

Abstract
Dilated cardiomyopathy is the most frequent form of myocardial disease. Many factors contribute to dilated cardiomyopathy, for instance, long-term use of doxorubicin, one of the anthracyclines clinically used for cancer chemotherapy, result in dilated cardiomyopathy and congestive heart failure. However, the mechanism underlining doxorubicin-induced cardiomyocyte is still not fully understood. In this study, we evaluate the effects and their mechanisms of PPARα and PGC-1α pathways in doxorubicin induced mice cardiomyocytes. In vitro, cardiomyocytes isolated from hearts of adult FVB/NJ mice were treated with doxorubicin, GW 6471 (PPARα inhibitors) and WY14643 (PPARα agonists). The expression of PPARα and PGC-1α were detected via western blotting and Quantitative Real-Time PCR methods. Changes in energy and substrate metabolism were analyzed. MTT and flow cytometry were used for cell proliferation and apoptosis analysis. We detected expression of PPARα and PGC-1α was significantly higher in control group than doxorubicin group. Mitochondrial dysfunction was found in doxorubicin group including lower content of high-energy phosphates, significantly decreased mitochondrial ANT transport activity and markedly reduced mitochondrial membrane potential compared with control group. Metabolic remodeling existed in doxorubicin group because of higher concentration of free fatty acid and glucose consumption than of control group. More accumulations of reactive oxygen species were detected in doxorubicin group. The decreased cell viability and increased cell apoptosis observed in doxorubicin group. Severe apoptosis in doxorubicin group was verified by a set of markers including Bax, Bcl-2, cytosolic cytochrome c and caspase-3 up-regulation expression. These findings indicate that the PPARα and PGC-1α are closely involved in energy metabolism remodeling and apoptosis in cardiomyocytes.
Keywords: PPARα, PGC-1α, doxorubicin, energy metabolism, in vitro
Introduction
Dilated cardiomyopathy (DCM) is the most frequent form of myocardial disease, characterized with progressive ventricular dilatation and systolic dysfunction [1]. Many factors contribute to DCM, for instance, long-term use of doxorubicin (DOX), one of the anthracyclines clinically used for cancer chemotherapy [2], can cause high activation of sympathetic nervous system and left ventricular dysfunction [3]. The major side effect of DOX is cardiac toxicity, which could further result in dilated cardiomyopathy and congestive heart failure [4]. It has been reported that DOX can increase oxidative stress, decrease ATP yield, suppress gene expression, and cause mitochondrial abnormality as well as cardiomyocyte apoptotic death [5-7]. However, the mechanism underlining DOX-induced cardiomyocyte is still not fully understood.
Heart is an organ with a very high and dynamic demand for ATP. In the adult human, most of energy comes from fatty acid oxidation responsible for 70-80% of myocardial ATP production [8]. Emerging evidences demonstrated that peroxisome proliferator activated receptor a (PPARα) in collaboration with PGC-1α (a co-activator of PPARα) modulates energy metabolism [9-11]. PPARα, one of the three subtypes of PPAR besides β/δ and γ subunits [12], regulates cardiac metabolism [13,14] and energy metabolism-related pathways [15] by affecting the activities of enzymes involved in free fatty acid oxidation [16]. PGC-1α, serving as a co-activator of PPARα, can enhance mitochondrial biogenesis and augment the oxidation of fatty acids through its synergistic effects with PPARα [17,18].
Our previous work showed that PPARα and PGC-1α are closely involved in energy metabolism in the DOX-induced mouse model of DCM [19]. Here, we reported the effect of PPARα/PGC-1α expression on the energy metabolism remodeling and apoptosis in DOX-induced cardiomyocytes in vitro.
Materials and methods
Cell culture and treatments
Cardiomyocytes were isolated by enzymatic digesting hearts of adult FVB/NJ mice. The isolated cardiomyocytes were cultured in DMEM medium containing 10% FBS and cultivated in 37°C, 50 ml/L CO2 atmosphere. After 24 h of plating, cardiomyocytes were divided into four groups, control group, DOX group, PPARα inhibition (PPARα-) group and PPARα activation (PPARα+) group. DOX group used 5 μM doxorubicin (Sigma-Aldrich), PPARα- group used 10 μM PPARα inhibitors GW 6471 (Sigma-Aldrich) and PPARα+ group used 100 μM PPARα agonists WY14643 (Sigma-Aldrich).
