

Abstract
Vasoactive intestinal peptide (VIP) mediates a broad range of biological responses by activating two related receptors, VIP receptor 1 and 2 (VIPR1 and VIPR2). Although the use of native VIP facilitates neuroprotection, clinical application of the hormone is limited due to VIP's rapid metabolism and inability to distinguish between VIPR1 and VIPR2 receptors. In addition, activation of both receptors by therapeutics may increase adverse secondary toxicities. Therefore, we developed metabolically stable and receptor-selective agonists for VIPR1 and VIPR2 to improve pharmacokinetic and pharmacodynamic therapeutic end points. Selective agonists were investigated for their abilities to protect mice against MPTP-induced neurodegeneration used to model Parkinson's disease (PD). Survival of tyrosine hydroxylase neurons in the substantia nigra was determined by stereological tests after MPTP intoxication in mice pretreated with either VIPR1 or VIPR2 agonist or after adoptive transfer of splenic cell populations from agonist-treated mice administered to MPTP-intoxicated animals. Treatment with VIPR2 agonist or splenocytes from agonist-treated mice resulted in increased neuronal sparing. Immunohistochemical tests showed that agonist-treated mice displayed reductions in microglial responses, with the most pronounced effects in VIPR2 agonist-treated, MPTP-intoxicated mice. In parallel studies, we observed reductions in proinflammatory cytokine release that included IL-17A, IL-6, and IFN-γ and increases in GM-CSF transcripts in CD4+ T cells recovered from VIPR2 agonist-treated animals. Moreover, a phenotypic shift of effector to regulatory T cells was observed. These results support the use of VIPR2-selective agonists as neuroprotective agents for PD treatment.
SIGNIFICANCE STATEMENT Vasoactive intestinal peptide receptor 2 can elicit immune transformation in a model of Parkinson's disease (PD). Such immunomodulatory capabilities can lead to neuroprotection by attenuating microglial activation and by slowing degradation of neuronal cell bodies and termini in MPTP-intoxicated mice. The protective mechanism arises from altering a Th1/Th2 immune cytokine response into an anti-inflammatory and neuronal sparing profile. These results are directly applicable for the development of novel PD therapies.
Keywords: adaptive immunity, inflammation, neuroprotection, Parkinson's disease, VIP, VPAC
Introduction
Aberrant innate and adaptive immune responses are known disease initiators for Parkinson's disease (PD) (Kosloski et al., 2010; Ha et al., 2012; Mosley et al., 2012). These result in chronic immune activation perpetuated by the extracellular accumulation of prion-like aggregated and post-translationally modified α-synuclein (α-syn) (Benner et al., 2008), which induces an inflammatory neurotoxic cascade affecting nigrostriatal degeneration (Mosley et al., 2006; Reynolds et al., 2007; Benner et al., 2008; Reynolds et al., 2008; Huang et al., 2009; Reynolds et al., 2010). Infiltrating CD4+ and CD8+ T cells, microglial activation, and dopaminergic cell loss affect PD pathobiology that is observed in mouse models and in postmortem human brain tissues (McGeer et al., 1988; Fiszer et al., 1994; Kurkowska-Jastrzebska et al., 1999; Bas et al., 2001; McLaughlin et al., 2006; Brochard et al., 2009). PD patients present higher frequencies of effector T-cell phenotypes with reduced regulatory T cell (Treg) function relative to controls (Saunders et al., 2012). T-cell transformation during progressive disease correlates with worsening movement clinical scores. Results from other laboratories confirm our observations of the presence of altered peripheral CD4+ T-cell phenotypes in PD and support a role of T-cell subsets in PD progression and disease control (Romero-Ramos et al., 2014).
Work performed in animal models demonstrates that adoptive transfer of effector T cells (Teffs) exacerbates neurodegeneration (Benner et al., 2008; Kroenke et al., 2008), whereas transfer of Tregs elicits neuroprotective responses (Reynolds et al., 2007; Reynolds et al., 2008; Huang et al., 2009; Kosloski et al., 2013). Treg-mediated protection from dopaminergic cell death is complemented by decreased microglial reactivity. Our work and the work of others has shown that granulocyte macrophage-colony stimulating factor (GM-CSF) is neuroprotective in neuronal lesions (Schäbitz et al., 2008; Kosloski et al., 2013; Kelso et al., 2015). GM-CSF changes a neurotoxic response to a protective regulatory response (Kosloski et al., 2013). Nonetheless, extended use of GM-CSF is associated with secondary toxicities including bone pain, fatigue, and nausea (Vial and Descotes, 1995). Therefore, a search for alternative immune-modulating treatment strategies that restore regulatory capacity is warranted.
One agent that could improve disease outcomes is vasoactive intestinal peptide (VIP). VIP is the natural 28-residue agonist of the G-protein-coupled receptors VIPR1 and VIPR2 (Reubi, 2003). It acts as a cytokine and neuropeptide by positively affecting immune responses (Delgado et al., 2001; Delgado et al., 2004b; Dickson and Finlayson, 2009). The immunomodulatory capabilities of VIP support its potential to transform T-cell phenotypes, leading to protection in models of inflammatory and autoimmune conditions (Delgado et al., 2000; Delgado et al., 2001; Abad et al., 2003; Abad et al., 2005; Chen et al., 2008; Abad et al., 2010; Deng et al., 2010). Therefore, we reasoned that VIP could also transform adaptive immune responses in PD. Indeed, prior studies show that VIP protects against neurotoxicity by attenuating microglial activation and degradation of neuronal cell bodies and termini in both MPTP- and 6-OHDA-induced injuries (Offen et al., 2000; Delgado and Ganea, 2003; Reynolds et al., 2010; Korkmaz et al., 2012; Tunçel et al., 2012). The protective mechanism arises from shifting the cytokine response into an anti-inflammatory profile (Vial and Descotes, 1995; Delgado et al., 1999; Delgado et al., 2004a; Delgado et al., 2005; Chen et al., 2008; Reynolds et al., 2010).
Based on these findings, we reasoned that VIP-induced neuroprotective responses could be harnessed for clinical benefit if the rapid proteolytic degradation and lack of VIPR selectivity of the native hormone itself could be overcome (Domschke et al., 1978). To this end, we developed backbone-modified analogs of VIP that resist protease degradation and display selective agonism of VIPR1 or VIPR2. Both VIPR1 and VIPR2 agonists caused reductions in the release of proinflammatory cytokines from stimulated CD4+ T cells. Transformation of Teff responses to anti-inflammatory and regulatory T-cell responses was also observed; however, the VIPR2-selective agonist better augmented Treg activity and elicited a greater neuroprotective response, thus providing evidence for its potential to ameliorate PD.
Materials and Methods
Peptide synthesis and purification.
Protected α-amino acids, resins, and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophostphate (HBTU) were from GL Biochem. Protected β-homo-amino acids were from PepTech. ACPC was from Chemimpex and Polypeptide; APC was from the organic chemistry facility at Fox Chase Cancer Center (Lee et al., 2001). Native VIP and [D-p-Cl-Phe6, Leu17]-VIP were from Genway Biotech and Tocris Bioscience. All other reagents and solvents were from Airgas, ChemImpex, Fisher Scientific, PharmcoAaper, or Sigma-Aldrich and used as received. Reverse-phase HPLC (RP-HPLC) was performed on Supelco and Phenomenex analytical or preparative scale C18 columns using gradients between 0.1% trifluoracetic acid (TFA) in water or in acetonitrile using Agilent 1260 and Varian Prostar systems. Peptides were synthesized by standard Fmoc-solid phase synthesis on Rink amide 4-methylbenzhydrylamine hydrochloride resin with norleucine. Microwave irradiation was used as previously described for peptide syntheses (Korendovych et al., 2010; Shandler et al., 2011). Briefly, protected amino acids were activated with HBTU and N-hydroxybenzotriazole in the presence of N,N-diisopropylethylamine (DIEA) in N-methyl-2-pyrrolidone for coupling reactions. Deprotections were effected using 20% piperidine in dimethylformamide (DMF). After the final deprotection, peptides were capped using acetic anhydride/DIEA in DMF. After synthesis was complete, peptides were cleaved from the resin using a solution of 95% TFA, 2.5% H2O, and 2.5% triisopropylsilane. Excess TFA was removed under a stream of nitrogen and crude peptide was precipitated by the addition of cold ether. Crude peptide solutions were purified using RP-HPLC on a preparative scale using C18 columns. The identity and purity of peptides were confirmed by mass spectrometry. After lyophilization, peptides were dissolved in TFE. An Agilent diode array, model 8453, was used to determine concentrations, where an extinction coefficient of 2980 M−1 cm−1 was applied. Aliquots for assays were dried under vacuum, resuspended in 20% acetonitrile/0.1% HCl, and lyophilized to exchange the counter ion. Dried peptide was resuspended in either DMSO or 1% DMSO in PBS.
Cell-based assays.
EC50 values for both VIPR1 and VIPR2 were determined in triplicate for each time point by monitoring intracellular cAMP concentrations of DiscoveRX's PathHunter CHO-K1 VIPR1 β-arrestin or CHO-K1 VIPR2 β-arrestin cell lines, respectively. To initiate assays, 2000 cells per well were plated into a 384-well plate and incubated 16 h in F-12K medium supplemented with 10% FBS. Both cell lines were used between two and five passages. The following day, each reconstituted compound was diluted by 12 3-fold serial dilutions and added to the plated cells. The final concentrations for each compound in the assay wells ranged between 1 μm and 5.6 pm in 1% DMSO/PBS. After a 90 min incubation period, the medium and compound were removed and intracellular cAMP concentrations were determined using the cAMP HiRange Kit available from Cisbio. All kit components were used at concentrations according to the manufacturer's recommendation. In brief, this kit functions by lysing the activated cells to release the intracellular cAMP, which is then available to disrupt a FRET pair. Homogenous time resolved fluorescence signals were monitored using a Tecan M1000 Pro in TR-FRET mode. All TR-FRET parameters, including excitation, emission, delay, and integration times, were set according to the manufacturer's recommendations and 100 flashes were used per well. The fluorescence intensity of each well at both 620 and 665 nm was collected and the ratio of these values was used to determined ΔF%, which is defined as [(standard or sample ratio − ratio of the negative control)/ratio of the negative control] × 100. The resulting data were calibrated to a negative control, baseline corrected, and then normalized using the average VIP response for each cell line as boundaries. This normalized log inhibitor response curve was fit to a four-parameter sigmoidal dose–response curve using least-squares fitting in the GraphPad Prism software package with a boundary constraint that each EC50 fit was <10−6 m.
Proteinase and pepsin assays.
The degradation rate of peptides was measured in the context of multiple aggressive proteases. All data were analyzed using analytical HPLC in triplicate for each time point. Pepsin assay proteinase stock was prepared fresh using 185 nm pepsin, 34 mm NaCl, and 84 mm HCl in deionized water. At time 0, either 2.8 nmol VIP or 7.0 nmol hybridtide was rapidly diluted into 350 μl of proteinase stock. Hybridtides were run at slightly higher concentrations to account for HPLC line broadening while detecting. At specified increments, 50 μl aliquots were removed and quenched with 10 μl of 1 m Tris, pH 9.5. Quenched samples were evaluated by HPLC. Peak integrals were determined using Chemstation software and normalized to the average integral value for the first time point for each dataset independently. These data were then fit to a single exponential decay using least-squares fitting through GraphPad Prism. Chymotrypsin and Proteinase K assays were performed equivalently to the pepsin assay with the following exceptions: all solutions were made in PBS, chymotrypsin was used at 40 nm, Proteinase K was used at 34.6 nm, and all time points were quenched with 10 μl of 99% acetonitrile/1% TFA.
Pharmacokinetic assays.
