

Abstract
The CXCR4 chemokine receptor plays an important role in cancer cell metastasis. The CXCR4 antagonist, AMD3100, has limited efficacy in controlling metastasis. HuR, an RNA-binding protein, regulates CXCR4 in cancer cells. We therefore investigated whether targeting HuR using a siRNA-based nanoparticle plus AMD3100 would suppress CXCR4 and inhibit lung cancer metastasis. We treated human H1299 lung cancer cell with HuR-specific siRNA contained in a folate-targeted lipid nanoparticle (HuR-FNP) plus AMD3100, and compared this with AMD3100 alone, HuR-FNP alone and no treatment. HuR-FNP plus AMD3100 treatment produced a G1 phase cell-cycle arrest and reduced cell viability above and beyond the effects of AMD3100 alone. HuR and CXCR4 mRNA and protein expression levels were markedly reduced in all treatment groups. Phosphorylated (p) AKTS473 protein was also reduced. P27 protein expression increased with HuR-FNP and combination treatment. Promoter-based reporter studies showed that the combination inhibited CXCR4 promoter activity more than did either treatment alone. Cell migration and invasion was significantly reduced with all treatment; the combination provided the most inhibition. Reduced matrix metalloprotease (MMP) -2 and -9 expression was associated with reduced invasion in all treatment groups. Thus, we found that combined HuR and CXCR4 targeting effectively controlled lung cancer metastasis.
Introduction
Lung cancer-related death is primarily due to disease recurrence and metastasis [1, 2]. Although molecularly-targeted therapies for lung cancer has demonstrated efficacy, the overall five-year survival of lung cancer patients continues to be dismal [1, 3]. Therefore, new and improved therapies that can effectively control metastasis will reduce the incidence of mortality and increase the disease-free survival of patients diagnosed with lung cancer.
Studies have shown that cellular signaling between the chemokine C-X-C receptor type 4 (CXCR4) and its ligand, the stromal cell derived factor (SDF)-1, also known as chemokine ligand (CXCL)-12, plays an important role in tumor growth, cell migration and invasion and metastases [4, 5]. High CXCR4 expression has previously been reported in several solid tumors, including non-small cell lung cancer (NSCLC) [6,7]. Further, the incidence of metastasis and increased risk of disease recurrence have been shown to be greater in high CXCR4-expressing lung tumors [8–11]. Results from these studies demonstrate that CXCR4 is a molecular target for cancer therapy and that suppressing the SDF-1/CXCR4 signaling axis will effectively inhibit tumor metastasis.
Preclinical studies have demonstrated that inhibiting CXCR4 or the SDF-1/CXCR4 signaling axis reduced tumor cell migration, invasion and metastasis [12–15]. Based on these results, a CXCR4 antagonist AMD3100 (Plerixafor, Mozobil™), is being tested as a cancer therapeutic for reducing metastasis [6–18]. Results from the studies have shown limited efficacy, warranting improved CXCR4-targeted therapeutic approaches [16–18].
HuR is an RNA binding protein that is ubiquitously expressed and belongs to the Drosophila embryonic lethal abnormal vision protein (ELAV) family [19]. HuR is a nucleo-cytoplasmic shuttling protein that binds to AU-rich (ARE) elements on the 3′ untranslated region of target messenger (m) RNAs and transports the mRNAs from the nucleus to the cytoplasm. Apart from shuttling the mRNA to the cytoplasm, HuR has been shown to play an important role in mRNA stability and protein translation [20]. mRNAs that have AREs at the 3′end and are targets of HuR include oncogenes, cytokines, chemokines, growth factors, and other factors that influence tumor cell growth, angiogenesis and metastasis [21–23]. Researchers have demonstrated high HuR expression in a variety of human cancers, including lung cancer [24–26]. Furthermore, high HuR expression in cancer tissues was correlated with metastasis, drug resistance, and poor survival [24–27]. More recently, CXCR4 was shown to be a target of, and regulated by HuR [28]. All of these reports suggest that HuR is a promising target for cancer therapy, and inhibiting HuR should produce anti-tumor and anti-metastatic effects.
In the present study, we investigated whether inhibiting HuR using a siRNA-based nanoparticle in combination with AMD3100 would effectively reduce CXCR4 and provide enhanced inhibition on lung cancer cell survival, cell migration and invasion.
Here, we demonstrate that combined targeting of HuR and CXCR4 has enhanced inhibitory effects on lung metastasis. Our results warrant further testing of the combination of HuR-FNP and AMD3100 as a means of controlling lung cancer metastasis.
Materials and Methods
Synthesis and characterization of nanoparticles
Cationic lipid nanoparticles (NPs) were synthesized using 20 mM DOTAP: cholesterol (Chol.) (Avanti Polar Lipids, Alabaster, AL) as previously described [29]. For preparation of DNA or siRNA containing NPs, DOTAP: Chol (20 mM) stock solution and DNA or siRNA solution diluted in 5% dextrose in water (D5W) were mixed in equal volumes to give a final concentration of 4 mM DOTAP:Chol- DNA (1μg) or siRNA (100nM). A stock solution of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[folate(polyethylene glycol)-5000 (Folate-PEG5000-DSPE) (17 μM; Avanti Polar Lipids) was synthesized by the thin film hydration method. Folate-PEG5000-DSPE was inserted into preformed DNA or siRNA containing NPs by the post insertion technique. Briefly, we mixed Folate-PEG5000-DSPE (0.03% of total lipid) with DNA or siRNA containing NP using vigorous pipetting, incubated the mixture at room temperature (RT) for 60 min. The NPs were then dialyzed against distilled water overnight at 4°C. The next day, the NP solution was removed from the dialysis bag, transferred to sterile 1-ml screw-cap tubes, and labeled as FNP. The FNP that contained HuR siRNA were labeled as HuR-FNP. Those containing control siRNA were labeled as C-FNP.
An aliquot of HuR-FNP and C-FNP was used to determine the particle size distribution and zeta potential using the Brookhaven ZetaPALS instrument as previously described [29, 30]. The shape and size of the NPs was analyzed with the transmission electron microscope (TEM) at the Oklahoma Medical Research Foundation (OMRF) core facility, as previously described [31].