Measurement of cell proliferation
Cell viability was determined by the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, cardiomyocytes were plated on 96-well plates at a density of 1×104 cells/well. After doxorubicin, GW 6471, or WY14643 treatments, 20 μL MTT (5 mg/mL) was added to each well and incubated for 4 h. The medium was then removed, and the formazan crystals were dissolved with dimethyl sulphoxide (DMSO). Absorbance was read at 570 nm on amicroplate reader (Multiskan Spectrum, Thermo Fisher).
Flow cytometry
Apoptotic cells can be recognized and distinguished from normal cells using an Annexin V-FITC/PI apoptosis kit for flow cytometry according to the manufacturer’s instructions (Invitrogen). After doxorubicin, GW 6471, or WY14643 treatments, cardiomyocytes were harvested and washed twice with cold PBS, and then incubated with 5 μl FITC-Annexin V and 1 μl PI working solution (100 μg/ml) for 15 min in the dark at room temperature. Cellular fluorescence was measured by flow cytometry analysis (FACStation, BD Biosciences) [20].
Reactive oxygen species (ROS) determination
The ROS generation from cardiomyocytes were measured by CM-H2DCFDA (chloromethyl-2’,7’-dichlorodihydro fluorescein diacetate) staining. For CM-H2DCFDA staining, cells at appropriate conditions were washed with ice cold PBS and treated with 10 μM CM-H2DCFDA (Invitrogen) in DMEM medium for 45 min. Then, cells were trypsinized and suspended in PBS, and the fluorescence was studied at 538 nm.
Western blotting
Total proteins were prepared from the cultured cardiomyocytes of each group. The protein concentration was determined using Bio-Rad protein assay system. Proteins were analyzed with SDS polyacrylamide gel electrophoresis. After electrophoresis, they were electro-transferred to the PVDF membrane. The membrane containing the proteins was used for immunoblotting with appropriate antibodies. The protein bands were scanned and quantified.
RNA isolation and quantitative real-time PCR analysis
Total RNA were extracted from the cultured cardiomyocytes of each group using TRIZOL Regent (Invitrogen). The cDNA Synthesis Kit (Takara) was used for the synthesis of cDNA according to the manufacturer’s instructions. Quantitative PCR was accomplished using the iCycler iQ Detection System (Bio-Rad, Richmond, CA) and interaction dye SYBR Green. Primer sequences are listed in Supplementary Table 1.
Analysis of adenylate in cardiomyocytes
The adenine nucleotide from cardiomyocytes of each group was determined according to the method of Yongyao Yang [19]. The wavelength of HPLC was 254 nm, and sensitivity was 0.01 AUFS. Chromatographic column was 4.6 nm ×250 nm, YWG-ODS C18 10 μm. The mobile phase was 2 mmol/L PBS (pH 5.5). The speed was 1 mL/min.
Determination of mitochondrial ANT transport activity in cardiomyocytes
We determined mitochondrial ANT transport activity use inhibitors termination method. 20 μl of 3H-ADP solution (0.3 μmol/L) was added into 50 μl of prepared mitochondrial suspension for 10 s, then 50 μl of ANT inhibitor ATR (3.2 nmol/L) was added and mixed immediately to terminate ANT transporter function. The mixture was centrifuged for 20 min with 12000 r/min speed at 4°C and the supernatant was discarded. 400 μl of H2O2 (8.8 mol/L) was added into the precipitation after being dissolved and cleaned with 1 ml separation medium, then it was digested for 40 min in 70°C water bath. The radioactivity was measured by liquid scintillation counting method.
Determination of free fatty acid in cardiomyocytes
Free fatty acid was separated from cardiomyocytes from each group and measured by HPLC method referring to Michael Puttmann [21].
Measurement of glucose consumption in cardiomyocytes
The glucose consumption of cardiomyocytes was measured in small glass spinner flasks refer to J.P. FREYER [22]. The concentration of glucose in complete medium was carried out d a standard enzyme reagent kit (Sigma) according to the manufacturer’s instructions.
Caspase 3 assay in cardiomyocytes
Caspase 3 activity was determined using the luminescent Caspase-Glo 3 assay (Promega). The assay was conducted according to the manufacturer’s protocol.
Statistical analysis
All the original experimental data were analyzed by SPSS19.0. Comparison among the groups was carried out by One-way ANOVA. The comparison between two groups was carried out by LSD. P<0.05 was considered as statistically significant differences.
Results
Expression of PPARα and PGC1-α in different groups
The results of q-PCR showed that the expression of PPARα and PGC-1α in DOX group were lower than that of control group (P<0.05). While the expression of PPARα and PGC-1α in PPARα+ group were higher compared with DOX and PPARα- group (P<0.05) (Figure 1A, 1B). The Western blotting results showed in Figure 1C-F.
