

Abstract
Rho GTPases are integral to the regulation of actin cytoskeleton-dependent processes, including mitosis. Rho and leukemia-associated Rho guanine-nucleotide exchange factor (LARG), also known as ARHGEF12, are involved in mitosis as well as diseases such as cancer and heart disease. Since LARG has a role in mitosis and diverse signalling functions beyond mitosis, it is important to understand the regulation of the protein through modifications such as phosphorylation. Our research provides further information about the mitotic phosphoregulation of the regulator of G protein signaling (RGS)-RhoGEF LARG. Here we report that LARG undergoes a mitotic-dependent and cyclin-dependent kinase 1 (Cdk1) inhibitor-sensitive phosphorylation. Additionally, LARG is phosphorylated at the onset of mitosis and dephosphorylated as cells exit mitosis, concomitant with Cdk1 activity. Furthermore, using an in vitro kinase assay, we show that LARG can be directly phosphorylated by Cdk1. Through expression of N- and C-terminal deletion and phosphonull mutants that contain non-phosphorylatable alanine mutations at Cdk1 S/TP sites, we demonstrate that LARG phosphorylation occurs in both termini. Using phosphospecific antibodies, we confirm that two sites, serine 190 and serine 1176, are phosphorylated during mitosis in a Cdk1-dependent manner. In addition, these phosphospecific antibodies show phosphorylated LARG at specific mitotic locations, namely the mitotic organizing centers and flanking the midbody. Lastly, RhoA activity assays reveal that phosphonull LARG is more active in cells than phosphomimetic LARG. Our data thus identifies LARG as a phosphoregulated RhoGEF during mitosis.
Keywords: Phosphorylation, RhoGEF, RhoA, mitosis, cytokinesis, Cdk1, subcellular localization
1. Introduction
Small GTPases of the Rho family are important regulators of actin dynamics and are involved in processes such as mitosis, embryogenesis, migration, and neurite extension [1]. Rho proteins, like all GTPases, function as molecular switches, cycling between inactive GDP-bound and active GTP-bound states. This cycle is under exquisite control by Rho guanine-nucleotide exchange factors (RhoGEFs) that promote activating GDP/GTP exchange by stimulating release of GDP from inactive GDP-bound Rho. Rho GTPase activating proteins (RhoGAPs) deactivate GTP-bound Rho by stimulating the GTP hydrolysis activity of the Rho protein.
Several Rho GTPases, including Rac1, Cdc42 and RhoA, have been demonstrated to function during mitosis. RhoA has been implicated in distinct activities at multiple phases of mitosis but most notably plays a crucial role in the regulation of the actomyosin contractile ring during cytokinesis [2, 3]. As with other cellular functions of RhoA, the mitotic function is regulated by RhoGAPs and RhoGEFs. Although more than 70 RhoGEFs exist in mammalian cells [4], only a very limited number have been shown to function in mitosis. Most prominently, the RhoGEF Ect2 is required for cytokinetic furrow ingression through the cortical activation and localization of RhoA [5]. GEF-H1 has also been shown to be important for activation of RhoA during furrow ingression [6], while MyoGEF functions earlier in mitosis by recruiting proteins, including Ect2, to the central spindle [7, 8]. Together, these RhoGEFs function to promote the proper assembly and ingression of the cytokinetic furrow.
Recent work from our laboratory identified the RhoGEF LARG as another mitotic RhoGEF [9]. LARG is one of three regulator of G protein signalling (RGS) domain-containing RhoGEFs and has been most studied for its role in directly connecting GPCR-activated heterotrimeric G protein α subunits of the G12/13 family to RhoA activation [10]. In addition, LARG has been implicated in many diseases, including cancer, heart disease, and in HIV viral replication [11-20]. In its newly recognized mitotic role, LARG was demonstrated to be required for abscission, the final cytokinetic event. LARG displayed mitotic-specific localization, residing at the spindle poles in metaphase, the central spindle in late anaphase, and, most prominently, at the midbody from furrow ingression until abscission. Moreover, LARG co-localized with RhoA during cytokinesis. Distinct from previously identified mitotic RhoGEFs, knockdown of LARG by siRNA resulted in a phenotype of delayed abscission with some cells that were unable to resolve their intercellular bridges undergoing apoptosis. Other RhoGEFs, particularly Ect2, have been clearly shown to regulate cleavage furrow ingression during cytokinesis [21, 22]; however, LARG is the only RhoGEF shown to have a later role in abscission.
Although it is not known how LARG is regulated during mitosis, phosphorylation is a common mechanism for tightly controlled regulation of mitotic proteins. The master mitotic kinase is cyclin-dependent kinase 1 (Cdk1), while additional serine/threonine kinases, such Aurora (A/B/C) and polo-like kinase 1 (Plk1), play key roles during mitosis. Cdk1’s role in the cell cycle is well established and the most studied of the mitotic kinases [23, 24]. To function as a mitotic kinase, Cdk1 must be bound to cyclin B, the protein levels of which rise during G2 and then abruptly fall after the anaphase transition as cells exit mitosis. Prior to mitosis, Cdk1, bound to cyclin B, is in a phosphorylated, inactive state; however, elevated activity of the phosphatase Cdc25 results in dephosphorylation and thus activation of Cdk1 at the G2/M transition. Cyclin B is degraded as cells progress through anaphase towards the end of mitosis, leading again to the inactivation Cdk1 [25]. Mitotic Cdk1/cyclin B phosphorylates and regulates a myriad of key mitotic proteins, including the mitotic RhoGEFs Ect2, GEF-H1, and MyoGEF [26]. For example, phosphorylation of GEF-H1 by Cdk1, along with Aurora A kinase, during early mitosis inhibits GEF-H1, while dephosphorylation of GEF-H1, temporally coinciding with Cdk1 inactivation, prior to cytokinesis activates GEF-H1, thus promoting activation of RhoA [6]. Thus, the purpose of our study was to determine whether LARG also undergoes mitotic-dependent phosphorylation and to examine the effect of phosphorylation.
Here we report that LARG is phosphorylated and dephosphorylated during mitosis and that Cdk1 is the mitotic kinase responsible for LARG phosphorylation. Results with phosphomimetic mutant and phosphonull mutants of LARG suggest that phosphorylation inhibits the GEF activity of LARG. These studies are the first to describe mitotic phosphoregulation of the RhoGEF LARG.