Cell culture
Human non-small cell lung cancer cells (H1299) were cultured in RPMI-1640 medium (GIBCO) as previously described [32, 33]. The cell line was authenticated at the Genetic Resource Core Resource Facility, Johns Hopkins University, Baltimore, MD. In all experiments, untreated cells served as controls.
Cellular uptake of FNP
H1299 cells (1×105 cells/well) that express high levels of folate receptor were seeded in a six-well plate. The following day, cells were transfected with FNPs loaded with fluorescently labelled siRNA (siGLO, Dharmacon; 100 nM) in serum-free medium and were compared with cells transfected with non-targeted NPs loaded with the siRNA. After transfection, the culture media was replaced with 2% serum-containing medium. The cells were harvested at various time points after transfection. The cellular uptake of FNP and NP was determined quantitatively by Perkin Elmer EnVision® Multilabel Reader (Waltham, MA, USA) and was qualitatively evaluated using the Operetta imaging system (Perkin Elmer). The results were calculated and FNP uptake was expressed as percent increase over NP uptake.
To determine the receptor-mediated cellular uptake, cells seeded in two six-well plates were transfected with FNP containing Td/tomato plasmid DNA (1 μg). After transfection, one six-well plate was incubated at 37°C, while a second six-well plate was incubated at 4°C. The plates were removed at 1 h and 4 h after incubation. Cells were harvested and the fluorescence activity was determined quantitatively using flow-cytometry (FACS Calibur; Becton Dickinson, San Jose, CA).
In a separate set of experiments, cells seeded in six-well plates were transfected with FNP containing Td/tomato plasmid DNA (1 μg) and were grown in folic-acid-containing RPMI-1640 medium or in folic-acid-deficient RPMI-1640 medium. Cells that were not transfected and grown in folic-acid-containing RPMI-1640 medium served as controls. At 24 h after transfection, the cells were harvested and the fluorescence activity was determined using flow cytometry.
Cell viability assay
Cells (1 × 105 cells/well) seeded in six-well plates were treated with HuR-FNP (100 nM siRNA). At 6 h following treatment, the culture media was replaced with 2% serum-containing culture media and the cells were either treated with AMD3100 (100 ng/ml) or not treated. Incubation continued at 37°C, 5% CO2. The number of viable cells at 24 h and 48 h after treatment was determined using the trypan blue exclusion assay method as described previously [32, 33]. The results were expressed as percentage inhibition over untreated control.
Cell cycle analysis
Cells (1 × 105) seeded in six-well plates were treated with HuR-FNP (100 nM), AMD3100 (100 ng/ml), or a combination. Untreated cells served as controls. Cells were collected at 24 h and 48 h after treatment and were subjected to flow cytometric analysis as previously described [32, 33].
Quantitative RT-PCR assay
H1299 cells (1 × 105cells/well) seeded in six-well plates were treated with HuR-FNP (100 nM siRNA) in serum-free medium. At 6 h after treatment, the culture media was replaced with 2% serum-containing media, with or without AMD3100 (100 ng/ml). Incubation continued at 37°C, 5% CO2. Untreated cells served as controls. At 24 h and 48 h after treatment, the cells were harvested and used for RNA isolation. Total RNA was isolated using Trizol reagent (Life Technologies, Grand Island, NY), as previously described [34]. RNA quantity was analyzed using a Denovix DS11 spectrophotometer. Using a Quant script cDNA synthesis kit (Bio-Rad, Richmond, CA), 2 μg of total RNA was subjected to reverse transcription, as previously described [34]. Briefly, 3 μl of the synthesized complementary DNA (cDNA) was used to perform real-time (RT)-PCR (Bio-Rad CFX96™ Touch Real-Time PCR Detection System) using the premix iQ SYBR green QRT-PCR kit (Bio-Rad) with human HuR-specific oligonucleotide primers (Forward- 5′ ATGAAGACCACATGGCCGAAGACT 3′-Sense; Reverse - 5′ TGTGGTCATGAGTCCTTCCACGAT 3′-Antisense) and human CXCR4-specific oligonucleotide primers (Forward-5′CCACCATCTACTCCATCATCTTC 3′-Sense, Reverse-5′ACTTGTCCGTCATGCTTCTC3′-AntiSense; Integrated DNA Technologies, Coralville, IA, USA). Thermal cycling was programmed as follows: 95°C for 30 s, followed by 40 cycles of 95°C for 20 s, alternating with 62°C for 20s and 72°C for 20 s. The crossing threshold (Ct) value assessed by RT-PCR was noted for the transcripts and normalized with human GAPDH (Forward-5′AGCCTCAAGATCATCAGCAATGCC 3′ and Reverse - 5′ TGTGGTCATGAGTCCTTCCACGAT 3′). The changes in mRNA expression levels were expressed as fold change relative to control ± the standard deviation (SD). Each sample was run in triplicate. The experiments were repeated at least twice for reproducibility and statistical calculation.
Western blotting
Cells (1 × 105/well) seeded in six-well plates were trypsinized and collected at 24 h and/or at 48 h after HuR-FNP (100 nM), AMD3100 (100 ng/ml), or a combination. Cell lysates were prepared and protein samples were subjected to western blotting [32–34]. Primary antibodies against HuR, p27, MMP-2 (Santa Cruz Biotechnology, Dallas, TX), CXCR4 (Abcam, Cambridge, MA, USA), MMP-9, phospho-AKTS473 and total AKT (Cell Signaling Technology, Inc., Beverly, MA, USA) were purchased and used. Beta-actin (Sigma Chemicals, St. Louis, MO, USA) was used as an internal loading control. Proteins were detected using the appropriate secondary antibodies, as previously described [32–34]. Protein expression levels were quantified using GeneTools (Syngene) software.