Figure 1.
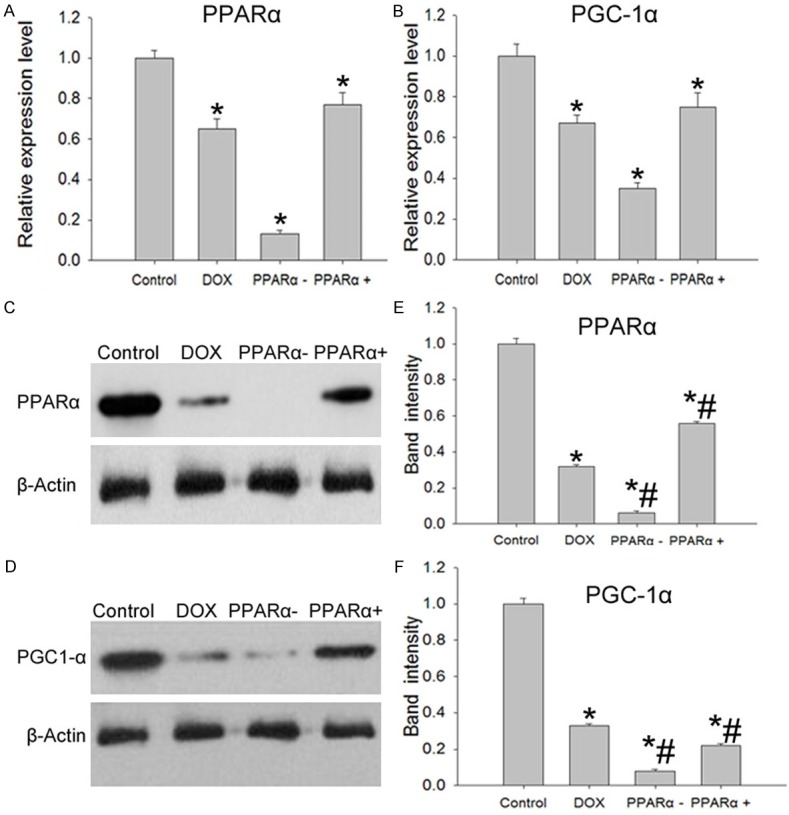
PPARα and PGC-1α expression in different groups. A. Q-PCR results of PPARα expression in different group. B. Q-PCR results of PGC-1α expression in different group. C and E. Western blotting results of PPARα expression in different group. D and F. Western blotting results of PGC-1α expression in different group. Data are the mean ± SEM (n=3), Each bar represents the mean of three independent experiments carried out in triplicate. *Compared with control group, P<0.05, #Compared with DOX group, P<0.05.
Mitochondrial dysfunction in different groups
The essential function of cardiomyocytes is energy production. Firstly, we measured concentration of adenylate in different groups using HPLC method. Results established that compared with control group, ATP, ADP and AMP decreased significantly in DOX, PPARα+ and PPARα- groups (P<0.05) (Table 1). The decline in the high-energy phosphate pool might be indicative for impaired mitochondrial function.
Table 1.
Concentration of adenylate in different groups
| Group | ATP (nmol/mg) | ADP (nmol/mg) | AMP (nmol/mg) |
|---|---|---|---|
| Control | 1.85 | 3.36 | 3.82 |
| DOX | 1.13* | 1.35* | 2.21* |
| PPARα- | 1.05* | 1.24* | 1.92* |
| PPARα+ | 1.33* | 1.51* | 2.39* |
The adenine nucleotide in cardiomyocytes was determined using HPLC method. Data are the mean ± SEM;
compared with control group, P<0.05.
The ATP produced in the mitochondrion is transported to the cytoplasm for important cellular work. Then, we analyzed mitochondrial ANT transport activity in different groups. Dates showed that compared with control group, the mitochondrial ANT transport activity decreased significantly in DOX, PPARα+ and PPARα- groups, the results were showed in Figure 2.
Figure 2.