For pharmacokinetic assays, 250 nmol/kg doses of LBT-3627 in 50 μl of 1% DMSO in PBS were injected subcutaneously into the neck scruff of six 25-week-old C57BL/6 mice across two study arms. This experimental design was followed to minimize blood sampling performed on each animal. Group 1 was sampled at 0, 30, 75, and 180 min and Group 2 was sampled at 15, 45, 120, and 300 min. At these designated time points, 20 μl of blood was removed from the tail vein using heparin-coated glass capillaries and mixed with mixtures of protease inhibitors (Thermo Scientific) and sodium heparin (Sagent Pharmaceuticals) at a 1× concentration and 1 IU/ml, respectively. Whole blood samples were centrifuged for 10 min at 7200 × g to isolate plasma, which was flash-frozen in liquid nitrogen and stored at −20°C. To prepare samples for LC-MS/MS analysis, an acetonitrile precipitation was performed by mixing 5 μl of thawed blood plasma with 15 μl of acetonitrile containing 1% formic acid; 13.3 nm LBT-3393 was used as an internal standard. This mixture was vortexed on a Vortex Genie 2 and then centrifuged for removal of precipitated proteins. Ten microliters of the supernatant was removed and transferred to an HPLC autosampler vial containing 23 μl of H2O of 5% formic acid in water. The sample was immediately vortexed before LC-MS/MS analysis.
For LC-MS/MS analysis, samples were transferred to an Agilent 1260 autosampler and held at 25°C until injection. Thirteen microliters of the sample was injected onto a Phenomenex Kinetex 2.6 μm C18 column (50 × 3.0 mm) heated to 50°C. An acetonitrile gradient of 20–35% formed over 2.5 min at 400 μl/min was used to isolated LBT-3627. Compound elution was detected with an AB Sciex 4000 mass spectrometer equipped with a Turbo V ion source. Two product ions were collected, summed, and integrated with Analyst version 1.6. Integral intensities were normalized to an internal standard. The data were fit to a single-compartment model using nonlinear regression within GraphPad Prism version 6.0f.
Animals, drug treatment, and MPTP intoxication.
Male C57BL/6J mice, 6–8 weeks old (The Jackson Laboratory) were used as donor and recipient mice in all studies. Donor and pretreated mice were administered either VIP (human ovine porcine rat; Genway Biotech), LBT-3393 (VIPR1 agonist), LBT-3627 (VIPR2 agonist), or scrambled peptide that were reconstituted using Dulbecco's PBS (DPBS) and given at a dosage of 15 μg intraperitoneally daily for 5 d before MPTP intoxication. For antagonist treatment, mice were administered with [D-p-Cl-Phe6,Leu17]-VIP at 8 μg intraperitoneally daily for 5 d. Recipient mice received four subcutaneous injections of vehicle (DPBS, 10 ml/kg body weight) or MPTP-HCl (Sigma-Aldrich) at 16 mg of MPTP (free base)/kg body weight in DPBS; each injection was given at 2 h intervals. Twelve hours after MPTP intoxication, splenocytes were harvested from donors and adoptively transferred to MPTP-intoxicated recipient mice (n = 5–8 mice per group per time point). MPTP safety precautions were followed in accordance with the determined safety and handling protocol (Jackson-Lewis and Przedborski, 2007) and all animal procedures were in agreement with National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center.
Isolation and adoptive transfer of CD4+ T cells.
After 5 d of peptide administration, donor mice were killed and single-cell suspensions were obtained from spleen and lymph nodes (brachial, axillary, cervical, and inguinal) and resuspended to 50 × 106 cells/0.25 ml for adoptive transfer. Recipient mice received 0.25 ml of cell suspension intravenously via the tail within 12 h of the final MPTP injection. CD4+ T cells were negatively selected using a CD4+ T-cell isolation kit II for mouse as per manufacturer's instructions (Miltenyi Biotech) and CD4+CD25+ T cells were selected using the CD4+CD25+ Treg isolation kit for mouse, with purity ranging from 65% to 90% depending on the assay. Isolated CD4+ T-cell populations were used for RNA isolation, genomic analysis, and cytometric bead assays. Freshly isolated CD4+CD25+ Tregs were used for carboxyfluorescein succinimidyl ester (CFSE) inhibition assays (Quah and Parish, 2010).
CD11b+ microglia isolation.
After MPTP intoxication or 5 d of peptide pretreatment, mice (n = 8) were killed and perfused with cold PBS. Ventral midbrain was collected from each mouse and dissociated into single-cell suspensions using a neural tissue dissociation kit (Miltenyi Biotech). Myelin was removed using a 30% Percoll discontinuous gradient in DPBS. CD11b+ cells were magnetically isolated using PE-conjugated anti-CD11b followed by supraparamagnetic bead-conjugated secondary antibodies. The labeled cells were passed through a magnetic MS column (Miltenyi Biotech), as described previously (Nikodemova and Watters, 2012).
Perfusions and immunohistochemistry.
Under terminal anesthesia (Fatal Plus, pentobarbital), mice were transcardially perfused with DPBS followed by 4% paraformaldehyde/DPBS (Sigma-Aldrich). Whole brains were harvested, processed, and flash-frozen to assess dopaminergic neurons in the substantia nigra (SN) and termini in the striatum (Benner et al., 2004). Frozen midbrain sections (30 μm) were immunostained for tyrosine hydroxylase (TH) (anti-TH, 1:2000; EMD/Millipore) and counterstained for Nissl substance. To assess microglial reactivity, midbrain sections (30 μm) were immunostained for macrophage antigen complex-1 (Mac-1) (anti-CD11b, 1:1000; AbD Serotech). To assess dopaminergic termini, striatal sections (30 μm) were labeled with anti-TH (1:1000; EMD Millipore), as described previously (Kosloski et al., 2013). Within the SN, total numbers of Mac-1+ cells, TH+Nissl+ (dopaminergic neurons), and TH−Nissl+ (nondopaminergic neurons) were estimated by stereological analysis using Stereo Investigator software with the optical fractionator module (MBF Bioscicence). Density of dopaminergic neuron termini in the striatum was determined by digital densitometry using ImageJ software as described previously (Benner et al., 2004).
MPTP metabolism and RP-HPLC.
Mice (n = 5) were intoxicated with MPTP alone or pretreated for 5 d with VIP, LBT-3393, or LBT-3627 followed by MPTP intoxication. Within 90 min after the last injection of MPTP, striatum and ventral midbrain were isolated and processed for MPTP and 1-methyl-4-phenylpyridinium (MPP+) levels (Jackson-Lewis and Przedborski, 2007). MPTP and MPP+ levels were determined using RP-HPLC analysis with UV illumination and detection at 245 and 295 nm, respectively.
RNA isolations and PCR.
Mice were pretreated with PBS, VIP, LBT-3393, or LBT-3627. CD4+ T cells or CD11b+ microglia were harvested and total RNA isolated with an RNeasy Mini Kit (Qiagen). All procedures were performed under RNase-free conditions. cDNA was generated from RNA using the RevertAid First Strand cDNA Synthesis kit (Thermo Scientific) and preamplification was performed using the appropriate primer mixes for RT2 PCR arrays for Mouse T Helper Cell Differentiation or Mouse Proinflammatory Response and Autoimmunity (Qiagen). Quantitative RT-PCR was performed on an Eppendorf Mastercycler Realplex EP as per the manufacturer's instructions (Eppendorf). Data analysis was performed using RT2 Profiler PCR Array web-based data analysis software, version 3.5 (Qiagen). Gene networks were generated using Ingenuity Pathway Analysis (IPA; Qiagen) and were designed using the pathway designer tool.
Flow cytometric tests and cytokine assessments.
Cell fractions were isolated from total splenocytes after peptide treatment. Samples were permeabilized using the FoxP3 staining buffer set kit (eBioscience); fluorescently labeled with monoclonal antibodies to CD4, CD25, CD8, and FoxP3 (eBioscience) to assess T-cell frequencies within the total population; and analyzed using a FACSCalibur flow cytometer (BD Biosciences). To assess cytokine production, CD4+ T cells were isolated, stimulated using anti-CD3/CD28 beads, and cell supernatant was collected at 12 h. Supernatants were assessed for cytokine levels using a multianalyte Th1/Th2/Th17 cytometric bead array (BD Biosciences) and data acquired using a FACSArray bioanalyzer (BD Biosciences) (Reynolds et al., 2010).
Statistical analyses.
All values are expressed as mean ± SEM. Differences in between-group means were analyzed using ANOVA followed by Fisher's least significant difference post hoc test (GraphPad Software). Comparisons of slope and elevation for CFSE inhibition assays were evaluated using linear regression. Slopes for all lines were significantly nonzero (p < 0.0005) and no line deviated from linearity as determined by a runs test (p ≥ 0.6667).
Results
Peptide stability
VIP elicits significant immune-based neuroprotection (Delgado and Ganea, 2003; Reynolds et al., 2010; Waschek, 2013). However, two properties of the peptide hormone are problematic from the perspective of therapeutic applications. First, VIP has a short half-life as a result of its rapid proteolysis (Domschke et al., 1978). Second, VIP activates two broadly distributed receptors, VIPR1 and VIPR2. Although both are members of the B-family of GPCRs (Reubi, 2003), it is not clear whether one or both receptors is responsible for VIP's neuroprotective activities. Therefore, we developed analogs of VIP that resist protease degradation and display receptor selectivity.
Our design centered on replacing α-amino acid with β-amino acid residues (Horne et al., 2008; Boersma et al., 2012; Cheloha et al., 2014; Johnson et al., 2014). These α→β substitutions alter the covalent spacing between amide groups along the peptidic backbone, which disrupts protease recognition while largely retaining native side chains and helical propensity (Johnson and Gellman, 2013). We focused on the impact of periodic α→β substitution in the C-terminal portion of VIP, which is presumed to be helical in the bound state. These efforts led to LBT-3393 (Table 1), an analog that contains five α→β replacements at residues Thr11, Lys15, Val19, Leu23, and Leu27. At each replacement site, the β residue bears the native side chain. In addition, LBT-3393 contains two α residue changes that are known to be well tolerated in VIP, Met17→Leu, and Ser25→Ala (Nicole et al., 2000; Igarashi et al., 2002a,b). LBT-3393 is a full agonist of VIPR1, although ∼100-fold less potent than VIP itself. LBT-3393 is highly selective for VIPR1 relative to VIPR2 (Fig. 1), exhibiting VIPR2 activity only at the highest concentrations tested.
Table 1.
Amino acid sequences for the peptides evaluated in these studies
| VIP | HSDAV FTDNY TRLRK QMAVK KYLNS ILN-NH2 |
| LBT-3393 | aAc-HSDAV FTDNY tRLRk QLAvK KYlNA IlN-NH2 |
| Ro 25-1553 | bAc-HSDAV FTENY TKLRK QnLAAK K*YLND* LKKGG T-NH2 |
| LBT-3627 | aAc-HxDAx FTExy TKLRK qLAAz KYxND LKkGg T-NH2 |
| LBT-SCR | Ac-Tyxtx EgLKx GFKTK RHzKA qYLxL ANADk D-NH2 |
aβ3-amino acids are in bold lowercase font, cyclic β-amino acids (X and Z) are indicated in bold, italic, lowercase font. Lowercase “L” is underlined.
bA lactam bridge between K* and D* exists in Ro25-1553 along with a single norleucine, indicated by nL.
Figure 1.

