

Abstract
Sporadic mast cell neoplasms and gastrointestinal stromal tumors (GISTs) often have various types of somatic gain-of-function mutations of the c-kit gene which encodes a receptor tyrosine kinase, KIT. Several types of germline gain-of-function mutations of the c-kit gene have been detected in families with multiple GISTs. All three types of model mice for the familial GISTs with germline c-kit gene mutations at exon 11, 13 or 17 show development of GIST, while they are different from each other in skin mast cell number. Skin mast cell number in the model mice with exon 17 mutation was unchanged compared to the corresponding wild-type mice. In the present study, we characterized various types of mast cells derived from the model mice with exon 17 mutation (KIT-Asp818Tyr) corresponding to human familial GIST case with human KIT-Asp820Tyr to clarify the role of the c-kit gene mutation in mast cells. Bone marrow-derived cultured mast cells (BMMCs) derived from wild-type mice, heterozygotes and homozygotes were used for the experiments. Immortalized BMMCs, designated as IMC-G4 cells, derived from BMMCs of a homozygote during long-term culture were also used. Ultrastructure, histamine contents, proliferation profiles and phosphorylation of various signaling molecules in those cells were examined. In IMC-G4 cells, presence of additional mutation(s) of the c-kit gene and effect of KIT inhibitors on both KIT autophosphorylation and cell proliferation were also analyzed. We demonstrated that KIT-Asp818Tyr did not affect ultrastructure and proliferation profiles but did histamine contents in BMMCs. IMC-G4 cells had an additional novel c-kit gene mutation of KIT-Tyr421Cys which is considered to induce neoplastic transformation of mouse mast cells and the mutation appeared to be resistant to a KIT inhibitor of imatinib but sensitive to another KIT inhibitor of nilotinib. IMC-G4 cells might be a useful mast cell line to investigate mast cell biology.
Keywords: Bone marrow-derived cultured mast cell, c-kit gene, exon 17, gastrointestinal stromal tumor, germline mutation, model mouse, histamine synthesis, mast cell neoplasm, imatinib, nilotinib
Introduction
The function of the c-kit gene product, KIT, is essential for the development of five cell lineages such as erythrocytes, melanocytes, germ cells, mast cells and interstitial cells of Cajal (ICCs) [1]. ICCs are considered to be a pacemaker of peristalsis in gastrointestinal tract. Since W mutant mice have loss-of-function mutations of the c-kit gene, they show five phenotypes of anemia, white coat color, infertility, deficiency of mast cells and abnormal gastrointestinal movement due to the impaired development of above-mentioned five cell lineages [1]. On the other hand, gain-of-function mutations of the c-kit gene are known to be detected in leukemia, malignant melanomas, seminomas, mast cell neoplasms and gastrointestinal stromal tumors (GISTs) at different frequency [1]. Since ICCs and GISTs express both KIT and CD34 in common and since ICCs are the only proper cells in gastrointestinal tract that express KIT, ICCs are considered to be the origin of GISTs [1,2].
Most of the sporadic GISTs have somatic gain-of-function mutations of the c-kit gene. The mutations are most frequently detected at exon 11 (70-80%), less frequently at exon 9 (approximately 10%) and rarely at exon 8, exon 13 and exon 17 (less than 2% each) [1-4]. Various types of exon 11 mutations are observed, while exon 9, exon 13 and exon 17 mutations usually show the particular types. In addition, several types of germline gain-of-function mutations of the c-kit gene have been detected in approximately 30 families with multiple GISTs [5-29]. Again, the mutations in the familial GISTs are most frequently detected at exon 11, but exon 8, exon 13 and exon 17 mutations are also reported [5-29]. Development of multiple GISTs with ICC hyperplasia is the essentially observed phenotype in the families [5-29], but some families have mast cell neoplasms [9,14] and/or hyperpigmentation of the digital, perioral and perineal regions [6,8,9,11,14,15,20,26].
Three types of mouse models for familial GISTs with germline gain-of-function mutations of the c-kit gene have been generated through the knock-in strategy. One has a deletion of codon 558 (valine) at exon 11 (KIT-del-Val558) corresponding to human familial GIST case with human KIT-del-Val559 [30]. Another has a substitution mutation of codon 641 from lysine to glutamic acid at exon 13 (KIT-Lys641Glu) corresponding to human familial GIST case with human KIT-Lys642Glu [31], and the other has a substitution of codon 818 from aspartic acid to tyrosine at exon 17 (KIT-Asp818Tyr) corresponding to human familial GIST case with human KIT-Asp820Tyr [32]. All types of model mice show development of a cecal GIST with ICC hyperplasia. As mentioned above, mast cell neoplasms and hyperpigmentation at the particular sites are sometimes observed in human multiple GIST families, and ectopic pigmentation at the lower esophagus is observed in some of the model mice [30,32]. On the other hand, mast cell numbers in the skin of the three model mice are different from each other as compared to respective wild type mice. Number of skin mast cells in the model mice with KIT-del-Val558 increases [30], that with KIT-Lys641Glu decreases [31], and that with KIT-Asp818Tyr is unchanged [32].
In sporadic human mast cell neoplasms, most of the c-kit gene mutations are present at exon 17 (KIT-Asp816Val or KIT-Asp816Tyr) [33,34]. In mouse neoplastic mast cell lines, moreover, KIT-Asp814Val or KIT-Asp814Tyr corresponding to human KIT-Asp816Val or KIT-Asp816Tyr has been reported [35]. In human mast cell neoplasms, several types of mutations of the c-kit gene have also been detected at exon 8, exon 9 or exon 11 [36].
In the present study, we characterized the various types of mast cells derived from the model mice with germline c-kit gene mutation at exon 17 (KIT-Asp818Tyr). In bone marrow-derived cultured mast cells (BMMCs) derived from wild-type (+/+) mice, heterozygotes (KIT-Asp818Tyr/+) and homozygotes (KIT-Asp818Tyr/KIT-Asp818Tyr) were used for the experiments. We also examined the biological features of the immortalized BMMCs, designated as IMC-G4 cells, derived from BMMCs of a KIT-Asp818Tyr/KIT-Asp818Tyr mouse during long-term culture.
Materials and methods
Mice
By cross-breeding the male and female KIT-Asp818Tyr/+ mice, we obtained +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice. Genotyping of the mice was determined by real-time PCR using DNA extracted from their tails as described previously [32]. All of the experimental procedures using animals were approved by the Hyogo College of Medicine Animal Experiment Committee.
Establishment of BMMCs and IMC-G4 cells
BMMCs were established from +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice. Briefly, bone marrow cells flushed out from the bilateral femoral bones were cultured in alpha-MEM (Life Technologies Corporation, Grand Island, NY) with 10% fetal bovine serum, 100 microgram/ml streptomycin, 100 U/ml penicillin and 5 ng/ml murine interleukin-3 (IL-3) at 37 degree Celsius in 5% CO2 in incubator. Since almost all cultured cells differentiated into mast cells in one month, the BMMCs were used for the experiments between 1 month and 3 months from the beginning of the culture. During long-term culture of the BMMCs derived from a KIT-Asp818Tyr/KIT-Asp818Tyr mouse, we obtained an immortalized clone, designated as IMC-G4, which do not need IL-3 for proliferation and could be cultured over 4 years. IMC-G4 cells were basically cultured without IL-3 and also used for the experiments.
