

Abstract
Ischemia induces extensive temporal changes in cerebral transcriptome that influences the neurologic outcome after stroke. In addition to protein-coding RNAs, many classes of noncoding RNAs, including long noncoding RNAs (LncRNAs), also undergo changes in the poststroke brain. We currently evaluated the functional significance of an LncRNA called Fos downstream transcript (FosDT) that is cogenic with Fos gene. Following transient middle cerebral artery occlusion (MCAO) in adult rats, expression of FosDT and Fos was induced. FosDT knockdown significantly ameliorated the postischemic motor deficits and reduced the infarct volume. Focal ischemia also increased FosDT binding to chromatin-modifying proteins (CMPs) Sin3a and coREST (corepressors of the transcription factor REST). Furthermore, FosDT knockdown derepressed REST-downstream genes GRIA2, NFκB2, and GRIN1 in the postischemic brain. Thus, FosDT induction and its interactions with REST-associated CMPs, and the resulting regulation of REST-downstream genes might modulate ischemic brain damage. LncRNAs, such as FosDT, can be therapeutically targeted to minimize poststroke brain damage.
SIGNIFICANCE STATEMENT Mammalian brain is abundantly enriched with long noncoding RNAs (LncRNAs). Functional roles of LncRNAs in normal and pathological states are not yet understood. This study identified that LncRNA FosDT induced after transient focal ischemia modulates poststroke behavioral deficits and brain damage. These effects of FosDT in part are due to its interactions with chromatin-modifying proteins Sin3a and coREST (corepressors of the transcription factor REST) and subsequent derepression of REST-downstream genes GRIA2, NFκB2, and GRIN1. Therefore, LncRNA-mediated epigenetic remodeling could determine stroke outcome.
Keywords: brain, gene expression, neuroprotection, noncoding RNA, stroke, transcriptional regulation
Introduction
Mammalian genomes are replete with thousands of genes that transcribe long noncoding RNAs (LncRNAs; >200 nt in length) from intergenic or intragenic locations (Xie et al., 2014). LncRNAs are thought to be crucial regulators of genomic imprinting, chromatin remodeling, transcription, and cell cycle, and their expression is spatially and temporally restricted to cell types and stages of development (Wapinski and Chang, 2011; Clark and Blackshaw, 2014; Maass et al., 2014). Although their function and mechanism of action are not clear, perturbations in LncRNA expression have been implicated in many diseases, including Alzheimer's disease, heart pathology, and multiple forms of cancer (Wahlestedt, 2013; Han et al., 2014; Yarmishyn and Kurochkin, 2015).
We recently showed that focal ischemia significantly alters the expression of many LncRNAs in adult rat brain (Dharap et al., 2012a). However, the significance of these changes to postischemic outcome is not known. To fill this void, we currently analyzed the role of an LncRNA called Fos downstream transcript (FosDT) after transient middle cerebral artery occlusion (MCAO) in adult rats. The rationale for selecting FosDT is that: (1) it was observed to be highly upregulated during the acute period after focal ischemia (Dharap et al., 2012a); (2) it is cogenic to Fos gene and rapid induction of Fos after brain injury is a marker of cellular stress (Shimazu et al., 1994); and (3) it binds to chromatin-modifying proteins (CMPs) Sin3 transcription regulatory family member a (Sin3a) and co-RE-1-silencing transcription factor (coREST) that are corepressors of the transcription factor REST, which is known to be induced after stroke (Noh et al., 2012; Dharap et al., 2013). REST plays a crucial role in apoptosis and cell differentiation during embryogenesis, and regulates neuronal function in the adult brain (Calderone et al., 2003; Noh et al., 2012; Baldelli and Meldolesi, 2015). REST complex (REST-coREST-Sin3a) has been shown to increase ischemic brain damage by modulating its downstream genes AMPA receptor subunit GluR2 (GRIA2), NFκ L chain enhancer of activated B cells 2 (NFκB2), and glutamate receptor, ionotropic, N-methyl d-aspartate 1 (GRIN1) (Calderone et al., 2003; Noh et al., 2012). We presently evaluated whether FosDT induction is a promoter of ischemic brain damage and if, yes, its mechanism of action is by modulating the REST-downstream gene expression.
Materials and Methods
Focal ischemia.
All the surgical procedures were approved by the Research Animal Resources and Care Committee of the University of Wisconsin-Madison, and the animals were cared for in accordance with the Guide for the Care and Use of Laboratory Animals (U.S. Department of Health and Human Services Publication 86-23, revised). All surgeries were performed under isoflurane anesthesia, and all efforts were made to minimize suffering. Transient MCAO of 1 h was induced in adult, male, spontaneously hypertensive rats (SHR; 280–300 g; Charles River) by an intraluminal suture method as described previously (Dharap et al., 2011, 2012a). Sham-operated rats served as control. Regional cerebral blood flow and physiological parameters (pH, Pao2, Paco2, hemoglobin, and blood glucose) were monitored, and rectal temperature was maintained at 37.0 ± 0.5°C during surgery.
