

Abstract
The resolution of inflammation is an active and dynamic process, mediated in large part by the innate immune system. Resolution represents not only an increase in anti-inflammatory actions, but also a paradigm shift in immune cell function to restore homeostasis. PPARγ, a ligand activated transcription factor, has long been studied for its anti-inflammatory actions, but an emerging body of literature is investigating the role of PPARγ and its ligands (including thiazolidinediones, prostaglandins, and oleanolic acids) in all phases of resolution. PPARγ can shift production from pro- to anti-inflammatory mediators by neutrophils, platelets, and macrophages. PPARγ and its ligands further modulate platelet and neutrophil function, decreasing trafficking, promoting neutrophil apoptosis, and preventing platelet-leukocyte interactions. PPARγ alters macrophage trafficking, increases efferocytosis and phagocytosis, and promotes alternative M2 macrophage activation. There are also roles for this receptor in the adaptive immune response, particularly regarding B cells. These effects contribute towards the attenuation of multiple disease states, including COPD, colitis, Alzheimer's disease, and obesity in animal models. Finally, novel specialized proresolving mediators—eicosanoids with critical roles in resolution—may act through PPARγ modulation to promote resolution, providing another exciting area of therapeutic potential for this receptor.
1. Introduction: Innate Immunity, Inflammation, and PPARγ
Identification of the cardinal signs of inflammation (calor, dolor, rubor, and tumor) dates all the way back to the 1st century. Identifying the resolution phase of inflammation (long thought to be a passive process) is a far more recent advancement. We now know that resolution is an active and dynamic process and critical to the prevention of chronic and/or excessive inflammation [1]. Proresolving actions are distinct from anti-inflammatory processes; while anti-inflammatory molecules and medications act to dampen and suppress proinflammatory cells and signals, resolution represents a phenotypic shift in immune cell function towards repair and homeostasis. Resolution is characterized by several distinct phases. First, there is an end to the production of proinflammatory cytokines and halting of inflammatory neutrophil influx. Second, neutrophils present at the inflammatory site undergo apoptosis. Third, macrophages demonstrate a phenotypic switch and enhanced efferocytosis of apoptotic cells; the second and third phases coincide with increased production of anti-inflammatory and proresolving molecules. Finally, there is a clearance of macrophages, promotion of wound healing, and tissue repair to mediate the end of the inflammatory response [1]. These phases are frequently overlapping and the specific aspects of resolution can vary based on the inflammatory stimuli, organ location, and individual host characteristics (Figure 1).
Figure 1.

Inflammation and resolution are active and dynamic processes. The initiation, progression, and resolution of inflammation are characterized by unique cellular signals and trafficking.
Many aspects of the resolution of inflammation are mediated by the innate immune system. The innate immune system is comprised of a collection of cells, including neutrophils, macrophages, dendritic cells, eosinophils, basophils, platelets, and natural killer cells. These cells are responsible for recruiting other immune cells to sites of injury and infection, initiating complement cascades, activating the adaptive immune system, and removing foreign invaders and apoptotic cells. Neutrophils and macrophages are the first responders to inflammatory stimuli and are the first cells to begin to signal the resolution process. Furthermore, macrophages have in recent years been shown to be polarized towards multiple phenotypes, allowing this diverse cell type to contribute to both the proinflammatory and proresolving phases of inflammation [2].
Peroxisome proliferator activated receptors (PPARs) are a family of nuclear hormone receptors with three isoforms—PPARα, PPARβ, and PPARγ. PPARγ has a wide variety of biological roles, including regulating fatty acid synthesis and storage and glucose metabolism, promoting adipogenesis, and inhibiting inflammatory signaling through NF-κB. In recent years, the role of PPARγ in mediating responses to inflammation has been of particular interest. PPARγ is expressed on numerous immune cells, including monocytes/macrophages, platelets, lymphocytes, and dendritic cells [3–5]. PPARγ usually exists as a heterodimer complexed with retinoid X receptor alpha (RXRα); these two molecules are typically bound to corepressors. Upon ligand stimulation, the corepressor molecules are displaced and ligand, PPARγ, RXRα, and coactivators (such as CBP and SRC1) form an active complex, binding to PPARγ response elements (PPRE). Alternatively, upon ligand stimulation PPARγ can bind with NF-κB to repress NF-κB target genes (Figure 2). A wide variety of PPARγ ligands have been identified which bind to and activate PPARγ (Table 1). These ligands include thiazolidinediones (TZDs), which are antidiabetic drugs, as well as several prostaglandins (prostaglandin D2 (PGD2) and its metabolite, 15-deoxy prostaglandin J2 (15d-PGJ2)), oleanolic acids, and other eicosanoids. Importantly, ligand activation of PPARγ has been shown to exert potent anti-inflammatory effects. In addition, several of these molecules have effects independent of PPARγ, many of which are also anti-inflammatory. The degree to which these ligands act in an independent manner varies, with compounds such as ibuprofen acting predominantly independent of PPARγ, prostaglandins exhibiting mixed dependent/independent activity, and TZDs acting in a heavily PPARγ-dependent manner; the PPARγ-independent effects of many of these ligands have been reviewed previously [6]. PPARγ research has begun to focus on these anti-inflammatory effects and to investigate the role that PPARγ and its ligands play in the resolution of inflammation.
Figure 2.
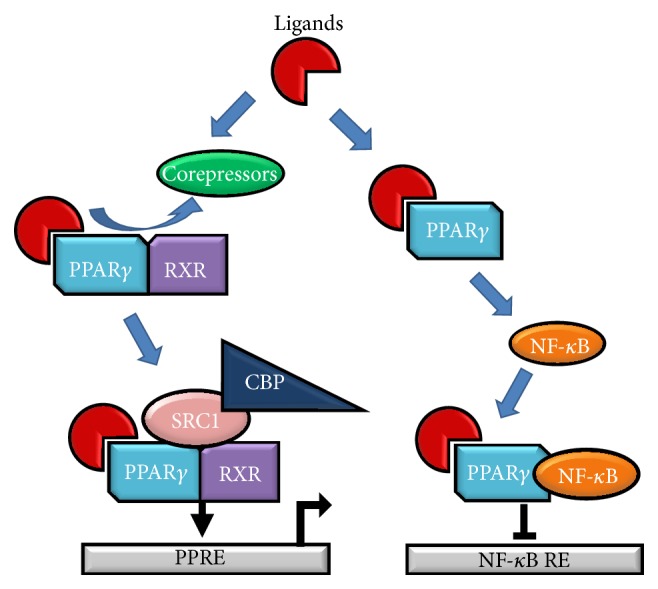
Overview of PPARγ activation. PPARγ typically exists as a heterodimer with RXRα, bound to corepressor molecules. Upon ligand stimulation, these corepressors are displaced and the ligand, PPARγ, RXRα, and coactivators (such as CBP and SRC1) form an active complex, binding to PPARγ response elements (PPRE). Alternatively, upon ligand stimulation PPARγ alone can bind with NF-κB to repress NF-κB target genes.
Table 1.
List of PPARγ ligands.
| Prostaglandins | Thiazolidinediones | Eicosanoids | Other lipids |
|---|---|---|---|
| Prostaglandin D2 ∗ | Rosiglitazone+ | Arachidonic acid∗ | Conjugated linoleic acids∗ |
| 15-Deoxy prostaglandin J2 ∗ | Pioglitazone+ | SPMs∗ | Oxidized LDLs+ |
| Prostaglandin A1∗ | Ciglitazone+ | 15-HETE∗ | |
| Troglitazone+ | DHA∗ | ||
| EPA∗ | |||
|
| |||
| Fibrates | Nutraceuticals | NSAIDs | Triterpenoids |
|
| |||
| Clofibrate+ | Genistein∗ | Indomethacin+ | Oleanolic acid∗ |
| Fenofibrate+ | Biochanin A∗ | Ibuprofen+ | CDDO+ |
| Gemfibrozil+ | Daidzein∗ | ||
| Ciprofibrate+ | Hesperidin∗ | ||
∗Synthetic ligand, +endogenous ligand.
SPMs: specialized proresolving mediators; HETE: hydroxyeicosatetraenoic acid; DHA: docosahexaenoic acid; EPA: eicosapentaenoic acid; LDLs: low density lipoproteins; CDDO: 2-cyano-3,12-dioxo-oleana-1,9(11)-dien-28-oic acid; NSAIDs: nonsteroidal anti-inflammatory drugs.
Many of PPARγ's established anti-inflammatory effects have been shown to occur through innate immune signaling, particularly in monocytes and macrophages [7]. These cells are furthermore capable of producing a number of PPARγ ligands, which can potentiate the anti-inflammatory and proresolving actions of this receptor on additional immune cells and other cells. Resolution is also mediated by a wide variety of signals, including cytokines and chemokines, apoptotic proteins, and eicosanoids. Eicosanoids, several of which are PPARγ ligands, are produced through lipid class switching. For instance, under resolution there may be increased production of 15d-PGJ2 rather than proinflammatory prostaglandins, both of which come from arachidonic acid precursors [8]. Prostaglandins are produced under strong temporal regulation, with distinct shifts in which mediators are produced depending on the phase of inflammation. Furthermore, prostaglandin precursors can be broken down into additional prostaglandin isoforms; PGD2 is rapidly metabolized to 15d-PGJ2 [9]. While both of these mediators have anti-inflammatory effects, 15d-PGJ2 is a much more potent activator of PPARγ [10]. Prostaglandins and other eicosanoids signal through multiple receptors to initiate resolution and appropriate cellular responses [11]. The roles of the PPARγ transcription factor, and PPARγ ligands, in resolution are beginning to be elucidated, and this receptor is emerging as an important player in all stages of the resolution of inflammation.
2. PPARγ and Altered Cytokine Production
PPARγ has been shown in numerous studies to affect the expression of proinflammatory cytokines. In this review, we have focused on cytokines and chemokines that are particularly important for their pro- and anti-inflammatory effects on innate immune cells. First, PPARγ has been shown to extensively affect expression of tumor necrosis factor-alpha (TNFα). TNFα is an important cytokine in regulating immune cell function and can act to induce fever, promote apoptosis, and stimulate other cytokines. TNFα can also act as a macrophage and neutrophil chemoattractant. While this cytokine has important roles in bacterial killing, excessive expression promotes chronic inflammation and poor health effects, such as rapid weight loss. In human neutrophils, TNFα actually increases mRNA and protein PPARγ expression, likely in a compensatory mechanism or a feedback loop [12]. In conjunction with this increased expression, PPARγ ligands, particularly TZDs, potently reduce TNFα expression [13]. Pioglitazone treated and lipopolysaccharide- (LPS-) exposed mice and guinea pigs have decreased TNFα expression. Indeed, both pioglitazone and rosiglitazone reduced TNFα expression in a wide range of inflammatory models, including sepsis, ischemia/reperfusion, colitis, gastric injury, and spinal trauma models [14–19]. These effects were independent of the route of administration, and the oral delivery of pioglitazone to decrease TNFα in mouse livers is particularly interesting from a therapeutic standpoint [20]. These effects were largely shown to be PPARγ-dependent, with the reduction of TNFα expression blocked by PPARγ antagonists [17, 19]. Other PPARγ ligands are also capable of dampening TNFα expression, including 15d-PGJ2 and oleanolic acid [21–23].
Along with TNFα, several interleukins (ILs) are produced in response to inflammatory stimuli. IL-6 is a component of the acute inflammatory response and a mediator of fever. Pioglitazone, rosiglitazone, and ciglitazone decrease local production of IL-6 in the intestine; colon; and lung, colon, and liver, respectively [14, 18, 22]. 15d-PGJ2 and PGD2 microspheres similarly reduced bacteria stimulated increases in IL-6 [22, 24]. IL-8 is a key chemokine for neutrophil trafficking. IL-8 is broadly expressed by a multitude of cell types, including macrophages, and induced by a variety of inflammatory stimuli. Rosiglitazone prevents tobacco smoke induced decreases in PPARγ, induction of leukotrienes, and IL-8 production [25]. IL-1β is also produced to promote acute inflammation and is reduced by rosiglitazone, 15d-PGJ2, and pioglitazone in a PPARγ-dependent manner [15, 17, 26]. The broad ranging effects of PPARγ ligands on proinflammatory cytokines may be due to PPARγ effects on the NF-κB pathway, as PPARγ has been shown to decrease NF-κB expression [16–18, 27].
Finally, PPARγ is also involved in the production of anti-inflammatory and proresolving cytokines, though these data remain controversial. IL-10, for instance, is a cytokine produced under both pro- and anti-inflammatory conditions. In the context of resolution, IL-10 can be produced by macrophages to mediate proresolving effects. Rosiglitazone induces IL-10 production in experimental colitis and Parkinson's models of disease [18, 26], but in a septic lung 15d-PGJ2 and pioglitazone reduced IL-10 expression [18, 22]. Whether these differences are due to differences in inflammatory stimuli, PPARγ ligand specific signaling, the time points chosen, or observed organ system requires additional studies to conclusively answer. Rosiglitazone similarly induced TGFβ expression, another proresolving cytokine, in microglia in Parkinson's model of disease [26]. This shift in cytokine production, rather than suppression of all cytokine signaling, implicates that PPARγ is simply acting in not only an anti-inflammatory manner, but a proresolving one as well.
3. PPARγ and Neutrophil Apoptosis and Clearance
Neutrophils are the first responders to most inflammatory stimuli. These cells are rapidly produced, with quick influx to a site of injury. Upon arrival at the injured site, they are responsible for phagocytosis of foreign invaders and cytokine/chemokine production to recruit other immune cells. An excessive influx of neutrophils, failure to shut down cellular influx, and a lack of clearance can lead to neutrophilia, a hallmark of many inflammatory diseases. Appropriate neutrophil apoptosis and clearance, as well as prevention of too many neutrophils trafficking to an inflammatory site, are a key component of resolution. PPARγ promotes apoptosis of a variety of cell types, and knockdown of PPARγ strongly decreases uptake of apoptotic cells [28]. Our focus here will be on the influx, apoptosis, and clearance of neutrophils.