2. Materials and Methods
2.1 Cell culture, plasmids, and transfections
Cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum and 2% HEPES. Mitotic cells were collected by shaking off following incubation for 16 h in media containing 0.5 μg/ml nocodazole (Sigma-Aldrich, St. Louis, Missouri). Cells remaining attached to the plate after mitotic shake off are referred to as G2 cells. For mitotic kinase inhibitor treatment, mitotic cells plated on poly-L-lysine (Sigma-Aldrich, St. Louis, Missouri) coated plates were incubated in media containing 0.5 μg/ml nocodazole along with 100 μM roscovitine (A.G. Scientific, San Diego, CA), 10 μM RO-3306 (Sigma-Aldrich, St. Louis, Missouri), 100 nM BI2536 (Axon Medchem BV, Groningen, The Netherlands), 50 nM ZM447439 (Biomol, Plymouth, PA) or 50 nM Calyculin A (Biomol, Plymouth, PA). For the phosphatase inhibition assay, cell lysates were treated with 50 U λ-protein phosphatase (New England Biolabs, Beverly, MA) at 30°C for 30 min according to manufacturer’s protocol. For synchronization and release at G1/S phase, 2 mM of thymidine (Sigma-Aldrich, St. Louis, Missouri) in media was added for 16 hours, released into thymidine-free media for 8 hours, thymidine-containing media again for 16 hours, and then released and collected over 15 hours.
The plasmid GFP-LARG, in which GFP is fused to the N-terminus of LARG, was described previously [27]. The phosphonull and phosphomimetic LARG mutants were made by synthesizing DNA (Genscript, Piscataway, NJ) and subcloning the mutation-containing fragments into GFP-LARG. The following amino acids were mutated to an alanine (A) for the phosphonull LARG mutants: N-terminal phosphonull: S28, S151, S170, S175, S178, S190, T230, T267, S309, and S341; C-terminal phosphonull: T1148, S1176, S1327, T1362, S1457, S1468, T1471, and S1492; N/C-terminal phosphonull: all 18 sites listed above were mutated to alanine; N/C phosphomimetic: all 18 sites listed above were mutated to glutamate (E).
For transfections, cells were plated in 10 cm or 6-well plates 24 h prior to transfection. Cells were transfected with Lipofectamine 2000 (Life Technologies, Carlsbad, CA) for 24-48 hours, unless otherwise noted. Knockdown with control and LARG siRNA was performed as previously described [9].
2.2 In vitro kinase assay
Full length-LARG (1-1544), RDPC-LARG (307-1544; called LARGΔN from hereon out), and RDP (307-1146; called LARGΔNΔC from hereon out) and their purification were previously described [28]. Purified LARG proteins (1 μM) from Sf9-baculovirus system were incubated with CDK1-cyclin B1 (13 nM) (New England Biolabs, Beverly, MA), composed of the catalytic subunit CDK1 and its positive regulatory subunit cyclin B1, in a final volume of 25 μl of incubation buffer (50 mM Tris, 10 mM Mg, 2 mM DTT, 0.1% EDTA, 0.01% Brij 35 containing 20 μM [gamma-32P] ATP (~200 cpm/pmol) for 5 min at 30°C. After incubation for 5 min, the products were separated by SDS-PAGE and subjected to autoradiography.
2.3 SRE Assays
HEK293 cells were plated 24 hours prior to transfection in a 24-well plate and then were transfected with GFP-LARG constructs or the control pEGFP, together with both SRE-firefly luciferase, and TK-renilla luciferase. After 24 hours, cells were lysed according to the manufacturer’s protocol, and luciferase activity was measured with a luminometer using the Dual Glo System (Promega, Madison Wisconsin). SRE-luciferase activity was normalized by the renilla luciferase, and then this normalized SRE-luciferase activity stimulated by the GFP-LARG constructs was divided by that measured for the control pEGFP construct to give fold over control (pEGFP). Further, slight variations in expression of the different GFP-LARG proteins were taken into account by densitometric quantitation of anti-GFP antibody immunoblots of cell lysates from the luciferase experiments; SRE-luciferase activity was then accordingly normalized.
2.4 RhoA Binding Assays
GST-G17A-RhoA was prepared and binding assays were performed as previously described [9]. Briefly, GFP-tagged LARG constructs were transfected in HEK293 cells, cell lysates were prepared, and active LARG was pulled down using the nucleotide-free form of RhoA, GST-G17A-RhoA, which preferentially binds to activated RhoGEFs [29]. Typically, 5% of the pull down and 0.5% of the cell lysate were analyzed by immunoblotting with a GFP monoclonal antibody (Covance), and this assay resulted in the affinity pull down of 10-20% of the total expressed GFP-LARG.
2.5 Phosphospecific Antibodies
Rabbit polyclonal antibodies directed against LARG phosphorylated at S190 or S1176 were generated by 21st Century Biochemicals, Inc. (Marlboro, MA) using two immunogen peptides together for each phosphosite. To generate the pS190 antibody the following peptides were used: C-Ahx-GNMERIT[pS]PVLMG-amide and acetyl-GNMERIT[pS]PVLMGEEN-Ahx-KC-amide. To generate the pS1176 antibody the following peptides were used: C-AhX-ISVTGLQ[pS]PDRDLGL-amide and acetyl-ISVTGLQ[pS]PDRDLGL-Ahx-KC-amide. Antibodies were purified via affinity purification with the phosphopeptide and immunodepleted using an affinity column containing a non-phosphorylated peptide.
2.6 Immunoblotting
Cells in 6-well or 10 cm plates were washed twice with ice cold PBS and lysed with sample buffer or lysis buffer (20 mM HEPES pH 7.4, 0.5% Triton X-100, 100 mM NaCl, 2.5 mM MgCl2, 1 mM EDTA, leupeptin and aprotinin (5mg/ml), 1 mM PMSF, 25 mM β-glycerophosphate, 1 mM sodium orthovanadate) on ice. LARG was detected with LARG H-70 antibody (Santa Cruz, Dallas, TX).
2.7 Cell Imaging
HeLa cells growing on coverslips were fixed with 10% trichloroacetic acid (TCA) in PBS for 10 min at 4°C. Following washes with PBS fixed cells were incubated in blocking buffer containing 2.5% non-fat milk or 1 % BSA in TBS-1 % Triton X-100. Detection of pS190 and pS1176 was done at 1:200. α-tubulin was detected by staining with a mouse monoclonal (clone DM1A) anti-α-tubulin antibody (Sigma, St. Louis, Missouri) followed by Alexa Fluor 594 goat anti-mouse (Molecular Probes, Eugene, OR). The coverslips were then washed with TBS/1% Triton X-100, rinsed with water and mounted on glass slides with 14 μl of Prolong Antifade reagent (Molecular Probes, Eugene, OR). For DAPI staining, coverslips were incubated with 0.1 μg/ml of DAPI (Molecular Probes, Eugene, OR) in PBS for 5 min following fixation or secondary antibody incubation and washes.
Representative images were acquired using an Olympus BX-61 upright microscope with an ORCA-ER (Hamamatsu, Bridgewater, NJ) cooled charge-coupled device camera controlled by Slidebook version 4.0 (Intelligent Imaging Innovations, Denver, CO). Images of fixed cells were captured with an Olympus PlanApo 60x/N.A.1.4 Oil objective.