Electron microscopic examination of cultured cells
BMMCs (1 × 108) derived from +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice were collected and centrifuged. IMC-G4 cells were also collected and centrifuged. The cell pellets were fixed with 1.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4), postfixed with 2% osmic acid in the same buffer, dehydrated and embedded in epoxy resin (Epon 812). Ultrathin sections, doubly stained with uranyl acetate-lead citrate, were examined with transmission electron microscope, Hitachi H-7100 (Hitachi, Tokyo, Japan).
Quantitative analysis of histamine
Histamine contents of BMMCs (8 × 106) derived from +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice were examined by colorimetric enzyme assay for the quantitative analysis of histamine using Histamine Test (Kikkoman, Tokyo, Japan) according to the manufacturer’s instructions. Those of IMC-G4 cells (8 × 106) were also examined both in the presence of IL-3 and in the absence of IL-3.
Proliferative and survival profiles in presence or absence of IL-3
Changes of the numbers of BMMCs derived from +/+, KIT-Asp8181Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice were examined in the presence of IL-3 (5 ng/ml) or in the absence of IL-3. Those of IMC-G4 cells were also examined in the absence of IL-3. For the experiment of proliferative profile, 5 × 104 cells per well were initially dispensed. For the experiment of survival profile, 5 × 105 cells per well were initially dispensed.
Western blotting of KIT and downstream signaling molecules
Lysis of BMMCs derived from +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice with the stimulation by IL-3 was done with CelLytic-M lysis buffer (Sigma-Aldrich, St. Louis, MO) with the addition of complete protease inhibitor cocktail tablets (Roche, Mannheim, Germany), phosphatase inhibitor cocktail (Sigma-Aldrich), and 1 microM Na3VO4. Lysis of IMC-G4 cells with or without the stimulation by IL-3 was carried out as well. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting were performed as described [32]. Antibodies (Abs) used were mouse anti-phospho-KIT Ab (pTyr823, Affinity BioReagents, Golden, CO), rabbit anti-KIT polyclonal Ab (A4502; DAKO Cytomation), phospho-p44/42 MAPK Ab (9101, Cell Signaling Technology, Beverly, MA), p44/42 MAPK Ab (9102, Cell Signaling Technology), phospho-Akt Ab (9271, Cell Signaling Technology), total Akt Ab (9272, Cell Signaling Technology), phospho-STAT1 Ab (9171, Cell Signaling Technology), STAT1 Ab (9172, Cell Signaling Technology), phpspho-STAT5 Ab (9351, Cell Signaling Technology), STAT5 Ab (9352, Cell Signaling Technology), phospho-Lyn Ab (2731, Cell Signaling Technology) and Lyn Ab (2732, Cell Signaling Technology). The binding of the Abs was detected by ECL Western Blotting Detection System (GE Healthcare, Buckinghamshire, UK).
RNA extraction, cDNA synthesis and sequencing
Total RNA was extracted from IMC-G4 cells by RNeasy Mini Kit (QIAGEN, Valencia, CA). Single-strand complementary DNA (cDNA) was synthesized using reverse transcriptase (Superscript III, Life Technologies Corporation, Carlsbad, CA) from total RNA, and polymerase chain reaction was performed for amplification of c-kit cDNA using primers as described previously [32]. Amplified products were purified with the QIAquick Gel Extraction Kit (QIAGEN), and direct sequencing was performed using ABI BigDye Terminator ver. 3.1 (Applied Biosystems, Foster City, CA) and an ABI Prism 3100-Avant Genetic Analyzer (Applied Biosystems).
Tumorigenesis of IMC-G4 cells
IMC-G4 cells cultured in IL-3 free medium were subcutaneously injected into the posterior flank of 4 nude mice (BALB/c-nu/nu, Japan SLC, Hamamatsu, Japan). As a control, BMMCs derived from KIT-Asp818Tyr/KIT-Asp818Tyr mice cultured in IL-3 containing medium were also transplanted subcutaneously at the posterior flank of another 4 nude mice. Number of the cells injected was 1 × 107 per one site. The tumor volume was calculated with the following formula: Tumor volume = 0.5 × a × b2, where a and b are the length and width in millimeters of the tumor mass, respectively.
Effect of KIT inhibitors on KIT autophosphorylation
To evaluate the effect of KIT inhibitors, imatinib and nilotinib, on KIT autophosphorylation in IMC-G4, BMMCs derived from KIT-Asp818Tyr/KIT-Asp818Tyr mice, Ba/F3 cells expressing KIT-Asp818Tyr, Western blotting of KIT was done. Ba/F3 cells expressing KIT-del-Val558&Val559 were used as a control of imatinib- and nilotinib-sensitive cells. Imatinib and nilotinib were generous gifts from Novartis (Basel, Switzerland). Briefly, 3 × 106 cells were cultured at various concentration of imatinib (0, 0.001, 0.01, 0.1, 1 and 10 microM) or nilotinib (0, 0.001, 0.01, 0.1, 1 and 10 microM). Then cells were lysed as described above, and sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting were performed using mouse anti-phospho-KIT Ab and rabbit anti-KIT polyclonal Ab.
Effect of KIT inhibitors on cell proliferation
To evaluate the effect of KIT inhibitors, imatinib and nilotinib, on proliferation of IMC-G4 cells, Ba/F3 cells expressing KIT-Asp818Tyr and Ba/F3 cells expressing KIT-del-Val558&Val559, MTS colorimetric assay was performed using CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI). Briefly, cells were plated in a 96-well plate at a concentration of 1.5 × 104/well and cultured with various concentrations of imatinib (0, 0.001, 0.01, 0.1, 1, and 10 microM) or nilotinib (0, 0.001, 0.01, 0.1, 1, and 10 microM) for 48 hours. Then cells were further cultured for 2 hours in the presence of MTS. The optical density was measured with a test wavelength of 490 nm and a reference wavelength of 650 nm. BMMCs derived from KIT-Asp818Tyr/KIT-Asp818Tyr mice were not examined because their proliferation is dependent on IL-3.
Results
Ultrastructure of various mast cells
We examined ultrastructure of BMMCs derived from +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice using transmission electron microscope. IMC-G4 cells derived from long-term cultured BMMCs of KIT-Asp818Tyr/KIT-Asp818Tyr mice were also examined. All types of cells were similar in appearance. They have a single nucleus, many secretory granules partially filled with an electron-dense core and many short villi (Figure 1A-D).
Figure 1.

Ultrastructure of various mast cells. Representative ultrastructure of BMMCs derived from +/+ mice (A), KIT-Asp818Tyr/+ mice (B) and KIT-Asp818Tyr/KIT-Asp818Tyr mice (C) and that of IMC-G4 cells (D) were examined with transmission electron microscope. All types of cells showed similar ultrastructural appearance such as single nucleus, a lot of granules in their cytoplasm and many short villi.