The effect of one TZD, pioglitazone, on neutrophil functions has been particularly well characterized. Pioglitazone treated mice and guinea pigs have decreased neutrophil numbers and myeloperoxidase (MPO) expression in an LPS model; MPO is frequently used as a marker of neutrophil presence [13, 29]. Other models of inflammation yielded similar effects, with pioglitazone decreasing MPO activity in mice on a high-fat diet and mice with bacterial sepsis [14, 20]. Pioglitazone also decreased neutrophil counts specifically in a mouse model of ischemia/reperfusion. Interestingly, there is less apoptosis in mouse hepatic cells in this model, suggesting either specific regulation of cellular apoptosis, wherein neutrophils are targeted but other cell types are impervious to PPARγ induced apoptosis, or a prevention of neutrophil influx in the first place. PPARγ expression increased immediately following injury, which could support a prevention of influx at this early time point [15]. Similar results were seen in a spinal trauma model, where another TZD, rosiglitazone, both increased PPARγ expression and prevented neural cell apoptosis. These effects were all prevented by GW9662, a PPARγ antagonist, thus implying a PPARγ-dependent mechanism [19]. Since rosiglitazone has been shown in other studies of colitis and gastric injury to reduce MPO activity [16–18, 27], TZDs could be acting to specifically target neutrophil apoptosis, though the exact mechanisms of this remain unclear.
Prostaglandins also play a highly important role in neutrophil trafficking and apoptosis. For example, 15d-PGJ2 both reduces MPO activity and directly reduces neutrophil numbers [30, 31]. However, 15d-PGJ2 is produced by alveolar macrophages before neutrophil infiltration, which could be an early feedback mechanism rather than induction of neutrophil apoptosis [32]. 15d-PGJ2 decreases in MPO activity and beneficial effects were associated with increased PPARγ DNA-binding, though a PPARγ antagonist did not block these effects, indicating they may be at least partially PPARγ-independent [22]. In 15d-PGJ2-treated mice with induced spinal cord injuries, the spinal cord MPO activity was significantly attenuated in comparison to vehicle treated mice. In this experiment, coadministration of GW9662 and 15d-PGJ2 significantly blocked the effect of the PPARγ agonist on neutrophil infiltration. There was an observed decrease in the number of apoptotic bodies, but this was not specifically tied to neutrophils [23]. The strongest evidence for 15d-PGJ2 induced neutrophil apoptosis is demonstrated by Gilroy and colleagues using a rat model of pleurisy. Cyclooxygenase-2 (Cox-2) was originally shown to promote neutrophil apoptosis and resolution, correlating with increases in PGD2 and 15d-PGJ2. Indomethacin (a nonspecific Cox inhibitor) and specific Cox-2 inhibitors prevented induction of neutrophil apoptosis, but addition of 15d-PGJ2 along with these inhibitors increases both neutrophil and macrophage apoptosis, showing specific actions of this prostaglandin [33].
PGD2 more clearly contributes to neutrophil trafficking and apoptosis, as increased PGD2 levels correspond to spontaneous neutrophil apoptosis. Incubating human neutrophils with PGD2 and 15d-PGJ2 also induced apoptosis in a dose-dependent manner. Furthermore, human macrophages incubated with opsonized and nonopsonized apoptotic neutrophils produce increased levels of PGD2. This correlates with clinical data, as certain patients fail to produce PGD2 in response to apoptotic neutrophils and have increased neutrophilia [34]. MPO is increased in correlation with PGD2 decreases in colonic inflammation, and exogenous addition of PGD2 decreases MPO activity and infiltration of neutrophils. While PPARγ expression does increase in this inflammatory model, whether or not PGD2's effects were PPARγ-independent or PPARγ-dependent was not evaluated [35]. There are clearly complex mechanisms at play regarding the PPARγ-independent and PPARγ-dependent effects of prostaglandins, but the overall evidence suggests that these mediators contribute to neutrophil influx, clearance, and apoptosis and that these regulatory pathways merit further investigation.
Several studies have examined both TZDs and PGs side-by-side. 15d-PGJ2 and pioglitazone both reduced neutrophil numbers and MPO activity in a caecal-ligation puncture model of sepsis. Importantly, while most studies see local effects of PPARγ ligands, this study showed decreased MPO activity in the lung, liver, and colon. These two ligands were able to rescue sepsis-induced decreases in PPARγ expression in the lung to normal constitutive levels [22]. In another study, treatment with either 15d-PGJ2 or troglitazone decreased human neutrophil chemotaxis in response to IL-8 but did not induce apoptosis. These results were PPARγ-dependent, as direct transfection with a constitutively active PPARγ gene also inhibited chemotaxis. These two ligands do appear to have unique pathways, though, as 15d-PGJ2 abolished ERK1/2 phosphorylation, while troglitazone dampened ERK-P but also inhibited neutrophil actin polymerization. This may not always be a beneficial response, as sepsis patients have higher expression of PPARγ, and blocking PPARγ reverses sepsis-induced inhibition of neutrophil chemotaxis [12].
Along with PGs and TZDs, oleanolic acid (OA) decreased neutrophils in bronchoalveolar lavage and decreased apoptosis of all cells across all other organ systems. Since this antiapoptotic effect was seen in all cell types, including neutrophils, OA is likely acting to prevent neutrophil influx [36]. Oleanolic acid-NO2, though, also decreased neutrophil counts in allergic airway disease and this reduction was due to induction of neutrophil apoptosis. OA-NO2 stimulated PPARγ binding and nuclear translocation, and differences in receptor binding may account for these different observed effects [21]. Another PPARγ ligand, CDDO, additionally decreased neutrophil numbers and MPO activity [37, 38].
There is additional evidence that neutrophils have an anti-inflammatory role and contribute to the resolution of inflammation. In a stroke model, rosiglitazone increased the numbers of neutrophils in the brain, and the protective effects of rosiglitazone were abolished after neutrophil depletion. These neutrophils express markers characteristic of M2 macrophages and are increased by rosiglitazone treatment. These “N2” neutrophils are preferentially phagocytosed by microglia [39]. While more extensive research is needed to fully characterize this potential new cell phenotype, the existence of multiple activation states for neutrophils is an interesting development in resolution biology, as is the role for PPARγ in modulating these cells.
4. PPARγ and Macrophage Trafficking
Monocytes and macrophages traffic to sites of injury in both the inflammatory and resolution phases of inflammation. Currently, studies regarding the effects of PPARγ and PPARγ ligands on monocyte trafficking are contradictory and complex. Multiple TZDs, including troglitazone and pioglitazone, have been shown to increase expression of monocyte chemoattractant protein-1 (MCP-1) and bone marrow monocyte/macrophage recruitment in the rat kidney, but these same TZDs suppressed expression of MCP-1 and the MCP-1 receptor CCR2, inhibited migration in human cells, and reduced macrophage counts in other mouse models [40–44]. When PPARγ is specifically knocked out in colon macrophages, mice had higher levels of CCR2 and MCP-1 [45].
These variable effects may be dependent on the inflammatory stimuli. Ciglitazone, for instance, dose-dependently inhibited monocyte chemotaxis due to plasmin stimulation, but not N-formyl-met-leu-phe- (FMLP-) induced influx, and may specifically target plasmin-induced chemotaxis pathways [46]. Rosiglitazone was further shown to decrease expression of CCR2 in a PPARγ-dependent manner in primary human blood monocytes in the absence of any inflammatory stimuli but did not further decrease expression levels after 48 hours [47]. PPARγ directly regulates CCR2 expression in mouse cells, leading to similar early decreases [48]. Taken together, these data imply that TZDs may be acting to “quiet” monocytes, preventing an early influx, but allowing alternative macrophage recruitment in the later stages of resolution to allow for cell clearance and tissue repair. Future studies should pay particular attention to PPARγ and its effects on the temporal regulation of macrophage chemotaxis.
In contrast to the complicated literature regarding TZDs, prostaglandins much more clearly decrease macrophage influx. 15d-PGJ2 inhibited macrophage chemotaxis towards zymosan-treated serum and specifically inhibited macrophage trafficking to the site of damage in chronic liver injury (T cell, dendritic cell, and neutrophil trafficking were not affected) [49, 50]. While 15d-PGJ2 does not affect MCP-1 expression, it decreases CCR2 expression and inhibits migration of monocytes/macrophages [40, 41]. Mice deficient in PGD2 synthase, and thereby deficient in PGD2 and 15d-PGJ2, have increased MCP-1 and increased macrophage accumulation. This was due in part to a lack of D and J series prostaglandins which would normally act to prevent macrophage influx, but also largely due to impaired clearance of leukocytes from draining lymphatics, indicating a role for prostaglandins in the end stage clearance of macrophages [31]. In contrast to the abilities of TZDs to enhance early monocyte/macrophage chemotaxis, prostaglandins act later in the inflammatory process to prevent excessive inflammatory monocyte/macrophage influx and to clear cells at the end of resolution.
Discrepancies in PPARγ ligand induced macrophage recruitment may also be due to differences in macrophage populations. PPARγ is expressed highly in lung and spleen macrophages but has very low expression in peritoneal macrophages. One particular study additionally showed that inflammatory monocytes recruited to the site of inflammation expressed increased levels of PPARγ as they differentiated to macrophages. PPARγ deficiency in these infiltrating monocytes did not impair the initiation of inflammation but inhibited the resolution of inflammation, with increased neutrophils and inflammatory cytokine production in macrophage specific PPARγ knockout mice. Furthermore, PPARγ deletion in lung macrophages, but not spleen macrophages, impaired resolution, though macrophage numbers were consistent in all groups [51]. These data would suggest that the effect of PPARγ monocyte and macrophage trafficking is secondary and that the more potent PPARγ-dependent actions occur through macrophages already present at the site of inflammation.
5. PPARγ Enhances Macrophage Phagocytosis
A key aspect of resolution is enhanced macrophage phagocytosis of neutrophils, bacterial components, and other cell debris. PPARγ and its agonists have been shown to reduce macrophage inflammatory activation while enhancing phagocytosis [7]. PPARγ is increased in macrophages in the presence of apoptotic cells and can act on expression of multiple proteins linked to phagocytosis [52]. For instance, PPARγ expression is necessary for basal expression of CD36, a major scavenger receptor [53, 54]. Induced increases in PPARγ-RXR expression by multiple agonists also increased expression of CD36 and phagocytosis of Plasmodium falciparum-parasitized erythrocytes. These effects were seen in both the human monocytic THP-1 cell line and primary human blood monocytes [55]. PPARγ may also act through IL-13 to enhance phagocytosis, since PPARγ is necessary for IL-13 induced production of 15d-PGJ2, alternative activation of macrophages, and enhanced phagocytosis of parasitized erythrocytes [54]. PPARγ is also involved in IL-13 induction of Dectin-1, which is required for Candida albicans clearance and resolution of yeast-induced inflammation [56].
Thiazolidinediones are the most potent group of PPARγ ligands for enhancing macrophage phagocytosis. In brain abscesses in C57BL/6 mice, ciglitazone dose-dependently decreased bacterial load, with greater decreases seen after multiple days of postinfection treatment. Primary microglia were further isolated and treated with ciglitazone; ciglitazone increased microbial uptake of Staphylococcus aureus. Additionally, the edge of the abscess areas showed enhanced PPARγ activity, demonstrating the contribution of PPARγ to this decreased bacterial burden [57]. Similarly, mice given intraperitoneal doses of ciglitazone following intranasal S. pneumonia infections had decreased bacterial burdens. In contrast to the brain, though, mouse alveolar macrophages treated in vitro with ciglitazone had no alteration of phagocytosis, demonstrating a difference in ligand-responsiveness between macrophage types [58]. Rosiglitazone also enhanced phagocytosis by macrophages. Peripheral blood monocytes had increased CD36 expression and enhanced uptake of P. falciparum following rosiglitazone treatment. These effects were mirrored in a mouse model of cerebral malaria. Mice that received rosiglitazone infused chow had reduced parasite levels, dependent on CD36 expression. This treatment was effective as late as 5 days after infection [59]. In other models of neural inflammation, rosiglitazone again induced expression of CD36, which enhanced phagocytosis of neutrophils and promoted resolution; these effects were blocked by PPARγ antagonists or PPARγ gene knockdown [60, 61]. Other TZDs including pioglitazone and troglitazone similarly increased macrophage or microglial phagocytosis and CD36 expression in a PPARγ-dependent manner [62, 63]. These phagocytic enhancements appear to be dependent on macrophage tissue origin. In one study, troglitazone and rosiglitazone were shown to enhance Fcγ receptor mediated phagocytosis of both beads and opsonized bacteria, but these increases were only seen in alveolar macrophages and not peritoneal macrophages [64]. These differences may be due to a difference in PPARγ expression between different macrophage types and responsiveness to specific TZD ligands [51].
Conjugated linoleic acids (CLAs) also play a role in modulating phagocytosis. RAW264.7 cells (a mouse macrophage cell line) treated with CLAs had a dose-dependent increase in uptake of latex beads which was attenuated by the PPARγ antagonist GW9662 [65]. Additionally, human monocytes incubated with CLAs and differentiated to macrophages had enhanced phagocytosis [66]. Another lipid molecule, OA-NO2, demonstrated similar actions and increased CD36 and macrophage phagocytosis [21]. While only a few studies exist examining the role of CLAs and OAs on phagocytosis, these data are promising, and low density lipoproteins and triterpenoids represent two under examined classes of PPARγ ligands that may have potent proresolving effects.
In contrast to other PPARγ ligands, PGD2 and 15d-PGJ2 both decrease macrophage phagocytic abilities. Human primary blood monocytes directly treated with PGD2 had mild dose-dependent inhibition of apoptotic neutrophil uptake [67]. While a study using PGD2 microspheres showed that these spheres were phagocytosed more efficiently than unloaded spheres, the PGD2 spheres also activated NF-κB inflammatory pathways to a greater extent, indicating that this enhanced uptake promotes inflammatory pathways and not resolution phagocytosis [24]. Similar to PGD2, 15d-PGJ2 inhibited phagocytosis of E. coli in isolated macrophages [49]. 15d-PGJ2 also dampened phagocytosis of Salmonella enterica by mouse macrophages, and infected macrophages actually produced higher levels of 15d-PGJ2 and other prostaglandins in a feed-forward loop [68]. 15d-PGJ2 further inhibited macrophage phagocytosis of latex beads by both mouse bone marrow derived macrophages and the mouse RAW264.7 cell line [50, 69]. There may be a temporal aspect to these studies that has not yet been conclusively evaluated, as prostaglandins are known to be produced under strong temporal regulation. Particularly, it appears that prostaglandins play a larger anti-inflammatory role early in the resolution process rather than acting to enhance macrophage functions.