Cell migration assay
Cells (5 × 104) suspended in 1 ml RPMI-1640 medium were seeded in the upper chambers (8 μm; BD Biosciences, Bedford, MA, USA ) and placed in a six-well plate filled with serum-free RPMI-1640 medium (lower chamber). After 24 h, the cells in the upper chamber were transfected with HuR-FNP in serum-free medium. After 6 h of transfection, the culture medium in the upper chamber was replaced with 2% serum-containing medium, with or without AMD3100 (100 ng/ml), while the lower chamber was replaced with 20% FBS-containing medium, and incubation continued. Cells that did not receive any treatment served as control. After incubation for 48 h, the inserts were removed, processed, and the number of migrated cells were counted using an inverted bright-field microscope as previously described [34–36].
Cell invasion assay
The cell invasion assay was performed using six-well Matrigel invasion chambers with 8-μm pore inserts (BD Biosciences). Cells (5 × 104) cells were seeded into the upper chambers. The lower chambers were filled with serum-free medium. After 24 h, the cells in the upper chamber were treated with HuR-FNP in serum-free medium. After 6 h of transfection, the culture medium in the upper chamber was replaced with 2% serum-containing medium with or without AMD3100 (100ng/ml), while the lower chamber was replaced with 20% FBS-containing medium and incubation continued. After an additional 48 h of incubation, the filters were removed, processed and the number of invaded cells counted as previously described [34–36]
CXCR4 promoter assay
H1299 cells (1 × 105cells/well) were transfected with 1 μg of reporter plasmid carrying the human CXCR4 promoter that drives the expression of Gaussia luciferase (GLuc) gene (GLuc–ON promotor reporter clone for CXCR4; GeneCopoeia, Rockville, MD). Following transfection, the cells were treated with C-FNP or HuR-FNP (100 nM siRNA) in serum-free medium. At 6 h after FNP treatment, the culture medium was replaced with 2% serum-containing medium, and incubation continued. After 24 h and 48 h of FNP treatment, 10 μl of the supernatant was taken out from each sample and the luciferase activity was measured using a Secrete pair Gaussia Luciferase Assay kit (GeneCopoeia) following the manufacturer’s protocol. The results from duplicate wells for each sample were calculated and represented as a percentage of luciferase expression [36].
To determine the ability of HuR-FNP to suppress CXCR4 promoter activity in the presence of SDF-1, cells (1 × 105cells/well) were transfected with CXCR4 promoter containing plasmid (1 μg; GLuc –ON plasmid), followed by treatment with HuR-FNP (100 nM siRNA) in serum-free medium. After 6 h of HuR-FNP treatment, the culture medium was replaced with 2% serum -containing medium and incubation continued overnight. The next day, SDF-1 (100 ng/ml) was added to the cells. Aliquots of culture supernatant were collected at 0.5 h, 1 h, 2 h, and 4 h after SDF-1 treatment and measured for luciferase activity.
For studies determining the inhibitory activity of the combination treatment of AMD3100 and HuR-FNP on CXCR4, cells were treated with AMD3100 (100 ng/ml), HuR-FNP (100 nM siRNA), or a combination, in the presence of CXCR4 promoter containing plasmid (1 μg; GLuc –ON plasmid). The luciferase activity was measured at 24 h and 48 h after treatment, as described above.
Statistical analysis
All experiments were performed at least twice. Unless otherwise stated, all data are shown as mean ± SD. Data were analyzed using two-tailed student’s t-test or one-way analysis of variance (ANOVA) using SPSS 12.0 (SPSS, Inc., Chicago, IL). P<0.05 was considered statistically significant.
Results
Physico-chemical characterization of the HuR-FNP
Physico-chemical characterization studies showed HuR-FNP was positively charged with a zeta potential of 4.3 mV, and 300±10 nm in size. Transmission electron microscopy (TEM) showed HuR-FNP was spherical in shape (Figure 1A). Next, we determined whether the siRNA was efficiently encapsulated and protected in the FNP. As shown in Figure 1B, agarose gel electrophoresis indicated that FNP efficiently encapsulated and protected the siRNA when compared with free siRNA that was used as a control.
Figure 1. Physico-chemical and functional characterization of HuR-FNP.


A, Transmission electron microscope (TEM) analysis showed that FNP were spherical in shape; B, Agarose gel electrophoresis showed that FNPs efficiently encapsulated and protected siRNA; lane 1, marker; lane 2, free siRNA; lane 3, siRNA encapsulated in NP; C, Upper panel, western blotting showing folate receptor expression in H1299 cells; KB cells served as positive control for folate receptor. Time-dependent uptake of siGlo-FNP was observed in folate-receptor positive H1299 cells; photograph panels on the right show siGlo-FNP uptake at 1 h and 4 h after transfection. D, FNP uptake was observed when siGLO-FNP-treated H1299 cells were incubated at 37°C. FNP uptake was reduced when the siGLO-FNP-treated cells were incubated at 4°C, indicating receptor-mediated entry of the nanoparticles; E, Increased fluorescence intensity of Td/tomato-FNP-treated cells grown in folic-acid-deficient tissue culture medium demonstrated specificity to folate receptor. Bars denote standard error (SE). Asterisk denote significance (p<0.05).
We next determined the siGLO-FNP uptake by folate receptor-positive H1299 lung cancer cells. A time-dependent increase in FNP uptake was observed in H1299 cells (p< 0.05; Figure 1C). That the FNP uptake was receptor-specific and was facilitated by receptor-mediated endocytosis was demonstrated by determining the fluorescence activity in siGLO-FNP-treated cells incubated at 37°C and 4°C. As shown in Figure 1D, the fluorescence activity increased over time when the FNP-treated cells were incubated at 37°C. In contrast, the fluorescence intensity was markedly reduced when the FNP-treated cells were incubated at 4°C. These results demonstrate that the FNP enters the cells by receptor-mediated endocytosis. To further test the specificity, we determined the uptake of FNP containing Td/tomato plasmid DNA in the presence and absence of folic-acid in the tissue culture medium. A significant increase in fluorescence was observed in FNP-treated cells growing in folic-acid-deficient RPMI-1640 culture medium compared with FNP-treated cells growing in folic-acid-containing RPMI-1640 culture medium (p<0.05; Figure 1E). The results demonstrate that our FNP are folate-targeted and can efficiently deliver plasmid DNA or siRNA into the cells.