2.8 Statistical Analysis
Statistics were conducted in GraphPad Prism (version 4.0b) using an unpaired t test (two tailed) or as otherwise stated.
3. Results
3.1 LARG is phosphorylated as cells enter mitosis and is dephosphorylated as cells exit mitosis
Known mitotic RhoGEFs such as Ect2 are phosphorylated specifically during mitosis [5, 30]. In order to investigate whether LARG is also phosphorylated during mitosis, mitotic, asynchronous, and interphase cells were assessed. HeLa cells were synchronized in prometaphase by treatment with nocodazole followed by mitotic shake-off, lysis, SDS-PAGE and immunoblot. LARG in cell lysates from asynchronous cells and from interphase/G2 cells, which were the cells left on the plate after shaking off mitotic cells, shows similar mobility via immunoblot (Fig. 1A). However, LARG in mitotic cells displays slower mobility in comparison to asynchronous and interphase LARG. To investigate whether this change in mobility was due to phosphorylation of LARG, lysate from nocodazole-arrested mitotic cells was treated with λ-phosphatase. The mitotic-dependent shift was abolished following phosphatase treatment of both HeLa and COS-7 cell lysates (Fig. 1B), confirming that the observed mobility shift reflects phosphorylation of LARG.
Figure 1. Mitotic-dependent phosphorylation of LARG.

(A) Mitotic-dependent mobility shift of LARG. HeLa cells were treated with nocodazole as described in Materials and Methods. Cells arrested in prometaphase (M) were collected by mitotic shake-off, and cells remaining on plates were collected and designated as G2 cells. Lysates from asynchronous (A), G2, or prometaphase-arrested (M) HeLa cells were immunoblotted with anti-LARG antibody. (B) Phosphatase treatment reverses mitotic-dependent phosphorylation of LARG. G2 or prometaphase-arrested (M) lysates from HeLa (left panels) and COS-7 (right panels) cells were prepared as above in A. Lysates from prometaphase-arrested (M) cells were then treated with or without λ protein phosphatase, followed by immunoblotting with anti-LARG antibody or anti-GAPDH antibody (loading control).
To examine the phosphorylation status, by gel mobility shift, of LARG as cells proceed through mitosis, several synchronization and release protocols were examined. Washing out nocodazole (nocodazole release) from mitotic shake-off collected cells releases cells from prometaphase arrest, thus allowing cells to resume and continue through mitosis. Cell lysates were prepared at various times after nocodazole release. Immunoblotting showed that LARG returned to the faster mobility form, consistent with dephosphorylation, as cells moved through and exited from mitosis (Fig. 2A). LARG appears to be fully dephosphorylated by 90-135 minutes after release from prometaphase arrest; this timing is coordinate with cyclin B1 degradation (Fig. 2A) and with initiation of furrow ingression, as observed by light microscopy (not shown). Next, HeLa cells were treated with the Cdk1 inhibitor RO-3306 to arrest cells in G2/M. This synchronization approach arrests cells earlier compared to nococazole-induced arrest, and this approach avoids the harsh poisoning of microtubules. Here, the slower mobility form of LARG transiently appeared strongly at 30 minutes post RO-3306 washout, remained at 60 minutes, and disappeared by 120 minutes release from G2/M (Fig. 2B). Previous work has demonstrated that Cdk1/cyclin B1 rapidly recovers activity after RO-3306 washout and that within 30-40 minutes 50% of cells have reached metaphase [31]. Since the slower mobility form of LARG is observed within 15-30 min of washout, these results suggest that LARG is phosphorylated at the G2/M boundary, or shortly thereafter, coordinate with recovery of Cdk1/cyclin B1 activity. Lastly, cells were synchronized at G1/S by double thymidine block, followed by collecting cell lysates at time points 3-15 hours after release. Again, a slower mobility form of LARG was detected as cells entered mitosis, and then this mobility shift was lost as cells exited mitosis; passage through mitosis was confirmed by the hallmark appearance and disappearance of cyclin B1 (Fig. 2C). The incomplete mobility shift of LARG after release from G2/M and G1/S arrest likely reflects partial synchronization and/or that only a fraction of a cell’s pool of LARG is phosphorylated during mitosis (Fig. 2 B,C). Taken together the studies in Figures 1 and 2 demonstrate that LARG undergoes phosphorylation at G2/M as cells enter mitosis and dephosphorylation with kinetics matching anaphase transition, coinciding with inactivation of Cdk1.
Figure 2. LARG is phosphorylated and dephosphorylated during mitosis.

(A) LARG is dephosphorylated as cells exit mitosis. HeLa cells arrested in prometaphase by nocodazole were collected by mitotic shake-off and re-plated on poly-L-lysine coated plates in fresh media without nocodazole to release the cells from mitotic arrest. Cells were lysed at the indicated times after release, and lysates were immunoblotted using anti-LARG, anti-cyclin B1 and anti-HSP90 (loading control) antibodies. (B) LARG is rapidly phosphorylated after release from Cdk1 inhibitor RO-3306. Cells were synchronized with RO-3306, released from G2/M block by washing and incubating in media without RO-3306, and lysed at indicated times. Cell lysates were immunoblotted using anti-LARG and anti-HSP90 (loading control) antibodies. (C) LARG is phosphorylated coordinate with Cyclin B1 accumulation and is dephosphorylated in late mitosis. HeLa cells were treated with 2 mM thymidine for 16 hrs to block the cells in S phase, followed by release in normal growth medium for 8 hrs and subsequent thymidine treatment for 16 hrs. HeLa cells were then released from S-phase block into normal growth media and collected at indicated time points. Cell lysates were immunoblotted using anti-LARG, anti-cyclin B1, anti-GAPDH (loading control) antibodies.
3.2 Mitotic kinase Cdk1 phosphorylates LARG
We next examined which mitotic kinase may be responsible for phosphorylating LARG. Cells were synchronized with nocodazole and then incubated with mitotic kinase inhibitors for Cdk1 (Roscovitine and RO-3306), Plk (BI2536), and Aurora (ZM447439), three kinases with well-documented roles during mitosis [32]. Only the inhibitors of Cdk1 were able to fully abolish the mitotic-dependent gel mobility shift of LARG (Fig. 3A). These data suggest that Cdk1 is responsible for the phosphorylation that causes a markedly slower form of LARG in SDS-PAGE. Calyculin A was also used, which prevents the removal of the phosphorylation through inhibiting phosphatases [33]. Calyculin A could potentially enhance the shift of mitotic LARG by allowing further accumulation of LARG phosphorylation; however, an enhanced shift was not detected. The Plk and Aurora inhibitors did not affect the mitotic-dependent mobility shift of LARG, suggesting that these kinases are not responsible for the phosphorylation shift of LARG; however, these experiments do not rule out that LARG could be a substrate for Plk or Aurora during mitosis. In summary, inhibition of the gel mobility shift with two inhibitors of Cdk1 provides evidence that Cdk1 is responsible for the mitotic-dependent phosphorylation of LARG.