LBT-3393 and LBT-3627 peptides are VIPR1 and VIPR2 agonists. cAMP dose–response assays comparing native VIP with LBT-3393 (top) and LBT-3627 (bottom). Data were generated by activating engineered CHO cells that overexpress either VIPR1 or VIPR2 with increasing concentrations of peptide and detecting intracellular cAMP activation using a two-component HTRF pair. The data are reported as normalized ΔF%, which is a measure of the ratio of nonfluorescent cAMP released by the cells being interrogated, which competes out the fluorescently labeled cAMP initially present at the beginning of the assay. Therefore, the ΔF% can be thought of as the inverse of a traditional dose–response curve. Higher concentrations of VIP (>10−8 m) were excluded from analysis as appropriate because they began to increase in relative value, likely a result of receptor internalization (data not shown). A best-fit dose-response curve could not be determined for LBT-3393 reactivity with VIPR2-expressing CHO cells.
Exploration of α→β substitutions based on the sequence of VIP did not deliver a VIPR2-selective agonist; therefore, we turned to the highly VIPR2-selective peptide Ro 25–1553 (Table 1) as a starting point (O'Donnell et al., 1994a,b). Analog LBT-3627 contains nine α→β substitutions (Table 1), some bearing the native side chains (Tyr10, Gln16, Lys28, and Gly30) and others having a cyclic structure. Dose–response assays showed that LBT-3627 is a potent agonist of VIPR2 and highly selective for VIPR2 versus VIPR1 (Fig. 1). LBT-3627 is superior to LBT-3393 in terms of resistance to proteinase activities (Fig. 2A–C), which translates to an improved in vivo pharmacokinetic profile (t½ = 24.33 min; Fig. 2D) of the unformulated peptide compared with previous human pharmacokinetic studies of VIP itself (t½ <1 min) (Domschke et al., 1978).
Figure 2.

LBT-3627 is resistant to protease activities. A–C, Relative stabilities of three peptides, VIP (solid line and circles), LBT-3393 (dotted lines and open circle), and LBT-3627 (dotted lines and open triangles) after exposures up to 120 min to different aggressive protease environments of pepsin (A), proteinase K (B), and chymotrypsin (C). Percentage of nondegraded peptide concentrations remaining from time 0 were determined by RP-HPLC. D, Pharmacokinetic (PK) profile of 250 nmol/kg unformulated LBT-3627 drug concentration using LC-MS/MS analysis, with t½ = 24.33 min.
VIPR agonists induce neuroprotective responses in MPTP-intoxicated mice
Our prior work demonstrated immune modulatory and neuroprotective activities for VIP (Reynolds et al., 2010). Indeed, previous work showed VIP induction of Treg with concomitant anti-inflammatory and neuroprotective responses in MPTP-intoxicated mice, as well as a broad number of animal models of inflammatory and neurodegenerative diseases (Delgado et al., 2001; Delgado and Ganea, 2003; Delgado et al., 2005; Chen et al., 2008; Reynolds et al., 2010). To extend such observations with an eye toward clinical translation, we assessed the abilities of the designed VIPR agonists to promote dopaminergic neuronal survival in our mouse model of PD. Mice were treated for 5 d with VIP, VIPR1 agonist (LBT-3393), or VIPR2 agonist (LBT-3627) and then intoxicated with MPTP. After MPTP treatment, total numbers of surviving dopaminergic neurons (TH+Nissl+) were observed in the SN (Fig. 3A). The numbers of dopaminergic neurons were determined by investigator-blinded stereological analysis and showed a significant increase in surviving neuron numbers associated with LBT-3627 treatment compared with PBS control (Fig. 3B). After MPTP intoxication, dopaminergic neuron numbers decreased from 9789 ± 1061 to 4411 ± 1228 (Fig. 3B). No differences in numbers of nondopaminergic neurons (TH−Nissl+) were observed because these are not susceptible to MPTP intoxication (Otto and Unsicker, 1993; Jackson-Lewis and Przedborski, 2007; Choi et al., 2008). After VIPR agonist pretreatments, MPTP-induced TH+ neuronal loss was attenuated for all treatment arms. Compared with MPTP alone, VIP, LBT-3393 and LBT-3627 pretreatment increased dopaminergic neuronal numbers to 5264 ± 1441, 4600 ± 945, and 7339 ± 2115, respectively. However, only LBT-3627 pretreatment showed significant protective capability. In contrast, striatal termini were not significantly spared by any peptide pretreatments (Fig. 3C). To determine whether the observed neuroprotective effects are linked to specific peptide-receptor interactions, a scrambled peptide (LBT-SCR) as a negative peptide control and a specific VIPR antagonist ([D-p-Cl-Phe6,Leu17]-VIP) were used (Pandol et al., 1986). These experiments were designed to inhibit binding of VIP, LBT-3393, or LBT-3627 to VIP receptors (Fig. 3D). Coadministration of [D-p-Cl-Phe6,Leu17]-VIP with VIP, LBT-3393, or LBT-3627 ameliorated neuroprotection previously observed by VIP or VIPR2 agonist. Under these conditions, the numbers of TH+ neurons were not significantly different compared with those from mice treated with MPTP alone or with MPTP and scrambled peptide. In addition, as expected, density of striatal termini remained decreased with antagonist treatment (Fig. 3E). To determine the neuroprotective efficacy, we administered graded doses of LBT-3627 (1.5, 5, 15, 45, or 90 μg/dose; Fig. 3F). Compared with MPTP treatment alone, the 1.5 μg/dose failed to elicit neuroprotection. Nonetheless, linear regression analysis suggests that there is a dose-dependent effect in TH+ neuronal sparing with treatment at higher doses (R2 = 0.4614, p = 0.001). The lack of increased protection observed at the 90 μg dose and the high variation observed at 45 μg suggested the attainment of a putative toxic threshold.
Figure 3.
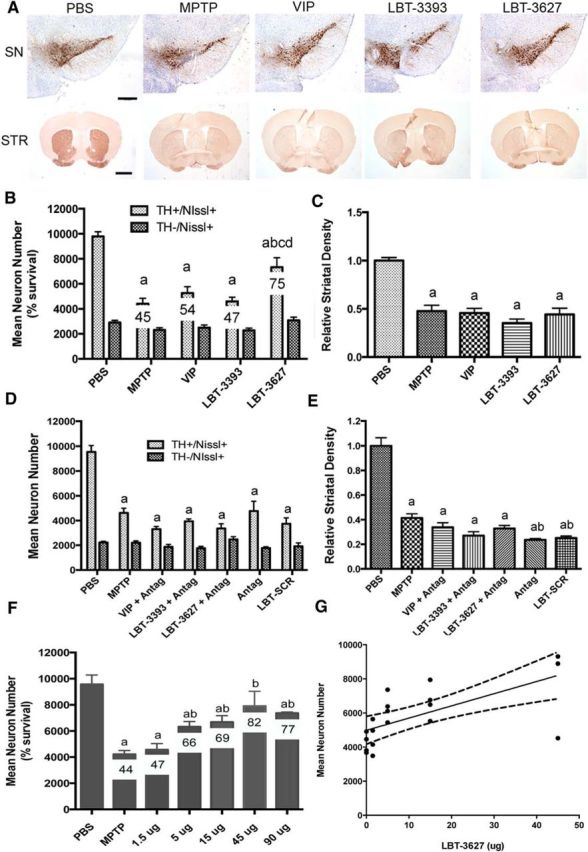
LBT-3627 pretreatment is neuroprotective in vivo. A, Photomicrographs of TH+Nissl+ neurons in the SN and TH+ striatal termini (STR) in mice treated with PBS, MPTP, or pretreated with VIP, LBT-3393, or LBT-3627 before MPTP intoxication (40× image; scale bar, 200 μm). Sections were immunostained with anti-TH and HRP-conjugated secondary antibody and visualized with DAB. SN sections were counterstained with thionin. B, Total numbers of surviving dopaminergic neurons (TH+Nissl+) and nondopaminergic neurons (TH−Nissl+) in the SN after MPTP treatment alone or pretreatment with VIP, LBT-3393, or LBT-3627. Percentages of spared dopaminergic neurons are included for each treatment (10× image; scale bar, 1000 μm). D, Total number of TH+Nissl+ and TH−Nissl+ neurons within the SN after MPTP intoxication alone or with pretreatment of [D-p-Cl-Phe6,Leu17]-VIP (Antag), scrambled peptide (LBT-SCR), or coadministration of VIP, LBT-3393, or LBT-3627 with antagonist. F, Dose–response for LBT-3627 at varying pretreatment doses of 1.5, 5, 15, 45, and 90 μg/dose followed by MPTP intoxication. G, Linear regression analysis of dose–response, R2 = 0.4614 p = 0.001. C, E, Relative TH densitometry of striatal dopaminergic termini after pretreatment. B–F, Differences in means (±SEM, n = 8) were determined where p < 0.05 compared with groups treated with PBS (a), MPTP (b), VIP (c), or LBT-3393 (d).
In a next series of experiments, we assessed the VIP-induced cellular response. Donor mice were treated with VIP, LBT-3393, or LBT-3627 for 5 d and splenocytes were isolated from donor mice and adoptively transferred to MPTP-intoxicated recipient mice. Dopaminergic neuronal numbers and striatal termini densities were assessed 7 d after MPTP intoxication by immunohistochemical analysis of TH+ nigral neurons and striatal termini (Fig. 4A). MPTP diminished TH+Nissl+ neuronal counts from 13011 ± 1025 to 3414 ± 382 (Fig. 4C). MPTP-induced neuronal loss was attenuated by adoptive transfer of splenocytes from nonintoxicated donor mice treated with the VIPR agonists showing significantly increased numbers of surviving dopaminergic neurons in mice receiving spleen cells from donors treated with LBT-3393 or LBT-3627, respectively, compared with those treated with MPTP alone. Transfer of splenocytes from VIP-treated donors increased dopaminergic neuronal survival by 14% and transfer using splenocytes from LBT-3393- or LBT-3627-treated mice increased dopaminergic neuronal survival by 25% and 54%, respectively. The numbers of nondopaminergic TH−Nissl+ neurons were not significantly different in any group treated with MPTP regardless of adoptive transfer. Compared with treatment with VIPR agonists before MPTP (Fig. 3C,E), striatal density analysis showed similar, nonsignificant effects for striatal sparing after adoptive transfer of splenocytes from mice treated with VIP or LBT-3393; however, in contrast, adoptive transfer of splenocytes from LBT-3627 treated donors showed significant sparing of striatal termini (Fig. 4D).
Figure 4.

Adoptive transfer of splenocytes from mice treated with VIPR agonists is neuroprotective. A, Photomicrographs of TH+Nissl+ neurons in the SN and STR in mice treated with PBS, MPTP, or MPTP followed by adoptive transfer of spleen cells from mice treated with VIP, LBT-3393, or LBT-3627 (40× image; scale bar, 200 μm). Sections were immunostained with anti-TH and HRP-conjugated secondary antibody and visualized with DAB. SN sections were counterstained with thionin. B, Representative photomicrographs of Mac-1+ microglia within the SN of mice treated with PBS alone, MPTP alone, or MPTP-treated mice receiving splenocytes from donors treated with VIP, LBT-3393, or LBT-3627 (40× image; scale bar, 200 μm; inset = 200× image). C, Total numbers of surviving dopaminergic neurons (TH+Nissl+) and nondopaminergic neurons (TH−Nissl+) in the SN after MPTP treatment and adoptive transfer (10× image; scale bar, 1000 μm). D, Relative TH densitometry of striatal dopaminergic termini. E, Quantification of reactive microglia taken from midbrains 2 d after MPTP treatment. Sections were stained for Mac-1+ microglia using an anti-Mac-1 antibody, HRP-conjugated secondary antibody, and DAB for color visualization. Numbers of reactive microglia (amoeboid Mac-1+) were determined by stereological analysis. C, D, Differences in means (±SEM, n = 8) were determined where p < 0.05 compared with groups treated with PBS (a), MPTP (b), VIP (c), or LBT-3393 (d). E, Differences in means (±SEM, n = 5) were determined where p < 0.05 compared with groups treated with PBS (a) or MPTP alone (b).