HuR-FNP inhibits H1299 cancer cell proliferation, but AMD3100 does not
The inhibitory activity of HuR-FNP and AMD3100 on H1299 cell proliferation was evaluated using conventional cell viability assay. Both HuR-FNP and AMD3100 treatment produced inhibitory activity (p<0.02; Figure 2) at 24 h and 48 h when compared with untreated control. However, HuR-FNP treatment produced more inhibition than the AMD3100 treatment at the two tested time-points (p<0.001). However, the combination of HuR-FNP and AMD3100 yielded no significant increase in the inhibition compared with HuR-FNP treatment alone.
Figure 2. HuR-FNP and HuR-FNP plus AMD3100 inhibit cell proliferation.

H1299 cells were treated with AMD3100, HuR-FNP, or a combination. Cell viability was determined at 24 h and 48 h. Untreated cells served as controls. A significant inhibition in cell proliferation was observed in all three treatment groups compared with control (p<0.05). However, HuR-FNP and HuR-FNP plus AMD3100 showed the highest inhibition. No significant difference was observed between HuR-FNP treatment and HuR-FNP plus AMD3100 treatment. Bars denote standard deviation (SD).
We next determined whether the FNP and AMD3100 treatments affected the target mRNA and protein expression. As shown in Figure 3A, CXCR4 mRNA expression was significantly reduced in all treatment groups at 24 h and 48 h, compared with control (p<0.002). HuR mRNA expression was also reduced all treatment groups at 24 h and 48 h, compared with control (p<0.02; Figure 3A). HuR-FNP and HuR-FNP plus AMD3100 demonstrated highest inhibition compared with control However, there was no significant difference (p=0.98) in the HuR mRNA inhibition between HuR-FNP and HuR-FNP plus AMD3100 at 24 h. Surprisingly, there was increased and significant suppression of HuR mRNA expression with combination treatment compared with HuR-FNP treatment at 48 h (p=0.01).
Figure 3. HuR-FNP plus AMD3100 treatment reduces HuR and CXCR4 mRNA and protein expression.

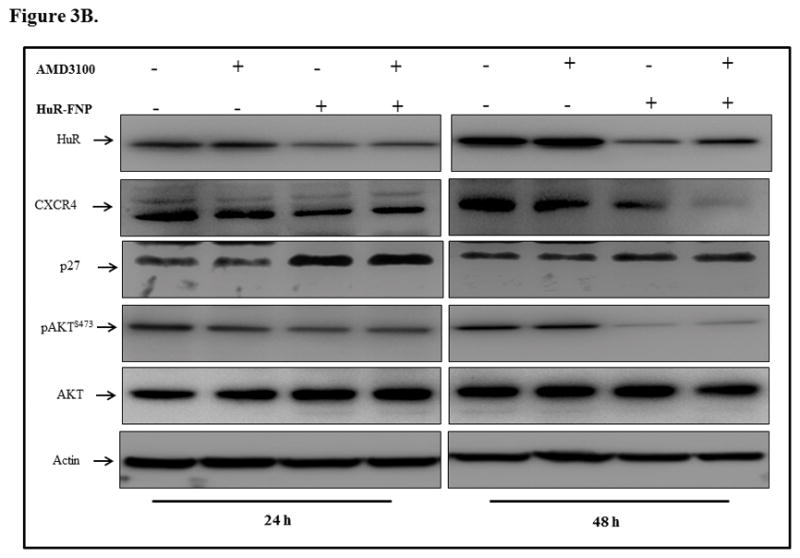

H1299 cells were treated with AMD3100, HuR-FNP, or a combination. Cells were harvested at 24 h and 48 h after treatment and analyzed for mRNA and protein expression using quantitative RT-PCR and western blot analysis, respectively. A, CXCR4 mRNA was markedly reduced in all three treatment groups, with the greatest reduction observed with HuR-FNP treatment (p< 0.05). HuR mRNA was significantly reduced with HuR-FNP and HuR-FNP plus AMD3100 treatments (p< 0.05). HuR mRNA levels were reduced in the AMD3100 group, but this reduction was not significant compared with control. B & C, Western blotting showed that all three treatments reduced CXCR4 protein expression, while only HuR-FNP and HuR-FNP plus AMD3100 reduced HuR protein expression. A slight reduction in HuR expression was observed in the AMD3100 treatment group, but this was not significant compared with control. Change in p27 and phosphorylated (p)AKTS473 expression was also observed in all three treatments compared with controls. Asterisk denote significance (p<0.05).
Since AMD3100, HuR-FNP, and the HuR-FNP and AMD3100 combination reduced CXCR4 and HuR mRNA expression, we next examined whether protein expression was also altered by the treatments. HuR-FNP treatment reduced HuR and CXCR4 protein expression at the two tested time points (p<0.01). AMD3100 treatment reduced CXCR4 protein expression at 48 h (p<0.01) but not at 24 h (p=0.21) and did not demonstrate significant reduction of HuR (p=0.21; Figure 3B, C). HuR-FNP plus AMD3100 treatment also reduced CXCR4 expression at 24 h and 48 h (p<0.01). However, the reduction in CXCR4 expression was greatest at 48 h with the combination treatment compared with individual treatments (p<0.001). HuR expression, although reduced was slightly lower after the combination treatment than observed following HuR-FNP alone (Figure 3B, C). Analysis for molecular downstream targets of HuR and CXCR4 showed a marked increase in p27 expression and reduced expression of phosphorylated (p) AKTS473in HuR-FNP, and HuR-FNP plus AMD3100 treatment groups compared with control (p<0.001; Figure 3B, C). In AMD3100 treatment group, no significant change in p27 expression was observed compared with control (p>0.5). However, pAKTS473 expression was reduced in AMD3100 treatment group compared with control (p<0.05; Figure 3B, C).