FosDT knockdown.
Three in vivo grade Silencer Select siRNAs targeting nonoverlapping regions of FosDT and nontargeting negative control siRNAs (Invitrogen) were injected intracerebrally as described previously (Satriotomo et al., 2006; Pandi et al., 2013). In brief, siRNA mix (8 nmol in 6 μl buffer + 2 μl invivofectamine) was incubated at room temperature for 15 min and injected with a Hamilton syringe (1 μl/5 min) into cerebral cortex (bregma; −0.2 mm posterior, 3 mm dorsoventral, 4.5 mm lateral) (Paxinos and Watson, 1998).
Motor function and infarct volume estimation.
Neurobehavioral deficits were determined by rotarod test, beam walk test, and adhesive removal test at 1, 3, 5, and 7 d of reperfusion (Schaar et al., 2010; Nakka et al., 2011). Rats were killed on day 7, and infarct volume was measured by National Institutes of Health ImageJ software using cresyl violet-stained serial coronal sections (6 sections/rat; 40 μm thick at an interval of 320 μm) as described previously (Tureyen et al., 2008; Dharap et al., 2009; Nakka et al., 2011). Injury volumes were corrected for edema and differential shrinkage using the Swanson formula (Swanson et al., 1990). Animals were randomly assigned to groups, and behavioral and histological analyses were performed by an investigator blinded to the study groups.
Real-time PCR.
Expression of FosDT (MRAK159688), Fos (NM_022197), REST (NM_031788.1), coREST (NM_001013994.1), Sin3a (NM_001108761.1), MeCP2 (NM_022673.2), GRIA2 (NM_017261.2), NFkB2 (NM_001008349.1), and GRIN1 (NM_017010.2) was evaluated with real-time PCR using the SYBR Green method as described previously (Dharap et al., 2009); 18s rRNA was used as an internal control.
Western blotting and RNA immunoprecipitation (RIP).
In brief, tissue from peri-infarct area was homogenized in buffer containing protease and phosphatase inhibitors, centrifuged at 14,000 × g, and the supernatant (40 μg) was electrophoresed, blotted onto nitrocellulose membranes, and probed with antibodies against REST (1:500; ARP32478P050, Aviva System Biology), coREST (1:5000; ab183711, Abcam), Sin3a (1:1000; ab129087, Abcam), and β-actin (1:000; 3700, Cell Signaling Technology) followed by HRP-conjugated anti-rabbit or anti-mouse IgG (Cell Signaling Technology). Protein bands were developed using enhanced chemiluminescence, visualized and quantitated with Image Studio software (LI-COR Biotechnology). RIP was performed using chromatin immunoprecipitation grade antibodies against Sin3a (ab3479; Abcam) and coREST (07-455; Millipore) and corresponding HRP-tagged secondary antibodies (Cell Signaling Technology) as described previously (Dharap et al., 2013).
Results
FosDT is cogenic with Fos
FosDT gene is downstream of Fos gene with a partial overlap (105 nt long region; Fig. 1A, box). These two genes are located in a gene desert with no other genes in a ∼260,000 nucleotide stretch of DNA. FosDT is highly conserved with ∼94% sequence similarity between rat, mouse, and human genomes (Fig. 1A). Whereas FosDT is 604 nt long, Fos mRNA is 1589 nt long (Fig. 1B).
Figure 1.