Figure 3. LARG is phosphorylated by Cdk1 during mitosis.

(A) Inhibition of Cdk1 abolishes the slower mobility of mitotic LARG. Mitotic cells, arrested after nocodazole treatment, were collected and re-plated on poly-L-lysine in the continued presence of nocodazole and in presence of the indicated mitotic kinase inhibitors or Calyculin A or vehicle control (DMSO) for 30 or 60 minutes. Cell lysates were immunoblotted using anti-LARG antibody. (B) Full-length LARG and ΔN LARG are phosphorylated in vitro by Cdk1. Purified proteins (1 μM) from Sf9-baculovirus system (right panel) were incubated with CDK1-cyclin B (13 nM) in incubation buffer (containing 20 μM [gamma-32P]ATP) for 5 min at 30°C. Samples were analyzed by SDS-PAGE followed by autoradiography (left panel). (C) Domain structure of LARG with potential Cdk1 Sites. Amino acid numbering is indicated in the above figure. Domains: PDZ, post synaptic density 95/disc-large/zona occludens; RGS, regulator of G protein signalling; DH, Dbl homology; PH, pleckstin homology; NLS, nuclear localization signal; CC, coiled-coil. The pink stars indicate potential Cdk1 sites. The dark blue stars indicate the sites that are the epitopes targeted by the pS190 and pS1176 phosphospecific antibodies. (D) Mitotic-dependent gel mobility shift of LARG is altered by N- and C-terminal phosphonull mutations. HEK293 cells were transfected with the indicated constructs, treated with nocodazole or vehicle control. Nocodazole-treated cells were subjected to mitotic shake-off, and cell lysates were prepared (M). Cell lysates were also prepared from asynchronously growing cells (A). Lysates were then immunoblotted using an anti-GFP antibody.
LARG contains 24 potential Cdk1 sites, based on the minimal consensus sequence of S/T-P [34], and many of these Cdk1 consensus sites are in the N and C terminus of LARG (Fig. 3C). To determine if Cdk1 could directly phosphorylate LARG, we conducted an in vitro kinase assay. We tested for phosphorylation using purified full length, ΔN (amino acids 307-1544), and ΔNΔC (amino acids 307-1146) LARG created using a baculovirus system and incubation with Cdk1/Cyclin B and radiolabeled ATP. This revealed that Cdk1 is indeed able to directly phosphorylate LARG. Moreover, these assays revealed the importance of the C-terminus of LARG as a region containing sites for Cdk1-dependent phosphorylation, as the ΔN, but not the ΔNΔC, was phosphorylated (Fig. 3B). Purified ΔC LARG was not available to be tested in in vitro kinase assays. In these experiments, we were not able to determine the molar stoichiometry of phosphorylation. Nonetheless, these experiments provide evidence that Cdk1, the master mitotic kinase, can directly phosphorylate LARG.
To further examine potential sites of Cdk1-dependent phosphorylation of LARG, we created phosphorylation mutants in GFP-LARG where the N-and or C-termini S/T-P potential Cdk1 sites were mutated from a serine or threonine to an alanine (Fig. 3C) and interrogated these phosphonull mutants in a mitotic-dependent gel mobility shift assay, after expression in HEK293 cells followed by nocodazole-induced mitotic arrest (Fig. 3D). The N-terminal phosphonull LARG (NpNull), containing ten S/T✧A mutations, displayed approximately half the shift of full-length LARG. The C-terminal phosphonull LARG (CpNull), which has eight S/T✧A mutations, also shows only a partial shift compared to full-length LARG. However, similar to the dual-termini deletion mutant of LARG in the in vitro Cdk1 kinase assay (Fig. 3B), the N/CpNull LARG displayed no mitotic-dependent gel mobility shift (Fig. 3D), further supporting that LARG has key sites of mitotic-dependent phosphorylation in both the N and C-termini.
3.3 Phosphospecific antibodies reveal that LARG is phosphorylated at S190 and S1176 during mitosis
To further investigate phosphorylation of LARG in a more endogenous setting as opposed to exogenous expression of mutated LARG, we set forth to create tools to probe phosphorylated sites of LARG. Since our data supports that phosphorylation occurs in the N-and C-termini, we chose two potential Cdk1 sites, one in each terminus, against which to develop phosphoantibodies: S190 and S1176. Additionally, Cdk1 tends to prefer clustering of Cdk1 consensus sites, and both S190 and S1176 exist in a cluster of S/TP sites [35]. Furthermore, previously published mass phosphoproteomic data identified both S190 [36, 37] and S1176 [37] as sites in LARG that are phosphorylated in mitosis.
In cell lysates from nocodazole-arrested HeLa cells, both pS190 and pS1176 antibodies recognize the slower migrating form of LARG; the signal is lost or diminished in cell lysates from asynchronous cells or when cell lysates from nocodazole-arrested cells are treated with λ-phosphatase (Fig. 4A). Detection with the pS190 and pS1176 antibodies is also lost when nocodazole-arrested HeLa cells are treated with the Cdk1 inhibitior RO-3306 (not shown). To further test the specificity of our antibodies, we used lysate from HeLa cells treated with non-specific (NS) siRNA or LARG siRNA (siL). With successful knockdown of LARG (Fig. 4C), the pS190 antibody and pS1176 recognize mitotic LARG in the NS siRNA treated mitotic lysate, but not in the siL treated mitotic lysate (Fig. 4B). To further characterize these phosphospecific antibodies, various point mutants of LARG were expressed in HeLa cells, followed by mitotic arrest and immunobotting of cell lysates containing the overexpressed LARG mutant. Confirming the antibody specificity, the phosphospecific antibodies only recognized LARG protein that contained non-mutated sites (Fig. 4D,E). The pS190 antibody detected mitotic GFP-LARG, mCherry-LARG-S1176A, and GFP-CpNull LARG, but it failed to recognize mitotic GFP-LARG-S190A, GFP-NpNull LARG, or the GFP-N/CpNull. Thus, the pS190 antibody only detects LARG mutants containing serine at position 190 and preferentially detects such overexpressed LARG mutants in mitotic cells; these results suggest that the pS190 antibody only recognizes mitotic LARG phosphorylated at S190. The pS1176 antibody detected mitotic GFP-LARG, GFP-LARG-S190A, and GFP-LARG-NpNull, but it failed to recognize mitotic mCherry-LARG-S1176A, GFP-LARG-CpNull, or the GFP-LARG-N/CpNull. Thus, the pS1176 antibody only detects LARG mutants containing serine at position 1176 and preferentially detects such overexpressed LARG mutants in mitotic cells; these results suggest that the pS1176 antibody only recognizes mitotic LARG phosphorylated at S1176. .All LARG mutants were expressed at similar levels in HeLa cells (Fig. 3B and not shown). Importantly, as described above, in all cases except for GFP-LARG-N/CpNull which is not detected by either of the phosphospecific antibodies, the mutant form of LARG is detected by one phosphospecific antibody, either pS190 or pS1176, but not the other one. Thus lack of detection by immunoblotting with a phosphospecific antibody (Fig. 4D,E) is not simply due to lack of protein expression, but instead represents loss of the cognate phosphorylatable site in LARG. Taken together, the data in Figure 4 support that these antibodies are detecting the phosphosite they are designed to recognize.