HuR-FNP alone and in combination with AMD3100 induce cell cycle arrest in the G1 phase
To determine whether HuR-FNP-, AMD3100-, and HuR-FNP plus AMD3100 altered the cell-cycle phases, cells were subjected to flow-cytometric analysis. HuR-FNP and HuR-FNP plus AMD3100 markedly induced cell cycle arrest in the G1 phase at 24 h and 48 h after treatment (Figure 4). AMD3100 treatment alone produced a slight increase in the G1 phase compared with untreated control, but this difference was not statistically significant.
Figure 4. HuR-FNP plus AMD3100 treatment induces G1 phase cell-cycle arrest.

H1299 cells were treated with AMD3100, HuR-FNP, or a combination. The cells were harvested at 24 h and 48 h after treatment and were analyzed for cell-cycle phases using flow cytometry. Untreated cells served as controls. HuR-FNP and HuR-FNP plus AMD3100 treatment induced G1 phase cell-cycle arrest at both time points. AMD3100 treatment alone showed no increase in G1 phase cell-cycle arrest compared with controls.
HuR-FNP and AMD3100 suppress CXCR4 at the promoter level
Since HuR-FNP and AMD3100 treatment suppressed CXCR4 mRNA and protein expression levels, we next used a CXCR4 promoter-based reporter plasmid to investigate whether the inhibition occurred at the promoter level. First, we determined the specificity of HuR-FNP inhibitory activity by comparing with an FNP-carrying control siRNA (C-FNP). HuR-FNP, but not C-FNP, significantly reduced luciferase expression at 24 h and 48 h after treatment compared with control (p<0.05; Figure 5A). Next, we tested the ability of HuR-FNP to inhibit CXCR4 in the presence of SDF-1. HuR-FNP significantly inhibited CXCR4 expression in both the presence and absence of SDF-1 at all tested-time points, compared with cells that expressed CXCR4 and were stimulated with SDF-1 (p<0.05; Figure 5B). These results showed that HuRsiRNA specifically inhibited CXCR4 expression and could override SDF-1-mediated CXCR4 activation. Finally, we tested the inhibitory activity of the combination of HuR-FNP and AMD3100, and compared this combination with AMD3100 and HuR-FNP alone (Figure 5C). CXCR4 activity was suppressed by all treatment groups compared with control. However, maximal inhibition was observed with the combination of HuR-FNP and AMD3100 (p<0.05), followed by HuR-FNP alone (p<0.05) and AMD3100 alone (p<0.05) when compared with control. In summary, our results demonstrated that both AMD3100 and HuR-FNP can inhibit CXCR4. However, combining HuR-FNP with AMD3100 provided the most inhibition of CXCR4.
Figure 5. HuR-FNP plus AMD3100 treatment suppress CXCR-4 at the promoter level.

A, H1299 cells were transfected with CXCR-4 promoter-based reporter plasmid (pCXCR4), which drives luciferase expression, followed by treatment with HuR-FNP or control (C)-FNP. Untreated cells served as controls. Analysis for luciferase expression showed that HuR-FNP significantly reduced luciferase expression compared with C-FNP and untreated controls (p< 0.05). B, pCXCR4-transfected cells were treated with HuR-FNP in the presence of SDF, and luciferase activity was determined at different time points. HuR-FNP significantly inhibited luciferase expression in the presence and absence of SDF at all tested-time points (p< 0.05). C, pCXCR4- transfected cells were treated with AMD3100, HuR-FNP, or HuR-FNP plus AMD3100. pCXCR4- transfected cells that did not receive any additional treatment served as controls. Luciferase activity was significantly inhibited by all three treatments at both 24 h and 48 h. However, the combination treatment produced the greatest inhibition (p< 0.05). Asterisk denote significance (p<0.05).
AMD3100, HuR-FNP, and HuR-FNP plus AMD3100 inhibit cell migration and invasion
CXCR4 has previously been shown to play an important role in cancer cell migration and invasion (13–15). We therefore investigated the effect(s) of AMD3100, HuR-FNP, and HuR-FNP plus AMD3100 on cell migration and invasion. All three treatment groups significantly inhibited cell migration at the two time points tested with the greatest inhibition observed in the combination treatment group (p< 0.05; Figure 6A). Similarly, cell invasion studies showed that HuR-FNP plus AMD3100 produced the most inhibition of cell invasion (p<0.05; Figure 6B). Both, AMD3100- and HuR-FNP-treatments also significantly inhibited cell invasion compared with control (p<0.05). However, there was no marked difference between these two treatment groups.
Figure 6. HuR-FNP plus AMD3100 treatment inhibits cell migration and invasion.


H1299 cells were treated with AMD3100, HuR-FNP, or a combination and were observed for cell migration and invasion. Untreated cells served as controls. A, Tumor cell migration and B, invasion was significantly inhibited by all three treatments compared with controls (p< 0.05). However, combination treatment produced a greater inhibitory effect on both cell migration and invasion than any individual treatment. Bars denote standard deviation (SD). Asterisk denote significance (p<0.05).
Since matrix metalloproteases (MMPs) are known to regulate cell invasion, and MMP-2 and-9 are highly expressed in lung cancer cells (37, 38), we analyzed for the expression of these proteins at 48 h after treatment. As shown in Figure 7A and B, MMP-2 and -9 expression was reduced in all three treatment groups compared with control. AMD3100 and HuR-FNP produced comparable MMP-9 inhibition which was greater than that observed with HuR-FNP plus AMD3100 combination. In contrast, HuR-FNP plus AMD3100 produced the highest inhibition of MMP-2, when compared with AMD3100 or HuR-FNP alone (Figure 7A, B). These results demonstrate that the inhibition on cell migration and invasion exerted by the three treatment is partly due to suppression of MMP-2 and -9.
Figure 7. HuR-FNP plus AMD3100 treatment inhibits matrix metalloproteases.

H1299 cells were treated with AMD3100, HuR-FNP, or a combination. Cell lysates were analyzed for MMP-2 and MMP-9 using western blotting. Untreated cells served as controls. A, Western blotting showing protein expression and B, semi-quantitative analysis showing the change in MMP-2 and -9 expression in different treatment groups. MMP-2 was significantly reduced in all three treatment groups compared with controls (p< 0.05). However, the greatest inhibition was observed with the combination treatment, and was significant compared with individual treatment (p< 0.05). MMP-9 levels were reduced in all three treatment groups compared with controls (p< 0.05). However, there was no significant difference among the three treatment groups. Bars denote standard deviation (SD). Asterisk denote significance (p<0.05).