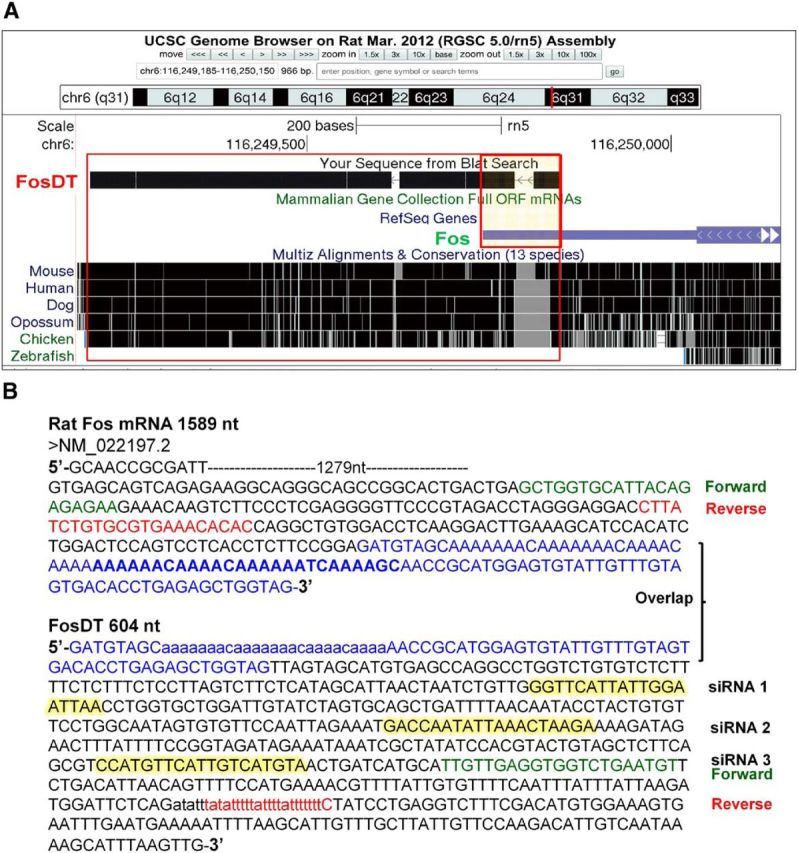
FosDT is downstream to Fos with an overlapping sequence of 105 nt (red box in A). In rat genome, FosDT and Fos are located in a gene desert of ∼260 kb in chromosome 6. BLAT alignment showed that FosDT is highly conserved among mammals (A). FosDT is 604 nt long and Fos is 1589 nt long (B). Primers used for amplification of FosDT and Fos were chosen from the non-overlapping regions (B). Three regions of the FosDT that are targeted by independent siRNAs used as a mixture are highlighted (B).
Focal ischemia-induced FosDT expression
Microarray analysis showed significantly increased FosDT expression at 3, 6, and 12 h of reperfusion (by 12- to 28-fold; p < 0.05) after transient MCAO compared with sham (Fig. 2A). At 12 h of reperfusion, real-time PCR also showed significantly increased expression of FosDT (by 17-fold; p < 0.05) and Fos (by 7-fold; p < 0.05) compared with sham control (Fig. 2B).
Figure 2.

Transient MCAO induced FosDT expression in the ipsilateral cortex at 3, 6, and 12 h of reperfusion compared with sham analyzed with LncRNA microarrays (A). Increased FosDT and Fos expression was observed at 12 h of reperfusion after transient MCAO with real-time PCR (B). Levels of a control LncRNA were not altered at this time point (B). Expression of REST and its corepressor CMPs coREST and MeCP2 measured by real-time PCR was observed to be increased at 12 h reperfusion following transient MCAO (C). Protein levels of REST and coREST were also observed to be increased at 12 h reperfusion following transient MCAO (D, E). RIP showed increased binding of coREST and Sin3a to FosDT at 6 h of reperfusion following transient MCAO (F). Values are mean ± SD (n = 3 or 4/group).*p < 0.05, compared with sham by Student's t test.
Focal ischemia induced the expression of REST and its corepressor partners
At 12 h of reperfusion following transient MCAO, mRNA levels of REST, coREST, and MeCP2 were observed to be significantly induced compared with sham (p < 0.05 in all cases) (Fig. 2C), whereas Sin3a expression was not altered in the ischemic brain (Fig. 2C). At 12 h of reperfusion following transient MCAO, protein levels of REST and coREST, but not of Sin3a, were also observed to be significantly increased compared with sham (p < 0.05 in all cases) (Fig. 2D,E). LncRNAs are known to bind to CMPs to guide them to the specific genomic loci (Wahlestedt, 2013). The CMPs coREST and Sin3a are essential components of the REST complex-mediated gene repression, and REST induction is known to promote postischemic brain damage (Calderone et al., 2003; Noh et al., 2012). RIP analysis showed that binding of FosDT to coREST and Sin3a increased significantly (by twofold to threefold; p < 0.05) at 6 h of reperfusion following transient MCAO compared with sham (Fig. 2F).
FosDT regulates the expression of REST-responsive genes
FosDT siRNA treatment significantly prevented the postischemic induction of FosDT and Fos, but not REST, compared with the control siRNA group (by 45%–50%, p < 0.05; Fig. 3A). This shows that FosDT alters REST complex by modulating the REST-associated CMPs, but not by a direct effect on REST expression. FosDT knockdown also derepressed REST-target genes GRIA2, NFκB2, and GRIN1 in the postischemic brain compared with the control siRNA group (Fig. 3B; p < 0.05 in all cases). This further shows the essential nature of FosDT in REST-mediated gene repression.
Figure 3.