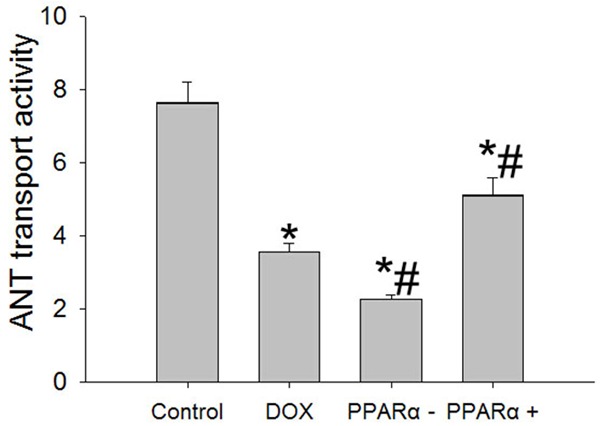
Changes of mitochondrial ANT transport activity in different groups. Data are the mean ± SEM (n=3), each bar represents the mean of three independent experiments carried out in triplicate. *Compared with control group, P<0.05; #Compared with DOX group, P<0.05.
In addition, we also found that mitochondrial membrane potential significantly reduced in DOX group, PPARα- group and PPARα+ group (Supplementary Figure 1).
Change in cardiac substrate utilization
The impaired mitochondrial function might be indicative for decline in oxidation of fatty acid and glucose. Free fatty acid content was detected and results revealed that palmitic acid, stearic acid, oleic acid, linoleic acid, linolenic acid, and arachidonic acid increased significantly in DOX group, PPARα- group and PPARα+ than control group (P<0.05) (Table 2).
Table 2.
Concentration of free fatty acid in different groups
| Group | C (FFAs) μmol/L | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Palmitic acid (c16:0) | Stearic acid (c18:0) | Oleic acid (c18:1) | Linoleic acid (c18:2) | Linolenic acid (c18:3) | Arachidonic acid (c20:4) | |
| Control | 298.73±10.01 | 179.63±6.65 | 322.5±19.84 | 514.23±14.52 | 29.12±2.56 | 91.23±6.54 |
| DOX | 364.98±12.45* | 239.01±9.14* | 385.2±17.89* | 555.62±19.54* | 59.6±3.89* | 118.94±8.79* |
| PPARα- | 412.01±15.96* | 264.53±8.78* | 411.2±19.24* | 599.87±18.74* | 77.08±4.52* | 132.25±5.96* |
| PPARα+ | 340.68±11.15* | 211.01±6.56* | 355.78±18.74* | 524.12±16.58* | 45.21±4.89* | 101.03±7.89* |
Free fatty acid was separated from cardiomyocytes and measured by HPLC method. Data are the mean ± SEM;
compared with control group, P<0.05.
To characterize whether free fatty acid metabolism was changed in different groups, we detected genes expression involved in free fatty acid oxidation pathway using real-time PCR method. The results showed that M-CPT1, ACOX1, ACOX3, and LBP expression markedly down-regulation expression in DOX group, PPARα- group and PPARα+ group respectively (Supplementary Figure 2).
Cardiomyocytes prior fatty acid to glucose for energy supporting, but in some heart disease marked shift in substrate preference away from fatty acids towards glucose. We conduct glucose consumption in different groups.
The data demonstrated that all the treatment groups possess higher glucose consumption regardless of 12 hr, 24 hr and 36 hr after treatment respectively than control group (Table 3).
Table 3.
Glucose consumption in different groups
| Group | 12 h | 24 h | 36 h |
|---|---|---|---|
| Control | 6.32±0.12 | 5.34±0.39 | 4.23±0.09 |
| DOX | 6.91±0.24* | 5.98±0.26* | 5.08±0.11* |
| PPARα- | 7.22±0.32* | 6.31±0.21* | 5.44±0.19* |
| PPARα+ | 6.69±0.27* | 5.71±0.29* | 4.78±0.27* |
The concentration of glucose in complete medium was carried out d a standard enzyme reagent kit. Data are the mean ± SEM,
compared with control group, P<0.05.
Collectively, these observations indicate that metabolic remodeling in DOX, PPARα- and PPARα+ groups.
Reactive oxygen species (ROS) accumulation in different groups
It is well known that higher concentration of glucose and free fatty acid have toxic effect on cells, and this toxicity is likely prompt more ROS production. ROS level was measured by CM-H2DCFDA staining in different groups. As shown in Figure 3, we observed more ROS accumulation in DOX group, PPARα- group and PPARα+ group than control group (P<0.05).
Figure 3.