Figure 4. Phosphospecific antibodies reveal that LARG is phosphorylated at S190 and S1176 during mitosis.

(A) Phosphoantibody specificity for phosphorylated LARG was determined by phosphatase treatment of mitotic cell lysates. Cell lysates were prepared from asynchronous HeLa cells (A) and nocodazole-arrested mitotic HeLa cells (M). As described in Materials and Methods, mitotic cell lysates were treated with λ protein phosphatase (+). Then lysates were immunoblotted with anti-LARG, anti-pS190 LARG, and anti-pS1176 antibodies, as indicated. (B) Phosphoantibody specificity for LARG was determined by siRNA knockdown of LARG. HeLa cells were treated with non-specific (ns) siRNA and siRNA specific for LARG (siLARG), as described previously [9]. After nocodazole treatment to arrest cells in prometaphase, mitotic cells were harvested by shake-off and lysates were prepared (M). Lysates were also prepared from the G2/interphase cells remaining on the culture dish (I). Lysates were then immunoblotted with anti-LARG, anti-pS190 LARG, and anti-pS1176 LARG antibodies, as indicated. (C) Immunoblot showing efficiency of LARG siRNA. Lysates were prepared from G2/interphase (G2) and mitotic cells (M) treated with Ns-siRNA or LARG siRNA, as described above and used in (B), followed by immunoblotting with an anti-LARG antibody. Ns lysates were used at 1, 0.5 and 0.25 equivalents compared to siLARG lysates to show efficient knockdown (≥90%) of endogenous LARG. (D and E) Phosphoantibodies were tested on LARG point and multi-site phosphomutants. HeLa cells were transfected with the indicated constructs, treated with nocodazole, and subjected to mitotic shake-off. Cell lysates from the mitotic cells (M) and asynchronous cells (A) were prepared and immunoblotted using anti-pS190 LARG, anti-pS1176 LARG and anti-HSP90 (loading control) antibodies, as indicated.
3.4 LARG, phosphorylated at sites S190 and S1176, shows distinct mitotic localization
Previously, endogenous LARG was shown to localize to the mitotic spindle poles, central spindle, and midbody [9]. Here we used pS190 and pS1176 antibodies to examine the localization of phosphorylated LARG during mitosis (Fig. 5). As expected, both the pS190 and pS1176 antibodies showed weak staining of interphase HeLa cells, consistent with immunoblots (Fig. 4B). Interestingly, in many interphase cells nuclear puncta were observed using the pS190 antibody, and a small fraction of the nuclei were stained in interphase cells using the p1176 antibody. Importantly, cells in prophase and metaphase showed a dramatic increase in the intensity of staining with both the pS190 and pS1176 antibodies, indicating that endogenous LARG undergoes phosphorylation at S190 and S1176 early in mitosis. These results confirm and extend the immunoblotting results with these phosphospecific antibodies that showed detection in lysates from nocodazole-arrested cells (Fig. 4B), and these results are consistent with the mitotic-dependent gel mobility shift immunoblots that showed phosphorylation of LARG in G2/M (Fig. 2). Moreover, it appears that S190- and S1176-phosphorylated LARG is localized primarily to the cytoplasm in early mitosis, consistent with the localization of the bulk of LARG in early mitosis [9].
Figure 5. LARG phosphorylated at sites S190 and S1176 localizes to the centrosomes and flanks the midbody in cytokinesis.


(A) HeLa cells were fixed and stained with anti-α-tubulin antibody (red), anti-pS190 LARG (green), and DNA was visualized by DAPI staining (blue). (B) Same as in left panel, except anti-pS1176 LARG is in green. Cells in different stages of mitosis were identified based on hallmark nuclear and microtubule staining.
The pS190 and pS1176 antibodies also detected phosphorylated LARG at specific mitotic structures throughout mitosis. Using the pS190 antibody, we observed localization to the mitotic organizing centers (MTOCs) in prophase, prometaphase, and anaphase (Fig. 5A), in addition to the cytoplasmic staining described above. In late mitosis, the pS190 antibody detects phosphorylated LARG flanking the midbody; previous results showed that a LARG-specific antibody detected endogenous LARG more centrally in the midbody in late mitosis [9]. Similar to the pS190 antibody, the pS1176 antibody detected phosphorylated LARG at the MTOCs in prophase, prometaphase, metaphase and anaphase (Fig. 5B). The pS1176 antibody also stained regions flanking the midbody in cytokinesis. Of particular interest, the bulk of staining with both pS190 and pS1176 antibodies decreases in cells containing intercellular bridges (Fig. 5A and 5B, late cytokinesis panels), although some midbody staining was still detected. The decreased overall staining with the pS190 and pS1176 antibodies in cytokinesis is consistent with the decreased phosphorylation of LARG observed by gel mobility shift as cells proceed from the anaphase transition to mitosis exit (Fig. 2). Thus, the pS190 and pS1176 LARG antibodies showed that phosphorylated LARG localizes to key mitotic structures during mitosis and that LARG is phosphorylated at S190 and S1176 as cells enter mitosis and dephosphosphorylated at these two amino acids as cells exit mitosis.
3.5 Activation of RhoA by wild type, phosphonull, and phosphomimetic LARG
To address the effect of mitotic phosphorylation of LARG activity, we examined phosphonull and phosphomimetic mutants of LARG (Fig. 6). The phosphonull mutant (N/CpNull), in which 18 serines/threonines found in minimal Cdk1 site motifs (S/T-P) were changed to alanines, was described above (Fig. 3C and D). In addition, we created a phosphomimetic LARG mutant (N/CpMim), in which the same 18 serines/threonines were substituted with glutamic acids to mimic the negative charge of multi-site mitotic phosphorylation of LARG. First, we assayed for serum response element (SRE)-luciferase activity in transfected cells, a widely used transcriptional reporter readout of RhoA-dependent activity in transfected cells. LARG-N/CpNull showed increased activity compared to wild-type LARG, while LARG-N/CpMim displayed lower SRE-luciferase acitivity compared to wild-type LARG (Fig. 6A). Next, we utilized an established GST-G17A-RhoA affinity pull down assay (Fig. 6B). In this assay nucleotide-empty G17A-RhoA binds to activated RhoGEFs, and thus this is a convenient assay for detecting relative amounts of particular LARG mutants that are active in terms of GEF activity [9, 29]. Here, we compared wild type LARG with the phosphonull and phosphomimetic mutants. Strikingly, LARG-N/CpMim showed a decreased GST-G17A-RhoA affinity pull down compared to LARG-N/CpNull and wild-type LARG. LARG-N/CpNull displayed a small increase in pull down, although not statistically significant. Taken together, the results in Figure 6 indicate that the Cdk1 site phosphomimetic LARG (N/CpMim) is a less active GEF in cells compared to wild type LARG or phosphonull LARG (N/CpNull), and by extension suggest that LARG phosphorylated by Cdk1 during mitosis has decreased activity toward RhoA.