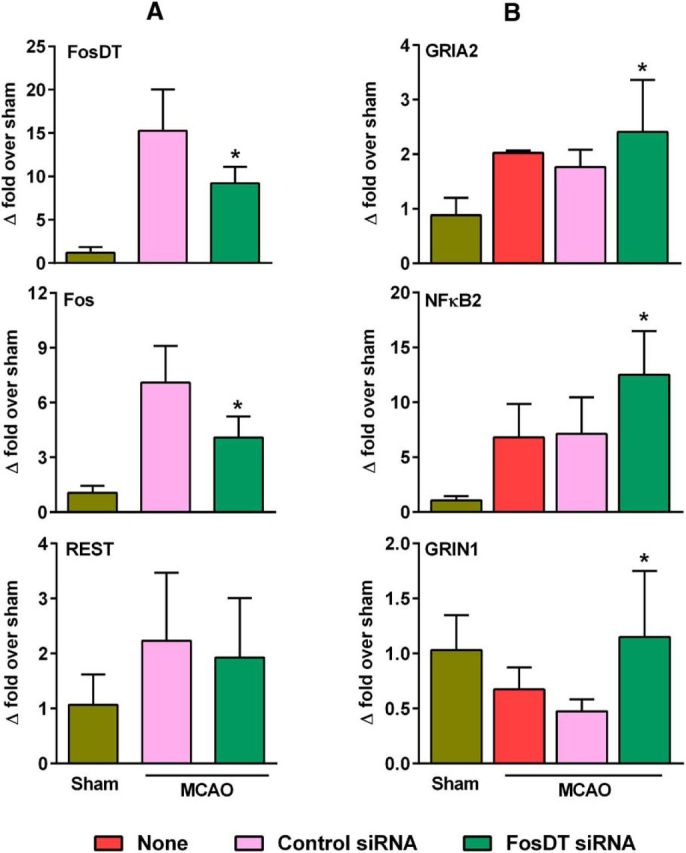
FosDT knockdown prevented postischemic induction of FosDT and Fos, without altering REST, measured at 12 h of reperfusion following transient MCAO (A). FosDT knockdown increased the expression of REST target genes GRIA2, NFκB2, and GRIN1, possibly by derepression (B). Values are mean ± SD (n = 4/group). *p < 0.05 versus control siRNA group by Student's t test.
FosDT silencing improved postischemic motor function and reduced infarction
Transient MCAO in adult rats resulted in significant motor dysfunction between day 1 and day 7 of reperfusion as measured by rotarod test, beam walk test, and adhesive removal test (Fig. 4A). FosDT knockdown significantly improved the motor recovery in all three functional tests compared with the control siRNA group (Fig. 4A). Furthermore, FosDT knockdown significantly decreased the infarct volume compared with the control siRNA group (by 37%; p < 0.05), measured at day 7 of reperfusion following transient MCAO (Fig. 4B,C).
Figure 4.
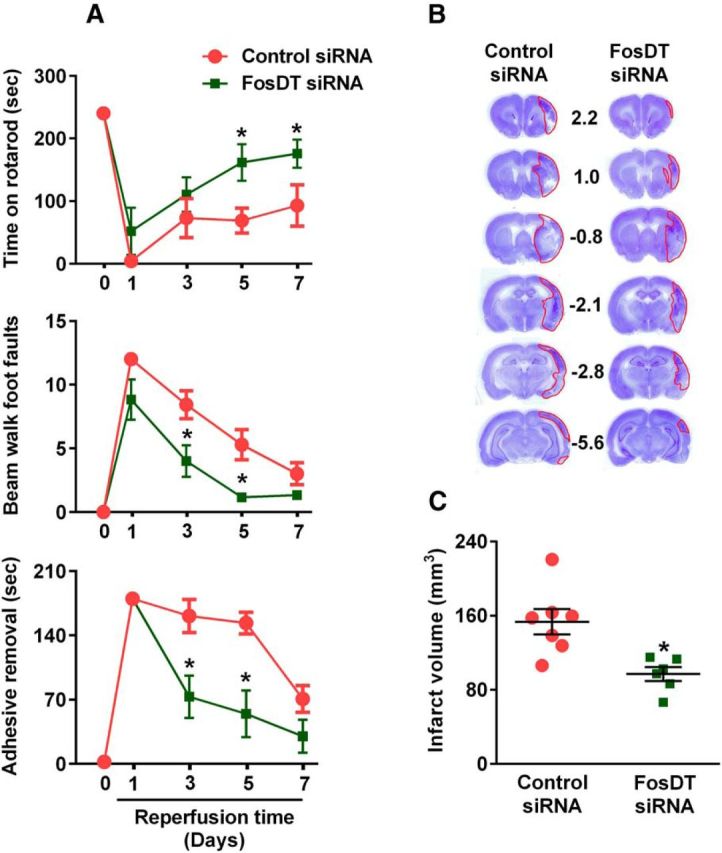
FosDT siRNA treatment significantly improved the postischemic motor function recovery compared with control siRNA-treated group measured by rotarod test (A, top), beam walk test (A, middle), and adhesive removal test (A, bottom) on days 1–7 of reperfusion following transient MCAO. Rats were tested by an investigator blinded to the study groups. The ability to stay on a beam (vestibulomotor activity) was tested in the beam balance test with a score range of 1–6. The motor coordination and motor learning were tested with the rotarod test, which measured the latency time to fall from a cylinder rotating at 8 RPM. Adhesive-removal test measured the time taken by rats to remove small adhesive tapes from forepaws. *p < 0.05, compared with respective reperfusion time point (one-way ANOVA followed by Neuman-Keul's multiple comparisons post test). Representative cresyl violet-stained serial sections shows a smaller infarct in the FosDT siRNA group compared with the control siRNA group at 7 d of reperfusion (B). The infarct volume measured from the serial sections is significantly lower in the FosDT siRNA group compared with the control siRNA group (C). Values are mean ± SEM (n = 6 or 7/group). *p < 0.05, compared with the control siRNA group (Student's t test).