Microglial reactivity is decreased with VIPR2 agonism
As previously discussed, microglial activation is associated with neuronal cell death in human PD and in mouse models of disease (Kurkowska-Jastrzebska et al., 1999; McLaughlin et al., 2006). Whether the neuroprotective effects upon either adoptive transfer from agonist-treated mice or agonist pretreatment of mice before MPTP intoxication were associated with changes in microglial morphology and proinflammatory responses was investigated. As described previously, donor animals were treated for 5 d before splenic cell isolation and adoptive transfer to recipient MPTP-intoxicated mice (Fig. 4B). Microglial responses were evaluated 2 d after MPTP intoxication, at the peak of the MPTP-induced neuroinflammatory response (Koziorowski et al., 2012). The SN was immunostained with Mac-1 antibody and reactive microglia were quantified. Microglia exhibiting amoeboid morphology and intense Mac-1 expression were scored as reactive microglia, whereas those with reduced Mac-1 expression and ramified structures were considered quiescent microglia. Adoptive transfer of splenic cells from agonist-treated donors was performed 12 hours after MPTP intoxication of recipient animals. Replicate datasets revealed reduced microglial reactivity in the SN after transfer of splenocytes from donors treated with VIP or either VIPR agonist (Fig. 4B). Compared with PBS-treated animals, MPTP intoxication increased the number of reactive microglia from 2.2 ± 0.45 to 29.55 ± 6.9 cells/mm2 (Fig. 4E). Adoptive transfer of cells from animals treated with VIP significantly reduced the numbers of activated microglia to 18.68 ± 7.6 cells/mm2. Comparable transfers from animals treated with either LBT-3393 or LBT-3627 scored at 23.95 ± 5.7 and 18.3 ± 2.9 cells/mm2, respectively. However, only VIP or LBT-3627 treatment elicited significant decreases in activated microglial numbers compared with MPTP alone.
Assessment of reactive microglia was also performed directly in mice receiving peptide pretreatment (Fig. 5A). Stereological analysis showed that the numbers of activated microglia increased from 2.8 ± 1.14 cells/mm2 in PBS-treated mice to 44.5 ± 6.9 cells/mm2 after MPTP intoxication (Fig. 5A). VIP pretreatment decreased reactive microglia to 25.05 ± 2.2 cells/mm2 and VIPR1 agonism (LBT-3393) showed similar numbers of activated microglia (28.9 ± 3.3 cells/mm2). The largest reduction in MPTP-induced reactive microglia was seen after pretreatment with the VIPR2 agonist LBT-3627, which significantly decreased the activated cell numbers to 18.05 ± 7.4 cells/mm2. Moreover, treatment with LBT-3627 diminished numbers of reactive microglia to levels below those from mice treated with MPTP alone, but also to levels below those from MPTP-intoxicated mice treated with VIP or LBT-3393. Together, these findings indicate that VIPR agonism can reduce reactive microgliosis associated with MPTP intoxication along two different treatment schemes. These favorable effects may be associated with reduced inflammatory responses induced by VIP and VIPR agonists that in turn result in neuroprotection.
Figure 5.
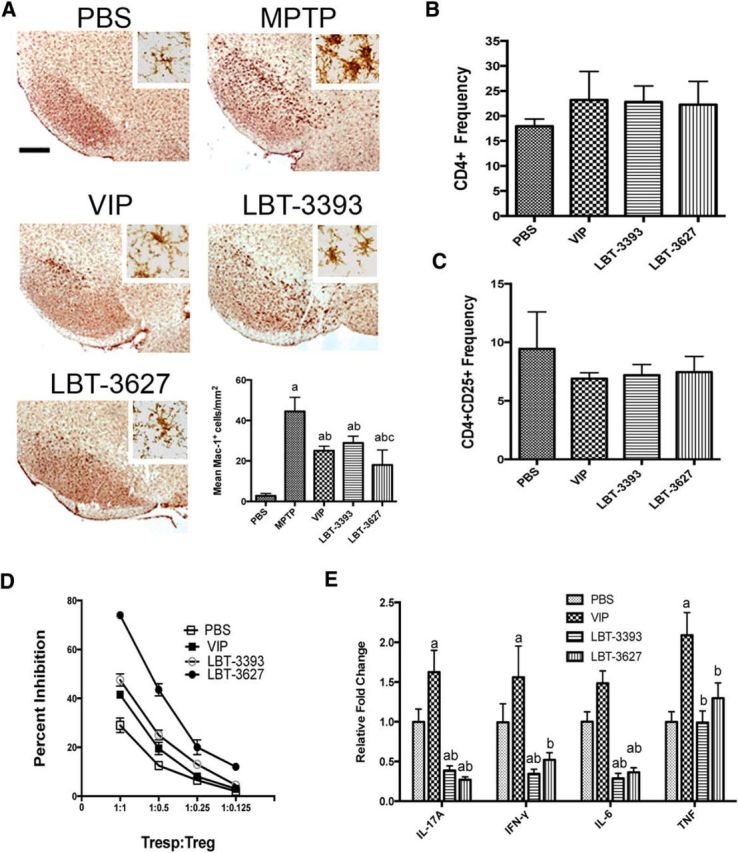
Pretreatment with VIPR agonists elicits changes in microglial reactivity and T-cell phenotypes during MPTP-induced inflammation. A, Photomicrographs of Mac-1+ microglia in the SN of PBS- or MPTP-treated mice pretreated with VIP, LBT-3393, or LBT-3627 (40× image; scale bar, 200 μm; inset = 200× image). Bottom right, Quantification of reactive microglia taken from midbrains 2 d after MPTP treatment, visualized as described in Figure 4. B, C, Mice were treated with PBS, VIP, LBT-3393, or LBT-3627 for 5 d and splenocytes assessed for frequencies of CD4+ T cells (B) and CD4+CD25+ Tregs (C). D, Assessment of Treg-mediated inhibition (±SEM) of CFSE-stained CD4+ Tresps that were stimulated for proliferation with anti-CD3/CD28. Tregs were isolated from PBS-, VIP-, LBT-3393-, or LBT-3627-treated mice. E, Relative concentration of proinflammatory cytokines in cell culture supernatants of anti-CD3-/CD28-stimulated CD4+ T cells after pretreatment (±SEM, n = 3). Cytokine concentrations were determined by cytokine bead array for pro-inflammatory and anti-inflammatory cytokines. A, Differences in means (± SEM, n = 6) were determined where p < 0.05 compared with groups treated with PBS (a), MPTP (b), or LBT-3393 (c). E, Differences in means (±SEM, n = 3) were determined where p < 0.05 compared with groups treated with PBS (a) or VIP (b).
Neuroprotective mechanisms for VIPR agonists
We showed that VIP treatment expands Treg numbers and elicits anti-inflammatory and immune-suppressive responses by modulating immune cell profiles and phenotypes (Reynolds et al., 2010). Therefore, to understand the immune-modulating potential of VIPR agonism, we assessed the ability of VIPR agonists to affect the levels of CD4+ T-cell populations and the function CD4+CD25+ Tregs and/or to modulate cytokine production. Flow cytometric analysis of total lymphocyte populations recovered from animals after 5 d of agonist treatment revealed no significant changes in either CD4+ (Fig. 5B) or CD4+CD25+ T-cell frequencies within the total lymphocyte population (Fig. 5C). We next evaluated the Treg function after peptide treatment as the capacity to inhibit CD3/CD28-stimulated proliferation of CD4+ T responder cells (Tresps) using a CFSE proliferation assay (Quah and Parish, 2010; Saunders et al., 2012). Tregs isolated from animals treated with LBT-3627 showed an increased functional capacity compared with Tregs isolated from animals treated with PBS, VIP, or LBT-3393 (Fig. 5D). Tregs from LBT-3627 treatment afforded a 74% inhibition of proliferation at a 1:1 Tresp:Treg ratio, whereas Treg-mediated inhibition was 29%, 41.5%, and 47.5% from mice treated with PBS, VIP, or LBT-3393, respectively. The inhibitory capacity of isolated Tregs decreased in a dose-dependent manner (R2 > 0.88, p < 0.0005, for all treatments); however, LBT-3627 Tregs were able to maintain enhanced suppressive capabilities even at the lowest dose compared with all other treatment arms. Linear regression analyses of Treg-mediated inhibition indicated that Treg dose responses from LBT-3627-treated mice were significantly enhanced (p < 0.03) over those from mice treated with PBS, VIP, or LBT-3393. Dose responses of Tregs from mice treated with LBT-3393 were significantly larger than those from PBS-treated controls (p = 0.016), whereas Treg responses from VIP-treated mice compared with PBS controls did not reach significance (p = 0.0788). Collectively, these data suggest that Treg frequencies are not affected by treatment with VIPR agonists, but functional properties of Tregs are enhanced upon these treatments, with the most pronounced enhancement resulting from the VIPR2-selective agonism.
Next, to determine the mechanism(s) by which VIPR agonists could enhance Tresp suppression and diminish inflammation with neuroprotective effects, we evaluated the effects of VIPR agonists on cytokine production after T-cell stimulation. For these studies, mice were treated with PBS, VIP, LBT-3393, or LBT-3627 for 5 d. CD4+ spleen cells from each treatment arm were isolated, stimulated with anti-CD3/CD28, cultured, and assessed for cytokine production by cytokine bead array. Relative to cytokine levels of stimulated CD4+ T cells from PBS-treated controls, LBT-3393 and LBT-3627 treatment significantly suppressed the production of the proinflammatory cytokines IL-17A, IFN-γ, and IL-6, but not TNF-α (Fig. 5E). Interestingly, VIP administration resulted in an opposite effect, with a significant upregulation of proinflammatory cytokines. This may be due to the fact that VIP is ∼10-fold more potent than the selective VIPR agonists, suggesting that increased potency may yield an undesirable effect on proinflammatory cytokine production. Together, these data demonstrated that selective agonism of either VIPR1 or VIPR2 induces downregulation of proinflammatory T-cell phenotypes and results in the enhanced immunosuppressive properties observed by Tregs. Observation of such T-cell shifts in phenotype within 12 h of stimulation also suggested that this VIPR-agonist-mediated shift may occur before stimulation via CD3/CD28.
Because Treg numbers or cytokine production alone could not readily explain differences in the preferential effects on Treg function and neuroprotection mediated by VIPR2 versus VIPR1 agonism, we next examined potential phenotypic shifts elicited by VIPR agonists. We investigated T-helper differentiation gene expression changes within the total CD4+ T-cell population. CD4+ T cells were isolated from animals treated for 5 d with PBS, VIP, LBT-3393, or LBT-3627; RNA was isolated; and gene expression evaluated by RT-PCR array for genes associated with T helper cell differentiation. Gene expression levels of T cells from mice treated with VIP, LBT-3393, or LBT-3627 were compared with those from animals treated with PBS alone. Significant fold changes in mRNA expression for each pretreatment are listed (Fig. 6A); each of the three VIPR agonists produces a distinct profile change. For VIP pretreatment, transmembrane emp24 protein transport domain containing 1 (Tmed1) was significantly upregulated, whereas Fas ligand (FasL), IL-18 receptor accessory protein (Il-18rap), IL-21, interferon regulatory factor 4 (Irf-4), Rel, and suppressor of cytokine signaling 5 (Socs5) were significantly downregulated. VIPR1 agonist pretreatment yielded a downregulation in Irf-4, Rel, and Tmed1. VIPR2 agonist pretreatment elicited a robust and significant increase in Gm-csf, as well as parallel increases in IL-17 receptor E (Il-17re), IL-18 receptor 1 (Il-18r1), RAR-related orphan receptor C (Rorc), Toll-like receptor 4 (Tlr4), and Tmed1. Il-21, inhibitor of DNA 2 (Id2), and Socs5 were increased as well, but these increases did not reach statistical significance. VIPR2 agonism did not lead to significant downregulation of any genes associated with T-cell differentiation.