Figure 6. Decreased activity of phosphomimetic mutant of LARG.

(A) LARG activity was assessed via serum response element (SRE) luciferase activity. HEK293 cells were transfected with expression vectors for GFP-LARG, GFP-LARG-N/CpNull, GFP-LARG-N/CpMim or control pEGFP, along with both SRE-luciferase and Renilla luciferase. SRE-luciferase activity was measured and normalized as described under Materials and Methods. The graph shows quantitation (average ± SEM, n=3) of multiple experiments. Statistical significance is represented as follows: LARG compared to N/CpNull (*, p<0.05, t-test); N/CpNull compared to N/CpMim: (**, p<0.01, t-test); and LARG compared to N/CpMim: (***, p<0.0001, t-test). (B) Pull-down of LARG using GST-G17A-RhoA. The indicated GFP-tagged LARG proteins were expressed in HEK293 cells. Cell lysates were prepared and active LARG was pulled down using GST-G17A-RhoA. LARG in the affinity pulldown (upper panel) and in the cell lysate (lower panel) was determined by immunoblotting with a GFP antibody. A representative blot is shown. The graph shows quantitation (average ± SEM, n=4) of multiple experiments, in which the amount of LARG in the GST-G17A-Rho pulldown was normalized by the total expressed LARG in the lysate. The asterisk indicates statistical significance (p<0.05, t-test) of the N/CpMim mutant compared to wt LARG and to the N/CpNull mutant.
4. Discussion
Here we demonstrate that LARG is phosphorylated as cells enter mitosis, dephosphorylated as cells exit mitosis, and that the master mitotic kinase Cdk1 can directly phosphorylate LARG. Phosphorylation of LARG, as observed by a mitotic-dependent gel mobility shift, requires multiple potential Cdk1 sites in both the N-terminal and C-terminal region of LARG. When all N- and C-terminal potential Cdk1 sites are mutated to alanines, as in LARG-N/CpNull, the mitotic-dependent gel mobility shift is abolished. Immunoblotting with phosphospecific antibodies reveal that S190 and S1176 of LARG are phosphorylated in a mitotic- and Cdk-1-dependent manner. Moreover, immunofluorescence microscopy using the pS190 and pS1176 antibodies indicates mitotic-specific localization of phosphorylated LARG to the MTOCs and flanking the midbody. Lastly, a phosphomimetic mutant of LARG (N/CpMim), in which multiple N- and C-terminal potential Cdk1 site serines or threonines are mutated to glutamic acids, displays decreased activity, as measured by both transcriptional readout of Rho-dependent signaling and by GST-G17A-RhoA affinity pull down. These results demonstrate mitotic-dependent phosphorylation of LARG, identify Cdk1 as the responsible kinase, and implicate a potential role for this phosphorylation in regulating the GEF activity of LARG.
Post-translational modification of LARG by phosphorylation has not been extensively studied to date. Previous work used general anti-phosphotyrosine antibodies to demonstrate tyrosine phosphorylation of LARG by focal adhesion kinase (FAK) and Tec tyrosine kinase [38, 39]. Chikumi, et al. showed that downstream of thrombin activation, FAK, a non-receptor tyrosine kinase, could tyrosine-phosphorylate LARG as well as PDZ-RhoGEF, another RGS-RhoGEF. While LARG is similar to PDZ-RhoGEF, only PDZ-RhoGEF was investigated further, and the results suggested that this modification may act in a positive feedback manner to potentiate PDZ-RhoGEF-mediated activation of RhoA [38]. In the other study, Suzuki, et al. demonstrated that the RhoGEF activity of LARG could not be activated by Gα12 unless LARG was phosphorylated by Tec tyrosine kinase; however, Gα13 could activate LARG regardless of tyrosine phosphorylation by Tec [39]. Both of these studies suggested that tyrosine phosphorylation of LARG can be an activating regulatory modification [38, 39]. In contrast, our work here demonstrates that LARG is phosphorylated on serine and/or threonine amino acids during mitosis and, as discussed further below, suggests that this phosphorylation attenuates LARG activity.
Previously, no antibodies were available to identify phosphorylation of LARG. In the course of these studies, we generated antibodies that detect mitotic phosphorylation of LARG at S190 and S1176. These novel tools further allowed us to identify the subcellular localization of phosphorylated LARG in mitotic cells. In a previous publication from our laboratory, it was shown that LARG localized to the spindle poles and the midbody during mitosis, and LARG colocalized with RhoA at the midbody after full ingression of the contractile ring [9]. Whereas our previous work showed that LARG and RhoA antibodies co-stained the midbody, we now show that the pS1176 and pS190 LARG antibodies only stained discrete regions flanking the midbody (Fig. 5). These data suggest that a spatially distinct population of phosphorylated LARG exists at midbody flanking regions, together with a population of non-phosphorylated LARG at the midbody. Perhaps in this midbody zone, non-phosphorylated LARG plays a key role in activating RhoA in the final scission of the cells (as discussed below, non-phosphorylated LARG may be more active than mitotic phosphorylated LARG), and yet there remains a discrete pool of less active, phosphorylated LARG in a region flanking the midbody. Alternatively, the LARG phospho-epitopes may be detected by the pS190 and pS1176 antibodies in the regions flanking the midbody but simply may not be available to be recognized in the protein dense midbody. As discussed further below, our results with the phosphomimetic LARG mutant (Fig. 6) suggests that mitotic phosphorylated LARG would be more active; an unknown role may exist for phosphorylated LARG that is retained at the midbody late in mitosis. Nonetheless, the phosphospecific antibodies developed here confirm S190 and S1176 as mitotic-dependent phosphorylation sites in LARG and provide a valuable tool for detecting phosphorylated LARG. Clearly additional sites of phosphorylation, beyond S190 and S1176, exist in LARG. In addition to the tyrosine phosphorylation of LARG discussed above [38, 39], mass phosphoproteomics studies have identified additional sites of phosphorylation of LARG [36, 37, 40]; such additional sites of phosphorylation remain to be identified and/or confirmed and characterized. In addition, it is anticipated that additional Cdk1 sites in LARG, beyond S190 and S1176, are phosphorylated in early mitosis, since LARG shows a substantial gel mobility shift (Figs. 1-4), likely due to multi-site phosphorylation and the fact that the single site substitutions, S190A or S1176, do not substantially reduce the mitotic-dependent gel mobility shift of LARG (Fig. 4D and 4E). It will be important in future work to define the spectrum of sites in LARG phosphorylated by Cdk1 and sites in LARG phosphorylated by other kinases.
