.Graham MF, Diegelmann RF, Elson CO, et al. Collagen content and types in the intestinal strictures of Crohn's disease. Gastroenterology. 1988;94:257–265. doi: 10.1016/0016-5085(88)90411-8. [DOI] [PubMed] [Google Scholar]
- 67.Borley NR, Mortensen NJ, Kettlewell MG, et al. Connective tissue changes in ileal Crohn's disease: relationship to disease phenotype and ulcer-associated cell lineage. Dis Colon Rectum. 2001;44:388–396. doi: 10.1007/BF02234738. [DOI] [PubMed] [Google Scholar]
- 68.Geboes K, El-Zine MY, Dalle I, et al. Tenascin and strictures in inflammatory bowel disease: an immunohistochemical study. Int J Surg Pathol. 2001;9:281–286. doi: 10.1177/106689690100900404. [DOI] [PubMed] [Google Scholar]
- 69.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fallowfield JA, Mizuno M, Kendall TJ, et al. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. 2007;178:5288–5295. doi: 10.4049/jimmunol.178.8.5288. [DOI] [PubMed] [Google Scholar]
- 71.Kisseleva T, Cong M, Paik Y, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A. 2012;109:9448–9453. doi: 10.1073/pnas.1201840109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Iredale JP. Hepatic stellate cell behavior during resolution of liver injury. Semin Liver Dis. 2001;21:427–436. doi: 10.1055/s-2001-17557. [DOI] [PubMed] [Google Scholar]
- 73.Troeger JS, Mederacke I, Gwak GY, et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. 2012;143:1073–1083 e1022. doi: 10.1053/j.gastro.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu X, Xu J, Brenner DA, et al. Reversibility of Liver Fibrosis and Inactivation of Fibrogenic Myofibroblasts. Curr Pathobiol Rep. 2013;1:209–214. doi: 10.1007/s40139-013-0018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saile B, Matthes N, Knittel T, et al. Transforming growth factor beta and tumor necrosis factor alpha inhibit both apoptosis and proliferation of activated rat hepatic stellate cells. Hepatology. 1999;30:196–202. doi: 10.1002/hep.510300144. [DOI] [PubMed] [Google Scholar]
- 76.Taimr P, Higuchi H, Kocova E, et al. Activated stellate cells express the TRAIL receptor-2/death receptor-5 and undergo TRAIL-mediated apoptosis. Hepatology. 2003;37:87–95. doi: 10.1053/jhep.2003.50002. [DOI] [PubMed] [Google Scholar]
- 77.Hammerich L, Bangen JM, Govaere O, et al. Chemokine receptor CCR6-dependent accumulation of gammadelta T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology. 2014;59:630–642. doi: 10.1002/hep.26697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Darby I, Skalli O, Gabbiani G. Alpha-smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab Invest. 1990;63:21–29. [PubMed] [Google Scholar]
- 79.Francoeur C, Bouatrouss Y, Seltana A, et al. Degeneration of the pericryptal myofibroblast sheath by proinflammatory cytokines in inflammatory bowel diseases. Gastroenterology. 2009;136:268–277 e263. doi: 10.1053/j.gastro.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 80.Smit JJ, Schinkel AH, Oude Elferink RP, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451–462. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 81.Garcia-Tsao G, Friedman S, Iredale J, et al. Now there are many (stages) where before there was one: In search of a pathophysiological classification of cirrhosis. Hepatology. 2010;51:1445–1449. doi: 10.1002/hep.23478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Olsen AL, Bloomer SA, Chan EP, et al. Hepatic stellate cells require a stiff environment for myofibroblastic differentiation. Am J Physiol Gastrointest Liver Physiol. 2011;301:G110–118. doi: 10.1152/ajpgi.00412.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Johnson LA, Luke A, Sauder K, et al. Intestinal fibrosis is reduced by early elimination of inflammation in a mouse model of IBD, impact of a “Top-Down” approach to intestinal fibrosis in mice. Inflamm Bowel Dis. 2012;18:460–471. doi: 10.1002/ibd.21812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Decaris ML, Gatmaitan M, Florcruz S, et al. Proteomic Analysis of Altered Extracellular Matrix Turnover in Bleomycin-Induced Pulmonary Fibrosis. Mol Cell Proteomics. 2014 doi: 10.1074/mcp.M113.037267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hernnas J, Nettelbladt O, Bjermer L, et al. Alveolar accumulation of fibronectin and hyaluronan precedes bleomycin-induced pulmonary fibrosis in the rat. Eur Respir J. 1992;5:404–410. [PubMed] [Google Scholar]
- 86.Iimuro Y, Nishio T, Morimoto T, et al. Delivery of matrix metalloproteinase-1 attenuates established liver fibrosis in the rat. Gastroenterology. 2003;124:445–458. doi: 10.1053/gast.2003.50063. [DOI] [PubMed] [Google Scholar]
- 87.Kokot A, Sindrilaru A, Schiller M, et al. alpha-melanocyte-stimulating hormone suppresses bleomycin-induced collagen synthesis and reduces tissue fibrosis in a mouse model of scleroderma: melanocortin peptides as a novel treatment strategy for scleroderma? Arthritis Rheum. 2009;60:592–603. doi: 10.1002/art.24228. [DOI] [PubMed] [Google Scholar]
- 88.Bettenworth DSA, Tepasse PR, Rijcken E, Apel M, Böhm M. Human Intestinal Fibroblasts Are Novel Target Cells for Alpha-Melanocyte-Stimulating Hormone -Possible Implications for the Treatment of Stricturing Crohn's Disease With Melanocortin Peptides and Derivatives. Gastroenterology. 2014;146:S–282. [Google Scholar]
- 89.Kisseleva T, Brenner DA. Anti-fibrogenic strategies and the regression of fibrosis. Best Pract Res Clin Gastroenterol. 2011;25:305–317. doi: 10.1016/j.bpg.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kisseleva T, Brenner DA. Hepatic stellate cells and the reversal of fibrosis. Journal of gastroenterology and hepatology. 2006;21 Suppl 3:S84–87. doi: 10.1111/j.1440-1746.2006.04584.x. [DOI] [PubMed] [Google Scholar]
- 91.Pellicoro A, Ramachandran P, Iredale JP. Reversibility of liver fibrosis. Fibrogenesis & tissue repair. 2012;5:S26. doi: 10.1186/1755-1536-5-S1-S26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bouhnik Y, Carbonnel F, Laharie D, et al. Efficacy of adalimumab in patients with Crohn's disease and symptomatic small bowel stricture: a multicentre, prospective, observational cohort study (CREOLE) ECCO abstract #DOP034. Journal of Crohns and Colitis. 2015;9 Suppl [Google Scholar]
- 93.Bettenworth D, Rieder F. Medical therapy of stricturing Crohn's disease: what the gut can learn from other organs - a systematic review. Fibrogenesis & tissue repair. 2014;7:5. doi: 10.1186/1755-1536-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]