Quantification of histamine contents in various mast cells
Histamine contents of BMMCs (8 × 106) derived from +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice were measured. Those of IMC-G4 cells (8 × 106) were also examined. BMMCs derived from KIT-Asp818Tyr/+ mice had similar amount of histamine compared to those from +/+ mice (Figure 2). However, those from KIT-Asp818Tyr/KIT-Asp818Tyr mice had approximately half amount of histamine as compared to those from +/+ mice (Figure 2). Moreover, IMC-G4 cells had approximately one-sixth amount of histamine both in the presence of IL-3 and in the absence of IL-3, compared to BMMCs derived from +/+ mice (Figure 2).
Figure 2.
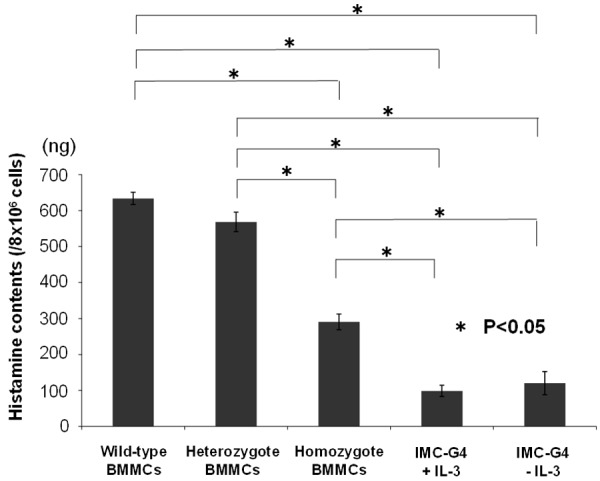
Quantification of histamine contents of various cultured mast cells by colorimetric enzyme assay. Histamine contents of BMMCs derived from +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice and IMC-G4 cells were examined. Those of BMMCs derived from KIT-Asp818Tyr/KIT-Asp818Tyr mice were approximately half those derived from +/+ and KIT-Asp818Tyr/+ mice. Those of IMC-G4 cells both in the presence of IL-3 and in the absence of IL-3 were further approximately half those derived from KIT-Asp818Tyr/KIT-Asp818Tyr mice.
Proliferation and survival profile of various mast cells
Proliferation profiles of BMMCs derived from +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice were examined both in the presence of IL-3 and in the absence of IL-3. Proliferation profile of IMC-G4 in the absence of IL-3 was also examined as a comparison. In the presence of IL-3, all types of BMMCs derived from +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice proliferated similarly (Figure 3A). On the other hand, IMC-G4 cells proliferated in the absence of IL-3, and did faster than all types of BMMCs in the presence of IL-3 (Figure 3A). In the absence of IL-3, numbers of all types of BMMCs decreased similarly (Figure 3B).
Figure 3.
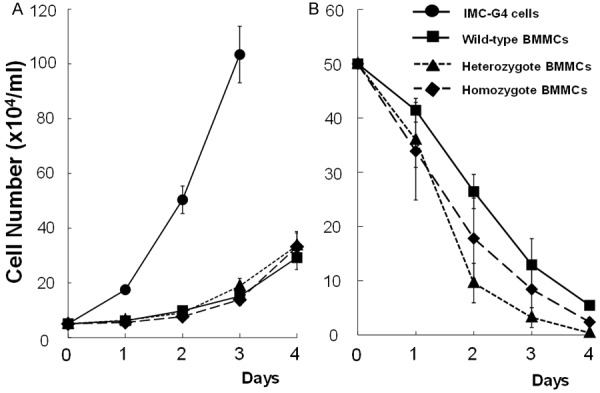
Proliferation and survival profiles of various mast cells in the presence or absence of IL-3. Proliferation of BMMCs derived from +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice was examined in the presence of 5 ng/ml of IL-3 (A). That of IMC-G4 was examined in the absence of IL-3 as a comparison (A). IMC-G4 cells proliferated autonomously even in the absence of IL-3 and their proliferation was the fastest. Although all types of BMMCs proliferated more slowly as compared to IMC-G4 cells, their proliferation profile was similar (A). When survival of BMMCs derived from +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice was examined in the absence of IL-3, they decreased similarly (B).
Activation status of KIT and its downstream signaling molecules
First, KIT autophosphorylation of BMMCs derived from +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice was examined without stimulation by stem cell factor (SCF). That of IMC-G4 cells was also examined. KIT of BMMCs derived from +/+ mice was barely phosphorylated, and that from KIT-Asp818Tyr/+ mice was slightly phosphorylated. On the other hand, KIT of BMMCs derived from KIT-Asp818Tyr/KIT-Asp818Tyr mice was apparently phosphorylated. KIT of IMC-G4 cells was more apparently phosphorylated under the conditions of both presence and absence of IL-3. Since SCF-KIT and Lyn-Syk-LAT systems are known to be possible major downstream signaling pathways in mast cells, we examined phosphorylation of STAT1, STAT5, MAPK, Akt and Lyn. Stat1 was slightly and similarly phosphorylated in all types of BMMCs and IMC-G4 cells. STAT5, MAPK and Lyn were apparently and similarly phosphorylated among them. Although phosphorylation of Akt was weak in all types of BMMCs derived from +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice, Akt in IMC-G4 cells was strongly phosphorylated under the conditions of both presence and absence of IL-3 (Figure 4).
Figure 4.

Expression and autophosphorylation of KIT and its downstream signaling molecules in various mast cells. All of BMMCs derived from +/+, KIT-Asp818Tyr/+ (Heterozygote) and KIT-Asp818Tyr/KIT-Asp818Tyr (Homozygote) mice strongly expressed KIT. However, KIT of BMMCs derived from +/+ mice was not apparently phosphorylated, KIT of BMMCs derived from KIT-Asp818Tyr/+ mice faintly phosphorylated, BMMC derived from KIT-Asp818Tyr/Asp818Tyr mice apparently phosphorylated. IMC-G4 cells strongly expressed KIT, and phosphorylation of KIT in them without IL-3 stimulation is stronger than that in BMMC derived from KIT-Asp818Tyr/Asp818Tyr mice. KIT expression and phosphorylation in IMC-G4 cells with IL-3 stimulation was also shown as a comparison. In spite of apparent phosphorylation of KIT in BMMC derived from KIT-Asp818Tyr/Asp818Tyr mice and KIT of IMC-G4 cells, no difference in phosphorylation status of downstream signaling molecules such as Stat1, Stat5, MAPK, Akt and Lyn was observed among BMMCs derived from +/+ mice, those derived from KIT-Asp818Tyr/+ mice and those derived from KIT-Asp818Tyr/Asp818Tyr mice. In IMC-G4 cells, on the other hand, magnitude of phosphorylation of signaling molecules such as Stat1, Stat5, MAPK and Lyn was not apparently different with that in those BMMCs. However, Akt was strongly phosphorylated as compared with that in those BMMCs.