The data comparing the phosphomimetic mutant (LARG-N/CpMim), the phosphonull mutant (LARG-N/CpNull) and wild type LARG (Fig. 6) suggest a model in which mitotic phosphorylation of LARG decreases its GEF activity toward RhoA while dephoshorylation of LARG relieves the early mitotic inhibition of RhoGEF activity (Fig. 7). This is a reasonable deduction that parallels with what is known about phosphorylation of other RhoGEFs in mitosis. GEF-H1, MyoGEF and Ect2 are all phosphorylated during mitosis, and the phosphorylation regulates activity, localization, subsequent phosphorylation, and interaction with other proteins. Phosphorylation of GEF-H1 by Cdk1 during early mitosis inhibits its GEF activity [6]. Conversely, phosphorylation of MyoGEF by Plk1 increases its activity toward RhoA and is also important for MyoGEF's localization to the central spindle [7]. Ect2 is phosphorylated by Cdk1 at three sites, which lead to changes in Ect2 conformation, allowing it to be more active, and priming Ect2 for interaction with Plk1 [5, 30]. Based on our data showing that phosphonull LARG has increased activity compared to the phosphomimetic LARG (Fig. 6) and that LARG is phosphorylated as cells enter mitosis and dephosphorylated late in mitosis (Fig. 2), we predict that mitotic phosphorylation of LARG functions similarly to what was shown for mitotic phosphorylation of GEF-H1. In the case of GEF-H1, phosphorylation early in mitosis serves to inhibit RhoGEF activity and subsequent dephosphorylation late in mitosis serves to activate the RhoGEF [6]. Therefore, we propose that the dephosphorylation of LARG that occurs as cells exit mitosis serves to relieve the early mitotic inhibition of RhoGEF activity (Fig. 7). Lastly, it is possible that Cdk1 phosphorylation of LARG is a priming event for subsequent phosphorylation by additional mitotic kinases. Another possibility, and potentially in combination with a priming event, is that phosphorylation of LARG allows LARG to complex with another protein(s) as occurs with Ect2 when Cdk1 phosphorylation at T412 is required for interaction with Plk1 [30]. It will be important to further explore how mitotic phosphorylation regulates LARG.
Figure 7. Proposed model of phosphoregulation of LARG in mitosis.
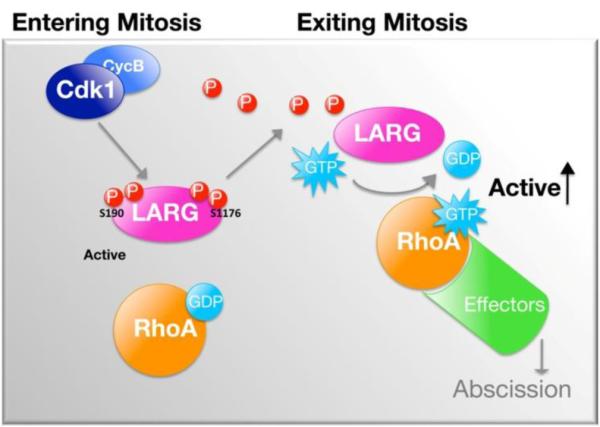
LARG is phosphorylated and dephosphorylated during mitosis, which regulates the GEF activity. Cdk1 phosphorylates LARG in early mitosis (G2/M) resulting in decreased GEF activity. LARG is then dephosphorylated, relieving the inhibition of GEF activity, as cells proceed past the anaphase transition towards mitotic exit. See text for further discussion.
5. Conclusions
The RGS-RhoGEF LARG is phosphorylated by Cdk1 in early mitosis and then dephosphorylated as cells exit mitosis. LARG is phosphorylated in early mitosis on multiple serines or threonines located in both the N- and C-termini, including serines 190 and 1176. Lastly, cell-based functional assays using LARG phosphomutants suggest a model in which mitotic phosphorylation decreases LARG’s GEF activity toward RhoA, while subsequent dephosphorylation relieves this inhibition.
Highlights.
- LARG, a RhoA specific RhoGEF that is required in late cytokinesis, is phosphorylated as cells enter mitosis and dephosphorylated as cells exit mitosis
- Cdk1, the master mitotic kinase, can directly phosphorylate LARG
- LARG is phosphorylated in the N- and C-termini, including at S190 and S1176
- LARG phosphorylated at S190 and S1176 exhibits localization to the spindle poles, mitotic spindle, and flanking the midbody in late cytokinesis
- A phosphomimetic mutant of LARG is less active compared to both wild type LARG and a phosphonull (Cdk1 phosphorylation deficient) mutant of LARG, suggesting that the mitotic phosphorylation of LARG inhibits activity and the subsequent dephosphorylation relieves the inhibition
Acknowledgements
This work was supported by NIH grant GM62884 (P.B.W.) and American Heart Association Grant 11PRE7260038 (M.C.H).