Discussion
Lung cancer-related deaths are primarily due to metastasis. The CXCR4 chemokine receptor plays an important role in cancer metastasis and is highly expressed in lung cancer [4, 5]. Thus, inhibiting CXCR4 interaction with its ligand SDF-1 is likely to reduce metastasis [12–15]. Among the several CXCR4-targeted drugs that are being developed and tested, only AMD3100 has been approved by the FDA for cancer treatment [16–18]. Studies using AMD3100 have shown that the SDF-1/CXCR4 axis can be effectively inhibited, resulting in anti-metastatic activity [39, 40]. However, clinical study results indicate that AMD3100 is not effective in controlling tumor metastasis, which warrants testing of innovative treatment approaches such as combination therapy that can effectively inhibit CXCR4 and reduce metastasis.
In the present study, we tested the combined inhibitory activity of HuR-FNP and AMD3100 on lung cancer growth, migration, and invasion. The rationale to target HuR was based on the report that showed that HuR regulated CXCR4 [41]. Thus, we hypothesized that suppressing HuR using a siRNA nanoparticle-based approach in combination with AMD3100 should produce greater inhibition on lung cancer metastasis. Furthermore, using a tumor-targeted nanoparticle would offer selectivity and reduce cytotoxicity to normal cells [42, 43]. Our present study showed that HuR-FNP and HuR-FNP plus AMD3100 markedly inhibited cell viability and induced G1 phase cell-cycle arrest compared with AMD3100 treatment alone (p<0.05). However, the inhibition produced by the combination treatment was comparable to that of HuR-FNP treatment alone. This observation is not surprising. A plausible explanation is that the inhibitory activity observed with the combination treatment is primarily due to HuR inhibition, as silencing HuR has previously been shown to inhibit cell proliferation and induce G1 arrest [42, 43]. AMD3100 is not known to produce direct cytotoxic activity. Molecular studies showed that all three treatments reduced HuR and CXCR4 protein expression to varying degree (p<0.05). Further, expressions of p27 and pAKTS473 proteins, which are downstream targets of HuR and CXCR4 respectively, were also regulated in all three treatments demonstrating that the individual and combination treatments effectively inhibited the downstream signaling that support cell survival.
HuR-FNP plus AMD3100, when tested in cell migration and invasion assays, demonstrated a profound inhibitory activity that was greater than that observed with individual treatments. Molecular studies indicated that the enhanced inhibition on cell migration and invasion was likely due to greater inhibition of CXCR4 promoter activity and reduced MMP-2 and MMP-9 expression. Our data are consistent with previous reports in which HuR silencing was shown to inhibit CXCR4 and MMPs [41, 44]. Similarly, AMD3100 has been demonstrated to inhibit AKT and CXCR4 expression [40, 45]. Our data also indicated that both HuR-FNP and AMD3100 inhibited the promoter activity of CXCR4. Thus, the inhibition of CXCR4, AKT, and MMPs that was produced by the combination of HuR-FNP and AMD3100 explains the significant reduction in cell migration and invasion. Our results clearly demonstrate that combination therapy for the treatment of metastatic lung cancer is likely to be more effective than individual treatments. However, additional combination therapy-based in-vitro and in-vivo studies are warranted to determine their efficacy in controlling metastasis.
In conclusion, we have demonstrated that HuR-targeted nanotherapy combined with AMD3100 produced an enhanced suppression of cell growth, migration, and invasion in lung cancer, and is an attractive treatment approach to control cancer metastasis.
Acknowledgments
The study was supported in part by a grant received from the National Institutes of Health (NIH) R01 CA167516, an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences (P20 GM103639) of the National Institutes of Health, and by funds received from the Jim and Christy Everest Endowed Chair in Cancer Developmental Therapeutics, the University of Oklahoma Health Sciences Center. We thank the Stephenson Cancer Center at the University of Oklahoma Health Sciences Center, Oklahoma City, OK, for the use of the cancer Functional Genomics Core and Biostatistics Core, which provided molecular analysis and statistical services. The authors thank Ms. Kathy Kyler at the office of Vice President of Research, OUHSC for editorial assistance. Rajagopal Ramesh is an Oklahoma TSET Research Scholar and holds the Jim and Christy Everest Endowed Chair in Cancer Developmental Therapeutics.
Footnotes
Author contributions
Conducted the experiments: RM, and JP; Synthesized and characterized the nanoparticle (HuR-FNP and C-FNP): RM; Conceived and designed the experiments: RM, JP, AC, YDZ, AM, and RR.; Wrote the paper: RM and RR.; Performed statistical analysis: AC, YDZ; Data collection, analysis and interpretation: RM, JP, AC, YDZ, AM, and RR; Critically reviewed, revised for intellectual content, and provided suggestions: RM, JP, AC, YDZ, AM, RR.; Supervised the project: RR.