Mutational analysis of the c-kit gene in IMC-G4 cells
Since autophosphorylation of KIT in IMC-G4 cells was apparently stronger than that in BMMCs derived from KIT-Asp818Tyr/KIT-Asp818Tyr mice as mentioned above, we speculated that some additional change in the c-kit gene might happen. To examine whether IMC-G4 cells have additional c-kit gene mutation(s) other than Asp818Tyr, sequencing of the c-kit cDNA of IMC-G4 cells was done. In addition to homozygous/hemizygous Asp818Tyr mutation, we found a nucleotide substitution of A to G at codon 421 resulting in homozygous/hemizygous Tyr421Cys mutation (Figure 5).
Figure 5.
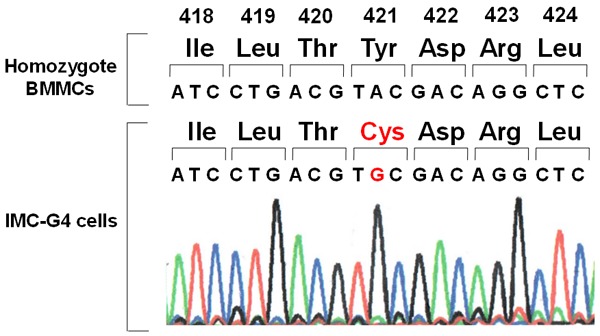
Mutational analysis of the c-kit gene in IMC-G4 cells. Sequencing of the c-kit cDNA derived from IMC-G4 cells revealed that they have a homozygous/hemizygous nucleotide substitution of A to C at codon 421 resulting in Tyr421Cys mutation in addition to homozygous/hemizygous Asp818Tyr mutation.
Tumorigenesis of IMC-G4 cells
To examine the biological behavior of immortalized IMC-G4 cells, they were subcutaneously injected into posterior flank of 4 nude mice. They autonomously proliferated and formed tumors (Figure 6). BMMCs derived from a KIT-Asp818Tyr/KIT-Asp818Tyr mouse were also injected into posterior flank of another 4 nude mice as a control, but they did not form tumors (Figure 6). Histological examination revealed that the tumors formed by injection of IMC-G4 cells were composed of mononuclear round cells (Figure 7A) whose cytoplasm was metachromatically stained purple with toluidine blue (Figure 7B), indicating that the tumor cells have characteristics of mast cell.
Figure 6.
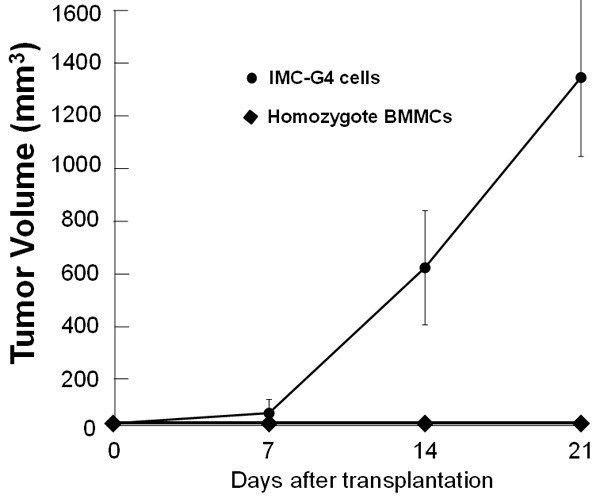
Tumorigenesis of IMC-G4 cells. IMC-G4 cells cultured in IL-3 free medium were subcutaneously injected into the posterior flank of 4 nude mice (BALB/c-nu/nu, Japan SLC, Hamamatsu, Japan). As a control, BMMCs derived from KIT-Asp818Tyr/KIT-Asp818Tyr mice cultured in IL-3 containing medium were also injected. IMC-G4 cells formed tumors but BMMCs derived from KIT-Asp818Tyr/KIT-Asp818Tyr mice did not.
Figure 7.
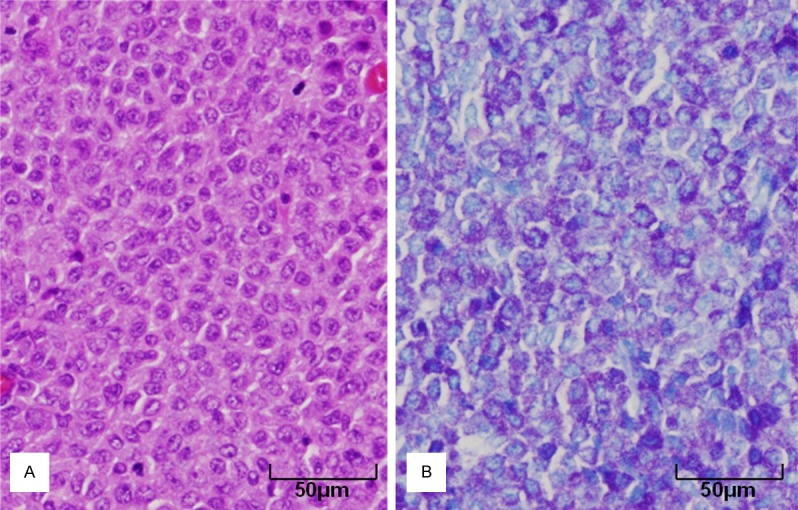
Histology of tumors formed by injection of IMC-G4 cells. Histological examination revealed that tumors formed by injection of IMC-G4 cells were composed of mononuclear round cells (A: H&E staining). The tumor cells were stained metachromatically dark purple by toluidine blue staining (B: toluidine blue staining).
Inhibitory effect of imatinib and nilotinib on KIT autophosphorylation
Imatinib almost completely inhibited KIT autophosphorylation of Ba/F3 cells expressing KIT-del-Val558&Val559 at the concentration of 0.1 microM. On the other hand, KIT autophosphorylation of Ba/F3 cells expressing KIT-Asp818Tyr, BMMCs derived from KIT-Asp818Tyr/KIT-Asp818Tyr mice and IMC-G4 cells was almost completely inhibited at the concentration of 10 microM (Figure 8A). Nilotinib almost completely inhibited KIT autophosphorylation of Ba/F3 cells expressing KIT-del-Val558&Val559 at the concentration of 0.1 microM. KIT autophospohrylation in Ba/F3 cells expressing KIT-Asp818Tyr, BMMCs derived from KIT-Asp818Tyr/KIT-Asp818Tyr mice and IMC-G4 cells was also almost inhibited at the concentration of 0.1 microM (Figure 8B).
Figure 8.

Effect of KIT inhibitors on KIT autophosphorylation. KIT autophosphorylation in BMMCs derived from KIT-Asp818Tyr/KIT-Asp818Tyr mice, IMC-G4, Ba/F3 cells expressing KIT-Asp818Tyr and Ba/F3 cells expressing KIT-del-Val558&Val559 was examined at various concentration of imatinib (0, 0.001, 0.01, 0.1, 1 and 10 microM) (A) or nilotinib (0, 0.001, 0.01, 0.1, 1 and 10 microM) (B). Imatinib inhibited KIT autophosphorylation in Ba/F3 cells expressing KIT-del-Val558&Val559 at the concentration of 0.1 microM, but did that in BMMCs derived from KIT-Asp818Tyr/Asp818Tyr mice, IMC-G4 cells and Ba/F3 cells expressing KIT-Asp818Tyr at the concentration of 10 microM (A). Nilotinib also inhibited KIT autophosphorylation in Ba/F3 cells expressing KIT-del-Val558&Val559 at the concentration of 0.1 microM, and did that in BMMCs derived from KIT-Asp818Tyr/KIT-Asp818Tyr mice and IMC-G4 cells and Ba/F3 cells expressing KIT-Asp818Tyr at the concentration of 0.1 microM (B).