Abbreviations
- LARG
leukemia-associated Rho guanine nucleotide exchange factor
- GTP
guanine-nucleotide triphosphate
- GEF
guanine-nucleotide exchange factor
- RhoGEF
Rho guanine-nucleotide exchange factor
- Cdk1
cyclin-dependent kinase
- pNull
phosphonull
- pMim
phosphomimetic
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References
- [1].Settleman J. Dev Cell. 2001;1(3):321–331. doi: 10.1016/s1534-5807(01)00053-3. [DOI] [PubMed] [Google Scholar]
- [2].Piekny A, Werner M, Glotzer M. Trends Cell Biol. 2005;15(12):651–658. doi: 10.1016/j.tcb.2005.10.006. [DOI] [PubMed] [Google Scholar]
- [3].Narumiya S, Yasuda S. Curr Opin Cell Biol. 2006;18(2):199–205. doi: 10.1016/j.ceb.2006.02.002. [DOI] [PubMed] [Google Scholar]
- [4].Rossman KL, Der CJ, Sondek J. Nat Rev Mol Cell Biol. 2005;6(2):167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- [5].Yuce O, Piekny A, Glotzer M. J Cell Biol. 2005;170(4):571–582. doi: 10.1083/jcb.200501097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Birkenfeld J, Nalbant P, Bohl BP, Pertz O, Hahn KM, Bokoch GM. Dev Cell. 2007;12(5):699–712. doi: 10.1016/j.devcel.2007.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Asiedu M, Wu D, Matsumura F, Wei Q. J Biol Chem. 2008;283(42):28392–28400. doi: 10.1074/jbc.M801801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wu D, Asiedu M, Adelstein RS, Wei Q. Cell Cycle. 2006;5(11):1234–1239. doi: 10.4161/cc.5.11.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Martz MK, Grabocka E, Beeharry N, Yen TJ, Wedegaertner PB. Mol Biol Cell. 2013;24(18):2785–2794. doi: 10.1091/mbc.E12-07-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fukuhara S, Chikumi H, Gutkind JS. FEBS Lett. 2000;485(2-3):183–188. doi: 10.1016/s0014-5793(00)02224-9. [DOI] [PubMed] [Google Scholar]
- [11].Kourlas PJ, Strout MP, Becknell B, Veronese ML, Croce CM, Theil KS, Krahe R, Ruutu T, Knuutila S, Bloomfield CD, Caligiuri MA. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(5):2145–2150. doi: 10.1073/pnas.040569197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shih LY, Liang DC, Fu JF, Wu JH, Wang PN, Lin TL, Dunn P, Kuo MC, Tang TC, Lin TH, Lai CL. Leukemia. 2006;20(2):218–223. doi: 10.1038/sj.leu.2404024. [DOI] [PubMed] [Google Scholar]
- [13].Dirse V, Bertasiute A, Gineikiene E, Zvirblis T, Dambrauskiene R, Gerbutavicius R, Juozaityte E, Malciute L, Paulsson K, Griskevicius L. Genes Chromosomes Cancer. 2015;54(5):326–333. doi: 10.1002/gcc.22246. [DOI] [PubMed] [Google Scholar]
- [14].Poppe B, Cauwelier B, Van Limbergen H, Yigit N, Philippe J, Verhasselt B, De Paepe A, Benoit Y, Speleman F. Haematologica. 2005;90(9):1179–1185. [PubMed] [Google Scholar]
- [15].Zheng R, Iwase A, Shen R, Goodman OB, Jr., Sugimoto N, Takuwa Y, Lerner DJ, Nanus DM. Oncogene. 2006;25(44):5942–5952. doi: 10.1038/sj.onc.1209586. [DOI] [PubMed] [Google Scholar]
- [16].Wang Q, Liu M, Kozasa T, Rothstein JD, Sternweis PC, Neubig RR. J Biol Chem. 2004 doi: 10.1074/jbc.C400105200. [DOI] [PubMed] [Google Scholar]
- [17].Bourguignon LY, Gilad E, Brightman A, Diedrich F, Singleton P. J Biol Chem. 2006;281(20):14026–14040. doi: 10.1074/jbc.M507734200. [DOI] [PubMed] [Google Scholar]
- [18].Ong DC, Ho YM, Rudduck C, Chin K, Kuo WL, Lie DK, Chua CL, Tan PH, Eu KW, Seow-Choen F, Wong CY, Hong GS, Gray JW, Lee AS. Oncogene. 2009;28(47):4189–4200. doi: 10.1038/onc.2009.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ariake K, Ohtsuka H, Motoi F, Douchi D, Oikawa M, Rikiyama T, Fukase K, Katayose Y, Egawa S, Unno M. Cancer Lett. 2012;325(1):99–107. doi: 10.1016/j.canlet.2012.06.012. [DOI] [PubMed] [Google Scholar]
- [20].Hodges A, Sharrocks K, Edelmann M, Baban D, Moris A, Schwartz O, Drakesmith H, Davies K, Kessler B, McMichael A, Simmons A. Nat Immunol. 2007;8(6):569–577. doi: 10.1038/ni1470. [DOI] [PubMed] [Google Scholar]
- [21].Tatsumoto T, Xie X, Blumenthal R, Okamoto I, Miki T. J Cell Biol. 1999;147(5):921–928. doi: 10.1083/jcb.147.5.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Su KC, Takaki T, Petronczki M. Dev Cell. 2011;21(6):1104–1115. doi: 10.1016/j.devcel.2011.11.003. [DOI] [PubMed] [Google Scholar]
- [23].Li JJ, Li SA. Pharmacol Ther. 2006;111(3):974–984. doi: 10.1016/j.pharmthera.2006.02.006. [DOI] [PubMed] [Google Scholar]
- [24].Nigg EA. Nat Rev Mol Cell Biol. 2001;2(1):21–32. doi: 10.1038/35048096. [DOI] [PubMed] [Google Scholar]
- [25].Lapenna S, Giordano A. Nat Rev Drug Discov. 2009;8(7):547–566. doi: 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]
- [26].Zuo Y, Oh W, Frost JA. Cell Signal. 2014;26(12):2998–3006. doi: 10.1016/j.cellsig.2014.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Grabocka E, Wedegaertner PB. Mol Pharmacol. 2007;72(4):993–1002. doi: 10.1124/mol.107.035162. [DOI] [PubMed] [Google Scholar]
- [28].Suzuki N, Tsumoto K, Hajicek N, Daigo K, Tokita R, Minami S, Kodama T, Hamakubo T, Kozasa T. J Biol Chem. 2009;284(8):5000–5009. doi: 10.1074/jbc.M804073200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Garcia-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, Burridge K. Methods Enzymol. 2006;406:425–437. doi: 10.1016/S0076-6879(06)06031-9. [DOI] [PubMed] [Google Scholar]
- [30].Niiya F, Tatsumoto T, Lee KS, Miki T. Oncogene. 2006;25(6):827–837. doi: 10.1038/sj.onc.1209124. [DOI] [PubMed] [Google Scholar]
- [31].Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, Chen L. Proc Natl Acad Sci U S A. 2006;103(28):10660–10665. doi: 10.1073/pnas.0600447103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Salaun P, Rannou Y, Prigent C. Adv Exp Med Biol. 2008;617:41–56. doi: 10.1007/978-0-387-69080-3_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ishihara H, Martin BL, Brautigan DL, Karaki H, Ozaki H, Kato Y, Fusetani N, Watabe S, Hashimoto K, Uemura D, et al. Biochem Biophys Res Commun. 1989;159(3):871–877. doi: 10.1016/0006-291x(89)92189-x. [DOI] [PubMed] [Google Scholar]
- [34].Blethrow JD, Glavy JS, Morgan DO, Shokat KM. Proc Natl Acad Sci U S A. 2008;105(5):1442–1447. doi: 10.1073/pnas.0708966105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Moses AM, Heriche JK, Durbin R. Genome Biol. 2007;8(2):R23. doi: 10.1186/gb-2007-8-2-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M. Sci Signal. 2010;3(104) doi: 10.1126/scisignal.2000475. ra3. [DOI] [PubMed] [Google Scholar]
- [37].Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA. Sci Signal. 2011;4(179) doi: 10.1126/scisignal.2001497. rs5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chikumi H, Fukuhara S, Gutkind JS. J Biol Chem. 2002;277(14):12463–12473. doi: 10.1074/jbc.M108504200. [DOI] [PubMed] [Google Scholar]
- [39].Suzuki N, Nakamura S, Mano H, Kozasa T. Proc Natl Acad Sci U S A. 2003;100(2):733–738. doi: 10.1073/pnas.0234057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. Proc Natl Acad Sci U S A. 2008;105(31):10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]