PPARγ ligand effects on phagocytosis appear to be broad ranging, as PPARγ ligands are able to enhance uptake of both opsonized and nonopsonized targets [64] and a broad range of pathogens including parasites and bacteria. These effects largely seem to be independent of the route of administration of ligands, as oral, intraperitoneal, and intravenous treatments all have demonstrated efficacy. Critical to further understanding of the ability of PPARγ ligands to enhance phagocytosis is evaluating their dependent and independent actions, since many of these studies have not yet addressed these questions.
6. PPARγ and Alternative Macrophage Activation
Recent studies have begun to distinguish two broad classes of macrophages: classically activated (M1) and alternatively activated (M2). Classically activated macrophages are associated with a proinflammatory phenotype. These cells are activated by a number of traditional inflammatory stimuli, including IFNγ and LPS. M1 macrophages produce numerous proinflammatory cytokines and chemokines, have increased levels of iNOS, and demonstrate enhanced abilities to kill intracellular pathogens. M2 macrophages are characterized by their roles in the resolution phase of inflammation and in tissue repair and are stimulated by IL-4, IL-13, IL-10, and TGFβ.
PPARγ is the principal, but not exclusive, member of the PPAR family in promoting M2 macrophages [70–72]. PPARγ response elements were identified in the promoter region of Arg-1, a key M2 marker, and confirmed by an electrophoretic mobility shift assay. Arg-1 expression is significantly decreased in macrophage specific PPARγ knockout mice. Further supporting the link between PPARγ and Arg-1, PPARγ activates an Arg-1 luciferase assay, and this activation is further potentiated by the addition of rosiglitazone [73].
PPARγ ligands play a particularly strong role in M2 polarization in the neural system. Pioglitazone promoted a shift from M1 to M2 macrophages and enhanced Aβ amyloid phagocytosis in the brain [74]. Rosiglitazone similarly induced CD206 expression (an M2 marker) in microglia in Parkinson's model of disease [26]. Along with modulating neuronal inflammation, TZDs can modulate pain responses through M1/M2 polarization. Local administration of rosiglitazone induced IL-10 production and M2 macrophage polarization and attenuated pain sensitivity responses. These effects were shown to be macrophage mediated [75, 76].
TZDs also promote M2 macrophages in other disease states. Pioglitazone enhanced M2 polarization in atherosclerotic lesions, diet-induced obesity, and a model of insulin resistance [77–79]. Rosiglitazone likewise enhanced expression of Arg-1 and CD206 in peritoneal and adipose tissue macrophages and in an in vitro model of COPD [80–82]. While rosiglitazone prevents specific M2c polarization (as defined by CD163 expression), troglitazone induced CD163 expression and dampened CD80 (M1) expression in human macrophages derived from blood monocytes [83, 84]. This wide range of actions is encouraging and points to common mechanisms of action and broader therapeutic use of these drugs. In general, TZDs appear to be the most potent PPARγ ligands regarding the induction of M2 polarization.
In contrast to the TZDs, oleanolic acid (OA) inhibited M2 polarization and IL-10 secretion from human macrophages. There is some evidence that M2 macrophages polarized under certain stimuli are associated with tumor growth and/or cancer; thus OA may be acting to prevent tumor-associated macrophage production [85]. Oxidized LDL also accumulated in M2 macrophages and enhanced their proinflammatory capabilities, generating a cell type with M2 markers but higher production of proinflammatory cytokines [86], but was shown in a different study to promote a traditional M2 phenotype with decreased inflammatory cytokine production in THP-1 cells [87]. In contrast, CLA promoted increased IL-10 and M2 polarization of bone marrow derived macrophages atherosclerotic lesions, suggesting that all fatty acids do not have the same PPARγ-dependent modulatory effects or that these ligands are acting in distinct PPARγ-independent manners [88].
Prostaglandins also have unique roles in promoting M2 macrophages. While PGD2 and 15d-PGJ2 have so far been shown to decrease phagocytic abilities in macrophages, they have both been shown to promote M2 polarization. Mouse peritoneal macrophages treated with 15d-PGJ2 had potent increases in numerous M2 markers, including Arg-1, CD206, and TGFβ [72]. PGD2 was also shown to promote an M2 phenotype, with increased levels of Arg-1 and CD206, though TNFα was also increased. Since PGD2 synthase is elevated in the differentiation of monocytes to macrophages, there may be mix of pro- and anti-inflammatory targets for this molecule, though it has been shown to be produced by macrophages to act in an autologous manner and promote M2 polarization [89]. Since M2 macrophages can be divided into further subclasses and mixed M1/M2 macrophage populations exist, further investigations are needed to fully elucidate the effects of prostaglandins on M2 phenotypes and functions.
Multiple other signals can also enhance PPARγ expression and thereby M2 signaling. For example, deficiency in Cx3Cr1 led to increased PPARγ expression on macrophages along with increased Arg-1 and decreased iNOS [90]. Netrin, adiponectin, and even exercise increased PPARγ expression and an array of M2 genes [91–94]. The effects of PPARγ may also be dependent on the type of macrophage assessed, as human alveolar macrophages have much higher PPARγ expression than blood monocytes and are thought to be more predisposed towards an M2 state [3].
Importantly, this PPARγ mediated induction of M2 macrophages plays a critical role in tissue repair and regeneration. These properties are highlighted in two diverse models, corneal scarring and diabetic wound healing. Alkali burn models are often used to induce optical wounds and scarring in mice, and PPARγ expression is increased following alkali injury. Overexpression of PPARγ in mouse corneal macrophages led to reduced proinflammatory cytokine and matrix metalloproteinase expression. These macrophages, in conjunction with fibroblasts and epithelial cells, contributed to reduced corneal scarring and faster reepithelialization and wound healing [95]. Similar effects were seen with administration of an ophthalmic pioglitazone solution, wherein treated mice had fewer myofibroblasts, decreased proinflammatory cytokine production, and more infiltrating M2 macrophages in the cornea compared to vehicle treated mice [96]. The inhibitory actions of PPARγ and PPARγ ligands (including TZDs and 15d-PGJ2) were observed in other optical scarring models, including TGFβ-induced scarring and scratch wounds [97, 98].
Along with optical scarring, M2 macrophages contribute to tissue repair in diabetics. In a study by Mirza and colleagues, diabetic mice were subjected to excisional wounding [99]. Diabetic mice had impaired wound healing which correlated with decreased PPARγ expression in mouse macrophages. Furthermore, loss of PPARγ from wild type macrophages led to delayed reepithelialization and impaired wound closure. 15d-PGJ2 and rosiglitazone both acted to reverse the diabetic proinflammatory environment and to promote wound healing [99]. The beneficial actions of PPARγ and its ligands in diabetic repair were modulated by M2 macrophage induction [73, 99]. These two disease states, along with multiple others, highlight the contribution of PPARγ and M2 macrophages to tissue repair and wound healing. In summary, PPARγ acts on multiple aspects of macrophages to mediate their key roles in resolution (Figure 3).
Figure 3.

PPARγ and macrophage activation. Proinflammatory macrophages are characterized by M1 activation markers, production of proinflammatory cytokines, and increased recruitment of immune cells. Upon stimulation with PPARγ ligands and/or increases in PPARγ expression, macrophages shift to an alternative M2 phenotype, with decreased proinflammatory actions, increased efferocytosis and phagocytosis, and production of anti-inflammatory cytokines.
7. PPARγ and Platelets
Platelets are anucleate cells in the innate immune system with important roles in hemostasis and inflammation. Platelets are produced by megakaryocytes and in turn produce microparticles; all three of these cells or cell components can modulate the function of other immune cells and release cytokines. Meg-01 cells (a human megakaryocyte cell line), primary human megakaryocytes, and primary human platelets were all shown by our lab to express PPARγ protein, though platelets did not contain PPARγ mRNA [4]. Platelets can package PPARγ in microparticles, which can then be taken up by leukocytes and other recipient cells [100]. PPARγ released in these microparticles retains DNA-binding ability and can thus alter target cell functionality [100]. In a study from our lab using microparticles engineered to express high levels of PPARγ, monocyte recipient cells had decreased production of proinflammatory mediators and increased CD36 expression following PPARγ-microparticle uptake [101]. PPARγ ligands are also capable of increasing platelet production, demonstrating a possible feed-forward loop for this receptor [102].
Platelets are capable of producing prothrombotic mediators that can alter the function of other immune cells. Plasminogen activator inhibitor 1 (PAI-1) is a serine proteinase inhibitor with multiple roles, including enhancement of leukocyte trafficking and recruitment. Patients given pioglitazone, troglitazone, or rosiglitazone in separate studies had significantly lower levels of PAI-1 in their serum [103–105]. TZDs have also been used therapeutically to reduce C-reactive protein (CRP), an acute-phase protein that increases following IL-6 secretion. These studies showed that patients who were given rosiglitazone or pioglitazone for 12–26 weeks have decreased serum levels of CRP and, where evaluated, IL-6 [106–109]. Troglitazone was further able to dampen thromboxane B2, a metabolite of prothrombotic thromboxane A2, in platelet-like human erythroleukemia cells and in human platelets [110].
Platelets can furthermore affect neutrophil trafficking by altering cellular adhesion and platelet/leukocyte interactions. Two particular molecules are key for mediating these effects, P-selectin and CD40L, both of which are highly expressed by platelets. P-selectin upregulation by platelets leads to enhanced chemokine synthesis and tethering of leukocytes in inflammatory sites, along with activation of NF-κB [111]. Diabetic mice treated with pioglitazone had lower levels of soluble and platelet P-selectin [112]. Complementing these results, rosiglitazone decreased the percentage of P-selectin expressing platelets in human diabetic patients [106]. Activated platelets are also the most important source of soluble CD40L, which binds to CD40 to mediate platelet/leukocyte interactions [113]. Furthermore, platelets from CD40L knockout mice fail to form these platelet/leukocyte aggregates and have decreased MCP-1 levels [113]. Diabetic patients treated with rosiglitazone also had decreased levels of circulating CD40L after 12 weeks or three months of treatment [114, 115]. Direct treatment of human platelets with 15d-PGJ2 or rosiglitazone also decreased levels of surface CD40L and soluble CD40L [4, 116]. The PPARγ antagonist GW9662 prevented these decreases, highlighting that these effects are PPARγ-dependent [116]. These results underscore that the function of platelets as immune cells is a crucial, yet understudied, area of research, particularly given the clear importance these cells have in modulating leukocyte function during inflammation and resolution (Figure 4).
Figure 4.
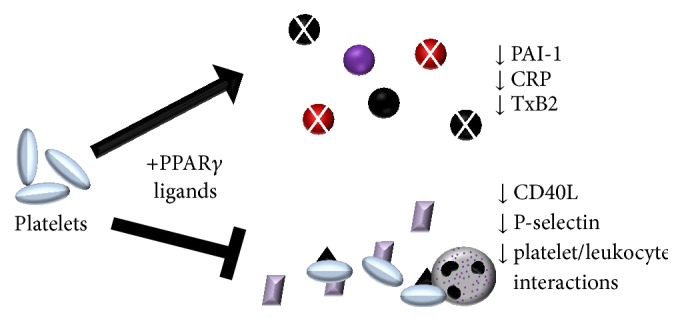
PPARγ and platelet function. Upon stimulation with PPARγ ligands, platelets decrease expression of multiple proinflammatory proteins and lipids, including PAI-1, CRP, and TxB2. PPARγ ligands also decrease expression of P-selectin and CD40L, thereby decreasing the number of proinflammatory platelet/leukocyte aggregates.
8. PPARγ and the Adaptive Immune Response
Along with the innate immune system, the adaptive immune system plays many roles in the resolution of inflammation. The effects of PPARγ on T cells have been reviewed elsewhere, and there is emerging evidence for the effects of PPARγ on B cells, which are known to express PPARγ [5]. Initial studies investigated the role of endogenous PPARγ agonists such as 15d-PGJ2 and synthetic ligands on B cell development and activation. High concentrations of PPARγ ligands (μM range) inhibit B cell proliferation and induce apoptosis in both normal and malignant B cells [5]. B cells treated with PPARγ ligands demonstrated characteristic markers of apoptosis, including reduction in mitochondria membrane potential, activation of caspases 3 and 9, and consequently increase in the cleavage of the substrate PARP [117, 118]. The ability of PPARγ agonists to induce apoptosis and suppress proliferation resulted from inhibition of NF-κB, thereby blocking the transcription of downstream prosurvival mediators [118]. PPARγ ligands can also directly prevent the activity of IKK, reducing the activation of NF-κB [119]. Another study has shown that MAPKs are involved in mediating the effects on PPARγ ligands to induce apoptosis in a mouse bone marrow pro-B cell line, indicating multiple mechanistic target pathways that may induce B cell apoptosis [120].
B lymphoma cells such as Burkitt lymphoma, Hodgkin and non-Hodgkin B cell lymphomas are known to have constitutively activated NF-κB which can lead to enhanced B cell proliferation and survival [119]. In this context, PPARγ agonists could be potential anticancer therapeutics in those tumors where NF-κB appears to play a unique survival role. However, many PPARγ agonists, especially 15d-PGJ2, have both PPARγ-dependent and PPARγ-independent effects which could be mediating the proresolving actions seen here [118]. Our lab used the Ramos B cell lymphoma cell line transfected with a dominant negative (DN) PPARγ construct to block the activity of endogenous PPARγ [121]. Surprisingly, 15d-PGJ2 maintained its antiproliferative effects on B cells regardless of the presence of DN PPARγ construct, suggesting PPARγ-independent actions of 15d-PGJ2 in lymphoma cells. The PPARγ-independent effects included enhanced ROS production and reduced glutathione-S levels, which in turn lead to cell apoptosis. On the other hand, ciglitazone, which does not possess the same reactive α,β-unsaturated carbonyl group as 15d-PGJ2, did not affect ROS and glutathione production, underscoring the differential actions of different PPARγ ligands.
Recently, studies started to focus on the role of PPARγ in B cell physiology. To investigate the intrinsic effects of PPARγ itself (rather than ligand induced activation), PPARγ expression was modulated using siRNA or a PPARγ-expressing lentiviral vector in Ramos B lymphoma cell line [122]. In B cells with PPARγ knockdown, cellular proliferation and NF-κB translocation were enhanced upon stimulation. Interestingly, B cells were less differentiated, with increased expression of CD19 and CD20 and reduction in CD38. This was the first evidence showing that PPARγ could be involved in regulating B cell differentiation. Similarly, PPARγ overexpression led to reduced B cell proliferation and a more differentiated phenotype. Moreover, B cells upregulate PPARγ expression upon stimulation, implying the regulatory role of PPARγ during B cell activation [123].