Figure 6.
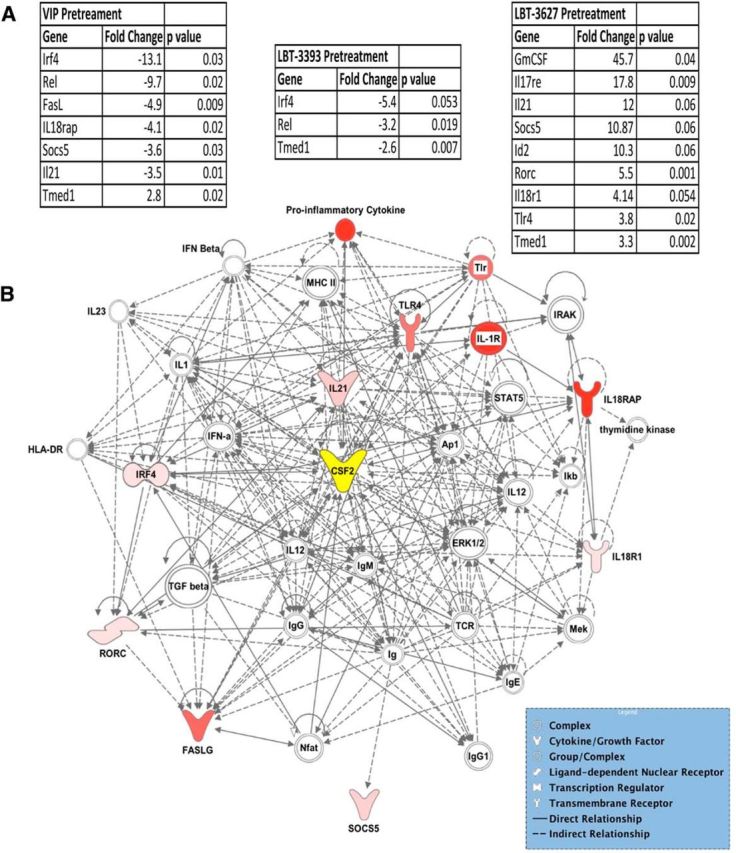
VIPR2 agonism induces dysregulation of genes associated with inflammatory responses and cell-to-cell signaling. PBS controls received no treatment, whereas VIP, LBT-3393, and LBT-3627 treatment groups were pretreated for 5 d followed by negative selection of the CD4+ T-cell subset. qRT-PCR data show gene expression changes by CD4+ T cells of mice pretreated with VIPR agonists (n = 3). Fold changes and p-values were determined using SABioscience RT2 Profiler PCR Array Data Analysis software, version 3.5. A, Fold changes and p-values for differentially regulated mRNA levels with VIP, LBT-3393, or LBT-3627 treatment normalized to PBS controls. B, Direct and indirect pathways using T-cell gene modulations that were significantly dysregulated with LBT-3627 treatment compared with the PBS treatment group. Resulting gene networks from each treatment group were analyzed using Qiagen IPA. Pink coloration indicates a modest increase in expression compared with PBS control and red indicates a profound increase in expression. Nodes lacking color indicate a molecule involved in the pathway, but not identified in the PCR dataset. The central node, CSF-2, or GM-CSF is indicated in yellow. Differences in fold change (n = 3) were determined where p < 0.05.
Because the majority of the detected neuroprotective and anti-inflammatory responses were associated with VIPR2 agonism, we focused on relationships among genes for which mRNA expression increased upon pretreatment with the VIPR2-selective agonist LBT-3627. Mapping the relationships by IPA among genes for which mRNA increased suggests that changes in two overlapping networks are induced by VIPR2 agonism (Fig. 6B). The changes implicate immunological disease and inflammatory response networks and cell-to-cell signaling and interaction networks. Changes in Gm-csf (CSF2) are central to these networks. Changes linked to both anti-inflammatory and proinflammatory genes, as well as innate and adaptive immunity, were observed, along with changes in T-cell transcription factors associated with T-cell differentiation. Furthermore, because T cells readily interact with microglial populations to elicit an immune response, we next sought to examine the effect of VIPR2 agonism on CD11b+ cell populations isolated from the ventral midbrain of MPTP-intoxicated mice. Investigation of the inflammatory response mediated by CD11b+ populations was performed after pretreatment with both LBT-3627 alone (Fig. 7A) and in combination with MPTP (Fig. 7B). The results suggest a downregulation in multiple innate and adaptive immune mediators with LBT-3627 treatment. Specifically, with LBT-3627 treatment alone, mRNA transcripts for genes associated with a proinflammatory and/or oxidative immune response, such as Il-1β, Il-23a, Ifn-γ, Ptgs2 (prostaglandin-endoperoxide synthase 2, also known as cyclooxygenase), Ltb (lymphotoxin B also known as TNF-C), Tnf, and Il-7, were profoundly decreased with a decrease of >10 fold compared with the inflammatory response associated with MPTP treatment alone (Fig. 7A). In combination with MPTP intoxication, LBT-3627 pretreatment yielded similar decreases in the proinflammatory response, showing moderate decreases in Ptgs2 and Ltb (at least a 2-fold decrease), as well as greater decreases in Tnf, Il-7, and NFκB expression (>10-fold decreases compared with MPTP intoxication alone). Therefore, the genomic analyses revealed that VIPR agonists, especially the agonist specific for VIPR2, can positively affect both innate and adaptive immune responses through modulation of gene expression in CD4+ T cells and CD11b+ microglial populations, with a coincident downregulation of proinflammatory cytokine production in vitro.
Figure 7.

Targeting VIPR2 with LBT-3627 changes inflammation-associated gene expression in microglial populations. CD11b+ microglial populations from the ventral midbrain of eight mice were collected after treatment with MPTP alone, LBT-3627 pretreatment + MPTP, or LBT-3627 treatment only. Gene expression changes within the CD11b+-enriched population were assessed using qRT-PCR (Inflammatory Response and Autoimmunity PCR profiler array) in each experimental group. Fold changes were determined using MPTP alone as the control. A, Fold changes for differentially regulated mRNA levels in response to LBT-3627 treatment only. B, Fold changes for mRNA levels in response to LBT-3627 pretreatment followed by MPTP intoxication normalized to MPTP alone. Dark red coloration indicates a profound increase compared with MPTP, pink indicates a modest increase, light green indicates a modest decrease, and dark green indicates a profound decrease in expression.
VIPR agonists do not affect MPTP metabolism
VIP can cross the blood–brain barrier (BBB) (Dogrukol-Ak et al., 2003) and possibly inhibit metabolism of the MPTP protoxin into the active MPP+ toxin, resulting in potential neuroprotection due to diminished intoxication. To rule out that possibility, we analyzed levels of MPTP and MPP+ by RP-HPLC within the midbrain and striatum of intoxicated mice and compared them with levels in intoxicated mice that received VIP, LBT-3393, or LBT-3627 before MPTP. Treatment with VIP or either VIPR agonist did not reduce the levels of MPP+ intoxicant in either the midbrain (Fig. 8A) or the striatum (Fig. 8B). Therefore, conversion from MPTP into MPP+ was achieved in all treatment arms and, in fact, MPP+ levels were greater in mice treated with either VIPR agonist before MPTP. Therefore, these data and the adoptive transfer data support the idea that VIPR2-agonist-mediated neuroprotective responses do not arise from effects on MPTP metabolism, which is consistent with a more direct immunomodulatory mechanism of action.
Figure 8.
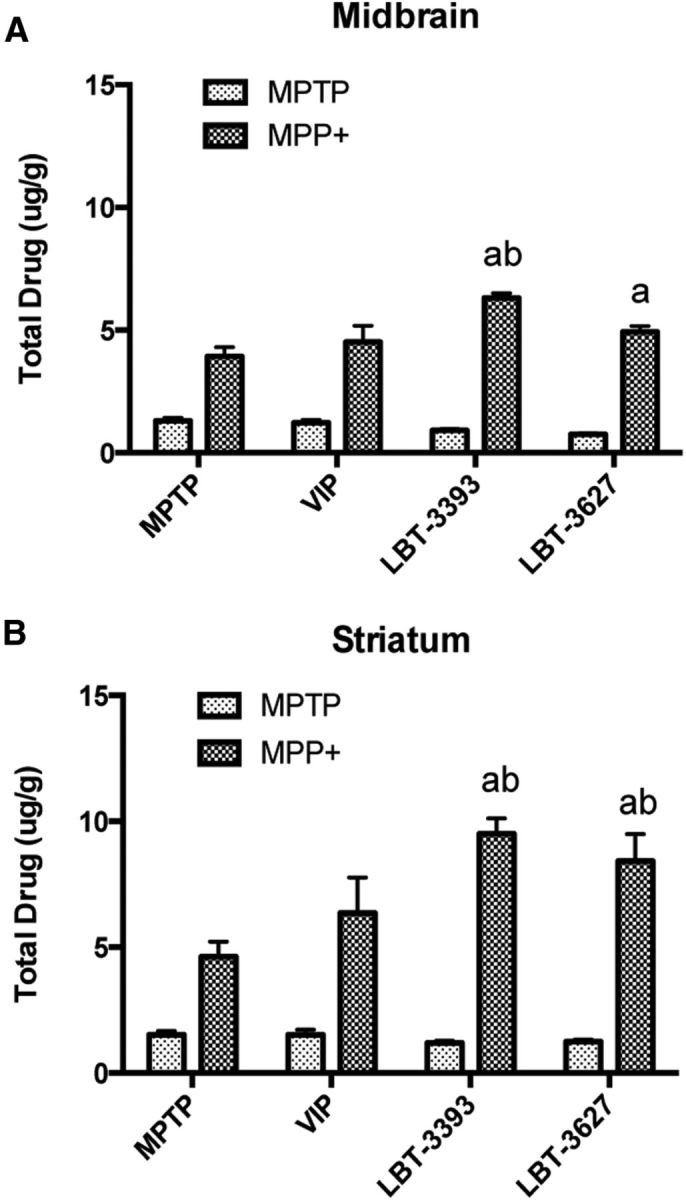
MPTP metabolism in the midbrain and striatum is not diminished by pretreatment with VIPR agonists. Quantification of total MPTP and MPP+ levels in μg/g tissue in the midbrain (A) and striatum (B) of mice pretreated with VIP, LBT-3393, or LBT-3627 followed by MPTP intoxication. Tissues were collected at 90 min after the final MPTP injection. Differences in means (±SEM, n = 5) were determined where p < 0.05 compared with groups treated with MPTP only (a) or VIP (b).