Discussion
In brief, the present study shows that focal ischemia upregulates the expression of LncRNA FosDT and its cogenic gene Fos. We further show that focal ischemia increases binding of FosDT to coREST and Sin3a, which are the essential corepressors of REST. Knockdown of FosDT derepressed REST-downstream genes and decreased postischemic infarction and motor dysfunction. Thus, FosDT induction might be a promoter of poststroke brain damage via modulation of REST.
In mammals, ∼98% of the transcriptional output is various classes of noncoding RNAs (ncRNAs), and perturbations in ncRNA levels and function are known to promote various pathologies (Esteller, 2011; Vemuganti, 2013). We and others recently showed that ischemia extensively alters the expression of many classes of ncRNAs, such as microRNAs (miRNAs), piRNAs, and LncRNAs (Dharap et al., 2009, 2011, 2012a; Dharap and Vemuganti, 2010). Although recent studies demonstrated the role of miRNAs in modulating ischemic brain damage (Siegel et al., 2011; Dharap et al., 2012b; Selvamani et al., 2012; Irmady et al., 2014; Lusardi et al., 2014; Stary et al., 2015; Yin et al., 2015), the functional significance of LncRNAs in this process is still unknown.
LncRNAs are transcribed from the intergenic or intragenic locations of the genome and are known to modulate transcription, translation, and epigenetics as they link DNA, histones, and CMPs, either acting as structural scaffolding or directly recruiting factors to chromatin (Wapinski and Chang, 2011; Wahlestedt, 2013; Clark and Blackshaw, 2014; Maass et al., 2014). LncRNA levels and function are known to be significantly altered and might play critical roles in many diseases, including neurodegenerative diseases, cardiac hypertrophy, and cancer (Han et al., 2014; Yarmishyn and Kurochkin, 2015).
Although LncRNA expression profiles are known to be altered after stroke (Dharap et al., 2012a), their functional significance to poststroke outcome is not known yet. As a proof of principle to show whether LncRNAs have a role in stroke, we tested the postischemic histological and functional outcome following knockdown of FosDT, an LncRNA that was significantly upregulated during the acute phase after transient MCAO in adult rats. The FosDT siRNA-treated rats developed significantly smaller infarcts and showed better neurological function recovery as measured by sensorimotor deficits (adhesive removal test), vestibulomotor function (beam balance test), and motor coordination/motor learning (rotarod test) compared with control siRNA-treated rats.