PPARγ ligands can also act to alter B cell differentiation and antibody production. 15d-PGJ2 enhanced antibody production, including IgM and IgG, and this was blunted with GW9662. While B cell proliferation or antibody production can contribute towards chronic inflammation, under certain contexts—like a viral infection or a vaccine response—enhanced antibody production may enhance viral clearance and speed a return to homeostasis. In addition to antibody production, the percentage of antibody-secreting cells (CD27+CD38+) and the expression of the transcription factor Blimp-1 (which is involved in B cell plasma cell differentiation) were also increased. These effects were mediated by Cox-2 and the addition of a Cox-2 selective inhibitor attenuated IgM and IgG production induced by 15d-PGJ2. The ability of PPARγ ligands to enhance B cell differentiation, though, was only partially PPARγ-dependent, as a PPARγ inhibitor blocked enhanced IgG production but not IgM production, suggesting that PPARγ might be involved in B cell class switching [123].
In our lab, the role of PPARγ in B cells has further been investigated using B cell-specific PPARγ knockout mice [124]. Even though some studies have shown the suppressive effects of PPARγ ligands on primary mouse bone marrow B cell proliferation in vitro [125], no significant differences were observed in splenic and bone marrow B cell numbers in our studies. In addition, serum antibody levels in B cell-specific PPARγ-deficient mice were similar to those in wild type mice. However, when PPARγ-deficient mice were challenged with the ovalbumin antigen, primary immune responses were impaired. Serum antigen-specific antibody levels, percentage of germinal center B cells, and CD138+ plasma cells were significantly lower than wild type mice. Moreover, the memory response was also impaired against reinjection with the antigen. Other investigators have studied B cells derived from PPARγ haploinsufficient (PPARγ +/−) mice with 50% reductions in PPARγ [126]. These mice have enhanced B cell proliferation and increased serum IgM and IgG production, but since the PPARγ knockout is not B cell specific, the loss of PPARγ in other immune cells, such as T cells, may cause immune response changes that in turn alter B cell function. The anti-inflammatory effects of 15d-PGJ2 on B cell IgE production in vitro were also reported; in Burkitt's lymphoma cell line, DND39, IL-4-induced transcription of epsilon germline transcript was suppressed by 15d-PGJ2, in turn inhibiting B cell class switching to IgE [127]. These contributions of the adaptive immune system are equally important for resolution and homeostasis, and PPARγ appears to work through B and T cells to mediate these effects.
9. PPARγ in Animal Models of Inflammation
Unchecked inflammation plays a role in the development and progression of many chronic diseases, with strong contributions from the innate immune system. PPARγ remains an attractive therapeutic target for treatment of many diseases with underlying inflammatory pathology. Unlike other PPAR isoforms, PPARγ knockout mice are embryonic, lethal [128]. Thus the majority of animal work investigating PPARγ in animal models has utilized pharmacologic activation using synthetic or endogenous PPARγ agonists, though conditional knockouts have been utilized to investigate tissue and cell type specific roles of PPARγ [129, 130]. Much work has been done looking at the effects of PPAR in metabolism, insulin sensitivity, and obesity, which has led to the use of PPARγ agonists as therapeutics for type 2 diabetes; these studies have been reviewed elsewhere. Here, we will highlight some of the other important inflammatory disease models in which PPARγ plays a role.
9.1. Lung
PPARγ is expressed in many cell types in the lung, leading to work investigating the potential for PPARγ agonists as treatments in inflammatory lung diseases. In a mouse model of allergic asthma, ciglitazone treatment was able to reduce airway inflammation and mucus production [131]. A similar PPARγ-dependent suppression of allergic inflammation was seen in a study using both pharmacologic activation of PPARγ and viral gene transfer of PPARγ cDNA [132]. These data support recent findings that asthmatics taking TZDs for treatment of diabetes had a reduced risk for asthma exacerbation [133]. Other nonallergic models of lung inflammation have highlighted the potential beneficial roles of PPARγ ligands. Inflammation from the profibrotic drug bleomycin is reduced in mice treated with the synthetic agonist me-CDDO [38]. A recent study showed that the loss of epithelial PPARγ in the lung enhanced inflammatory mediator production, immune cell recruitment, and exacerbating emphysematous changes following chronic smoke exposure [129]. Pharmacological activation in wild type mice with PPARγ agonists further protected against inflammation following smoke exposure. Another report showed treatment with rosiglitazone, both prophylactically and therapeutically, reduced inflammatory cell counts in the bronchoalveolar lavage fluid in response to four weeks of cigarette exposure. However, inflammatory mediator levels were only reduced with prophylactic treatment, suggesting that the therapeutic effects of PPARγ may be independent of changes in inflammatory mediators [134]. When an exacerbatory infection with nontypeable Haemophilus influenzae was added, rosiglitazone reduced neutrophil influx into the lung without compromising bacterial clearance, though no differences in the inflammatory signal IL-1α, MCP-1, or CXCL5 were detected in the BALF, further supporting the concept that PPARγ therapeutic effects are not due to simply regulating inflammatory mediator production [134]. Interestingly, in a mouse model of influenza infection, treatment with 15d-PGJ2 starting one day after infection dampened inflammatory mediator gene transcription and increased mouse survival and decreased weight loss [135]. No protection was seen if 15d-PGJ2 treatment started on day 0. In conclusion, the ability of PPARγ to regulate pulmonary inflammatory mediators seems to be important in protecting against future insult, whereas the mechanisms of PPARγ mediated therapeutic benefits are less clear.
9.2. Colon
The role of PPARγ in inflammation in other organ systems has also been investigated. Colonic tissue has high expression levels of PPARγ. Indeed, synthetic [136, 137] and endogenous [130, 138] PPARγ ligands have been shown to be efficacious in reducing inflammation in mouse and rat models of experimental colitis. Similar findings have been seen in a pig model of bacterial colitis, where conjugated linoleic acid (CLA) supplementation reduced inflammatory-induced mucosal damage [139]. Loss of function studies have shown that mice that are heterozygous deficient for PPARγ are more susceptible to inflammation from intestinal ischemia/reperfusion injury [140] and colitis [141]. In support of this, tissue specific knockout of PPARγ in the colon prevents the ability of CLA to protect against dextran sodium sulfate induced colitis [130]. Another study found that during colitis PPARγ RNA levels are decreased in the intestinal lamina propria and peritoneal exudate and that adenoviral gene transfer of PPARγ rescued sensitivity to PPARγ ligands and reduced markers of inflammation and improved mouse survival [142].
9.3. Central Nervous System
Inflammation contributes to many neurodegenerative diseases, including Alzheimer's disease and Parkinson's disease, and indeed PPARγ activation has shown beneficial effects in many animal models [143]. A study using a transgenic model of Alzheimer's disease showed that rosiglitazone treatment reduced the appearance of Aβ plaques in multiple areas of the brain [144]. This was accompanied by the reduction of the RNA levels of the proinflammatory markers Cox-2 and TNFα. In a model of Parkinson's disease, chronic treatment with rosiglitazone reduced microglial activation and neuronal loss which corresponded with improved behavioral function [145]. Similar results have been seen with pioglitazone and with a novel PPARγ ligand, MDG548 [146–148]. PPARγ has been tested in experimental models of experimental allergic encephalomyelitis (EAE) as model for multiple sclerosis. 15d-PGJ2 and ciglitazone treatment were able to reduce inflammation and demyelination in mice immunized with mouse spinal cord homogenate [149]. Similarly, troglitazone treatment reduced EAE lesion size and clinical score which correlated with decreased transcripts of TNFα and IL-1β [150].
PPARγ has shown protective effects in many models of disease and shows both the ability to protect against inflammation and stimulate recovery. Although most work shows regulation of inflammation, the role of PPARγ requires further study as beneficial actions can be seen even after inflammation has been established, suggesting therapeutic actions for PPARγ in promoting resolution that may be independent of the regulation of proinflammatory mediators.
10. Specialized Proresolving Mediators: Emerging Players in Resolution
The consumption of dietary omega-3 polyunsaturated fatty acids (ω-3 PUFAs), such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), confers numerous reported health benefits, including an improved prognosis in a wide variety of chronic inflammatory diseases [151]. While the mechanisms underlying these benefits remain largely unknown, EPA and DHA are natural PPARγ ligands [152, 153], and a growing body of in vitro and in vivo evidence indicates that these ω-3 PUFAs exert at least some of their anti-inflammatory and proresolving effects via PPARγ activation. Specialized proresolving mediators (SPMs) constitute a novel and growing array of endogenously produced, lipid-derived compounds that actively promote the resolution of inflammation; many SPMs are metabolites of ω-3 PUFAs, and an exciting biological circuitry connecting SPMs and PPARγ is now beginning to emerge [1].
ω-3 PUFAs exert anti-inflammatory and proresolving effects in multiple organs; growing evidence shows the importance of PPARγ in mediating these effects. For instance, in an immortalized human proximal renal tubular cell line (i.e., human kidney 2 (HK-2) cells), both EPA and DHA increase PPARγ mRNA and protein expression and attenuate LPS-induced activation of NF-κB and expression of monocyte chemoattractant protein-1. Importantly, treatment with the PPARγ agonist/antagonist bisphenol A diglycidyl ether (BADGE) inhibits the activation of PPARγ by EPA and DHA and abolishes both ω-3 PUFAs' inhibitory effects on LPS-induced NF-κB activation in HK-2 cells [154].
Unsurprisingly, the anti-inflammatory and proresolving effects of ω-3 PUFAs are also mediated in part by influences on innate immune cells, and many of these influences appear to be PPARγ-dependent. For instance, in human bone marrow derived dendritic cells (DCs), exposure to DHA inhibits expression of proinflammatory IL-12 via a mechanism that is in part dependent on activation of PPARγ [155]. Similarly, in human monocyte-derived DCs, DHA diminishes both the expression of IL-12 and the capacity of DCs to activate autologous T cells; these effects are also abolished by cotreatment with GW9662, indicating PPARγ dependence [156]. In murine macrophage-like RAW264.7 cells, DHA increases PPARγ mRNA expression and nuclear translocation, enhances phagocytosis and efferocytosis, induces M2 polarization (as indicated by mRNA levels of CD36, IL-10, and TGFβ; surface expression of CD36 protein; and secretion of IL-10 and TGFβ), and inhibits LPS-induced M1 polarization (as indicated by production of proinflammatory cytokines TNFα, IL-1β, and IL-6). Knockdown of PPARγ using siRNA abolishes the stimulatory effects of DHA on efferocytosis and M2 polarization [157].
Human clinical studies have begun to link ω-3 PUFAs and PPARs. In a recent study, patients consumed a moderately high dose of ω-3 PUFAs (3.4 g/day of EPA and DHA ethyl esters) for 2-3 weeks prior to undergoing elective cardiac surgery. Compared to that of untreated controls, the atrial myocardium of patients given ω-3 PUFA had greater transactivation of PPARγ and higher mRNA levels of several genes known to be activated by PPARγ, including CD36, heart-type fatty acid binding protein, and long-chain acyl-CoA dehydrogenase; furthermore, atrial tissue from ω-3 PUFA-treated patients had enhanced mitochondrial respiration supported by palmitoyl-carnitine [158]. More work remains to elucidate the extent to which PPARγ activation mediates ω-3 PUFA-induced enhancement of mitochondrial fatty acid oxidation and antioxidant capacity in human atrial myocardium; yet, this translational research underscores the growing recognition of the link between ω-3 PUFAs and PPARγ activation.
Studies demonstrating PPARγ-dependent anti-inflammatory effects of ω-3 PUFAs inspire the hypothesis that active EPA or DHA metabolites, including SPMs, may act as PPARγ ligands to exert anti-inflammatory effects. Because (1) SPMs and PPARγ ligands share many overlapping anti-inflammatory and proresolving functions, (2) many SPMs are derived from known PPARγ ligands such as EPA and DHA, and (3) PPARs have wide binding sites that can accommodate a variety of ligands, several researchers have postulated that SPMs could act as direct PPAR ligands. Krishnamoorthy and colleagues used cell-based luciferase reporters to test ability of RvD1 and RvE1 to activate PPARα, PPARδ, PPARγ, and RXRα; neither RvD1 nor RvE1 induced strong activation of any of the PPARs and RXRs tested. [159]. On the other hand, there is mounting evidence that some SPMs may act as PPARγ activators and that some of the effects of SPMs are at least in part PPAR dependent. For example, one group administered LPS intratracheally to BALB/c mice to model acute lung injury (ALI). Administration of RvD1 intravenously prior to LPS exposure significantly abrogated LPS-induced inflammation, as indicated by histologic ALI score, and blunted elevations in BALF neutrophils, TNFα, and IL-6; RvD1 treatment also significantly inhibited LPS-induced IκBα degradation, NF-κB p65 subunit nuclear translocation, and DNA-binding activity of NF-κB. Furthermore, RvD1 increased protein levels of PPARγ in the nucleus of lung tissues. Importantly, all of these effects of RvD1 were partially reversed by pretreating mice intravenously with the PPARγ antagonist GW9662, suggesting that RvD1 attenuates LPS-induced ALI via a mechanism that is at least somewhat PPARγ-dependent [160].
Of the SPMs studied to date, protectins have shown the strongest associations with PPARγ. Using an adipogenesis assay and a PPARγ transactivation reporter, Bazan and colleagues demonstrated that neuroprotection D1 (NPD1) is a direct PPARγ activator. Furthermore, the group reported PPARγ-dependent effects of NPD1 in in vitro and 3x-Tg-AD mouse models of Alzheimer's disease. Decreases in hippocampal levels of neuroprotective DHA and NPD1 were detected by mass spectrometry in older wild type and Alzheimer's mice. NPD1 repressed proamyloidogenic processing of β-amyloid polypeptide via the β-secretase pathway and enhanced nonamyloidogenic processing via α-secretase in primary human neuronal glia. In contrast, rosiglitazone treatment or transient PPARγ overexpression only inhibited the β-secretase pathway with no effects on the α-secretase pathway. Accordingly, the PPARγ antagonist GW9662 abrogated NPD1's modulation of the β-secretase pathway but not its stimulatory effect on the α-secretase pathway. These results indicate that NPD1's antiamyloidogenic effects are partly dependent on its ability to activate PPARγ [161].