Discussion
VIP has potential for the treatment of neuroinflammatory conditions based on its ability to transform T cells (Gonzalez-Rey et al., 2007). An obstacle to the clinical use of VIP is the hormone's lack of specificity for VIPR1 and VIPR2, as well as its rapid degradation (Usdin et al., 1994; Reubi, 2003). Our work aimed to better define therapeutic potential in this arena by developing protease-resistant VIPR agonists that target each receptor independently. We hypothesized that targeting specific VIP receptors individually would elicit a robust neuroprotective response connected to changes in innate and adaptive immunity. To achieve these goals, peptide modifications were made based on prior studies showing that periodic α→β replacements in the C-terminal portions of other peptide hormones that act on B-family GPCRs, such as glucagon-like peptide-1 (GLP-1) (7–37) and parathyroid hormone (PTH) (1–34), can yield potent agonists with prolonged activity in vivo relative to the endogenous proteins (Cheloha et al., 2014; Johnson et al., 2014). To this end, we made a metabolically stable and VIPR2-specific agonist, LBT-3627, and showed that it was an effective immunomodulatory agent in a disease-relevant PD model. Treatment of MPTP-intoxicated mice with LBT-3627 significantly spared dopaminergic neuronal cell bodies, decreased the amount of reactive microgliosis, decreased levels of proinflammatory gene expression associated with the inflammatory response in CD11b+ microglia populations, downregulated proinflammatory cytokine production, and modulated T-cell phenotypes with treatment. In contrast, treatment with the stable, VIPR1-selective agonist LBT-3393 yielded only lesser neuroprotective responses. This observation may be explained by increased proteolysis during administration compared with LBT-3627 alone, as observed in Figure 2. Therefore, in our model of PD, VIPR2 agonism elicits a link between neuroprotection and modulation of the immune response with systemic treatment. Interestingly, significant sparing of striatal termini was only observed after adoptive transfer, not during pretreatment. We posit that the lack of termini survival may be due to increased MPP+ levels associated with VIPR pretreatment, whereas the increased MPTP conversion would not have been observed with adoptive transfer because no drug interaction would have been possible. The other possibility is that MPTP/MPP+ has been shown to be toxic to lymphoid cell populations and MPTP treatment of pretreated mice may partially diminish VIPR-agonist-induced T cells, whereas adoptively transferred T cells would not meet the same fate being transferred 12 h after intoxication (Benner et al., 2004).
Specific targeting of VIPR2 has been shown to mediate anti-inflammatory and therapeutic effects in rheumatoid arthritis (Juarranz et al., 2008) and models of spinal muscular atrophy (Hadwen et al., 2014), ultimately leading to enhanced production of Th2-type transcription factors such as c-Maf and JunB, anti-inflammatory cytokines such as IL-4 and IL-5 (Voice et al., 2004) and IL-10 (Larocca et al., 2007), and decreased macrophage-derived proinflammatory cytokine production (Delgado et al., 2000). In addition, the loss of VIPR2 leads to enhanced and exacerbated disease states in experimental autoimmune encephalitis and experimental colitis, suggesting that VIPR2 activity is important for mediating disease processes (Yadav et al., 2011; Tan et al., 2015). This loss was associated with increased Th1/Th17 responses and decreased Th2/Treg responses (Yadav et al., 2008; Tan et al., 2015). These previous findings are consistent with our observation of enhanced neuroprotective activity of a selective VIPR2 agonist relative to a selective VIPR1 agonist or the nonselective native hormone VIP. Our work highlights the potential of a VIPR2-selective agonist to modulate the adaptive immune response in therapeutically favorable ways.
Initially, we hypothesized that VIPR2 agonism would yield an increase in the CD4+CD25+ Treg population, resulting in increased neuroprotection. Even though prior work has shown VIP-mediated increases in frequencies of Tregs (Fernandez-Martin et al., 2006; Reynolds et al., 2010; Fraccaroli et al., 2015), we were unable to demonstrate this response at the dose we used, so we began to assess other avenues of regulatory function that promote dopaminergic neuron survival upon MPTP intoxication. Therefore, we assessed the ability of Tregs isolated from VIPR agonist-treated mice to suppress the proliferation of Tresp in vitro. We found that coculture of Tresps with Tregs from animals treated with the VIPR2-selective agonist LBT-3627 elicited an enhanced suppressive effect compared with Tregs from animals subjected to other treatments. This suppressive effect corresponded with downregulation of proinflammatory cytokine production associated with Th1/Th17 T-cell phenotypes. The regulatory and immune-suppressive roles of LBT-3627 are likely a result of T-cell phenotype modulation leading to a change in the adaptive immune response associated with inflammation.
The impact of VIPR agonists on MPTP-intoxicated mice led us to use gene expression analysis for further exploration of T-cell differentiation and possible phenotypic shifts induced by agonist treatment. We observed increases in genes associated with both proinflammatory and anti-inflammatory responses. The most abundant change was a 45-fold upregulation of Gm-csf transcript. Previously, we and others have demonstrated the potent and robust neuroprotective responses associated with GM-CSF treatment, such as increased neuronal survival, decreased microglial reactivity, induction of Tregs, and changes in innate and adaptive immune responses associated with inflammation (Schäbitz et al., 2008; Kosloski et al., 2013). Although genes associated with multiple T-cell subsets were altered upon LBT-3627 treatment, many were associated with anti-inflammatory subsets including Tregs and Th2 populations. Id2 was upregulated 10-fold with treatment. This gene has been associated with maintaining Treg populations in inflammatory disease to enhance suppressive capabilities (Miyazaki et al., 2014). Likewise, Tmed1 was upregulated 3.3-fold compared with PBS treatment. Tmed1 has been shown to be involved in IL-33 signaling (Connolly et al., 2013); IL-33 is an IL-1-like cytokine that induces Th2 type cytokines such as IL-4, IL-5, and IL-13 (Schmitz et al., 2005). Although mRNA levels for these Th2-associated cytokines were upregulated >2-fold with LBT-3627, the modest changes were not significant for the current sample size. Small changes in cytokine levels can ultimately affect T-cell subsets depending on the microenvironment at the sites of inflammation. The expression level changes we observed may suggest that LBT-3627 induces a shift toward an anti-inflammatory response associated with regulatory subsets rather than a proinflammatory, Th1/Th17-mediated response. This possibility is supported by the downregulation of Th1/Th17 cytokine production after treatment. Upon cytometric bead analysis, we found a significant downregulation of IFN-γ, IL-6, and IL-17A with LBT-3627 treatment. The VIPR2-selective agonist also caused a >2-fold decrease in IFN-γ gene transcripts, although the effect did not reach significance. Collectively, we showed alterations in inflammatory disease response and cell–cell signaling pathways after VIPR2 agonism. Gm-csf is a central component for both networks and its upregulation can elicit changes linked to both innate and adaptive immunity (Kosloski et al., 2013; Kelso et al., 2015). This ability to modulate the immune response was strengthened with the observed changes in inflammatory response transcripts in microglia populations isolated from the ventral midbrain. Treatment with LBT-3627 alone elicited profound downregulation of mRNA levels of potent proinflammatory mediators such as IL-1β, TNF-α, TNF-c, COX, and IFN-γ. Given the enhanced inflammatory cascade that occurs during MPTP intoxication, it is important to note that pretreatment with LBT-3627 followed by MPTP intoxication was also able to elicit changes in some of the same detrimental mediators, such as decreased COX, TNF-c, and TNF-α. Collectively, these findings could be indicative of cross talk between CD4+ T-cell populations and their interactions with CD11b+ microglia populations, creating a shift in responses that ultimately leads to enhanced neuroprotective capabilities.
VIP can cross the BBB (Dogrukol-Ak et al., 2003), which may allow direct interaction with resident cells within the CNS. Both glia cells and neurons express VIP receptors (Chneiweiss et al., 1985; Hösli and Hösli, 1989) and VIPRs are expressed within the SN (Joo et al., 2004). Activation of VIPR1 and/or VIPR2 within the CNS could lead to other protective effects that are not directly associated with the modulation of T-cell responses and/or phenotypes. VIP binding to its receptors on microglia and other antigen-presenting cells has been associated with downregulation of costimulatory molecules, possibly resulting in desensitization of an inflammatory response (Ganea et al., 2003), as well as inhibition of proinflammatory cytokine production by microglia (Kim et al., 2000). VIP interaction with astrocytes results in increased neurotrophin production leading to decreased cellular toxicities and increased neuronal survival (Gozes and Brenneman, 1996). VIP interaction with neurons has been documented as well, causing VIP to be widely accepted as a neuropeptide for neuronal signaling and regulation of reactive gliosis (Brenneman and Foster, 1987; Waschek, 2013).
We have not determined whether LBT-3393 or LBT-3627 can cross the BBB. It is possible that these agonists share the ability of VIP to cross the BBB and act directly on microglia, astrocytes, and neurons. Further exploration of the effects of LBT-3393 and LBT-3627 on cell types within and outside the CNS will be necessary to elucidate the immunomodulatory and neuroprotective effects manifested by these compounds in the MPTP mouse model of PD.
Due to its influence on the immune system, we used the MPTP acute inflammatory model of PD. Alternatives could include the preformed α-synuclein model that allows Lewy body aggregate formation, a clinical hallmark of PD (Volpicelli-Daley et al., 2011; Dehay, 2012). MPTP intoxication causes nigrostriatal degeneration with an accompanying neuroinflammatory response associated with dopaminergic neuronal loss with the opportunity to effectively study immunomodulatory neuroprotective therapies that were relevant in the current study. Indeed, the results reported in this MPTP model show that systemic administration of a VIPR2-selective agonist elicited profound neuroprotective and anti-inflammatory responses, supporting further exploration from a clinical perspective. Our evaluation has been limited to an animal model, but we hypothesize that such agents would elicit similar anti-inflammatory responses and T-cell phenotypic shifts in humans. Such agonists might counteract the imbalanced neuroinflammatory response associated with neurodegeneration in PD. By enhancing the suppressive function of Tregs and downregulating proinflammatory cytokine production, a long-acting VIPR2-selective agonist might restore proinflammatory and anti-inflammatory responses to a homeostatic state, ultimately sparing dopaminergic neurons. Overall, we provide strong evidence that VIPR2 agonism has the potential to slow the pathogenesis of PD through modulation of the inflammatory response.
Footnotes
This work was supported by the National Institutes of Health Grants P01 DA028555, R01 NS036126, P01 NS031492, 2R01 NS034239, P01 MH064570, P01 NS043985, P30 MH062261, and R01 AG043540 to H.E.G; R01 NS070190 to R.L.M.; DOD Grant 421-20-09A to H.E.G.; the Michael J. Fox Foundation to S.J.S. and H.E.G.; K.E.O. was supported by a Graduate Student Fellowship from the University of Nebraska Medical Center, Graduate Studies Office. We thank the University of Nebraska Medical Center Flow Cytometry Research Facility for flow cytometric data acquisition and technical support and Diana Palandri for excellent technical support and data acquisition using the RP-HPLC, Peter Berget in the Biological Sciences Department at USciences for use of his TECAN infinite M1000, and Lisa Davis in the Pharmacology Department at USciences for use of her AB Science 4000.
B.E.C., B.J.D., J.M.G., and S.J.S. are employees of Longevity Biotech, Inc., which is developing LBT-3627 as a clinical candidate. The remaining authors declare no competing financial interests.