The mechanism of action of FosDT is not known; however, we observed that FosDT binds to CMPs coREST and Sin3a; and more importantly, this binding increased following ischemia. REST is a transcription factor that represses gene expression in CNS, and REST induction is a known promoter of ischemic neuronal death (Calderone et al., 2003; Noh et al., 2012; Pandi et al., 2013). Both coREST and Sin3a are essential partners of REST in orchestrating epigenetic reprogramming of neuronal genes in developing and adult brain (Calderone et al., 2003; Noh et al., 2012; Pandi et al., 2013). Following stroke, Sin3a levels were unaltered, but its binding to FosDT increased, indicating that normal levels of Sin3a might be sufficient to bind to FosDT to induce REST-mediated gene suppression. Our studies indicate that FosDT scaffolds coREST and Sin3a to enable the formation of REST complex. Recent studies also suggest that LncRNAs recruit REST-containing polycomb repressive complex 2 to target genes to alter their expression and function (Tsai et al., 2010); and using ChIP-on-Chip analysis, Noh et al. (2012) showed that REST binds to a discrete subset of target genes that contain RE-1 binding sites in their promoters. Of those, GRIA2, NFκB2, and GRIN1 are known to modulate ischemic neuronal death (Calderone et al., 2003; Wu and Xie, 2006; Noh et al., 2012). GRIA2 gene encodes GluR2 subunit that prevents Ca2+ entry through AMPA receptors, and GluR2-lacking AMPA receptors are highly permeable to Ca2+, which elevates to toxic levels in ischemic neurons (Calderone et al., 2003), and GRIA2 expression determines the sensitivity of neurons to ischemic insults (Noh et al., 2012). REST activated after stroke is known to suppress GluR2 expression, leading to Ca2+ entry and neuronal death (Calderone et al., 2003; Noh et al., 2012). NFκB2 gene encodes NFκB2 p100/p52 protein, which exhibits IκB properties due to a cluster of ankyrin repeats in the C-terminal domains. NFκB2 p100 mediates cytoplasmic retention of NFκB RelA/p65 to prevent its nuclear translocation, and thus minimizes inflammation and cell death after ischemia (Schneider et al., 1999; Zhang et al., 2005). Hence, REST-mediated repression of NFκB2 might be a promoter of neuronal death after ischemia. GRIN1 codes for GluN1 subunit of NMDA receptor and, together with GluN2 subunit, forms channels that are responsible for synaptic plasticity (Hasan et al., 2013). The present study shows that FosDT knockdown derepressed the REST target genes (GRIA2, NFκB2, and GRIN1), indicating that FosDT might play a role in REST-mediated repression by scaffolding coREST and Sin3a. Following global cerebral ischemia, expression of GRIA2 was shown to be repressed in the hippocampus (Calderone et al., 2003; Noh et al., 2012). However, we presently did not observe altered GRIA2 expression in cerebral cortex after focal ischemia. These previous studies used a global ischemia model that kills hippocampal neurons, whereas we used a focal ischemia model that mainly damages cerebral cortex and striatum. Furthermore, the neuronal death after global ischemia is apoptosis while a mixture of necrosis and apoptosis occurs after focal ischemia. Hence, the mechanisms controlled by FosDT and/or REST might not be exactly same after focal and global ischemia. It was also suggested that the majority of REST-responsive gene promoters do not appear to interact with REST expressed at the physiological levels, yet retain the ability to bind REST when its expression levels are increased and thus poised for REST-mediated repression (Bruce et al., 2004). It was further suggested that these effects are not attributable to an endogenous REST deficiency; however, the presence of multiple RE1 sites might allow repression to occur at different threshold concentrations or over a greater range of REST concentrations (Bruce et al., 2004). Furthermore, REST can target various other genes, including Na+/Ca2+ exchanger 1 and mu opioid receptor 1 (Formisano et al., 2007, 2013; Noh et al., 2012).
We presently show that FosDT interacts with coREST and Sin3a, which are important corepressors of REST. FosDT might function by guiding these chromatin modifiers to specific genomic loci (Dharap et al., 2013; Wahlestedt, 2013). However, future studies are needed to show whether FosDT modulates any other REST target genes. Interestingly, FosDT knockdown reduced the levels of FosDT and Fos without altering REST, indicating that FosDT modulates formation of REST protein complex but does not directly influence REST expression. Both FosDT knockdown that disrupts REST complex and direct REST knockdown are neuroprotective after stroke; hence, both manipulations can be used to develop future therapies. A surprising finding is that FosDT knockdown prevented postischemic Fos induction. This is not due to Fos knockdown as siRNAs used are exclusive for FosDT and does not span the overlapping area of the FosDT and Fos sequences. This indicates that intragenic LncRNAs, such as FosDT, can modulate the expression of their host genes.
Overall, this is the first study that shows that an LncRNA has a functional role in mediating ischemic brain damage. However, this is only a proof-of-principle mechanistic study that encourages further studies to evaluate the potential of LncRNA-based therapies for poststroke neuroprotection. As LncRNAs do not translate any proteins, developing small molecules to modulate their function is difficult. However, siRNAs and synthetic LncRNA mimics can be used to prevent or increase LncRNA levels and function, respectively. They can be therapeutically translated if formulated to deliver peripherally together with transfection agents that are not toxic. The pharmacodynamics/pharmacokinetics also need to be tested. To use them for stroke therapy, preclinical studies are also needed to test the window of therapeutic opportunity, effect of age, sex, and comorbid conditions to satisfy Stroke Treatment Academic Industry Roundtable criteria (Albers et al., 2011). Overall, the present study shows that LncRNAs are candidates for developing novel stroke therapies.
Footnotes
This work was supported by American Heart Association Grants 15IRG23050015 and 14PRE20150039, and National Institute of Health Grants RO3NS083007, R21NS082957, R21NS079585, and R21NS095192.
The authors declare no competing financial interests.