Additional evidence for PPARγ activation by protectins came from Marette and colleagues, who studied the visceral adipose tissue of fat-1 transgenic mice. These mice convert endogenous ω-6 to ω-3 PUFA; when fed a high-fat diet (HFD) rich in ω-6 PUFA, fat-1 mice maintain an adipose tissue ω-3 : ω-6 ratio of 1 : 1, compared to 50 : 1 in wild type counterparts. A microarray of epididymal adipose tissue revealed upregulation of PPARγ and RXRγ in HFD-fed fat-1 mice. Additionally, compared with HFD-fed wild type mice, HFD-fed fat-1 mice synthesized much higher epididymal adipose tissue levels of protectin DX (PDX), which along with PD1 was identified as a PPARγ agonist [162].
Another set of experiments suggests not only that some SPMs can activate PPARγ, but also that some in vivo actions of PPARγ agonists may be mediated by effects on SPM biosynthesis. In addition to its proresolving effects described above, rosiglitazone induced the expression of 5-lipoxygenase (5-LO), the dioxygenase that (a) catalyzes the conversion of arachidonic acid to 5-HPETE, the precursor to the proinflammatory leukotrienes, and (b) catalyzes the biosynthesis of proresolving lipoxins, in a rodent model of stroke using middle cerebral artery occlusion (MCAO) [163]. Notably, pharmacologic inhibition of 5-LO using BWA4C dose-dependently diminished the neuroprotective and anti-inflammatory effects of rosiglitazone in the ischemic brain. Moreover, rosiglitazone amplified PPARγ transcriptional activity and induced the synthesis of LxA4 in the ischemic cortex of MCAO-treated rats; both of these effects were abolished by the 5-LO inhibitor BWA4C. Based on this finding, the authors administered LxA4 intracerebroventricularly to rats undergoing MCAO and noted neuroprotection as indicated by reduced infarct volume and improved neurological deficit scores; surprisingly, administration of the PPARγ antagonist T0070907 reversed the beneficial effects of LxA4, suggesting its effects are partially PPARγ-dependent. Indeed, in isolated nuclei from rat cerebral cortex, incubation with LxA4 increased PPARγ transcriptional activity [163].
Taken together, these studies demonstrate a complex link between SPMs and PPARγ and suggest the existence of feed-forward amplification loops wherein PPARγ agonists activate PPARγ, which in turn activates biosynthetic pathways that produce SPMs. This increase in SPMs can further augment PPARγ activation in order to mediate anti-inflammatory, proresolving effects. Much more work remains to better understand the implications of context-dependent crosstalk between ω-3 PUFAs, SPMs, and PPARγ in health, disease, and the development of safe, effective, novel therapeutics.
11. Conclusions and Remaining Questions: PPARγ and the Resolution of Inflammation
It is clear that PPARγ plays critical roles in all phases of inflammation (Figure 5). This evidence is largely drawn from a broad evaluation of PPARγ literature, but some studies comprehensively evaluated the role of PPARγ throughout the inflammatory and resolving phases. In a model of murine chronic granulomatous disease pioglitazone increased TGFβ and IL-10 while decreasing KC (mouse homologue to IL-8), IL-6, and TNFα expression. Pioglitazone treated mice had a decrease in the total number of neutrophils and enhanced efferocytosis of apoptotic neutrophils. This clearance was mediated by macrophages, which had increased PPARγ and CD36 expression following treatment. Importantly, these results were seen when pioglitazone was administered as a pretreatment and when pioglitazone was given after the onset of inflammation, indicating that it is truly during the resolution phase of inflammation that PPARγ is playing a role [164].
Figure 5.

PPARγ and the resolution of inflammation. PPARγ and PPARγ ligands (denoted by green diamond) play roles in all stages of inflammation. Early on, PPARγ and its ligands decrease neutrophil recruitment and proinflammatory cytokine production. PPARγ and its ligands then act to promote neutrophil apoptosis and efferocytosis and induction from proinflammatory to anti-inflammatory production. Finally, macrophages move to an M2 phenotype and tissue repair is initiated to return to homeostasis.
Several important questions remain regarding PPARγ's role in modulating inflammation. First and foremost, the dependent versus independent effects of PPARγ ligands need further elucidation. Many studies have made use of a wide variety of techniques to block PPARγ signaling, including PPARγ knockout or knockdown, antagonists, and siRNAs; these methods, particularly antagonists, can have off-target effects which should be closely evaluated. Other studies have evaluated induction of PPARγ expression or PPARγ-dependent luciferase reporters, but few studies show direct PPARγ binding. Furthermore, many studies have used putative PPARγ ligands without evaluating the dependency on PPARγ at all. All work using these ligands should more carefully evaluate the specificity of these effects, as more complete knowledge of PPARγ ligand binding and activation is critical for therapeutic use.
Additionally, there is a lack of evidence regarding the temporal aspects of PPARγ regulation. Most studies conducted involved a pretreatment or single postexposure dose of PPARγ ligands, but given the variable effects between ligands (i.e., TZDs versus prostaglandins in enhancing or decreasing macrophage phagocytosis), these activators may have the most beneficial effects when administered at different phases of the inflammatory and resolving processes. Overall, the emerging literature regarding the proresolving roles of PPARγ, particularly in the context of innate immunity, is encouraging and opens up new questions for investigation and new opportunities for therapeutic use of PPARγ ligands.
Acknowledgments
This work was funded by NIEHS T32ES007026; NIH P30ES01247, R21HL128129, RO1HL120908, and T32HL066988; CTSI Incubator (NIH UL1RR024160); CTSI 8UL1TR000042; the US Army Medical Department; and the PhRMA Foundation.
Abbreviations
- 15d-PGJ2:
15-Deoxy prostaglandin J2
- ALI:
Acute lung injury
- BADGE:
Bisphenol A diglycidyl ether
- Cox-2:
Cyclooxygenase-2
- CRP:
C-reactive protein
- DHA:
Docosahexaenoic acid
- EPA:
Eicosapentaenoic acid
- HFD:
High-fat diet
- LDLs:
Low density lipoproteins
- CDDO:
2-Cyano-3,12-dioxo-oleana-1,9(11)-dien-28-oic acid
- CLA:
Conjugated linoleic acid
- IL:
Interleukin
- LPS:
Lipopolysaccharide
- MCAO:
Middle cerebral artery occlusion
- MCP-1:
Monocyte chemoattractant protein-1
- MPO:
Myeloperoxidase
- NPD1:
Neuroprotectin D1
- OA:
Oleanolic acid
- ω-3 PUFAs:
Omega-3 polyunsaturated fatty acids
- PAI-1:
Plasminogen activator inhibitor 1
- PDX:
Protectin DX
- PPARs:
Peroxisome proliferator activated receptors
- PGD2:
Prostaglandin D2
- RXRα:
Retinoid X receptor alpha
- SPMs:
Specialized proresolving mediators
- TNFα:
Tumor necrosis factor-alpha
- TZDs:
Thiazolidinediones.
Conflict of Interests
Shannon H. Lacy is an employee of the US Government and this work was prepared as part of his official duties. He declares that the views expressed herein are those of the author and do not reflect the official policy or position of the Department of the Army, Department of Defense, or the US Government. All authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Serhan C. N., Chiang N., Van Dyke T. E. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nature Reviews Immunology. 2008;8(5):349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cassetta L., Cassol E., Poli G. Macrophage polarization in health and disease. The Scientific World Journal. 2011;11:2391–2402. doi: 10.1100/2011/213962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asada K., Sasaki S., Suda T., Chida K., Nakamura H. Antiinflammatory roles of peroxisome proliferator-activated receptor gamma in human alveolar macrophages. American Journal of Respiratory and Critical Care Medicine. 2004;169(2):195–200. doi: 10.1164/rccm.200207-740oc. [DOI] [PubMed] [Google Scholar]
- 4.Akbiyik F., Ray D. M., Gettings K. F., Blumberg N., Francis C. W., Phipps R. P. Human bone marrow megakaryocytes and platelets express PPARγ, and PPARγ agonists blunt platelet release of CD40 ligand and thromboxanes. Blood. 2004;104(5):1361–1368. doi: 10.1182/blood-2004-03-0926. [DOI] [PubMed] [Google Scholar]
- 5.Padilla J., Leung E., Phipps R. P. Human B lymphocytes and B lymphomas express PPAR-γ and are killed by PPAR-γ agonists. Clinical Immunology. 2002;103(1):22–33. doi: 10.1006/clim.2001.5181. [DOI] [PubMed] [Google Scholar]
- 6.Kulkarni A. A., Woeller C. F., Thatcher T. H., Ramon S., Phipps R. P., Sime P. J. Emerging PPARγ-independent role of PPARγ ligands in lung diseases. PPAR Research. 2012;2012:13. doi: 10.1155/2012/705352.705352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reddy R. C. Immunomodulatory role of PPAR-gamma in alveolar macrophages. Journal of Investigative Medicine. 2008;56(2):522–527. doi: 10.2310/JIM.0b013e3181659972. [DOI] [PubMed] [Google Scholar]
- 8.Colas R. A., Shinohara M., Dalli J., Chiang N., Serhan C. N. Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. American Journal of Physiology—Cell Physiology. 2014;307(1):C39–C54. doi: 10.1152/ajpcell.00024.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giles H., Leff P. The biology and pharmacology of PGD2. Prostaglandins. 1988;35(2):277–300. doi: 10.1016/0090-6980(88)90093-7. [DOI] [PubMed] [Google Scholar]
- 10.Scher J. U., Pillinger M. H. The anti-inflammatory effects of prostaglandins. Journal of Investigative Medicine. 2009;57(6):703–708. doi: 10.231/JIM.0b013e31819aaa76. [DOI] [PubMed] [Google Scholar]
- 11.Buckley C. D., Gilroy D. W., Serhan C. N. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity. 2014;40(3):315–327. doi: 10.1016/j.immuni.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reddy R. C., Narala V. R., Keshamouni V. G., Milam J. E., Newstead M. W., Standiford T. J. Sepsis-induced inhibition of neutrophil chemotaxis is mediated by activation of peroxisome proliferator-activated receptor-γ . Blood. 2008;112(10):4250–4258. doi: 10.1182/blood-2007-12-128967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma R., Kaundal R. K., Sharma S. S. Amelioration of pulmonary dysfunction and neutrophilic inflammation by PPAR gamma agonist in LPS-exposed guinea pigs. Pulmonary Pharmacology and Therapeutics. 2009;22(3):183–189. doi: 10.1016/j.pupt.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 14.Gao M., Jiang Y., Xiao X., Peng Y., Xiao X., Yang M. Protective effect of pioglitazone on sepsis-induced intestinal injury in a rodent model. Journal of Surgical Research. 2015;195(2):550–558. doi: 10.1016/j.jss.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 15.Akahori T., Sho M., Hamada K., et al. Importance of peroxisome proliferator-activated receptor-γ in hepatic ischemia/reperfusion injury in mice. Journal of Hepatology. 2007;47(6):784–792. doi: 10.1016/j.jhep.2007.07.030. [DOI] [PubMed] [Google Scholar]
- 16.Villegas I., Martín A. R., Toma W., De La Lastra C. A. Rosiglitazone, an agonist of peroxisome proliferator-activated receptor gamma, protects against gastric ischemia-reperfusion damage in rats: role of oxygen free radicals generation. European Journal of Pharmacology. 2004;505(1–3):195–203. doi: 10.1016/j.ejphar.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 17.Cuzzocrea S., Pisano B., Dugo L., et al. Rosiglitazone and 15-deoxy-Δ 12,14-prostaglandin J 2, ligands of the peroxisome proliferator-activated receptor-γ (PPAR-γ), reduce ischaemia/reperfusion injury of the gut. British Journal of Pharmacology. 2003;140(2):366–376. doi: 10.1038/sj.bjp.0705419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Celinski K., Dworzanski T., Korolczuk A., et al. Effects of peroxisome proliferator-activated receptors-γ ligands on dextran sodium sulphate-induced colitis in rats. Journal of Physiology and Pharmacology. 2011;62(3):347–356. [PubMed] [Google Scholar]
- 19.Zhang Q., Hu W., Meng B., Tang T. PPARγ agonist rosiglitazone is neuroprotective after traumatic spinal cord injury via anti-inflammatory in adult rats. Neurological Research. 2010;32(8):852–859. doi: 10.1179/016164110x12556180206112. [DOI] [PubMed] [Google Scholar]
- 20.Collino M., Aragno M., Castiglia S., et al. Pioglitazone improves lipid and insulin levels in overweight rats on a high cholesterol and fructose diet by decreasing hepatic inflammation. British Journal of Pharmacology. 2010;160(8):1892–1902. doi: 10.1111/j.1476-5381.2010.00671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reddy A. T., Lakshmi S. P., Dornadula S., Pinni S., Rampa D. R., Reddy R. C. The nitrated fatty acid 10-nitro-oleate attenuates allergic airway disease. Journal of Immunology. 2013;191(5):2053–2063. doi: 10.4049/jimmunol.1300730. [DOI] [PubMed] [Google Scholar]
- 22.Zingarelli B., Sheehan M., Hake P. W., O'Connor M., Denenberg A., Cook J. A. Peroxisome proliferator activator receptor-γ ligands, 15-deoxy-Δ12,14-prostaglandin J2 and ciglitazone, reduce systemic inflammation in polymicrobial sepsis by modulation of signal transduction pathways. Journal of Immunology. 2003;171(12):6827–6837. doi: 10.4049/jimmunol.171.12.6827. [DOI] [PubMed] [Google Scholar]
- 23.Genovese T., Esposito E., Mazzon E., et al. Effect of cyclopentanone prostaglandin 15-deoxy-Δ12, 14PGJ2 on early functional recovery from experimental spinal cord injury. Shock. 2008;30(2):142–152. doi: 10.1097/SHK.0b013e31815dd381. [DOI] [PubMed] [Google Scholar]