References
- Abad C, Martinez C, Juarranz MG, Arranz A, Leceta J, Delgado M, Gomariz RP. Therapeutic effects of vasoactive intestinal peptide in the trinitrobenzene sulfonic acid mice model of Crohn's disease. Gastroenterology. 2003;124:961–971. doi: 10.1053/gast.2003.50141. [DOI] [PubMed] [Google Scholar]
- Abad C, Juarranz Y, Martinez C, Arranz A, Rosignoli F, Garcia-Gómez M, Leceta J, Gomariz RP. cDNA array analysis of cytokines, chemokines, and receptors involved in the development of TNBS-induced colitis: homeostatic role of VIP. Inflamm Bowel Dis. 2005;11:674–684. doi: 10.1097/01.MIB.0000171872.70738.58. [DOI] [PubMed] [Google Scholar]
- Abad C, Tan YV, Lopez R, Nobuta H, Dong H, Phan P, Feng JM, Campagnoni AT, Waschek JA. Vasoactive intestinal peptide loss leads to impaired CNS parenchymal T-cell infiltration and resistance to experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2010;107:19555–19560. doi: 10.1073/pnas.1007622107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bas J, Calopa M, Mestre M, Molleví DG, Cutillas B, Ambrosio S, Buendia E. Lymphocyte populations in Parkinson's disease and in rat models of parkinsonism. J Neuroimmunol. 2001;113:146–152. doi: 10.1016/S0165-5728(00)00422-7. [DOI] [PubMed] [Google Scholar]
- Benner EJ, Mosley RL, Destache CJ, Lewis TB, Jackson-Lewis V, Gorantla S, Nemachek C, Green SR, Przedborski S, Gendelman HE. Therapeutic immunization protects dopaminergic neurons in a mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2004;101:9435–9440. doi: 10.1073/pnas.0400569101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, Nemachek C, Ciborowski P, Przedborski S, Mosley RL, Gendelman HE. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS One. 2008;3:e1376. doi: 10.1371/journal.pone.0001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma MD, Haase HS, Peterson-Kaufman KJ, Lee EF, Clarke OB, Colman PM, Smith BJ, Horne WS, Fairlie WD, Gellman SH. Evaluation of diverse alpha/beta-backbone patterns for functional alpha-helix mimicry: analogues of the Bim BH3 domain. J Am Chem Soc. 2012;134:315–323. doi: 10.1021/ja207148m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneman DE, Foster GA. Structural specificity of peptides influencing neuronal survival during development. Peptides. 1987;8:687–694. doi: 10.1016/0196-9781(87)90044-1. [DOI] [PubMed] [Google Scholar]
- Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, Duyckaerts C, Flavell RA, Hirsch EC, Hunot S. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119:182–192. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloha RW, Maeda A, Dean T, Gardella TJ, Gellman SH. Backbone modification of a polypeptide drug alters duration of action in vivo. Nat Biotechnol. 2014;32:653–655. doi: 10.1038/nbt.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Hao J, Xi Y, Wang W, Wang Z, Li N, Li W. The therapeutic effect of vasoactive intestinal peptide on experimental arthritis is associated with CD4+CD25+ T regulatory cells. Scand J Immunol. 2008;68:572–578. doi: 10.1111/j.1365-3083.2008.02178.x. [DOI] [PubMed] [Google Scholar]
- Chneiweiss H, Glowinski J, Prémont J. Vasoactive intestinal polypeptide receptors linked to an adenylate cyclase, and their relationship with biogenic amine- and somatostatin-sensitive adenylate cyclases on central neuronal and glial cells in primary cultures. J Neurochem. 1985;44:779–786. doi: 10.1111/j.1471-4159.1985.tb12883.x. [DOI] [PubMed] [Google Scholar]
- Choi WS, Kruse SE, Palmiter RD, Xia Z. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP+, or paraquat. Proc Natl Acad Sci U S A. 2008;105:15136–15141. doi: 10.1073/pnas.0807581105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly DJ, O'Neill LA, McGettrick AF. The GOLD domain-containing protein TMED1 is involved in interleukin-33 signaling. J Biol Chem. 2013;288:5616–5623. doi: 10.1074/jbc.M112.403899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B. New experimental approach for modeling Parkinson's disease. Mov Disord. 2012;27:344. doi: 10.1002/mds.24933. [DOI] [PubMed] [Google Scholar]
- Delgado M, Ganea D. Neuroprotective effect of vasoactive intestinal peptide (VIP) in a mouse model of Parkinson's disease by blocking microglial activation. FASEB J. 2003;17:944–946. doi: 10.1096/fj.02-0799fje. [DOI] [PubMed] [Google Scholar]
- Delgado M, Munoz-Elias EJ, Gomariz RP, Ganea D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide enhance IL-10 production by murine macrophages: in vitro and in vivo studies. J Immunol. 1999;162:1707–1716. [PubMed] [Google Scholar]
- Delgado M, Gomariz RP, Martinez C, Abad C, Leceta J. Anti-inflammatory properties of the type 1 and type 2 vasoactive intestinal peptide receptors: role in lethal endotoxic shock. Eur J Immunol. 2000;30:3236–3246. doi: 10.1002/1521-4141(200011)30:11%3C3236::AID-IMMU3236%3E3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Delgado M, Abad C, Martinez C, Leceta J, Gomariz RP. Vasoactive intestinal peptide prevents experimental arthritis by downregulating both autoimmune and inflammatory components of the disease. Nat Med. 2001;7:563–568. doi: 10.1038/87887. [DOI] [PubMed] [Google Scholar]
- Delgado M, Gonzalez-Rey E, Ganea D. VIP/PACAP preferentially attract Th2 effectors through differential regulation of chemokine production by dendritic cells. FASEB J. 2004a;18:1453–1455. doi: 10.1096/fj.04-1548fje. [DOI] [PubMed] [Google Scholar]
- Delgado M, Pozo D, Ganea D. The significance of vasoactive intestinal peptide in immunomodulation. Pharmacol Rev. 2004b;56:249–290. doi: 10.1124/pr.56.2.7. [DOI] [PubMed] [Google Scholar]
- Delgado M, Chorny A, Gonzalez-Rey E, Ganea D. Vasoactive intestinal peptide generates CD4+CD25+ regulatory T cells in vivo. J Leukoc Biol. 2005;78:1327–1338. doi: 10.1189/jlb.0605299. [DOI] [PubMed] [Google Scholar]
- Deng S, Xi Y, Wang H, Hao J, Niu X, Li W, Tao Y, Chen G. Regulatory effect of vasoactive intestinal peptide on the balance of Treg and Th17 in collagen-induced arthritis. Cell Immunol. 2010;265:105–110. doi: 10.1016/j.cellimm.2010.07.010. [DOI] [PubMed] [Google Scholar]
- Dickson L, Finlayson K. VPAC and PAC receptors: From ligands to function. Pharmacol Ther. 2009;121:294–316. doi: 10.1016/j.pharmthera.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Dogrukol-Ak D, Banks WA, Tunçel N, Tunçel M. Passage of vasoactive intestinal peptide across the blood-brain barrier. Peptides. 2003;24:437–444. doi: 10.1016/S0196-9781(03)00059-7. [DOI] [PubMed] [Google Scholar]
- Domschke S, Domschke W, Bloom SR, Mitznegg P, Mitchell SJ, Lux G, Strunz U. Vasoactive intestinal peptide in man: pharmacokinetics, metabolic and circulatory effects. Gut. 1978;19:1049–1053. doi: 10.1136/gut.19.11.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Martin A, Gonzalez-Rey E, Chorny A, Ganea D, Delgado M. Vasoactive intestinal peptide induces regulatory T cells during experimental autoimmune encephalomyelitis. Eur J Immunol. 2006;36:318–326. doi: 10.1002/eji.200535430. [DOI] [PubMed] [Google Scholar]
- Fiszer U, Mix E, Fredrikson S, Kostulas V, Olsson T, Link H. gamma delta+ T cells are increased in patients with Parkinson's disease. J Neurol Sci. 1994;121:39–45. doi: 10.1016/0022-510X(94)90154-6. [DOI] [PubMed] [Google Scholar]
- Fraccaroli L, Grasso E, Hauk V, Paparini D, Soczewski E, Mor G, Pérez Leirós C, Ramhorst R. VIP boosts regulatory T cell induction by trophoblast cells in an in vitro model of trophoblast-maternal leukocyte interaction. J Leukoc Biol. 2015;98:49–58. doi: 10.1189/jlb.1A1014-492RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganea D, Rodriguez R, Delgado M. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide: players in innate and adaptive immunity. Cell Mol Biol. 2003;49:127–142. [PubMed] [Google Scholar]
- Gonzalez-Rey E, Varela N, Chorny A, Delgado M. Therapeutical approaches of vasoactive intestinal peptide as a pleiotropic immunomodulator. Curr Pharm Des. 2007;13:1113–1139. doi: 10.2174/138161207780618966. [DOI] [PubMed] [Google Scholar]
- Gozes I, Brenneman DE. Activity-dependent neurotrophic factor (ADNF): an extracellular neuroprotective chaperonin? J Mol Neurosci. 1996;7:235–244. doi: 10.1007/BF02737061. [DOI] [PubMed] [Google Scholar]
- Ha D, Stone DK, Mosley RL, Gendelman HE. Immunization strategies for Parkinson's disease. Parkinsonism Relat Disord. 2012;18:S218–S221. doi: 10.1016/S1353-8020(11)70067-0. [DOI] [PubMed] [Google Scholar]
- Hadwen J, MacKenzie D, Shamim F, Mongeon K, Holcik M, MacKenzie A, Farooq F. VPAC2 receptor agonist BAY 55–9837 increases SMN protein levels and moderates disease phenotype in severe spinal muscular atrophy mouse models. Orphanet J Rare Dis. 2014;9:4. doi: 10.1186/1750-1172-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne WS, Price JL, Gellman SH. Interplay among side chain sequence, backbone composition, and residue rigidification in polypeptide folding and assembly. Proc Natl Acad Sci U S A. 2008;105:9151–9156. doi: 10.1073/pnas.0801135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hösli E, Hösli L. Autoradiographic localization of binding sites for vasoactive intestinal peptide and angiotensin II on neurons and astrocytes of cultured rat central nervous system. Neuroscience. 1989;31:463–470. doi: 10.1016/0306-4522(89)90388-6. [DOI] [PubMed] [Google Scholar]
- Huang X, Reynolds AD, Mosley RL, Gendelman HE. CD 4+ T cells in the pathobiology of neurodegenerative disorders. J Neuroimmunol. 2009;211:3–15. doi: 10.1016/j.jneuroim.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi H, Ito T, Pradhan TK, Mantey SA, Hou W, Coy DH, Jensen RT. Elucidation of the vasoactive intestinal peptide pharmacophore for VPAC(2) receptors in human and rat and comparison to the pharmacophore for VPAC(1) receptors. J Pharmacol Exp Ther. 2002a;303:445–460. doi: 10.1124/jpet.102.038075. [DOI] [PubMed] [Google Scholar]
- Igarashi H, Ito T, Hou W, Mantey SA, Pradhan TK, Ulrich CD, 2nd, Hocart SJ, Coy DH, Jensen RT. Elucidation of vasoactive intestinal peptide pharmacophore for VPAC(1) receptors in human, rat, and guinea pig. J Pharmacol Exp Ther. 2002b;301:37–50. doi: 10.1124/jpet.301.1.37. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Przedborski S. Protocol for the MPTP mouse model of Parkinson's disease. Nat Protoc. 2007;2:141–151. doi: 10.1038/nprot.2006.342. [DOI] [PubMed] [Google Scholar]
- Johnson LM, Gellman SH. alpha-Helix mimicry with alpha/beta-peptides. Methods Enzymol. 2013;523:407–429. doi: 10.1016/B978-0-12-394292-0.00019-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LM, Barrick S, Hager MV, McFedries A, Homan EA, Rabaglia ME, Keller MP, Attie AD, Saghatelian A, Bisello A, Gellman SH. A potent alpha/beta-peptide analogue of GLP-1 with prolonged action in vivo. J Am Chem Soc. 2014;136:12848–12851. doi: 10.1021/ja507168t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo KM, Chung YH, Kim MK, Nam RH, Lee BL, Lee KH, Cha CI. Distribution of vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide receptors (VPAC1, VPAC2, and PAC1 receptor) in the rat brain. J Comp Neurol. 2004;476:388–413. doi: 10.1002/cne.20231. [DOI] [PubMed] [Google Scholar]