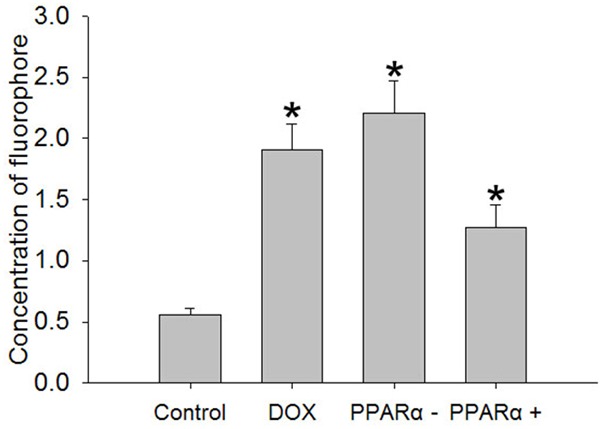
Reactive oxygens accumulate in different groups. The ROS generation from cardiomyocytes was measured by CM-H2DCFDA. Data are the mean ± SEM (n=3), Each bar represents the mean of three independent experiments carried out in triplicate. *Compared with control group, P<0.05.
Cell viability and apoptosis in different groups
To determine the effect of ROS on cardiomyocytes, MTT method was used to detect cell viability in different groups. Experiments exhibited that DOX group, PPARα- group and PPARα+ group, cell viability decrease significantly compared with control group (Figure 4A).
Figure 4.
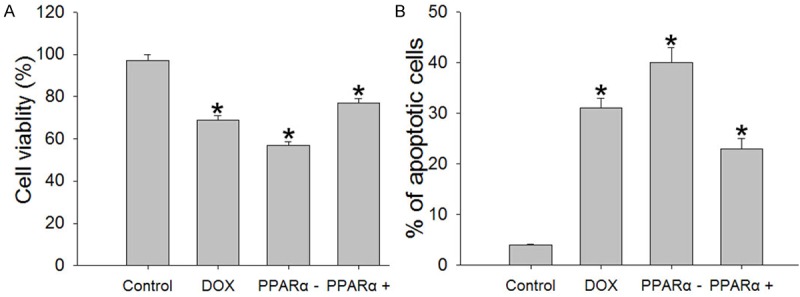
Cell viability and apoptosis in different groups. A. Cell viability was measured by the MTT colorimetric assay. B. Cell apoptosis in different groups. Apoptotic cells can be recognized and distinguished using an Annexin V-FITC/PI apoptosis kit for flow cytometry. Data are the mean ± SEM (n=3), each bar represents the mean of three independent experiments carried out in triplicate. *Compared with control group, P<0.05.
Lower cell viability in treated groups made us to characterize whether cell apoptosis was changed in different groups by means of flow cytometry. As shown in Figure 4B, DOX significantly increased the number of apoptotic cells compared with control group (P<0.05).
Markers for apoptosis in different groups
To confirm the reliability of apoptosis result, we performed western blotting analysis of a set of markers for apoptosis, including Bax, Bcl-2, cytosolic cytochrome c and caspase-3. We found that a strong increase in the expression of Bax and a reduced expression of Bcl-2 in DOX group, PPARα- group and PPARα+ group compared with control group. Furthermore, Cytosolic cytochrome c and caspase-3 expression level also showed markedly increased in DOX group, PPARα- group and PPARα+ group (Figure 5). In addition, we found that caspase-3 activity significantly higher in DOX group, PPARα- group and PPARα+ group (Figure 6).
Figure 5.