- 24.Pereira P. A., Bitencourt Cda S., dos Santos D. F., Nicolete R., Gelfuso G. M., Faccioli L. H. Prostaglandin D2-loaded microspheres effectively activate macrophage effector functions. European Journal of Pharmaceutical Sciences. 2015;78:132–139. doi: 10.1016/j.ejps.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 25.Yin Y., Hou G., Li E., Wang Q., Kang J. PPARgamma agonists regulate tobacco smoke-induced toll like receptor 4 expression in alveolar macrophages. Respiratory Research. 2014;15, article 28 doi: 10.1186/1465-9921-15-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pisanu A., Lecca D., Mulas G., et al. Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-gamma agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson's disease. Neurobiology of Disease. 2014;71(1):280–291. doi: 10.1016/j.nbd.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 27.Sánchez-Hidalgo M., Martín A. R., Villegas I., Alarcón De La Lastra C. Rosiglitazone, an agonist of peroxisome proliferator-activated receptor gamma, reduces chronic colonic inflammation in rats. Biochemical Pharmacology. 2005;69(12):1733–1744. doi: 10.1016/j.bcp.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 28.Konopleva M., Elstner E., McQueen T. J., et al. Peroxisome proliferator-activated receptor gamma and retinoid X receptor ligands are potent inducers of differentiation and apoptosis in leukemias. Molecular Cancer Therapeutics. 2004;3(10):1249–1262. [PubMed] [Google Scholar]
- 29.Honda T., Kaikita K., Tsujita K., et al. Pioglitazone, a peroxisome proliferator-activated receptor-γ agonist, attenuates myocardial ischemia-reperfusion injury in mice with metabolic disorders. Journal of Molecular and Cellular Cardiology. 2008;44(5):915–926. doi: 10.1016/j.yjmcc.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 30.Liu D., Geng Z. L., Zhu W. K., Wang H. W., Chen Y., Liang J. 15-deoxy-Δ12, 14-prostaglandin J2 ameliorates endotoxin-induced acute lung injury in rats. Chinese Medical Journal. 2014;127(5):815–820. doi: 10.3760/cma.j.issn.0366-6999.20131079. [DOI] [PubMed] [Google Scholar]
- 31.Rajakariar R., Hilliard M., Lawrence T., et al. Hematopoietic prostaglandin D2 synthase controls the onset and resolution of acute inflammation through PGD2 and 15-deoxyΔ12–14 PGJ2 . Proceedings of the National Academy of Sciences of the United States of America. 2007;104(52):20979–20984. doi: 10.1073/pnas.0707394104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernandez-Bustamante A., Klawitter J., Wilson P., et al. Early increase in alveolar macrophage prostaglandin 15d-PGJ2 precedes neutrophil recruitment into lungs of cytokine-insufflated rats. Inflammation. 2013;36(5):1030–1040. doi: 10.1007/s10753-013-9635-x. [DOI] [PubMed] [Google Scholar]
- 33.Gilroy D. W., Colville-Nash P. R., McMaster S., Sawatzky D. A., Willoughby D. A., Lawrence T. Inducible cyclooxygenase-derived 15-deoxyΔ12–14 PGJ2 brings about acute inflammatory resolution in rat pleurisy by inducing neutrophil and macrophage apoptosis. The FASEB Journal. 2003;17(15):2269–2271. doi: 10.1096/fj.02-1162fje. [DOI] [PubMed] [Google Scholar]
- 34.Brown J. R., Goldblatt D., Buddle J., Morton L., Thrasher A. J. Diminished production of anti-inflammatory mediators during neutrophil apoptosis and macrophage phagocytosis in chronic granulomatous disease (CGD) Journal of Leukocyte Biology. 2003;73(5):591–599. doi: 10.1189/jlb.1202599. [DOI] [PubMed] [Google Scholar]
- 35.Ajuebor M. N., Singh A., Wallace J. L. Cyclooxygenase-2-derived prostaglandin D(2) is an early anti-inflammatory signal in experimental colitis. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2000;279(1):G238–G244. doi: 10.1152/ajpgi.2000.279.1.G238. [DOI] [PubMed] [Google Scholar]
- 36.Santos R. S., Silva P. L., de Oliveira G. P., et al. Oleanolic acid improves pulmonary morphofunctional parameters in experimental sepsis by modulating oxidative and apoptotic processes. Respiratory Physiology and Neurobiology. 2013;189(3):484–490. doi: 10.1016/j.resp.2013.08.019. [DOI] [PubMed] [Google Scholar]
- 37.Lin M. H., Chen M., Chen T., Chang H., Chou T. Magnolol ameliorates lipopolysaccharide-induced acute lung injury in rats through PPAR-γ-dependent inhibition of NF-κB activation. International Immunopharmacology. 2015;28(1):270–278. doi: 10.1016/j.intimp.2015.05.051. [DOI] [PubMed] [Google Scholar]
- 38.Kulkarni A. A., Thatcher T. H., Hsiao H.-M., et al. The triterpenoid CDDO-Me inhibits bleomycin-induced lung inflammation and fibrosis. PLoS ONE. 2013;8(5) doi: 10.1371/journal.pone.0063798.e63798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cuartero M. I., Ballesteros I., Moraga A., et al. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the pparγ agonist rosiglitazone. Stroke. 2013;44(12):3498–3508. doi: 10.1161/strokeaha.113.002470. [DOI] [PubMed] [Google Scholar]
- 40.Panzer U., Schneider A., Guan Y., et al. Effects of different PPARgamma-agonists on MCP-1 expression and monocyte recruitment in experimental glomerulonephritis. Kidney International. 2002;62(2):455–464. doi: 10.1046/j.1523-1755.2002.00476.x. [DOI] [PubMed] [Google Scholar]
- 41.Tanaka T., Fukunaga Y., Itoh H., et al. Therapeutic potential of thiazolidinediones in activation of peroxisome proliferator-activated receptor γ for monocyte recruitment and endothelial regeneration. European Journal of Pharmacology. 2005;508(1–3):255–265. doi: 10.1016/j.ejphar.2004.10.056. [DOI] [PubMed] [Google Scholar]
- 42.Koppaka S., Kehlenbrink S., Carey M., et al. Reduced adipose tissue macrophage content is associated with improved insulin sensitivity in thiazolidinedione-treated diabetic humans. Diabetes. 2013;62(6):1843–1854. doi: 10.2337/db12-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Momoi A., Murao K., Imachi H., et al. Inhibition of monocyte chemoattractant protein-1 expression in cytokine-treated human lung epithelial cells by thiazolidinedione. Chest. 2001;120(4):1293–1300. doi: 10.1378/chest.120.4.1293. [DOI] [PubMed] [Google Scholar]
- 44.Venegas-Pont M., Sartori-Valinotti J. C., Maric C., et al. Rosiglitazone decreases blood pressure and renal injury in a female mouse model of systemic lupus erythematosus. American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 2009;296(4):R1282–R1289. doi: 10.1152/ajpregu.90992.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shah Y. M., Morimura K., Gonzalez F. J. Expression of peroxisome proliferator-activated receptor-γ in macrophage suppresses experimentally induced colitis. The American Journal of Physiology—Gastrointestinal and Liver Physiology. 2007;292(2):G657–G666. doi: 10.1152/ajpgi.00381.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Syrovets T., Schüle A., Jendrach M., Büchele B., Simmet T. Ciglitazone inhibits plasmin-induced proinflammatory monocyte activation via modulation of p38 MAP kinase activity. Thrombosis and Haemostasis. 2002;88(2):274–281. [PubMed] [Google Scholar]
- 47.Han K. H., Chang M. K., Boullier A., et al. Oxidized LDL reduces monocyte CCR2 expression through pathways involving peroxisome proliferator-activated receptor γ . Journal of Clinical Investigation. 2000;106(6):793–802. doi: 10.1172/jci10052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Y., Green S. R., Ho J., Li A., Almazan F., Quehenberger O. The mouse CCR2 gene is regulated by two promoters that are responsive to plasma cholesterol and peroxisome proliferator-activated receptor γ ligands. Biochemical and Biophysical Research Communications. 2005;332(1):188–193. doi: 10.1016/j.bbrc.2005.04.110. [DOI] [PubMed] [Google Scholar]
- 49.Azuma Y., Shinohara M., Wang P. L., Ohura K. 15-Deoxy-Δ12,14-prostaglandin J2 is a negative regulator of macrophage functions. International Immunopharmacology. 2001;1(12):2101–2108. doi: 10.1016/s1567-5769(01)00133-3. [DOI] [PubMed] [Google Scholar]
- 50.Han Z., Zhu T., Liu X., et al. 15-deoxy-Δ12,14-prostaglandin J2 reduces recruitment of bone marrow-derived monocyte/macrophages in chronic liver injury in mice. Hepatology. 2012;56(1):350–360. doi: 10.1002/hep.25672. [DOI] [PubMed] [Google Scholar]
- 51.Gautier E. L., Chow A., Spanbroek R., et al. Systemic analysis of PPARγ in mouse macrophage populations reveals marked diversity in expression with critical roles in resolution of inflammation and airway immunity. The Journal of Immunology. 2012;189(5):2614–2624. doi: 10.4049/jimmunol.1200495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johann A. M., von Knethen A., Lindemann D., Brüne B. Recognition of apoptotic cells by macrophages activates the peroxisome proliferator-activated receptor-γ and attenuates the oxidative burst. Cell Death and Differentiation. 2006;13(9):1533–1540. doi: 10.1038/sj.cdd.4401832. [DOI] [PubMed] [Google Scholar]
- 53.Moore K. J., Rosen E. D., Fitzgerald M. L., et al. The role of PPAR-γ in macrophage differentiation and cholesterol uptake. Nature Medicine. 2001;7(1):41–47. doi: 10.1038/83328. [DOI] [PubMed] [Google Scholar]
- 54.Berry A., Balard P., Coste A., et al. IL-13 induces expression of CD36 in human monocytes through PPARγ activation. European Journal of Immunology. 2007;37(6):1642–1652. doi: 10.1002/eji.200636625. [DOI] [PubMed] [Google Scholar]
- 55.Serghides L., Kain K. C. Peroxisome proliferator-activated receptor γ-retinoid X receptor agonists increase CD36-dependent phagocytosis of Plasmodium falciparum-parasitized erythrocytes and decrease malaria-induced TNF-α secretion by monocytes/macrophages. Journal of Immunology. 2001;166(11):6742–6748. doi: 10.4049/jimmunol.166.11.6742. [DOI] [PubMed] [Google Scholar]
- 56.Galès A., Conduché A., Bernad J., et al. PPARγ controls Dectin-1 expression required for host antifungal defense against Candida albicans . PLoS Pathogens. 2010;6(1) doi: 10.1371/journal.ppat.1000714.e1000714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kielian T., Syed M. M., Liu S., et al. The synthetic peroxisome proliferator-activated receptor-α agonist ciglitazone attenuates neuroinflammation and accelerates encapsulation in bacterial brain abscesses. Journal of Immunology. 2008;180(7):5004–5016. doi: 10.4049/jimmunol.180.7.5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stegenga M. E., Florquin S., De Vos A. F., Van Der Poll T. The thiazolidinedione ciglitazone reduces bacterial outgrowth and early inflammation during Streptococcus pneumoniae pneumonia in mice. Critical Care Medicine. 2009;37(2):614–618. doi: 10.1097/ccm.0b013e31819599b6. [DOI] [PubMed] [Google Scholar]
- 59.Serghides L., Patel S. N., Ayi K., et al. Rosiglitazone modulates the innate immune response to Plasmodium falciparum infection and improves outcome in experimental cerebral malaria. Journal of Infectious Diseases. 2009;199(10):1536–1545. doi: 10.1086/598222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ballesteros I., Cuartero M. I., Pradillo J. M., et al. Rosiglitazone-induced CD36 up-regulation resolves inflammation by PPARγ and 5-LO-dependent pathways. Journal of Leukocyte Biology. 2014;95(4):587–598. doi: 10.1189/jlb.0613326. [DOI] [PubMed] [Google Scholar]
- 61.Zhao X., Sun G., Zhang J., et al. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Annals of Neurology. 2007;61(4):352–362. doi: 10.1002/ana.21097. [DOI] [PubMed] [Google Scholar]
- 62.Shimizu K., Kobayashi M., Tahara J., Shiratori K. Cytokines and peroxisome proliferator-activated receptor γ ligand regulate phagocytosis by pancreatic stellate cells. Gastroenterology. 2005;128(7):2105–2118. doi: 10.1053/j.gastro.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 63.Yamanaka M., Ishikawa T., Griep A., Axt D., Kummer M. P., Heneka M. T. PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. Journal of Neuroscience. 2012;32(48):17321–17331. doi: 10.1523/jneurosci.1569-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aronoff D. M., Serezani C. H., Carstens J. K., et al. Stimulatory effects of peroxisome proliferator-activated receptor-γ on Fcγ receptor-mediated phagocytosis by alveolar macrophages. PPAR Research. 2007;2007:8. doi: 10.1155/2007/52546.52546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Song D.-H., Kang J.-H., Lee G.-S., Jeung E.-B., Yang M.-P. Upregulation of tumor necrosis factor-α expression by trans10-cis12 conjugated linoleic acid enhances phagocytosis of RAW macrophages via a peroxisome proliferator-activated receptor γ-dependent pathway. Cytokine. 2007;37(3):227–235. doi: 10.1016/j.cyto.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 66.Stachowska E., Baśkiewicz-Masiuk M., Dziedziejko V., et al. Conjugated linoleic acids can change phagocytosis of human monocytes/macrophages by reduction in Cox-2 expression. Lipids. 2007;42(8):707–716. doi: 10.1007/s11745-007-3072-2. [DOI] [PubMed] [Google Scholar]
- 67.Rossi A. G., McCutcheon J. C., Roy N., Chilvers E. R., Haslett C., Dransfield I. Regulation of macrophage phagocytosis of apoptotic cells by cAMP. The Journal of Immunology. 1998;160(7):3562–3568. [PubMed] [Google Scholar]
- 68.Buckner M. M. C., Antunes L. C. M., Gill N., Russell S. L., Shames S. R., Finlay B. B. 15-Deoxy-Δ12,14-Prostaglandin J2 inhibits macrophage colonization by Salmonella enterica serovar Typhimurium. PLoS ONE. 2013;8(7) doi: 10.1371/journal.pone.0069759.e69759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu X., Yu H., Yang L., Li C., Li L. 15-Deoxy-Δ12,14-prostaglandin J2 attenuates the biological activities of monocyte/macrophage cell lines. European Journal of Cell Biology. 2012;91(8):654–661. doi: 10.1016/j.ejcb.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 70.Bouhlel M. A., Brozek J., Derudas B., et al. Unlike PPARγ, PPARα or PPARβ/δ activation does not promote human monocyte differentiation toward alternative macrophages. Biochemical and Biophysical Research Communications. 2009;386(3):459–462. doi: 10.1016/j.bbrc.2009.06.047. [DOI] [PubMed] [Google Scholar]