Inhibitory effect of imatinib and nilotinib on cell proliferation
Imatinib almost completely inhibited cell proliferation of Ba/F3 cells expressing KIT-del-Val558&Val559 at the concentration of 0.1 microM. On the other hand, proliferation of Ba/F3 cells expressing KIT-Asp818Tyr and IMC-G4 cells was almost completely inhibited at 10 microM of imatinib (Figure 9A). Nilotinib completely inhibited cell proliferation of Ba/F3 cells expressing KIT-del-Val558&Val559, Ba/F3 cells expressing KIT-Asp818Tyr and IMC-G4 cells at the concentration of 1 microM in a similar manner (Figure 9B). Ba/F3 cells expressing KIT-del-Val558&Val559 were apt to be more easily inhibited than IMC-G4 cells and Ba/F3 cells expressing KIT-Asp818Tyr (Figure 9B).
Figure 9.
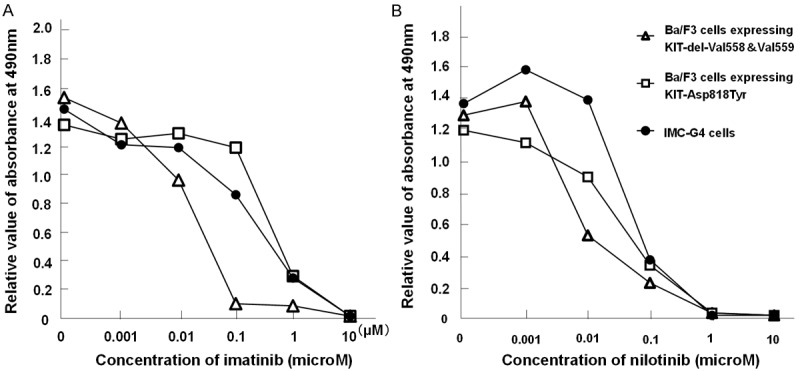
Effect of KIT inhibitors on cell proliferation. Proliferation of IMC-G4 cells, Ba/F3 cells expressing KIT-Asp818Tyr, those expressing KIT-del-Val558&Val559 (exon 11) was examined with various concentrations of imatinib (0, 0.001, 0.01, 0.1, 1, and 10 microM) (A) or nilotinib (0, 0.001, 0.01, 0.1, 1, and 10 microM) (B) using MTS colorimetric assay. Imatinib completely inhibited cell proliferation of Ba/F3 cells expressing KIT-del-Val558&Val559 at the concentration of 0.1 microM (A). Proliferation of IMC-G4 cells and Ba/F3 cells expressing KIT-Asp818Tyr was almost inhibited at 10 microM (A). Nilotinib inhibited cell proliferation of Ba/F3 cells expressing KIT-del-Val558&Val559, IMC-G4 cells and Ba/F3 cells expressing KIT-Asp818Tyr similarly at the concentration of 1 microM (B).
Discussion
Most of the mast cell neoplasms and GISTs have various types of c-kit gene mutations at various exons [1,34]. Some types of mutations are observed in mast cell neoplasms and GISTs in common, but some types of them are mutually exclusive between mast cell neoplasms and GISTs. For example, KIT-del-Val559, KIT-Lys642Glu and KIT-Asp820Tyr are detected in GISTs but not in mast cell neoplasms. In model mice for familial GISTs with KIT-del-Val558, KIT-Lys641Glu and KIT-Asp818Tyr corresponding to human KIT-del-Val559, KIT-Lys642Glu and KIT-Asp820Tyr respectively, the numbers of mast cells in the skin of the three model mice are different from each other when compared to the respective wild type mice. The number of skin mast cells in the model mice with KIT-del-Val558 mutation increases [30], that with KIT-Lys641Glu mutation decreased [31], and that with KIT-Asp818Tyr mutation is unchanged [32]. These results suggest that the particular types of gain-of-function mutations of the c-kit gene could activate downstream signaling molecules of SCF-KIT systems in ICCs but not in mast cells resulting in development of GISTs but not in proliferation of mast cells or mast cell neoplasms. In the present study, we explored the influence of KIT-Asp818Tyr mutation in mast cells using various types of BMMCs derived from knock-in mice with KIT-Asp818Tyr mutation. We examined ultrastructure, histamine contents, proliferation profiles, phosphorylation of various signaling molecules in BMMCs derived from +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice. We also examined those in immortalized BMMCs, IMC-G4 cells, derived from a KIT-Asp818Tyr/KIT-Asp818Tyr mouse. In IMC-G4 cells, the presence of an additional c-kit gene mutation and effect of KIT inhibitors on KIT autophosphorylation and cell proliferation were also examined.
In the present study, all types of BMMCs derived from +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice had similar ultrastructures. All of BMMCs derived from +/+, KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice also showed similar proliferation and survival profiles. On the other hand, histamine contents of BMMCs derived from KIT-Asp818Tyr/KIT-Asp818Tyr mice were approximately half compared to those from +/+ and KIT-Asp818Tyr/+ mice. These results suggested that KIT-Asp818Tyr mutation did not appear to affect cell structure and cell proliferation but appear to recessively influence histamine synthesis in BMMCs. The mechanism of different effect of KIT-Asp818Tyr mutation on histamine synthesis and cell structure/cell proliferation remained unclear.
BMMCs derived from +/+ mice did not show apparent KIT phosphorylation, but those from KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice demonstrated apparent phosphorylation of KIT. However, phosphorylation of SCF-KIT downstream signaling molecules such as MAPK, Akt, Stat1 and Stat5 was not stronger in BMMCs derived from KIT-Asp818Tyr/+ and KIT-Asp818Tyr/KIT-Asp818Tyr mice than in those from +/+ mice. Thus, phosphorylation of KIT-Asp818Tyr did not result in activation of downstream signaling molecules of SCF-KIT system in BMMCs. Since the patients with germline KIT-Asp820Tyr mutation do not show mast cell disorders [10,17,24], this type of mutation appears not to influence mast cell proliferation in both humans and mice. More precise mechanism why KIT-Asp818Tyr mutation cannot activate downstream signaling molecules of SCF-KIT system has to be clarified.
In Ba/F3 cells, permanent expression of KIT-Asp818Tyr could induce autonomous proliferation of the cells [10]. On the other hand, BMMCs expressing KIT-Asp818Tyr could not proliferate without IL-3 in the present study. Since Ba/F3 cells are a pro-B-cell line and BMMCs are mast cells, KIT-Asp818Tyr could induce autonomous proliferation in lymphocytic cells as in the case of ICCs but not in mast cells. Different signal transduction molecules expressed in different cell types might be associated with different effects of KIT-Asp818Tyr in different cell types. The mechanism remains to be clarified.