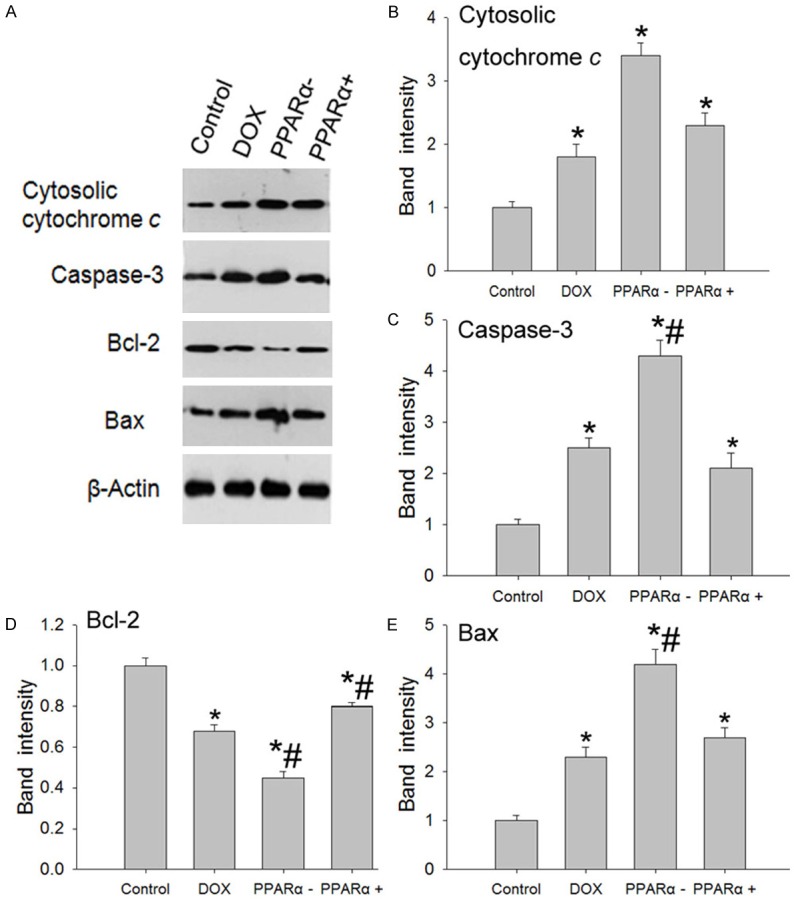
Expression level of markers for apoptosis in different groups. A. Western blotting results of markers for apoptosis expression indifferent groups. B-E. The graph represents a quantitative measure of band intensities of cytosolic cytochrome c, caspase-3, Bcl-2 and Bax, normalizing to β-actin in each lane. Data are the mean ± SEM (n=3), each bar represents the mean of three independent experiments carried out in triplicate. *Compared with control group, P<0.05, #compared with DOX group, P<0.05.
Figure 6.
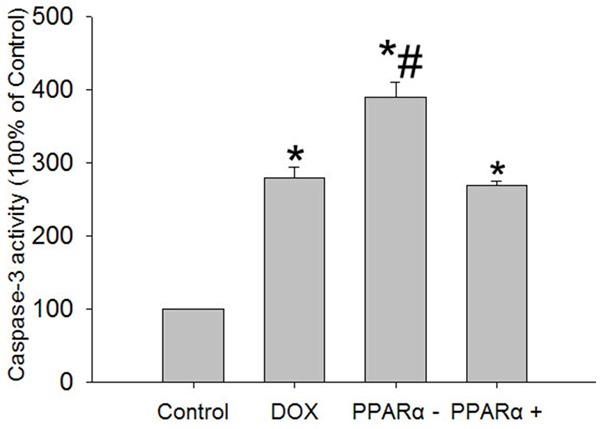
Enzymatic assay of caspase-3 activity. Activity of caspase-3 was measured by using a caspase-3 colorimetric protease assay kit. Data are the mean ± SEM (n=3), each bar represents the mean of three independent experiments carried out in triplicate, *compared with control group, P<0.05.
Discussion
Doxorubicin (DOX) was widely used as effective anticancer drug. However, DOX is a double-edged sword due to its use could cause a cardiomyopathy which lead to a refractory form of heart failure. DOX was used to induce the model of congestive cardiomyopathy these years [23,24]. Dilated cardiomyopathy (DCM) is the most frequent form of myocardial disease. However, the mechanism of DCM induced by DOX still not fully understood until now. Generally, mitochondria are the main target organs in the pathogenesis of DOX and lead to mitochondrial dysfunction occurred and energy imbalance, such as inhibiting the oxidative phosphorylation, blocking ATP generation [19].
Emerging evidence demonstrates the importance of PPARα and PGC-1α molecules in the transcriptional activation of genes expression involved in fatty acid oxidation [25,26]. At present, the changes and regulations of myocardial energy metabolism in DCM are poorly understood, especially the transcriptional regulation of PPARα and PGC-1α on energy metabolism of drug-induced cardiomyopathy is rarely reported. Our previous work showed that PPARα and PGC-1α were closely involved in energy metabolism in the DOX-induced mouse model of DCM [19]. Here, we investigate the effect of PPARα/PGC-1α expression on the energy metabolism in DOX-induced cardiomyocytes.
Doxorubicin treatment lead to the expression of PPARα and PGC-1α decreased significantly in DOX group than that of control group. Lower content of high-energy phosphates and significantly decreased mitochondrial ANT transport activity implied that abnormal energy metabolism in DOX group compared with control group. These imbalances maybe further prompt higher concentration of free fatty acid and lower glucose consumption detected in DOX group. More ROS molecules, might from the disorder energy metabolism, were harmful to cells. The ROS will inflict mitochondrial damage and make the dysfunction of the respiratory chains. And this possible were the mainly reasons for lower cell viability and higher apoptosis in DOX, PPARα- and PPARα+ groups.