- 71.Odegaard J. I., Ricardo-Gonzalez R. R., Red Eagle A., et al. Alternative M2 activation of Kupffer cells by PPARδ ameliorates obesity-induced insulin resistance. Cell Metabolism. 2008;7(6):496–507. doi: 10.1016/j.cmet.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Penas F., Mirkin G. A., Vera M., et al. Treatment in vitro with PPARalpha and PPARgamma ligands drives M1-to-M2 polarization of macrophages from T. cruzi-infected mice. Biochimica et Biophysica Acta—Molecular Basis of Disease. 2015;1852(5):893–904. doi: 10.1016/j.bbadis.2014.12.019. [DOI] [PubMed] [Google Scholar]
- 73.Odegaard J. I., Ricardo-Gonzalez R. R., Goforth M. H., et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature. 2007;447(7148):1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mandrekar-Colucci S., Karlo J. C., Landreth G. E. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer's disease. Journal of Neuroscience. 2012;32(30):10117–10128. doi: 10.1523/jneurosci.5268-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hasegawa-Moriyama M., Kurimoto T., Nakama M., et al. Peroxisome proliferator-activated receptor-gamma agonist rosiglitazone attenuates inflammatory pain through the induction of heme oxygenase-1 in macrophages. Pain. 2013;154(8):1402–1412. doi: 10.1016/j.pain.2013.04.039. [DOI] [PubMed] [Google Scholar]
- 76.Hasegawa-Moriyama M., Ohnou T., Godai K., Kurimoto T., Nakama M., Kanmura Y. Peroxisome proliferator-activated receptor-gamma agonist rosiglitazone attenuates postincisional pain by regulating macrophage polarization. Biochemical and Biophysical Research Communications. 2012;426(1):76–82. doi: 10.1016/j.bbrc.2012.08.039. [DOI] [PubMed] [Google Scholar]
- 77.Yamamoto S., Zhong J., Yancey P. G., et al. Atherosclerosis following renal injury is ameliorated by pioglitazone and losartan via macrophage phenotype. Atherosclerosis. 2015;242(1):56–64. doi: 10.1016/j.atherosclerosis.2015.06.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shaul M. E., Bennett G., Strissel K. J., Greenberg A. S., Obin M. S. Dynamic, M2-like remodeling phenotypes of CD11c+ adipose tissue macrophages during high-fat diet—induced obesity in mice. Diabetes. 2010;59(5):1171–1181. doi: 10.2337/db09-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spencer M., Yang L., Adu A., et al. Pioglitazone treatment reduces adipose tissue inflammation through reduction of mast cell and macrophage number and by improving vascularity. PLoS ONE. 2014;9(7) doi: 10.1371/journal.pone.0102190.e102190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lefèvre L., Galès A., Olagnier D., et al. PPARγ ligands switched high fat diet-induced macrophage M2b polarization toward M2a thereby improving intestinal Candida elimination. PLoS ONE. 2010;5(9) doi: 10.1371/journal.pone.0012828.e12828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lea S., Plumb J., Metcalfe H., et al. The effect of peroxisome proliferatoractivated receptor-γ ligands on in vitro and in vivo models of COPD. European Respiratory Journal. 2014;43(2):409–420. doi: 10.1183/09031936.00187812. [DOI] [PubMed] [Google Scholar]
- 82.Ming Y., Hu X., Song Y., et al. CMHX008, a novel peroxisome proliferator-activated receptor gamma partial agonist, enhances insulin sensitivity in vitro and in vivo. PLoS ONE. 2014;9(7) doi: 10.1371/journal.pone.0102102.e102102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zizzo G., Cohen P. L. The PPAR-gamma antagonist GW9662 elicits differentiation of M2c-like cells and upregulation of the MerTK/Gas6 axis: a key role for PPAR-gamma in human macrophage polarization. Journal of Inflammation. 2015;12, article 36 doi: 10.1186/s12950-015-0081-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bullers S. J., Baker S. C., Ingham E., Southgate J. The human tissue-biomaterial interface: a role for PPARγ-dependent glucocorticoid receptor activation in regulating the CD163+ M2 macrophage phenotype. Tissue Engineering Part A. 2014;20(17-18):2390–2401. doi: 10.1089/ten.tea.2013.0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fujiwara Y., Komohara Y., Kudo R., et al. Oleanolic acid inhibits macrophage differentiation into the M2 phenotype and glioblastoma cell proliferation by suppressing the activation of STAT3. Oncology Reports. 2011;26(6):1533–1537. doi: 10.3892/or.2011.1454. [DOI] [PubMed] [Google Scholar]
- 86.van Tits L. J. H., Stienstra R., van Lent P. L., Netea M. G., Joosten L. A. B., Stalenhoef A. F. H. Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages: a crucial role for Krüppel-like factor 2. Atherosclerosis. 2011;214(2):345–349. doi: 10.1016/j.atherosclerosis.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 87.Rios F. J., Koga M. M., Pecenin M., Ferracini M., Gidlund M., Jancar S. Oxidized LDL induces alternative macrophage phenotype through activation of CD36 and PAFR. Mediators of Inflammation. 2013;2013:8. doi: 10.1155/2013/198193.198193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McCarthy C., Duffy M. M., Mooney D., et al. IL-10 mediates the immunoregulatory response in conjugated linoleic acid-induced regression of atherosclerosis. The FASEB Journal. 2013;27(2):499–510. doi: 10.1096/fj.12-215442. [DOI] [PubMed] [Google Scholar]
- 89.Virtue S., Masoodi M., de Weijer B. A., et al. Prostaglandin profiling reveals a role for haematopoietic prostaglandin D synthase in adipose tissue macrophage polarisation in mice and humans. International Journal of Obesity. 2015;39(7):1151–1160. doi: 10.1038/ijo.2015.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ran L., Yu Q., Zhang S., et al. Cx3cr1 deficiency in mice attenuates hepatic granuloma formation during acute schistosomiasis by enhancing the M2-type polarization of macrophages. Disease Models and Mechanisms. 2015;8(7):691–700. doi: 10.1242/dmm.018242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mao X., Xing H., Mao A., et al. Netrin-1 attenuates cardiac ischemia reperfusion injury and generates alternatively activated macrophages. Inflammation. 2014;37(2):573–580. doi: 10.1007/s10753-013-9771-3. [DOI] [PubMed] [Google Scholar]
- 92.Ranganathan P. V., Jayakumar C., Ramesh G. Netrin-1-treated macrophages protect the kidney against ischemia-reperfusion injury and suppress inflammation by inducing M2 polarization. The American Journal of Physiology—Renal Physiology. 2013;304(7):F948–F957. doi: 10.1152/ajprenal.00580.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yakeu G., Butcher L., Isa S., et al. Low-intensity exercise enhances expression of markers of alternative activation in circulating leukocytes: roles of PPARγ and Th2 cytokines. Atherosclerosis. 2010;212(2):668–673. doi: 10.1016/j.atherosclerosis.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 94.Lovren F., Pan Y., Quan A., et al. Adiponectin primes human monocytes into alternative anti-inflammatory M2 macrophages. The American Journal of Physiology—Heart and Circulatory Physiology. 2010;299(3):H656–H663. doi: 10.1152/ajpheart.00115.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Saika S., Yamanaka O., Okada Y., et al. Effect of overexpression of pparγ on the healing process of corneal alkali burn in mice. The American Journal of Physiology—Cell Physiology. 2007;293(1):C75–C86. doi: 10.1152/ajpcell.00332.2006. [DOI] [PubMed] [Google Scholar]
- 96.Uchiyama M., Shimizu A., Masuda Y., Nagasaka S., Fukuda Y., Takahashi H. An ophthalmic solution of a peroxisome proliferator-activated receptor gamma agonist prevents corneal inflammation in a rat alkali burn model. Molecular Vision. 2013;19:2135–2150. [PMC free article] [PubMed] [Google Scholar]
- 97.Yamanaka O., Miyazaki K.-I., Kitano A., Saika S., Nakajima Y., Ikeda K. Suppression of injury-induced conjunctiva scarring by peroxisome proliferator-activated receptor gamma gene transfer in mice. Investigative Ophthalmology and Visual Science. 2009;50(1):187–193. doi: 10.1167/iovs.08-2282. [DOI] [PubMed] [Google Scholar]
- 98.Jeon K. I., et al. Inhibitory effects of PPARγ ligands on TGF-β1—induced corneal myofibroblast transformation. The American Journal of Pathology. 2014;184(5):1429–1445. doi: 10.1016/j.ajpath.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mirza R. E., Fang M. M., Novak M. L., et al. Macrophage PPARγ and impaired wound healing in type 2 diabetes. The Journal of Pathology. 2015;236(4):433–444. doi: 10.1002/path.4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ray D. M., Spinelli S. L., Pollock S. J., et al. Peroxisome proliferator-activated receptor γ and retinoid X receptor transcription factors are released from activated human platelets and shed in microparticles. Thrombosis and Haemostasis. 2008;99(1):86–95. doi: 10.1160/th07-05-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sahler J., Woeller C. F., Phipps R. P. Microparticles engineered to highly express peroxisome proliferator-activated receptor-γ decreased inflammatory mediator production and increased adhesion of recipient monocytes. PLoS ONE. 2014;9(11) doi: 10.1371/journal.pone.0113189.0113189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.O'Brien J. J., Spinelli S. L., Tober J., et al. 15-Deoxy-delta12,14-PGJ2 enhances platelet production from megakaryocytes. Blood. 2008;112(10):4051–4060. doi: 10.1182/blood-2008-05-158535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Derosa G., Cicero A. F. G., Gaddi A., et al. A comparison of the effects of pioglitazone and rosiglitazone combined with glimepiride on prothrombotic state in type 2 diabetic patients with the metabolic syndrome. Diabetes Research and Clinical Practice. 2005;69(1):5–13. doi: 10.1016/j.diabres.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 104.Kato K., Yamada D., Midorikawa S., Sato W., Watanabe T. Improvement by the insulin-sensitizing agent, troglitazone, of abnormal fibrinolysis in type 2 diabetes mellitus. Metabolism. 2000;49(5):662–665. doi: 10.1016/s0026-0495(00)80045-1. [DOI] [PubMed] [Google Scholar]
- 105.Fonseca V. A., Reynolds T., Hemphill D., et al. Effect of troglitazone on fibrinolysis and activated coagulation in patients with non-insulin-dependent diabetes mellitus. Journal of Diabetes and its Complications. 1998;12(4):181–186. doi: 10.1016/S1056-8727(97)00109-8. [DOI] [PubMed] [Google Scholar]
- 106.Sidhu J. S., Cowan D., Tooze J. A., Kaski J.-C. Peroxisome proliferator-activated receptor-gamma agonist rosiglitazone reduces circulating platelet activity in patients without diabetes mellitus who have coronary artery disease. American heart journal. 2004;147(6, article e25) doi: 10.1016/j.ahj.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 107.Haffner S. M., Greenberg A. S., Weston W. M., Chen H., Williams K., Freed M. I. Effect of rosiglitazone treatment on nontraditional markers of cardiovascular disease in patients with type 2 diabetes mellitus. Circulation. 2002;106(6):679–684. doi: 10.1161/01.CIR.0000025403.20953.23. [DOI] [PubMed] [Google Scholar]
- 108.Schöndorf T., Musholt P. B., Hohberg C., et al. The fixed combination of pioglitazone and metformin improves biomarkers of platelet function and chronic inflammation in type 2 diabetes patients: results from the PIOfIx study. Journal of Diabetes Science and Technology. 2011;5(2):426–432. doi: 10.1177/193229681100500233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Samaha F. F., Szapary P. O., Iqbal N., et al. Effects of rosiglitazone on lipids, adipokines, and inflammatory markers in nondiabetic patients with low high-density lipoprotein cholesterol and metabolic syndrome. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(3):624–630. doi: 10.1161/01.atv.0000200136.56716.30. [DOI] [PubMed] [Google Scholar]
- 110.Hishinuma T., Yamazaki T., Mizugaki M. Troglitazone has a reducing effect on thromboxane production. Prostaglandins and Other Lipid Mediators. 2000;62(2):135–143. doi: 10.1016/S0090-6980(00)00059-9. [DOI] [PubMed] [Google Scholar]
- 111.Østerud B. A global view on the role of monocytes and platelets in atherogenesis. Thrombosis Research. 1997;85(1):1–22. doi: 10.1016/S0049-3848(96)00205-8. [DOI] [PubMed] [Google Scholar]
- 112.Bodary P. F., Vargas F. B., King S. A. D., Jongeward K. L., Wickenheiser K. J., Eitzman D. T. Pioglitazone protects against thrombosis in a mouse model of obesity and insulin resistance. Journal of Thrombosis and Haemostasis. 2005;3(10):2149–2153. doi: 10.1111/j.1538-7836.2005.01551.x. [DOI] [PubMed] [Google Scholar]
- 113.Lievens D., Zernecke A., Seijkens T., et al. Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood. 2010;116(20):4317–4327. doi: 10.1182/blood-2010-01-261206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chu C.-S., Lee K.-T., Lee M.-Y., et al. Effects of Rosiglitazone alone and in combination with Atorvastatin on nontraditional markers of cardiovascular disease in patients with type 2 diabetes mellitus. The American Journal of Cardiology. 2006;97(5):646–650. doi: 10.1016/j.amjcard.2005.09.101. [DOI] [PubMed] [Google Scholar]
- 115.Marx N., Imhof A., Froehlich J., et al. Effect of rosiglitazone treatment on soluble CD40L in patients with type 2 diabetes and coronary artery disease. Circulation. 2003;107(15):1954–1957. doi: 10.1161/01.CIR.0000069272.06194.91. [DOI] [PubMed] [Google Scholar]
- 116.Rao F., Yang R.-Q., Chen X.-S., et al. PPARγ ligands decrease hydrostatic pressure-induced platelet aggregation and proinflammatory activity. PLoS ONE. 2014;9(2) doi: 10.1371/journal.pone.0089654.e89654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ray D. M., Bernstein S. H., Phipps R. P. Human multiple myeloma cells express peroxisome proliferator-activated receptor gamma and undergo apoptosis upon exposure to PPARgamma ligands. Clinical Immunology. 2004;113(2):203–213. doi: 10.1016/j.clim.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 118.Ray D. M., Akbiyik F., Bernstein S. H., Phipps R. P. CD40 engagement prevents peroxisome proliferator-activated receptor γ agonist-induced apoptosis of B lymphocytes and B lymphoma cells by an NF-κB-dependent mechanism. Journal of Immunology. 2005;174(7):4060–4069. doi: 10.4049/jimmunol.174.7.4060. [DOI] [PubMed] [Google Scholar]