We obtained an immortalized cell line designated as IMC-G4 cells derived from long-term culture of BMMCs of KIT-Asp818Tyr/KIT-Asp818Tyr. They formed tumors when they were injected into the posterior flank of nude mice, confirming that they are autonomously proliferative neoplastic cells. The neoplastic cells were metachromatically stained purple with toluidine blue. They had many secretory granules in their cytoplasm, although histamine contents was approximately one-sixth compared to those in BMMCs from +/+ mice. These results indicate that IMC-G4 cells could be neoplastic mast cells which have less histamine contents compared to normal mast cells. To further characterize the mast cell properties of IMC-G4 cells, expression of mast cell specific proteins such as high-affinity receptor for IgE and mouse mast cell proteases should be examined.
Sequencing of the c-kit cDNA revealed that IMC-G4 cells have an additional mutation of KIT-Tyr421Cys at exon 8. KIT in IMC-G4 cells was strongly phosphorylated. Moreover, phosphorylation of Akt in IMC-G4 cells was apparently stronger than that in BMMCs derived from KIT-Asp818Tyr/KIT-Asp818Tyr mice while phosphorylation of MAPK, Stat1 and Stat5 was similar in IMC-G4 cells and BMMCs of KIT-Asp818Tyr/KIT-Asp818Tyr mice. These results suggested that KIT-Tyr421Cys but not KIT-Asp818Tyr plays a role of the driver mutation in autonomous proliferation and immortalization of the IMC-G4 cells probably through Akt activation. However, there is a possibility that double mutations of KIT-Asp818Tyr and KIT-Tyr421Cys are required for the immortal mast cell proliferation. Less histamine contents in IMC-G4 cells might be also associated with the addition of KIT-Tyr421Cys mutation. These possibilities need to be further clarified.
As described above, Akt activation appeared to result in immortalization of mast cell, i.e. development of IMC-G4 cells. We previously demonstrated that development of the cecal GIST in the model mice with KIT-Asp818Tyr is strongly associated with activation of MAPK, STAT1 and STAT5 [32]. These results suggest that Akt activation is crucial for autonomous proliferation of mast cells and MAPK, STAT1 and STAT5 are important for GIST development. We have to further examine the mechanism intimately.
Imatinib could not sufficiently inhibit KIT autophosphorylation in IMC-G4 cells as well as in BMMCs of KIT-Asp818Tyr/KIT-Asp818Tyr mice when compared to Ba/F3 cells expressing imatinib-sensitive KIT-exon 11 mutation. Proliferation of IMC-G4 cells was not also effectively inhibited by imatinib as observed in BMMCs of KIT-Asp818Tyr/KIT-Asp818Tyr mice. On the other hand, nilotinib could inhibit KIT autophosphorylation similarly in all types of cells examined. Proliferation of all types of cells examined was also similarly and effectively inhibited by nilotinib. Imatinib-resistant property of IMC-G4 cells suggests that KIT-Tyr421Cys mutation might be imatinib-resistant. Nilotinib-sensitive characteristic of IMC-G4 cells appears to indicate nilotinib-sensitivity of KIT-Tyr421Cys mutation. For clarification of sensitivity of Tyr421Cys mutation for imatinib and nilotinib, KIT only with Tyr421Cys mutation should be examined.
In various species of mast cell neoplasms, most of them have c-kit gene mutations at exon 17 corresponding to murine KIT-Asp814Val or KIT-Asp814Tyr [34,35]. In human mastcytomas, some other c-kit gene mutaions at exon 8, 9 or 11 have been reported. In human pediatric mastocytosis, Bodemer et al reported that approximately half of them had exon 8 or 9 c-kit gene mutations [36]. However, the KIT-Tyr418Cys at exon 8 corresponding to murine KIT-Tyr421Cys at exon 8 observed in IMC-G4 cells has not been reported yet in any species of mast cell neoplasms. On the other hand, KIT-Tyr418Asn but not KIT-Tyr418Cys was detected in a patient with childhood core-binding factor acute myeloid leukemia [37]. There is a possibility that the mutation might be a cause of human pediatric mast cell neoplasms or acute myeloid leukemia.
In the present study, we showed insufficiency or unnecessity of KIT-Asp818Tyr mutation in autonomous proliferation of mast cells. Kosmider et al reported that KIT-Asp818Tyr have an important role on development of erythroleukemia using Spi-1/PU.1 transgenic mice [38]. In those mice, acquisition of KIT-Asp818Tyr gave malignant transformation (development of erythroleukemia from non-malignant proerythroblast) with early arrest of differentiation [38]. These results also suggested that KIT-Asp818Tyr mutation has some role in erythroblast differentiation/proliferation as in the case of ICC differentiation/proliferation but not in mast cell differentiation/proliferation. In knock-in mice with KIT-Asp818Tyr mutation, we did not notice apparent abnormality in bone marrow cells by histological examination. Further biological examinations of erythroid cells in this mouse model are needed.
In summary, we showed that KIT-Asp818Tyr mutation is not sufficient or necessary for ultrastructural and proliferative changes but for alteration of histamine synthesis in mast cells. We also showed that an addition of the novel mutation of KIT-Tyr421Cys could induce neoplastic transformation of mast cells in mice. KIT-Tyr421Cys might be an imatinib-resistant and nilotinib-sensitive mutation. There is a possibility that KIT-Tyr418Cys is detected in human mast cell neoplasms or acute myeloid leukemia. IMC-G4 cells might be a useful mast cell line for various examinations in mast cell biology including the effect of molecular target drugs on human KIT-Tyr418Cys.
Disclosure of conflict of interest
None.