The prognosis is significant for heart failure patients diagnosed. Few special new drugs have been designed to aim myocardial metabolic pathways in spite of more and more evidences from fundamental researches reveal that heart failure because of an imbalance between myocardial energy demand and supply [27]. For heart failure, energy metabolism defect was closely correlates with heart dysfunction which incapable of generation sufficient amounts of ATP. Down-regulation expression of fatty acid oxidation and glucose consumption likely urge the severity of this phenotype [28]. From aspect of energy metabolism, our results exhibit that lower capacity in producing energy maybe break normal cell life cycle, and cause cell apoptosis. An attempt to improve substrate oxidation and adenylate generation, maybe a new pharmaceutical therapies for heart failure patients induced by doxorubicin.
Acknowledgements
This work was supported by The National Natural Science Fund (81260053).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Jefferies JL, Towbin JA. Dilated cardiomyopathy. Seminar. 2010;375:27. doi: 10.1016/S0140-6736(09)62023-7. [DOI] [PubMed] [Google Scholar]
- 2.Takemura G, Fujiwara H. Doxorubicin-Induced Cardiomyopathy From the Cardiotoxic Mechanisms to Management. Prog Cardiovasc Dis. 2007;49:330–352. doi: 10.1016/j.pcad.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Baryshnikova OK, Robertson IM, Pascal Mercier, Brian D. Sykes: The dilated cardiomyopathy G159D mutation in cardiac troponin C weakens the anchoring interaction with troponin I. Biochemistry. 2008;47:10950–10960. doi: 10.1021/bi801165c. [DOI] [PubMed] [Google Scholar]
- 4.Shaddy RE, Olsen SL, Bristow MR, Taylor DO, Bullock EA, Tani LY, Renlund DG. Efficacy and safety of metoprolol in the treatment of doxorubicin-induced cardiomyopathy in pediatric patients. Am Heart J. 1995;129:197–199. doi: 10.1016/0002-8703(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 5.Monti E, Prosperi E, Supino R, Bottiroli G. Free radical dependent DNA lesions are involved in the delayed cardiotoxicity induced by adriamycin in the rat. Anticancer Res. 1995;15:193–197. [PubMed] [Google Scholar]
- 6.Gosalvez M, van Rossum GD, Blanco MF. Inhibition of sodium potassium activated adenosine 5’-triphosphatase and ion transport by adriamycin. Cancer Res. 1979;39:257–261. [PubMed] [Google Scholar]
- 7.Singal PK, Panagia V. Direct effects of adriamycinon the rat heart sarcolemma. Res Commun Chem Pathol Pharmacol. 1984;43:67–77. [PubMed] [Google Scholar]
- 8.Dávila-Román VG, Vedala G, Herrero P, de las Fuentes L, Rogers JG, Kelly DP, Gropler RJ. Altered Myocardial Fatty Acid and Glucose Metabolism in Idiopathic Dilated Cardiomyopathy. J Am Coll Cardiol. 2002;40:271–277. doi: 10.1016/s0735-1097(02)01967-8. [DOI] [PubMed] [Google Scholar]
- 9.Liang H, Ward WF. PGC-1α: a key regulator of energy metabolism. Adv Physiol Educ. 2006;30:145–151. doi: 10.1152/advan.00052.2006. [DOI] [PubMed] [Google Scholar]
- 10.Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, Rybkin II, Shelton JM, Manieri M, Cinti S, Schoen FJ, Bassel-Duby R, Rosenzweig A, Ingwall JS, Spiegelman BM. Transcriptional coactivator PGC-1α controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Benton CR, Holloway GP, Han XX, Yoshida Y, Snook LA, Lally J, Glatz JF, Luiken JJ, Chabowski A, Bonen A. Increased levels of peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1α) improve lipid utilization, insulin signalling and glucose transport in skeletal muscle of lean and insulin-resistant obese Zucker rats. Diabetologia. 2010;53:2008–2019. doi: 10.1007/s00125-010-1773-1. [DOI] [PubMed] [Google Scholar]
- 12.Handschin C. Peroxisome proliferator activated receptor γ coactivator 1β (PGC-1β) improves skeletal muscle mitochondrial function and insulin sensitivity. Commentary. 2011;54:1270–1272. doi: 10.1007/s00125-011-2135-3. [DOI] [PubMed] [Google Scholar]