- 119.Piva R., Gianferretti P., Ciucci A., Taulli R., Belardo G., Santoro M. G. 15-Deoxy-delta 12,14-prostaglandin J2 induces apoptosis in human malignant B cells: an effect associated with inhibition of NF-kappa B activity and down-regulation of antiapoptotic proteins. Blood. 2005;105(4):1750–1758. doi: 10.1182/blood-2004-04-1360. [DOI] [PubMed] [Google Scholar]
- 120.Schlezinger J. J., Emberley J. K., Sherr D. H. Activation of multiple mitogen-activated protein kinases in pro/pre-B cells by GW7845, a peroxisome proliferator-activated receptor γ agonist, and their contribution to GW7845-induced apoptosis. Toxicological Sciences. 2006;92(2):433–444. doi: 10.1093/toxsci/kfl003. [DOI] [PubMed] [Google Scholar]
- 121.Ray D. M., Akbiyik F., Phipps R. P. The peroxisome proliferator-activated receptor γ (PPARγ) ligands 15-deoxy-Δ12,14-prostaglandin J2 and ciglitazone induce human B lymphocyte and B cell lymphoma apoptosis by PPARγ-independent mechanisms. The Journal of Immunology. 2006;177(8):5068–5076. doi: 10.4049/jimmunol.177.8.5068. [DOI] [PubMed] [Google Scholar]
- 122.Garcia-Bates T. M., Peslak S. A., Baglole C. J., Maggirwar S. B., Bernstein S. H., Phipps R. P. Peroxisome proliferator-activated receptor gamma overexpression and knockdown: impact on human B cell lymphoma proliferation and survival. Cancer Immunology, Immunotherapy. 2009;58(7):1071–1083. doi: 10.1007/s00262-008-0625-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Garcia-Bates T. M., Baglole C. J., Bernard M. P., Murant T. I., Simpson-Haidaris P. J., Phipps R. P. Peroxisome proliferator-activated receptor γ ligands enhance human B cell antibody production and differentiation. Journal of Immunology. 2009;183(11):6903–6912. doi: 10.4049/jimmunol.0900324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ramon S., Bancos S., Thatcher T. H., et al. Peroxisome proliferator-activated receptor γ b cell-specific-deficient mice have an impaired antibody response. Journal of Immunology. 2012;189(10):4740–4747. doi: 10.4049/jimmunol.1200956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schlezinger J. J., Howard G. J., Hurst C. H., et al. Environmental and endogenous peroxisome proliferator-activated receptor γ agonists induce bone marrow B cell growth arrest and apoptosis: interactions between mono(2-ethylhexyl)phthalate, 9-cis-retinoic acid, and 15-deoxy-Δ12,14-prostaglandin J2 . The Journal of Immunology. 2004;173(3):3165–3177. doi: 10.4049/jimmunol.173.5.3165. [DOI] [PubMed] [Google Scholar]
- 126.Setoguchi K., Misaki Y., Terauchi Y., et al. Peroxisome proliferator-activated receptor-γ haploinsufficiency enhances B cell proliferative responses and exacerbates experimentally induced arthritis. The Journal of Clinical Investigation. 2001;108(11):1667–1675. doi: 10.1172/jci200113202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Miyazaki Y., Tachibana H., Yamada K. Inhibitory effect of peroxisome proliferator-activated receptor-gamma ligands on the expression of IgE heavy chain germline transcripts in the human B cell line DND39. Biochemical and Biophysical Research Communications. 2002;295(2):547–552. doi: 10.1016/s0006-291x(02)00709-x. [DOI] [PubMed] [Google Scholar]
- 128.Barak Y., Nelson M. C., Ong E. S., et al. PPARγ is required for placental, cardiac, and adipose tissue development. Molecular Cell. 1999;4(4):585–595. doi: 10.1016/s1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]
- 129.Solleti S. K., Simon D. M., Srisuma S., et al. Airway epithelial cell PPARγ modulates cigarette smoke induced chemokine expression and emphysema susceptibility in mice. American Journal of Physiology—Lung Cellular and Molecular Physiology. 2015;309(3):L293–L304. doi: 10.1152/ajplung.00287.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bassaganya-Riera J., Reynolds K., Martino-Catt S., et al. Activation of PPARγ and δ by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology. 2004;127(3):777–791. doi: 10.1053/j.gastro.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 131.Mueller C., Weaver V., Vanden Heuvel J. P., August A., Cantorna M. T. Peroxisome proliferator-activated receptor gamma ligands attenuate immunological symptoms of experimental allergic asthma. Archives of Biochemistry and Biophysics. 2003;418(2):186–196. doi: 10.1016/j.abb.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 132.Lee K. S., Park S. J., Hwang P. H., et al. PPAR-gamma modulates allergic inflammation through up-regulation of PTEN. The FASEB Journal. 2005;19(8):1033–1035. doi: 10.1096/fj.04-3309fje. [DOI] [PubMed] [Google Scholar]
- 133.Rinne S. T., Feemster L. C., Collins B. F., et al. Thiazolidinediones and the risk of asthma exacerbation among patients with diabetes: a cohort study. Allergy, Asthma & Clinical Immunology. 2014;10(1):p. 34. doi: 10.1186/1710-1492-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Morissette M. C., Shen P., Thayaparan D., Stämpfli M. R. Impacts of peroxisome proliferator-activated receptor-γ activation on cigarette smoke-induced exacerbated response to bacteria. European Respiratory Journal. 2015;45(1):191–200. doi: 10.1183/09031936.00004314. [DOI] [PubMed] [Google Scholar]
- 135.Cloutier A., Marois I., Cloutier D., Verreault C., Cantin A. M., Richter M. V. The prostanoid 15-deoxy-Δ 12,14-prostaglandin-J2 reduces lung inflammation and protects mice against lethal influenza infection. Journal of Infectious Diseases. 2012;205(4):621–630. doi: 10.1093/infdis/jir804. [DOI] [PubMed] [Google Scholar]
- 136.Su C. G., Wen X., Bailey S. T., et al. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. The Journal of Clinical Investigation. 1999;104(4):383–389. doi: 10.1172/jci7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tanaka T., Kohno H., Yoshitani S., et al. Ligands for peroxisome proliferator-activated receptors alpha and gamma inhibit chemically induced colitis and formation of aberrant crypt foci in rats. Cancer Research. 2001;61(6):2424–2428. [PubMed] [Google Scholar]
- 138.Cuzzocrea S., Ianaro A., Wayman N. S., et al. The cyclopentenone prostaglandin 15-deoxy-Δ12,14-PGJ2 attenuates the development of colon injury caused by dinitrobenzene sulphonic acid in the rat. British Journal of Pharmacology. 2003;138(4):678–688. doi: 10.1038/sj.bjp.0705077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hontecillas R., Wannemeulher M. J., Zimmerman D. R., et al. Nutritional regulation of porcine bacterial-induced colitis by conjugated linoleic acid. Journal of Nutrition. 2002;132(7):2019–2027. doi: 10.1093/jn/132.7.2019. [DOI] [PubMed] [Google Scholar]
- 140.Nakajima A., Wada K., Miki H., et al. Endogenous PPARγ mediates anti-inflammatory activity in murine ischemia-reperfusion injury. Gastroenterology. 2001;120(2):460–469. doi: 10.1053/gast.2001.21191. [DOI] [PubMed] [Google Scholar]
- 141.Rousseaux C., Lefebvre B., Dubuquoy L., et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. Journal of Experimental Medicine. 2005;201(8):1205–1215. doi: 10.1084/jem.20041948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Katayama K., Wada K., Nakajima A., et al. A novel PPARγ gene therapy to control inflammation associated with inflammatory bowel disease in a murine model. Gastroenterology. 2003;124(5):1315–1324. doi: 10.1016/s0016-5085(03)00262-2. [DOI] [PubMed] [Google Scholar]
- 143.Skerrett R., Malm T., Landreth G. Nuclear receptors in neurodegenerative diseases. Neurobiology of Disease. 2014;72(part A):104–116. doi: 10.1016/j.nbd.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Escribano L., Simón A.-M., Gimeno E., et al. Rosiglitazone rescues memory impairment in Alzheimer's transgenic mice: mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology. 2010;35(7):1593–1604. doi: 10.1038/npp.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Schintu N., Frau L., Ibba M., et al. PPAR-gamma-mediated neuroprotection in a chronic mouse model of Parkinson's disease. European Journal of Neuroscience. 2009;29(5):954–963. doi: 10.1111/j.1460-9568.2009.06657.x. [DOI] [PubMed] [Google Scholar]
- 146.Breidert T., Callebert J., Heneka M. T., Landreth G., Launay J. M., Hirsch E. C. Protective action of the peroxisome proliferator-activated receptor-gamma agonist pioglitazone in a mouse model of Parkinson's disease. Journal of Neurochemistry. 2002;82(3):615–624. doi: 10.1046/j.1471-4159.2002.00990.x. [DOI] [PubMed] [Google Scholar]
- 147.Dehmer T., Heneka M. T., Sastre M., Dichgans J., Schulz J. B. Protection by pioglitazone in the MPTP model of Parkinson's disease correlates with IκBα induction and block of NFκB and iNOS activation. Journal of Neurochemistry. 2004;88(2):494–501. doi: 10.1046/j.1471-4159.2003.02210.x. [DOI] [PubMed] [Google Scholar]
- 148.Lecca D., Nevin D., Mulas G., et al. Neuroprotective and anti-inflammatory properties of a novel non-thiazolidinedione PPARγ agonist in vitro and in MPTP-treated mice. Neuroscience. 2015;302:23–35. doi: 10.1016/j.neuroscience.2015.04.026. [DOI] [PubMed] [Google Scholar]
- 149.Natarajan C., Bright J. J. Peroxisome proliferator-activated receptor-gamma agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes & Immunity. 2002;3(2):59–70. doi: 10.1038/sj.gene.6363832. [DOI] [PubMed] [Google Scholar]
- 150.Niino M., Iwabuchi K., Kikuchi S., et al. Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-gamma. Journal of Neuroimmunology. 2001;116(1):40–48. doi: 10.1016/s0165-5728(01)00285-5. [DOI] [PubMed] [Google Scholar]
- 151.Simopoulos A. P. Omega-3 fatty acids in inflammation and autoimmune diseases. Journal of the American College of Nutrition. 2002;21(6):495–505. doi: 10.1080/07315724.2002.10719248. [DOI] [PubMed] [Google Scholar]
- 152.Kliewer S. A., Sundseth S. S., Jones S. A., et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ . Proceedings of the National Academy of Sciences of the United States of America. 1997;94(9):4318–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Xu H. E., Lambert M. H., Montana V. G., et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Molecular Cell. 1999;3(3):397–403. doi: 10.1016/s1097-2765(00)80467-0. [DOI] [PubMed] [Google Scholar]
- 154.Li H., Ruan X. Z., Powis S. H., et al. EPA and DHA reduce LPS-induced inflammation responses in HK-2 cells: evidence for a PPAR-γ-dependent mechanism. Kidney International. 2005;67(3):867–874. doi: 10.1111/j.1523-1755.2005.00151.x. [DOI] [PubMed] [Google Scholar]
- 155.Kong W., Yen J.-H., Vassiliou E., Adhikary S., Toscano M. G., Ganea D. Docosahexaenoic acid prevents dendritic cell maturation and in vitro and in vivo expression of the IL-12 cytokine family. Lipids in Health and Disease. 2010;9, article 12 doi: 10.1186/1476-511x-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zapata-Gonzalez F., Rueda F., Petriz J., et al. Human dendritic cell activities are modulated by the omega-3 fatty acid, docosahexaenoic acid, mainly through PPARγ:RXR heterodimers: comparison with other polyunsaturated fatty acids. Journal of Leukocyte Biology. 2008;84(4):1172–1182. doi: 10.1189/jlb.1007688. [DOI] [PubMed] [Google Scholar]
- 157.Chang H. Y., Lee H.-N., Kim W., Surh Y.-J. Docosahexaenoic acid induces M2 macrophage polarization through peroxisome proliferator-activated receptor γ activation. Life Sciences. 2015;120:39–47. doi: 10.1016/j.lfs.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 158.Anderson E. J., Thayne K. A., Harris M., et al. Do fish oil omega-3 fatty acids enhance antioxidant capacity and mitochondrial fatty acid oxidation in human atrial myocardium via PPARγ activation? Antioxidants and Redox Signaling. 2014;21(8):1156–1163. doi: 10.1089/ars.2014.5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Krishnamoorthy S., Recchiuti A., Chiang N., et al. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(4):1660–1665. doi: 10.1073/pnas.0907342107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Liao Z., Dong J., Wu W., et al. Resolvin D1 attenuates inflammation in lipopolysaccharide-induced acute lung injury through a process involving the PPARγ/NF-κB pathway. Respiratory Research. 2012;13, article 110 doi: 10.1186/1465-9921-13-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhao Y., Calon F., Julien C., et al. Docosahexaenoic acid-derived neuroprotectin D1 induces neuronal survival via secretase- and PPARγ-mediated mechanisms in Alzheimer's disease models. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0015816.e15816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.White P. J., Mitchell P. L., Schwab M., et al. Transgenic ω-3 PUFA enrichment alters morphology and gene expression profile in adipose tissue of obese mice: potential role for protectins. Metabolism. 2015;64(6):666–676. doi: 10.1016/j.metabol.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 163.Sobrado M., Pereira M. P., Ballesteros I., et al. Synthesis of lipoxin A4 by 5-lipoxygenase mediates PPARgamma-dependent, neuroprotective effects of rosiglitazone in experimental stroke. Journal of Neuroscience. 2009;29(12):3875–3884. doi: 10.1523/jneurosci.5529-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Fernandez-Boyanapalli R., Frasch S. C., Riches D. W. H., Vandivier R. W., Henson P. M., Bratton D. L. PPARγ activation normalizes resolution of acute sterile inflammation in murine chronic granulomatous disease. Blood. 2010;116(22):4512–4522. doi: 10.1182/blood-2010-02-272005. [DOI] [PMC free article] [PubMed] [Google Scholar]