Conflict of interest
The authors report no conflicts of interest.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Liu L, Zhao E, Li C, Huang L, Xiao L, Cheng L, et al. TRIM28, a new molecular marker predicting metastasis and survival in early-stage non-small cell lung cancer. Cancer Epidemiol. 2013;37:71–78. doi: 10.1016/j.canep.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 3.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 4.Phillips RJ, Burdick MD, Lutz M, Belperio JA, Keane MP, Streiter RM, et al. The stromal derived factor-1/CXCL12-CXC chemokine receptor 4 biological axis in non-small cell lung cancer metastases. Am J Respir Crit Care Med. 2003;167:1676–1686. doi: 10.1164/rccm.200301-071OC. [DOI] [PubMed] [Google Scholar]
- 5.Vandercappellen J, Van Damme J, Struyf S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 2008;267:226–244. doi: 10.1016/j.canlet.2008.04.050. [DOI] [PubMed] [Google Scholar]
- 6.Su L, Zhang J, Xu H, Wang Y, Chu Y, Liu R, et al. Differential expression of CXCR4 is associated with the metastatic potential of human non-small cell lung cancer cells. Clin Cancer Res. 2005;11:8273–8280. doi: 10.1158/1078-0432.CCR-05-0537. [DOI] [PubMed] [Google Scholar]
- 7.Oonakahara K, Matsuyama W, Higashimoto I, Kawabata M, Arimura K, Osame M, et al. Stromal-derived factor-1alpha/CXCL12-CXCR 4 axis is involved in the dissemination of NSCLC cells into pleural space. Am J Respir Cell Mol Biol. 2004;30:671–677. doi: 10.1165/rcmb.2003-0340OC. [DOI] [PubMed] [Google Scholar]
- 8.Minamiya Y, Saito H, Takahashi N, Ito M, Imai K, Ono T, et al. Expression of the chemokine receptor CXCR4 correlates with a favorable prognosis in patients with adenocarcinoma of the lung. Lung Cancer. 2010;68:466–471. doi: 10.1016/j.lungcan.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 9.Wagner PL, Hyjek E, Vazquez MF, Meherally D, Liu YF, Chadwick PA, et al. CXCL12 and CXCR4 in adenocarcinoma of the lung: association with metastasis and survival. J Thorac Cardiovasc Surg. 2009;137:615–621. doi: 10.1016/j.jtcvs.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 10.Na IK, Scheibenbogen C, Adam C, Stroux A, Ghadjar P, Thiel E, et al. Nuclear expression of CXCR4 in tumor cells of non-small cell lung cancer is correlated with lymph node metastasis. Hum Pathol. 2008;39:1751–1755. doi: 10.1016/j.humpath.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 11.Otsuka S, Klimowicz AC, Kopciuk K, Petrillo SK, Konno M, Hao D, et al. CXCR4 overexpression is associated with poor outcome in females diagnosed with stage IV non-small cell lung cancer. J Thorac Oncol. 2011;6:1169–1178. doi: 10.1097/JTO.0b013e3182199a99. [DOI] [PubMed] [Google Scholar]
- 12.Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16:2927–2931. doi: 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- 13.D’Alterio C, Barbieri A, Portella L, Palma G, Polimeno M, Riccio A, et al. Inhibition of stromal CXCR4 impairs development of lung metastases. Cancer Immunol Immunother. 2012;61:1713–1720. doi: 10.1007/s00262-012-1223-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uchida D, Onoue T, Kuribayashi N, Tomizuka Y, Tamatani T, Nagai H, et al. Blockade of CXCR4 in oral squamous cell carcinoma inhibits lymph node metastases. Eur J Cancer. 2011;147:452–459. doi: 10.1016/j.ejca.2010.09.028. [DOI] [PubMed] [Google Scholar]
- 15.Ramsey DM, McAlpine SR. Halting metastasis through CXCR4 inhibition. Bioorg Med Chem Lett. 2013;23:20–25. doi: 10.1016/j.bmcl.2012.10.138. [DOI] [PubMed] [Google Scholar]
- 16.Uy GL, Rettig MP, Motabi IH, McFarland K, Trinkaus KM, Hladnik LM, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119:3917–3924. doi: 10.1182/blood-2011-10-383406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nervi B, Ramirez P, Rettig MP, Uy GL, Holt MS, Ritchey JK, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113:6206–6214. doi: 10.1182/blood-2008-06-162123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peled A, Wald O, Burger J. Development of novel CXCR4-based therapeutics. Expert Opin Investig Drugs. 2012;21:341–353. doi: 10.1517/13543784.2012.656197. [DOI] [PubMed] [Google Scholar]
- 19.Srikantan S, Gorospe M. HuR function in disease. Front Biosci (Landmark Ed) 2012;17:189–205. doi: 10.2741/3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abdelmohsen K, Gorospe M. Post transcriptional regulation of cancer traits by HuR. Wiley Interdiscip Rev RNA. 2010;1:214–229. doi: 10.1002/wrna.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Guo Y, Chu H, Guan Y, Bi Ji, Wang B. Multiple Functions of the RNA-Binding Protein HuR in Cancer Progression, Treatment Responses and Prognosis. Int J Mol Sci. 2013;14:10015–10041. doi: 10.3390/ijms140510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.López de Silanes I, Lal A, Gorospe M. HuR: post-transcriptional paths to malignancy. RNA Biol. 2005;2:11–13. doi: 10.4161/rna.2.1.1552. [DOI] [PubMed] [Google Scholar]
- 23.Wang WG, Caldwell MC, Lin SK, Furneaux H, Gorospe M. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J. 2000;19:2340–2350. doi: 10.1093/emboj/19.10.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lauriola L, Granone P, Ramella S, Lanza P, Ranelletti FO. Expression of the RNA-binding protein HuR and its clinical significance in human stage I and II lung adenocarcinoma. Histol Histopathol. 2012;27:617–626. doi: 10.14670/HH-27.617. [DOI] [PubMed] [Google Scholar]
- 25.Denkert C, Koch I, von Keyserlingk N, Noske A, Niesporek S, Dietel M, et al. Expression of the ELAV-like protein HuR in human colon cancer: association with tumor stage and cyclooxygenase-2. Mod Pathol. 2006;19:1261–1269. doi: 10.1038/modpathol.3800645. [DOI] [PubMed] [Google Scholar]