- Juarranz Y, Gutiérrez-Cañas I, Santiago B, Carrión M, Pablos JL, Gomariz RP. Differential expression of vasoactive intestinal peptide and its functional receptors in human osteoarthritic and rheumatoid synovial fibroblasts. Arthritis Rheum. 2008;58:1086–1095. doi: 10.1002/art.23403. [DOI] [PubMed] [Google Scholar]
- Kelso ML, Elliott BR, Haverland NA, Mosley RL, Gendelman HE. Granulocyte-macrophage colony stimulating factor exerts protective and immunomodulatory effects in cortical trauma. J Neuroimmunol. 2015;278:162–173. doi: 10.1016/j.jneuroim.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WK, Kan Y, Ganea D, Hart RP, Gozes I, Jonakait GM. Vasoactive intestinal peptide and pituitary adenylyl cyclase-activating polypeptide inhibit tumor necrosis factor-alpha production in injured spinal cord and in activated microglia via a cAMP-dependent pathway. J Neurosci. 2000;20:3622–3630. doi: 10.1523/JNEUROSCI.20-10-03622.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korendovych IV, Kim YH, Ryan AH, Lear JD, Degrado WF, Shandler SJ. Computational design of a self-assembling beta-peptide oligomer. Org Lett. 2010;12:5142–5145. doi: 10.1021/ol102092r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkmaz O, Ay H, Ulupinar E, Tunçel N. Vasoactive intestinal peptide enhances striatal plasticity and prevents dopaminergic cell loss in Parkinsonian rats. J Mol Neurosci. 2012;48:565–573. doi: 10.1007/s12031-012-9781-x. [DOI] [PubMed] [Google Scholar]
- Kosloski LM, Ha DM, Hutter JA, Stone DK, Pichler MR, Reynolds AD, Gendelman HE, Mosley RL. Adaptive immune regulation of glial homeostasis as an immunization strategy for neurodegenerative diseases. J Neurochem. 2010;114:1261–1276. doi: 10.1111/j.1471-4159.2010.06834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosloski LM, Kosmacek EA, Olson KE, Mosley RL, Gendelman HE. GM-CSF induces neuroprotective and anti-inflammatory responses in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxicated mice. J Neuroimmunol. 2013;265:1–10. doi: 10.1016/j.jneuroim.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziorowski D, Tomasiuk R, Szlufik S, Friedman A. Inflammatory cytokines and NT-proCNP in Parkinson's disease patients. Cytokine. 2012;60:762–766. doi: 10.1016/j.cyto.2012.07.030. [DOI] [PubMed] [Google Scholar]
- Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurkowska-Jastrzebska I, Wrońska A, Kohutnicka M, Czlonkowski A, Czlonkowska A. The inflammatory reaction following 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine intoxication in mouse. Exp Neurol. 1999;156:50–61. doi: 10.1006/exnr.1998.6993. [DOI] [PubMed] [Google Scholar]
- Larocca L, Calafat M, Roca V, Franchi AM, Leirós CP. VIP limits LPS-induced nitric oxide production through IL-10 in NOD mice macrophages. Int Immunopharmacol. 2007;7:1343–1349. doi: 10.1016/j.intimp.2007.05.017. [DOI] [PubMed] [Google Scholar]
- Lee HS, LePlae PR, Porter EA, Gellman SH. An efficient route to either enantiomer of orthogonally protected trans-3-aminopyrrolidine-4-carboxylic acid. J Org Chem. 2001;66:3597–3599. doi: 10.1021/jo001534l. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Akiyama H, McGeer EG. Rate of cell death in parkinsonism indicates active neuropathological process. Ann Neurol. 1988;24:574–576. doi: 10.1002/ana.410240415. [DOI] [PubMed] [Google Scholar]
- McLaughlin P, Zhou Y, Ma T, Liu J, Zhang W, Hong JS, Kovacs M, Zhang J. Proteomic analysis of microglial contribution to mouse strain-dependent dopaminergic neurotoxicity. Glia. 2006;53:567–582. doi: 10.1002/glia.20294. [DOI] [PubMed] [Google Scholar]
- Miyazaki M, Miyazaki K, Chen S, Itoi M, Miller M, Lu LF, Varki N, Chang AN, Broide DH, Murre C. Id2 and Id3 maintain the regulatory T cell pool to suppress inflammatory disease. Nat Immunol. 2014;15:767–776. doi: 10.1038/ni.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosley RL, Benner EJ, Kadiu I, Thomas M, Boska MD, Hasan K, Laurie C, Gendelman HE. Neuroinflammation, oxidative Stress and the pathogenesis of Parkinson's disease. Clin Neurosci Res. 2006;6:261–281. doi: 10.1016/j.cnr.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosley RL, Hutter-Saunders JA, Stone DK, Gendelman HE. Inflammation and adaptive immunity in Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a009381. doi: 10.1101/cshperspect.a009381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicole P, Lins L, Rouyer-Fessard C, Drouot C, Fulcrand P, Thomas A, Couvineau A, Martinez J, Brasseur R, Laburthe M. Identification of key residues for interaction of vasoactive intestinal peptide with human VPAC1 and VPAC2 receptors and development of a highly selective VPAC1 receptor agonist: alanine scanning and molecular modeling of the peptide. J Biol Chem. 2000;275:24003–24012. doi: 10.1074/jbc.M002325200. [DOI] [PubMed] [Google Scholar]
- Nikodemova M, Watters JJ. Efficient isolation of live microglia with preserved phenotypes from adult mouse brain. J Neuroinflammation. 2012;9:147. doi: 10.1186/1742-2094-9-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell M, Garippa RJ, Rinaldi N, Selig WM, Tocker JE, Tannu SA, Wasserman MA, Welton A, Bolin DR. Ro 25–1553: a novel, long-acting vasoactive intestinal peptide agonist. Part II: Effect on in vitro and in vivo models of pulmonary anaphylaxis. J Pharmacol Exp Ther. 1994a;270:1289–1294. [PubMed] [Google Scholar]
- O'Donnell M, Garippa RJ, Rinaldi N, Selig WM, Simko B, Renzetti L, Tannu SA, Wasserman MA, Welton A, Bolin DR. Ro 25–1553: a novel, long-acting vasoactive intestinal peptide agonist. Part I: In vitro and in vivo bronchodilator studies. J Pharmacol Exp Ther. 1994b;270:1282–1288. [PubMed] [Google Scholar]
- Offen D, Sherki Y, Melamed E, Fridkin M, Brenneman DE, Gozes I. Vasoactive intestinal peptide (VIP) prevents neurotoxicity in neuronal cultures: relevance to neuroprotection in Parkinson's disease. Brain Res. 2000;854:257–262. doi: 10.1016/S0006-8993(99)02375-6. [DOI] [PubMed] [Google Scholar]
- Otto D, Unsicker K. FGF-2-mediated protection of cultured mesencephalic dopaminergic neurons against MPTP and MPP+: specificity and impact of culture conditions, non-dopaminergic neurons, and astroglial cells. J Neurosci Res. 1993;34:382–393. doi: 10.1002/jnr.490340403. [DOI] [PubMed] [Google Scholar]
- Pandol SJ, Dharmsathaphorn K, Schoeffield MS, Vale W, Rivier J. Vasoactive intestinal peptide receptor antagonist [4Cl-D-Phe6, Leu17] VIP. Am J Physiol. 1986;250:G553–G557. doi: 10.1152/ajpgi.1986.250.4.G553. [DOI] [PubMed] [Google Scholar]
- Quah BJ, Parish CR. The use of carboxyfluorescein diacetate succinimidyl ester (CFSE) to monitor lymphocyte proliferation. J Vis Exp. 2010;pii:2259. doi: 10.3791/2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reubi JC. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr Rev. 2003;24:389–427. doi: 10.1210/er.2002-0007. [DOI] [PubMed] [Google Scholar]
- Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson's disease. J Leukoc Biol. 2007;82:1083–1094. doi: 10.1189/jlb.0507296. [DOI] [PubMed] [Google Scholar]
- Reynolds AD, Glanzer JG, Kadiu I, Ricardo-Dukelow M, Chaudhuri A, Ciborowski P, Cerny R, Gelman B, Thomas MP, Mosley RL, Gendelman HE. Nitrated alpha-synuclein-activated microglial profiling for Parkinson's disease. J Neurochem. 2008;104:1504–1525. doi: 10.1111/j.1471-4159.2007.05087.x. [DOI] [PubMed] [Google Scholar]
- Reynolds AD, Stone DK, Hutter JA, Benner EJ, Mosley RL, Gendelman HE. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson's disease. J Immunol. 2010;184:2261–2271. doi: 10.4049/jimmunol.0901852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Ramos M, von Euler Chelpin M, Sanchez-Guajardo V. Vaccination strategies for Parkinson disease: induction of a swift attack or raising tolerance? Hum Vaccin Immunother. 2014;10:852–867. doi: 10.4161/hv.28578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders JA, Estes KA, Kosloski LM, Allen HE, Dempsey KM, Torres-Russotto DR, Meza JL, Santamaria PM, Bertoni JM, Murman DL, Ali HH, Standaert DG, Mosley RL, Gendelman HE. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson's disease. J Neuroimmune Pharmacol. 2012;7:927–938. doi: 10.1007/s11481-012-9402-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäbitz WR, Krüger C, Pitzer C, Weber D, Laage R, Gassler N, Aronowski J, Mier W, Kirsch F, Dittgen T, Bach A, Sommer C, Schneider A. A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony stimulating factor (GM-CSF) J Cereb Blood Flow Metab. 2008;28:29–43. doi: 10.1038/sj.jcbfm.9600496. [DOI] [PubMed] [Google Scholar]
- Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, Bazan JF, Kastelein RA. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- Shandler SJ, Korendovych IV, Moore DT, Smith-Dupont KB, Streu CN, Litvinov RI, Billings PC, Gai F, Bennett JS, DeGrado WF. Computational design of a beta-peptide that targets transmembrane helices. J Am Chem Soc. 2011;133:12378–12381. doi: 10.1021/ja204215f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan YV, Abad C, Wang Y, Lopez R, Waschek JA. VPAC2 (vasoactive intestinal peptide receptor type 2) receptor deficient mice develop exacerbated experimental autoimmune encephalomyelitis with increased Th1/Th17 and reduced Th2/Treg responses. Brain Behav Immun. 2015;44:167–175. doi: 10.1016/j.bbi.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunçel N, Korkmaz OT, Tekin N, Şener E, Akyüz F, Inal M. Antioxidant and anti-apoptotic activity of vasoactive intestinal peptide (VIP) against 6-hydroxy dopamine toxicity in the rat corpus striatum. J Mol Neurosci. 2012;46:51–57. doi: 10.1007/s12031-011-9618-z. [DOI] [PubMed] [Google Scholar]
- Usdin TB, Bonner TI, Mezey E. Two receptors for vasoactive intestinal polypeptide with similar specificity and complementary distributions. Endocrinology. 1994;135:2662–2680. doi: 10.1210/endo.135.6.7988457. [DOI] [PubMed] [Google Scholar]
- Vial T, Descotes J. Clinical toxicity of cytokines used as haemopoietic growth factors. Drug Saf. 1995;13:371–406. doi: 10.2165/00002018-199513060-00006. [DOI] [PubMed] [Google Scholar]
- Voice J, Donnelly S, Dorsam G, Dolganov G, Paul S, Goetzl EJ. c-Maf and JunB mediation of Th2 differentiation induced by the type 2 G protein-coupled receptor (VPAC2) for vasoactive intestinal peptide. J Immunol. 2004;172:7289–7296. doi: 10.4049/jimmunol.172.12.7289. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waschek JA. VIP and PACAP: neuropeptide modulators of CNS inflammation, injury, and repair. Br J Pharmacol. 2013;169:512–523. doi: 10.1111/bph.12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav M, Rosenbaum J, Goetzl EJ. Cutting edge: vasoactive intestinal peptide (VIP) induces differentiation of Th17 cells with a distinctive cytokine profile. J Immunol. 2008;180:2772–2776. doi: 10.4049/jimmunol.180.5.2772. [DOI] [PubMed] [Google Scholar]
- Yadav M, Huang MC, Goetzl EJ. VPAC1 (vasoactive intestinal peptide (VIP) receptor type 1) G protein-coupled receptor mediation of VIP enhancement of murine experimental colitis. Cell Immunol. 2011;267:124–132. doi: 10.1016/j.cellimm.2011.01.001. [DOI] [PubMed] [Google Scholar]