References
- Albers GW, Goldstein LB, Hess DC, Wechsler LR, Furie KL, Gorelick PB, Hurn P, Liebeskind DS, Nogueira RG, Saver JL. Stroke Treatment Academic Industry Roundtable (STAIR) recommendations for maximizing the use of intravenous thrombolytics and expanding treatment options with intra-arterial and neuroprotective therapies. Stroke. 2011;42:2645–2650. doi: 10.1161/STROKEAHA.111.618850. [DOI] [PubMed] [Google Scholar]
- Baldelli P, Meldolesi J. The transcription repressor REST in adult neurons: physiology, pathology, and diseases. eNeuro. 2015;2 doi: 10.1523/ENEURO.0010-15.2015. piiENEURO.0010-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce AW, Donaldson IJ, Wood IC, Yerbury SA, Sadowski MI, Chapman M, Göttgens B, Buckley NJ. Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc Natl Acad Sci U S A. 2004;101:10458–10463. doi: 10.1073/pnas.0401827101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderone A, Jover T, Noh KM, Tanaka H, Yokota H, Lin Y, Grooms SY, Regis R, Bennett MV, Zukin RS. Ischemic insults derepress the gene silencer REST in neurons destined to die. J Neurosci. 2003;23:2112–2121. doi: 10.1523/JNEUROSCI.23-06-02112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BS, Blackshaw S. Long non-coding RNA-dependent transcriptional regulation in neuronal development and disease. Front Genet. 2014;5:164. doi: 10.3389/fgene.2014.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharap A, Vemuganti R. Ischemic pre-conditioning alters cerebral microRNAs that are upstream to neuroprotective signaling pathways. J Neurochem. 2010;113:1685–1691. doi: 10.1111/j.1471-4159.2010.06735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharap A, Bowen K, Place R, Li LC, Vemuganti R. Transient focal ischemia induces extensive temporal changes in rat cerebral microRNAome. J Cereb Blood Flow Metab. 2009;29:675–687. doi: 10.1038/jcbfm.2008.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharap A, Nakka VP, Vemuganti R. Altered expression of PIWI RNA in the rat brain after transient focal ischemia. Stroke. 2011;42:1105–1109. doi: 10.1161/STROKEAHA.110.598391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharap A, Nakka VP, Vemuganti R. Effect of focal ischemia on long noncoding RNAs. Stroke. 2012a;43:2800–2802. doi: 10.1161/STROKEAHA.112.669465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharap A, Nakka VP, Vemuganti R. microRNAs in ischemic brain: the fine-tuning specialists and novel therapeutic targets. In: Lapchak PA, Zhang JH, editors. Translational stroke research: from target selection to clinical trials. New York: Springer; 2012b. pp. 335–352. [Google Scholar]
- Dharap A, Pokrzywa C, Vemuganti R. Increased binding of stroke-induced long non-coding RNAs to the transcriptional corepressors Sin3A and coREST. ASN Neuro. 2013;5:283–289. doi: 10.1042/AN20130029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- Formisano L, Noh KM, Miyawaki T, Mashiko T, Bennett MV, Zukin RS. Ischemic insults promote epigenetic reprogramming of mu opioid receptor expression in hippocampal neurons. Proc Natl Acad Sci U S A. 2007;104:4170–4175. doi: 10.1073/pnas.0611704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formisano L, Guida N, Valsecchi V, Pignataro G, Vinciguerra A, Pannaccione A, Secondo A, Boscia F, Molinaro P, Sisalli MJ, Sirabella R, Casamassa A, Canzoniero LM, Di Renzo G, Annunziato L. NCX1 is a new rest target gene: role in cerebral ischemia. Neurobiol Dis. 2013;50:76–85. doi: 10.1016/j.nbd.2012.10.010. [DOI] [PubMed] [Google Scholar]
- Han P, Li W, Lin CH, Yang J, Shang C, Nurnberg ST, Jin KK, Xu W, Lin CY, Lin CJ, Xiong Y, Chien HC, Zhou B, Ashley E, Bernstein D, Chen PS, Chen HS, Quertermous T, Chang CP. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102–106. doi: 10.1038/nature13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan MT, Hernández-González S, Dogbevia G, Treviño M, Bertocchi I, Gruart A, Delgado-García JM. Role of motor cortex NMDA receptors in learning-dependent synaptic plasticity of behaving mice. Nat Commun. 2013;4:2258. doi: 10.1038/ncomms3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irmady K, Jackman KA, Padow VA, Shahani N, Martin LA, Cerchietti L, Unsicker K, Iadecola C, Hempstead BL. Mir-592 regulates the induction and cell death-promoting activity of p75NTR in neuronal ischemic injury. J Neurosci. 2014;34:3419–3428. doi: 10.1523/JNEUROSCI.1982-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusardi TA, Murphy SJ, Phillips JI, Chen Y, Davis CM, Young JM, Thompson SJ, Saugstad JA. MicroRNA responses to focal cerebral ischemia in male and female mouse brain. Front Mol Neurosci. 2014;7:11. doi: 10.3389/fnmol.2014.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass PG, Luft FC, Bähring S. Long non-coding RNA in health and disease. J Mol Med. 2014;92:337–346. doi: 10.1007/s00109-014-1131-8. [DOI] [PubMed] [Google Scholar]