- 26.Niesporek S, Kristiansen G, Thoma A, Weichert W, Noske A, Buckendahl AC, et al. Expression of the ELAV-like protein HuR in human prostate carcinoma is an indicator of disease relapse and linked to COX-2 expression. Int J Oncol. 2008;32:341–347. [PubMed] [Google Scholar]
- 27.Wang D, Wang M, Hu C, Shuang T, Zhou Y, Yan X. Expression of the ELAV-like protein HuR in the cytoplasm is associated with endometrial carcinoma progression. Tumour Biol. 2014;35:11939–11947. doi: 10.1007/s13277-014-2485-9. [DOI] [PubMed] [Google Scholar]
- 28.Al-Souhibani N, Al-Ghamdi M, Al-Ahmadi W, Khabar KS. Posttranscriptional control of the chemokine receptor CXCR4 expression in cancer cells. Carcinogenesis. 2014;35:1983–1992. doi: 10.1093/carcin/bgu080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramesh R, Saeki T, Templeton NS, Ji L, Stephens LC, Ito I, et al. Successful treatment of primary and disseminated human lung cancers by systemic delivery of tumor suppressor genes using an improved liposome vector. Mol Ther. 2001;3:337–350. doi: 10.1006/mthe.2001.0266. [DOI] [PubMed] [Google Scholar]
- 30.Began G, Ito I, Branch CD, Clifton Stephens L, Roth JA, Ramesh R. Nanoparticle based systemic gene therapy for lung cancer: Molecular mechanisms, and strategies to suppress nanoparticle-mediated inflammatory response. Technol Cancer Res Treat. 2004;3:647–657. doi: 10.1177/153303460400300615. [DOI] [PubMed] [Google Scholar]
- 31.Babu A, Wang Q, Muralidharan R, Shanker M, Munshi A, Ramesh R. Chitosan coated poly(lactic acid) polymeric nanoparticle-mediated combinatorial delivery of cisplatin and siRNA/plasmid DNA chemosensitizes cisplatin-resistant human ovarian cancer cells. Mol Pharm. 2014;11:2720–2733. doi: 10.1021/mp500259e. [DOI] [PubMed] [Google Scholar]
- 32.Saeki T, Mhashilkar A, Chada S, Branch C, Roth JA, Ramesh R. Tumor-suppressive effects by adenovirus-mediated mda-7 gene transfer in non-small cell lung cancer cell in vitro. Gene Ther. 2000;7:2051–2057. doi: 10.1038/sj.gt.3301330. [DOI] [PubMed] [Google Scholar]
- 33.Ramesh R, Mhashilkar AM, Tanaka F, Saito Y, Branch CD, Sieger K, et al. Melanoma differentiation-associated gene 7/interleukin (IL)-24 is a novel ligand that regulates angiogenesis via the IL-22 receptor. Cancer Res. 2003;63:5105–5113. [PubMed] [Google Scholar]
- 34.Panneerselvam J, Shanker M, Jin J, Branch CD, Muralidharan R, Zhao DY, et al. Phosphorylation of interleukin (IL)-24 is required for mediating its anti-cancer activity. Oncotarget. 2015;18:16271–16286. doi: 10.18632/oncotarget.3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramesh R, Ito I, Gopalan B, Saito Y, Mhashilkar AM, Chada S. Ectopic production of MDA-7/IL-24 inhibits invasion and migration of human lung cancer cells. Mol Ther. 2004;9:510–518. doi: 10.1016/j.ymthe.2004.01.019. [DOI] [PubMed] [Google Scholar]
- 36.Panneerselvam J, Jin J, Shanker M, Lauderdale J, Bates J, Wang Q, et al. Disruption of the CXCR-4/SDF axis by interleukin (IL)-24 reduces tumor cell migration and invasion in lung cancer cells. PLoS One. 2015;10:e0122439. doi: 10.1371/journal.pone.0122439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu J, Ping W, Zu Y, Sun W. Correlations of lysyl oxidase with MMP2/MMP9 expression and its prognostic value in non-small cell lung cancer. Int J Clin Exp Pathol. 2014;7:6040–6047. [PMC free article] [PubMed] [Google Scholar]
- 38.Pritchard SC, Nicolson MC, Lloret C, McKay JA, Ross VG, Kerr KM, et al. Expression of matrix metalloproteinases 1, 2, 9 and their tissue inhibitors in stage II non-small cell lung cancer: implications for MMP inhibition therapy. Oncol Rep. 2001;8:421–424. [PubMed] [Google Scholar]
- 39.Darash-Yahana M, Pikarsky E, Abramovitch R, Zeira E, Pal B, Karplus R, et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 2004;18:1240–1242. doi: 10.1096/fj.03-0935fje. [DOI] [PubMed] [Google Scholar]
- 40.Chinni SR, Sivalogan S, Dong Z, Filho JC, Deng X, Bonfil RD, et al. CXCL12/CXCR4 signaling activates Akt-1 and MMP-9 expression in prostate cancer cells: the role of bone microenvironment-associated CXCL12. Prostate. 2006;66:32–48. doi: 10.1002/pros.20318. [DOI] [PubMed] [Google Scholar]
- 41.Al-Souhibani N, Al-Ghamdi M, Al-Ahmadi W, Khabar KS. Posttranscriptional control of the chemokine receptor CXCR4 expression in cancer cells. Carcinogenesis. 2014;35:1983–1992. doi: 10.1093/carcin/bgu080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muralidharan R, Babu A, Basalingappa K, Mehta M, Munshi A, Ramesh R. Designing of tumor-targeted HuRsiRNA nanoparticle as a therapeutic for lung cancer. In: Gandhi V, Mehta K, Grover RK, Pathak S, Aggarwal BB, editors. Multitargeted approach to the treatment of cancer. Springer International Publishing; Switzerland: 2015. pp. 277–294. [Google Scholar]
- 43.Babu A, Amreddy N, Muralidharan R, Munshi A, Ramesh R. Nanoparticle-mediated siRNA delivery for lung cancer treatment. In: Naik J, editor. Nano based drug delivery. IAPC Publishing; Croatia: 2015. pp. 195–215. [Google Scholar]
- 44.Akool el-S, Kleinert H, Hamada FM, Abdelwahab MH, Förstermann U, Pfeilschifter J, et al. Nitric oxide increases the decay of matrix metalloproteinase 9 mRNA by inhibiting the expression of mRNA-stabilizing factor HuR. Mol Cell Biol. 2003;23:4901–4916. doi: 10.1128/MCB.23.14.4901-4916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen H, Xu X, Teng J, Cheng S, Bunjhoo H, Cao Y, et al. CXCR4 inhibitor attenuates allergen-induced lung inflammation by down-regulating MMP-9 and ERK1/2. Int J Clin Exp Pathol. 2015;8:6700–6777. [PMC free article] [PubMed] [Google Scholar]