- 13.Bajaj M, Suraamornkul S, Hardies LJ, Glass L, Musi N, DeFronzo RA. Effects of peroxisome proliferator activated receptor (PPAR) α and PPAR γ agonists on glucose and lipid metabolism in patients with type 2 diabetes mellitus. Diabetologia. 2007;50:1723–1731. doi: 10.1007/s00125-007-0698-9. [DOI] [PubMed] [Google Scholar]
- 14.Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, Gonzalez FJ. Altered constitutive expression of fatty Acid metabolizing enzymes in mice lacking the peroxisome proliferator activated receptor (PPARα) J Biol Chem. 1998;273:5678–5684. doi: 10.1074/jbc.273.10.5678. [DOI] [PubMed] [Google Scholar]
- 15.Barger PM, Kelly DP. PPAR Signaling in the Control of Cardiac Energy Metabolism. TCM. 2000;10:238–245. doi: 10.1016/s1050-1738(00)00077-3. [DOI] [PubMed] [Google Scholar]
- 16.Barger PM, Leone TC, Weinheimer CJ, Kelly DP. Deactivation of peroxisome proliferator activated receptor α during cardiac hypertrophic growth. J Clin Invest. 2000;105:1723–1730. doi: 10.1172/JCI9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol Improves Mitochondrial Function and Protects against Metabolic Disease by Activating SIRT1 and PGC-1α. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 18.Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam NA, Bernal-Mizrachi C, Chen ZJ, Holloszy JO, Medeiros DM, Kelly DP. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Y, Zhang H, Li X, Yang T, Jiang Q. Effects of PPARα/PGC-1α on the myocardial energy metabolism during heart failure in the doxorubicin induced dilated cardiomyopathy in mice. Int J Clin Exp Med. 2014;7:2435–2442. [PMC free article] [PubMed] [Google Scholar]
- 20.Schmid I, Uittenbogaart CH, Giorgi JV. Sensitive method for measuring apoptosis and cell surface phenotype in human thymocytes by flow cytometry. Cytometry. 1994;15:12–20. doi: 10.1002/cyto.990150104. [DOI] [PubMed] [Google Scholar]
- 21.Püttmann M, Krug H, von Ochsenstein E, Kattermann R. Fast HPLC determination of serum free fatty acids in the picomole range. Clin Chem. 1993;39:825–832. [PubMed] [Google Scholar]
- 22.Freyer JP, Sutherland RM. Reduction in the In Situ Rates of Oxygen and Glucose Consumption of Cells in EMT6/Ro Spheroids During Growth. J Cell Physiol. 1985;124:516–524. doi: 10.1002/jcp.1041240323. [DOI] [PubMed] [Google Scholar]
- 23.Lefrak EA, Pitha J, Rosenheim S, Gottlieb JA. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer. 1973;32:302–314. doi: 10.1002/1097-0142(197308)32:2<302::aid-cncr2820320205>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 24.Gilladoga AC, Manuel C, Tan CT, Wollner N, Sternberg SS, Murphy ML. Thecardiotoxicity of adriamycin and daunomycin in children. Cancer. 1976;37:1070–1078. doi: 10.1002/1097-0142(197602)37:2+<1070::aid-cncr2820370814>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 25.Disch DL, Rader TA, Cresci S, Leone TC, Barger PM, Vega R, Wood PA, Kelly DP. Transcriptional control of a nuclear gene encoding a mitochondrial fatty acid oxidation enzyme in transgenic mice: role for nuclear receptors in cardiac and brown adipose expression. Mol Cell Biol. 1996;16:4043–4051. doi: 10.1128/mcb.16.8.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator activated receptor alpha (PPARα) in the cellular fasting response: the PPARα-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci U S A. 1999;96:7473–7478. doi: 10.1073/pnas.96.13.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turakhia S, Venkatakrishnan CD, Dunsmore K, Wong H, Kuppusamy P, Zweier JL, Ilangovan G. Doxorubicin-induced cardiotoxicity: direct correlation of cardiac fibroblast and H9c2 cell survival and aconitase activity with heat shock protein. Am J Physiol Heart Circ Physiol. 2007;293:H3111–H3121. doi: 10.1152/ajpheart.00328.2007. [DOI] [PubMed] [Google Scholar]
- 28.Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML, McDonald JA, Kelly DP. Cardiac-Specific Induction of the Transcriptional Coactivator Peroxisome Proliferator-Activated Receptor γ Coactivator-1α Promotes Mitochondrial Biogenesis and Reversible Cardiomyopathy in a Developmental Stage Dependent Manner. Circ Res. 2004;94:525–533. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.