- Nakka VP, Lang BT, Lenschow DJ, Zhang DE, Dempsey RJ, Vemuganti R. Increased cerebral protein ISGylation after focal ischemia is neuroprotective. J Cereb Blood Flow Metab. 2011;31:2375–2384. doi: 10.1038/jcbfm.2011.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh KM, Hwang JY, Follenzi A, Athanasiadou R, Miyawaki T, Greally JM, Bennett MV, Zukin RS. Repressor element-1 silencing transcription factor (REST)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proc Natl Acad Sci U S A. 2012;109:E962–E971. doi: 10.1073/pnas.1121568109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandi G, Nakka VP, Dharap A, Roopra A, Vemuganti R. MicroRNA miR-29c down-regulation leading to de-repression of its target DNA methyltransferase 3a promotes ischemic brain damage. PLoS One. 2013;8:e58039. doi: 10.1371/journal.pone.0058039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 4. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Satriotomo I, Bowen KK, Vemuganti R. JAK2 and STAT3 activation contributes to neuronal damage following transient focal cerebral ischemia. J Neurochem. 2006;98:1353–1368. doi: 10.1111/j.1471-4159.2006.04051.x. [DOI] [PubMed] [Google Scholar]
- Schaar KL, Brenneman MM, Savitz SI. Functional assessments in the rodent stroke model. Exp Transl Stroke Med. 2010;2:13. doi: 10.1186/2040-7378-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- Selvamani A, Sathyan P, Miranda RC, Sohrabji F. An antagomir to microRNA Let7f promotes neuroprotection in an ischemic stroke model. PLoS One. 2012;7:e32662. doi: 10.1371/journal.pone.0032662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu M, Mizushima H, Sasaki K, Arai Y, Matsumoto K, Shioda S, Nakai Y. Expression of c-fos in the rat cerebral cortex after focal ischemia and reperfusion. Brain Res Bull. 1994;33:689–697. doi: 10.1016/0361-9230(94)90234-8. [DOI] [PubMed] [Google Scholar]
- Siegel C, Li J, Liu F, Benashski SE, McCullough LD. miR-23a regulation of X-linked inhibitor of apoptosis (XIAP) contributes to sex differences in the response to cerebral ischemia. Proc Natl Acad Sci U S A. 2011;108:11662–11667. doi: 10.1073/pnas.1102635108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stary CM, Xu L, Sun X, Ouyang YB, White RE, Leong J, Li J, Xiong X, Giffard RG. MicroRNA-200c contributes to injury from transient focal cerebral ischemia by targeting Reelin. Stroke. 2015;46:551–556. doi: 10.1161/STROKEAHA.114.007041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tureyen K, Brooks N, Bowen K, Svaren J, Vemuganti R. Transcription factor early growth response-1 induction mediates inflammatory gene expression and brain damage following transient focal ischemia. J Neurochem. 2008;105:1313–1324. doi: 10.1111/j.1471-4159.2008.05233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuganti R. All's well that transcribes well: non-coding RNAs and poststroke brain damage. Neurochem Int. 2013;63:438–449. doi: 10.1016/j.neuint.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlestedt C. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat Rev Drug Discov. 2013;12:433–446. doi: 10.1038/nrd4018. [DOI] [PubMed] [Google Scholar]
- Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354–361. doi: 10.1016/j.tcb.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Wu J, Xie X. Comparative sequence analysis reveals an intricate network among REST, CREB and miRNA in mediating neuronal gene expression. Genome Biol. 2006;7:R85. doi: 10.1186/gb-2006-7-9-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Yuan J, Li H, Li M, Zhao G, Bu D, Zhu W, Wu W, Chen R, Zhao Y. NONCODEv4: exploring the world of long non-coding RNA genes. Nucleic Acids Res. 2014;42:D98–D103. doi: 10.1093/nar/gkt1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarmishyn AA, Kurochkin IV. Long noncoding RNAs: a potential novel class of cancer biomarkers. Front Genet. 2015;6:145. doi: 10.3389/fgene.2015.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin KJ, Hamblin M, Chen YE. Angiogenesis-regulating microRNAs and ischemic stroke. Curr Vasc Pharmacol. 2015;13:352–365. doi: 10.2174/15701611113119990016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Potrovita I, Tarabin V, Herrmann O, Beer V, Weih F, Schneider A, Schwaninger M. Neuronal activation of NF-kappaB contributes to cell death in cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:30–40. doi: 10.1038/sj.jcbfm.9600004. [DOI] [PubMed] [Google Scholar]