References
- 1.Hirota S, Isozaki K. Pathology of gastrointestinal stromal tumors. Pathol Int. 2006;56:1–9. doi: 10.1111/j.1440-1827.2006.01924.x. [DOI] [PubMed] [Google Scholar]
- 2.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Tunio GM, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 3.Huss S, Künstlinger H, Wardelmann E, Kleine MA, Binot E, Merkelbach-Bruse S, Rüdiger T, Mittler J, Hartmann W, Büttner R, Schildhaus HU. A subset of gastrointestinal stromal tumors previously regarded as wild-type tumors carries somatic activating mutations in KIT exon 8 (p. D419del) Mod Pathol. 2013;26:1004–1012. doi: 10.1038/modpathol.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito T, Yamamura M, Hirai T, Ishikawa T, Kanda T, Nakai T, Ohkouchi M, Hashikura Y, Isozaki K, Hirota S. Gastrointestinal stromal tumors with exon 8 c-kit gene mutation might occur at extragastric sites and have metastasis-prone nature. Int J Clin Exp Pathol. 2014;7:8024–8031. [PMC free article] [PubMed] [Google Scholar]
- 5.Nishida T, Hirota S, Taniguchi M, Hashimoto K, Isozaki K, Nakamura H, Kanakura Y, Tanaka T, Takabayashi A, Matsuda H, Kitamura Y. Familial gastrointestinal stromal tumors with germline mutation of the KIT gene. Nat Genet. 1998;19:323–324. doi: 10.1038/1209. [DOI] [PubMed] [Google Scholar]
- 6.Hirota S, Okazaki T, Kitamura Y, O’Brien P, Kapusta L, Dardick I. Cause of familial and multiple gastrointestinal autonomic nerve tumors with hyperplasia of interstitial cells of Cajal is germline mutation of the c-kit gene. Am J Surg Pathol. 2000;24:326–327. doi: 10.1097/00000478-200002000-00045. [DOI] [PubMed] [Google Scholar]
- 7.Isozaki K, Terris B, Belghiti J, Schiffmann S, Hirota S, Vanderwinden JM. Germline-activating mutation in the kinase domain of KIT gene in familial gastrointestinal stromal tumors. Am J Pathol. 2000;57:1581–1585. doi: 10.1016/S0002-9440(10)64795-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maeyama H, Hidaka E, Ota H, Minami S, Kajiyama M, Kuraishi A, Mori H, Matsuda Y, Wada S, Sodeyama H, Nakata S, Kawamura N, Hata S, Watanabe M, Iijima Y, Katsuyama T. Familial gastrointestinal stromal tumor with hyperpigmentation: association with a germline mutation of the c-kit gene. Gastroenterology. 2001;120:210–215. doi: 10.1053/gast.2001.20880. [DOI] [PubMed] [Google Scholar]
- 9.Beghini A, Tibiletti MG, Roversi G, Chiaravalli AM, Serio G, Capella C, Larizza L. Germline mutation in the juxtamembrane domain of the kit gene in a family with gastrointestinal stromal tumors and urticaria pigmentosa. Cancer. 2001;92:657–662. doi: 10.1002/1097-0142(20010801)92:3<657::aid-cncr1367>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 10.Hirota S, Nishida T, Isozaki K, Taniguchi M, Nishikawa K, Ohashi A, Takabayashi A, Obayashi T, Okuno T, Kinoshita K, Chen H, Shinomura Y, Kitamura Y. Familial gastrointestinal stromal tumors associated with dysphagia and novel type germline mutation of KIT gene. Gastroenterology. 2002;122:1493–1499. doi: 10.1053/gast.2002.33024. [DOI] [PubMed] [Google Scholar]
- 11.Robson ME, Glogowski E, Sommer G, Antonescu CR, Nafa K, Maki RG, Ellis N, Besmer P, Brennan M, Offit K. Pleomorphic characteristics of a germ line KIT mutation in a large kindred with gastrointestinal stromal tumors, hyperpigmentation, and dysphagia. Clin Cancer Res. 2004;10:1250–1254. doi: 10.1158/1078-0432.ccr-03-0110. [DOI] [PubMed] [Google Scholar]
- 12.Hartmann K, Wardelmann E, Ma Y, Merkelbach-Bruse S, Preussner LM, Woolery C, Baldus SE, Heinicke T, Thiele J, Buettner R, Longley BJ. Novel germline mutaion of KIT associated with familial gastrointestinal stromal tumors and mastocytosis. Gastroenterology. 2005;129:1042–1046. doi: 10.1053/j.gastro.2005.06.060. [DOI] [PubMed] [Google Scholar]
- 13.Kim HJ, Lim SJ, Park K, Yuh YJ, Jang SJ, Choi J. Multiple gastrointestinal stromal tumors with a germline c-kit mutation. Pathol Int. 2005;55:655–659. doi: 10.1111/j.1440-1827.2005.01885.x. [DOI] [PubMed] [Google Scholar]
- 14.Li FP, Fletcher JA, Heinrich MC, Garber JE, Sallan SE, Curiel-Lewandrowski C, Duensing A, van de Rijn M, Schnipper LE, Demetri GD. Familial gastrointestinal stromal tumor syndrome: phenotypic and molecular features in a kindred. J. Clin. Oncol. 2005;23:2735–2743. doi: 10.1200/JCO.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 15.Carballo M, Roig I, Aguilar F, Pol MA, Gamundi MJ, Hernan I, Martinez-Gimeno M. Novel c-KIT germline mutation in a family with gastrointestinal stromal tumor tumors and cutaneous hyperpigmentation. Am J Med Genet A. 2005;32:361–364. doi: 10.1002/ajmg.a.30388. [DOI] [PubMed] [Google Scholar]
- 16.Tarn C, Merkel E, Canutescu AA, Shen W, Skorobogatko Y, Heslin MJ, Eisenberg B, Birbe R, Patchefsky A, Dunbrack R, Arnoletti JP, von Mehren M, Godwin AK. Analysis of KIT mutations in sporadic and familial gastrointestinal stromal tumors: therapeutic implications through modeling. Clin Cancer Res. 2005;11:3668–3677. doi: 10.1158/1078-0432.CCR-04-2515. [DOI] [PubMed] [Google Scholar]
- 17.O’Riain C, Corless CL, Heinrich MC, Keegan D, Vioreanu M, Maguire D, Sheahan K. Gastrointestinal stromal tumors: insights from a new familial GIST kindred with unusual genetic and pathologic features. Am J Surg Pathol. 2005;29:1680–1683. doi: 10.1097/01.pas.0000173024.79852.08. [DOI] [PubMed] [Google Scholar]
- 18.Lasota J, Miettinen M. A new familial GIST identified. Am J Surg Pathol. 2006;30:1342. doi: 10.1097/01.pas.0000213364.56498.3b. [DOI] [PubMed] [Google Scholar]
- 19.Graham J, Debiec-Rychter M, Corless CL, Reid R, davidson R, White JD. Imatinib in the management of multiple gastrointestinal stromal tumors associated with a germline KIT K642E mutaion. Arch Pathol Lab Med. 2007;131:1393–1396. doi: 10.5858/2007-131-1393-IITMOM. [DOI] [PubMed] [Google Scholar]
- 20.Kang DY, Park CK, Choi JS, Jin SY, Kim HJ, Joo M, Kang MS, Moon WS, Yun KJ, Yu ES, Kang H, Kim KM. Multiple gastrointestinal stromal tumors: clinicopathologic and genetic analysis of 12 patients. Am J Surg Pathol. 2007;31:224–232. doi: 10.1097/01.pas.0000213318.66800.94. [DOI] [PubMed] [Google Scholar]
- 21.Wozniak A, Rutkowski P, Sciot R, Ruka W, Michej W, Debiec-Rychter M. Rectal gastrointestinal stromal tumors associated with a novel germline KIT mutation. Int J Cancer. 2008;122:2160–2164. doi: 10.1002/ijc.23338. [DOI] [PubMed] [Google Scholar]
- 22.Kleinbaum EP, Lazar AJ, Tamborini E, Mcauliffe JC, Sylvestre PB, Sunnenberg TD, Strong L, Chen LL, Choi H, Benjamin RS, Zhang W, Trent JC. Clinical, histopathologic, molecular and therapeutic findings in a large kindred with gastrointestinal stromal tumor. Int J Cancer. 2008;122:711–718. doi: 10.1002/ijc.23137. [DOI] [PubMed] [Google Scholar]
- 23.Thalheimer A, Schlemmer M, Bueter M, Merkelbach-Bruse S, Schildhaus HU, Buettner R, Hartung E, Thiede A, Meyer D, Fein M, Maroske J, Wardelmann E. Familial gastrointestinal stromal tumors caused by the novel KIT exon 17 germline mutaion N822Y. Am J Surg Pathol. 2008;32:1560–1565. doi: 10.1097/PAS.0b013e318172ce6f. [DOI] [PubMed] [Google Scholar]
- 24.Veiga I, Silva M, Vieira J, Pinto C, Pinheiro M, Torres L, Soares M, Santos L, Duarte H, Bastos AL, Coutinho C, Dinis J, Lopes C, Teixeria MR. Hereditary gastrointestinal stromal tumors sharing the KIT exon 17 germline mutation p. Asp820Tyr develop through different cytogenetic progression pathways. Genes Chromosomes Cancer. 2010;49:91–98. doi: 10.1002/gcc.20720. [DOI] [PubMed] [Google Scholar]
- 25.Kuroda N, Tanida N, Hirota S, Daum O, Hes O, Michal M, Lee GH. Familial gastrointestinal stromal tumor with germ line mutation of the juxtamembrane domain of the KIT gene observed in relatively young women. Ann Diagn Pathol. 2011;15:358–361. doi: 10.1016/j.anndiagpath.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 26.Vilain RE, Dudding T, Braye SG, Groombridge C, Meldrum C, Spigelman AD, Ackland S, Ashman L, Scott RJ. Can a familial gastrointestinal tumour syndrome be alleic with Waardenburg syndrome. Clin Genet. 2011;79:554–560. doi: 10.1111/j.1399-0004.2010.01489.x. [DOI] [PubMed] [Google Scholar]
- 27.Nakai M, Hashikura Y, Ohkouchi M, Yamamura M, Akiyama T, Shiba K, Kajimoto N, Tsukamoto Y, Hao H, Isozaki K, Hirai T, Hirota S. Characterization of novel germline c-kit gene mutation, KIT-Tyr553Cys, observed in a family with multiple gastrointestinal stromal tumors. Lab Invest. 2012;92:451–457. doi: 10.1038/labinvest.2011.165. [DOI] [PubMed] [Google Scholar]
- 28.Neuhann TM, Mansmann V, Merkelbach-Bruse S, Klink B, Hellinger A, Höffkes HG, Wardelmann E, Schildhaus HU, Tinschert S. A novel germline KIT mutation (p. L576P) in a family presenting with juvenile onset of multiple gastrointestinal stromal tumors, skin hyperpigmentations, and esophageal stenosis. Am J Surg Pathol. 2013;37:898–905. doi: 10.1097/PAS.0b013e31827bc071. [DOI] [PubMed] [Google Scholar]
- 29.Bamba S, Hirota S, Inatomi O, Ban H, Nishimura T, Shioya M, Imaeda H, Nishida A, Sasaki M, Murata S, Andoh A. Familial and multiple gastrointestinal stromal tumors with fair response to a half-dose of imatinib. Intern Med. 2015;54:759–764. doi: 10.2169/internalmedicine.54.3585. [DOI] [PubMed] [Google Scholar]
- 30.Sommer G, Agosti V, Ehlers I, Rossi F, Corbacioglu S, Farkas J, Moore M, Manova K, Antonescu CR, Besmer P. Gastrointestinal stromal tumors in a mouse model by target mutation of the Kit receptor tyrosine kinase. Proc Natl Acad Sci U S A. 2003;100:6706–6711. doi: 10.1073/pnas.1037763100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubin BP, Antonescu CR, Scott-Browne JP, Comstock ML, Gu Y, Tanas MR, Ware CB, Woodell J. A knock-in mouse model of gastrointestinal stromal tumor harboring KIT K641E. Cancer Res. 2005;65:6631–6639. doi: 10.1158/0008-5472.CAN-05-0891. [DOI] [PubMed] [Google Scholar]
- 32.Nakai N, Ishikawa T, Nishitani A, Liu NN, Shincho M, Hao H, Isozaki K, Kanda T, Nishida T, Fujimoto J, Hirota S. A mouse model of a human multiple GIST family with KIT-Asp820tyr mutation generated by a knock-in strategy. J Pathol. 2008;214:302–311. doi: 10.1002/path.2296. [DOI] [PubMed] [Google Scholar]
- 33.Longley BJ Jr, Metcalfe DD, Tharp M, Wang X, Tyrrell L, Lu SZ, Heitjan D, Ma Y. Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc Natl Acad Sci U S A. 1999;96:1609–1614. doi: 10.1073/pnas.96.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valent P, Akin C, Sperr WR, Mayerhofer M, Födinger M, Fritsche-Polanz R, Sotlar K, Escribano L, Arock M, Horny HP, Metcalfe DD. Mastocytosis: pathology, genetics, and current options for therapy. Leuk Lymphoma. 2005;46:35–48. doi: 10.1080/10428190400010775. [DOI] [PubMed] [Google Scholar]
- 35.Tsujimura T, Furitsu T, Morimoto M, Isozaki K, Nomura S, Matsuzawa Y, Kitamura Y, Kanakura Y. Ligand-independent activation of c-kit receptor tyrosine kinase in a murine mastocytoma cell line P-815 generated by a point mutation. Blood. 1994;83:2619–2626. [PubMed] [Google Scholar]
- 36.Bodemer C, Hermine O, Palmerini F, Yang Y, Grandpeix-Guyodo C, Leventhal PS, Hadj-Rabia S, Nasca L, Georgin-Lavialle S, Cohen-Akenine A, Launay JM, Barete S, Feger F, Arock M, Catteau B, Sans B, Stalder JF, Skowron F, Thomas L, Lorette G, Plantin P, Bordigoni P, Lortholary O, de Prost Y, Moussy A, Sobol H, Dubreuil P. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol. 2010;130:804–815. doi: 10.1038/jid.2009.281. [DOI] [PubMed] [Google Scholar]
- 37.Shih LY, Liang DC, Huang CF, Chang YT, Lai CL, Lin TH, Yang CP, Hung IJ, Liu HC, Jaing TH, Wang LY, Yeh TC. Cooperating mutations of receptor tyrosine kinases and Ras genes in childhood core-binding factor acute myeloid leukemia and a comparative analysis on paired diagnosis and relapse samples. Leukemia. 2008;22:303–307. doi: 10.1038/sj.leu.2404995. [DOI] [PubMed] [Google Scholar]
- 38.Kosmider O, Denis N, Lacout C, Vainchenker W, Dubreuil P, Moreau-Gachelin F. Kit-activating mutations cooperate with Spi-1/PU. 1 overexpression to promote tumorigenic progression during erythroleukemia in mice. Cancer Cell. 2005;8:467–478. doi: 10.1016/j.ccr.2005.11.009. [DOI] [PubMed] [Google Scholar]