

Abstract
MicroRNAs (miRNAs) are classes of small, non-coding RNAs that regulate the translation of target mRNA transcripts. In this study, we demonstrated that miR-592 was downregulated in human hepatocellular carcinoma (HCC) and could suppress growth of the human HCC cell line HepG2. A tumor oncogene, DEK, was identified as a direct target of miR-592. Luciferase report assay indicated miR-592 regulates DEK expression though bind to its 3’UTR. Furthermore, knockdown of DEK also suppressed cell proliferation of HepG2 cells, which was consist with miR-592. At last, we suggested that DEK was upregulated in HCC tissues inversely with miR-592. These results demonstrated that miR-592 targets DEK transcript and suppresses HCC cell growth, and may provide potential therapeutic target in human HCC.
Keywords: DEK, hepatocellular carcinoma, miR-592, cell growth
Introduction
MicroRNAs are a new group of molecules, which are non-coding RNAs that are approximately 22 nt in length, were identified and shown to function as post-transcriptional regulators [1]. Studies have been reported that miRNAs are involved in many physiological and pathological progresses [2-4]. It has been indicates that many miRNAs function as oncogenes or tumor suppressors and play an important role in cancer initiation and progression by regulating their target genes negatively, including hepatocellular carcinoma (HCC), breast, ovarian, and lung cancers [5-8] as well as gastric adenocarcinoma [9]. By using Taqman miRNAs array and reverse transcription polymerase chain reaction method, the previous study confirmed a set of miRNAs deregulated in HCC tissues compared to the adjacent normal liver tissues, including ten up-regulated miRNAs (miR-217, miR-518b, miR-517c, miR-520g, miR-519a, miR-522, miR-518e, miR-525-3p, miR-512-3p, and miR-518a-3p) and 11 down-regulated miRNAs (miR-138, miR-214, miR-214*, miR-199a-5p, miR-433, miR-511, miR-592, miR-483-3p, miR-483-5p, miR-708 and miR-1275) [1]. Among these dysregulated miRNAs, miR-592 was largely unexplored in HCC. Therefore, the aim of this study was to detect the detailed mechanism of this differentially expressed miRNAs in the pathogenesis of HCC.
DEK, which encode by DEK gene, is a 43-kDa phosphoprotein. It has been indicated to be expressed in the proliferating cells and connect with chromatin reconstruction, gene transcription and cell apoptosis, which was originally identified in a subset of acute myeloid leukemia patients carrying the t(6;9) translocation [10,11]. Previous study has been demonstrated that its’ expression levels were significantly increased during the progression of colorectal cancers [12]. DEK is also a biomarker of cell proliferation in breast cancer [13]. Furthermore, in lung neuroendocrine carcinomas, the levels of DEK are inversely correlated with survival rates [14,15].
However, few studies was about the regulation mechanism of DEK during cancer pathogenesis despite its description as one of the most deregulated transcripts involved in cellular proliferation in HCC [16]. In this study, we found miR-592 could inhibit the HepG2 cell proliferation. Then we found that the miR-592 can directly target the 3’-UTR region of the DEK mRNA by repressing its translation, and consequently suppressing cell growth in HCC. In addition, knockdown of DEK has the same effect on HepG2 cell proliferation. These suggested miR-592/DEK axis play on important role in HCC cell tumorigenesis, and may serve as a target for HCC therapeutic.
Materials and methods
Clinical species and RNA isolation
Nine pairs of human HCC tissues and matched adjacent normal tissues used in this study were collected from the affiliated hospital of Changchun University of Chinese Medicine. The matched adjacent normal liver tissues were taken from the distal end of the operative excisions, far from the tumor. All of the samples were obtained with the patients’ informed consent and approved by the Ethics Committee of affiliated hospital of Changchun University of Chinese Medicine. Large and small RNAs were extracted from the tissue samples and purified using the mirVana miRNA Isolation Kit (Ambion, Austin, TX, USA) according to the manufacturer’s instructions.
Cell culture and transfection
The human HCC cell line HepG2 was grown in RPMI 1640 (GIBCO) supplemented with 10% fetal bovine serum, 100 IU/ml penicillin and 100 lg/ml streptomycin and was incubated at 37°C in a humidified chamber supplemented with 5% CO2. The transfection was performed using the Lipofectamine 2000 Reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions.
Cell proliferation assay
HepG2 cells were seeded in 96-well plate at a density of 4000 cells per well and transfected with miR-592 or control 24 hour later after seeding. MTT assay was used to measure the number of viable, proliferating cells at 24 h and 48 h after transfection. The absorbance at 570 nm was measured using an lQuant Universal Microplate Spectrophotometer (Bio-tek Instruments).
Colony formation assay
After transfection, the cells were counted and seeded in 12-well plates (in triplicate) at 150 cells per well. Fresh culture medium was replaced every 3 days. Colonies were counted only if they contained more than 50 cells, and the number of colonies was counted from the 10th day after seeding and then the cells were stained using crystal violet.
Fluorescent report assay
The luciferase expression vector pGL3/luciferase was constructed as previously described [2]. The 3’-UTR fragment of DEK containing the miR-592 binding site was amplified by PCR using the following primers: DEK sense, 5’-CGCGAATTCAGAGAGCTAAACCAAGTACTT-3’ and DEK antisense, 5’-GGATTTACTGATACGTACATGTACTTCTAGG-3’. The resulting PCR product was cloned into pGL3/luciferase at the BamHI and EcoRI sites. The RFP expression vector pDs-Red2-N1 (Clontech, Mountain View, CA, USA) was spiked in and used for normalization. The transfected cells were lysed with radio immunoprecipitation assay (RIPA) lysis buffer (DingGuo bioconpany, Beijing, China), and the proteins were harvested. The intensities of the luciferase fluorescence were detected with the Fluorescence Spectrophotometer F-4500 (HITACHI, Tokyo, Japan).
Quantitative RT-PCR
Small RNA (5 μg) was reverse transcribed into cDNA using M-MLV reverse transcriptase (Promega, Madison, WI, USA) with the following specific primers: miR-592-RT, 5’-GTCGTATCCAGTGCAGGGTCCGAGGTGCACTGGATACGAACATCATC-3’; and U6-RT, 5’-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAAAATATGGAAC-3’. The cDNA was used as template to amplify either mature miR-592 or an endogenous control U6 snRNA by PCR. The PCR was performed as follows: 94°C for 3 min, followed by 40 cycles of 94°C for 30 s, 50°C for 30 s and 72°C for 30 s. The real-time PCR was performed using SYBR Premix Ex Taq (TaKaRa, Otsu, Shiga, Japan) on the iQ5 Real-Time PCR Detection system (Bio-Rad). Real-time RT-PCR was performed to detect the relative level of DEK mRNA. Briefly, a cDNA library was generated through reverse transcription using M-MLV reversetranscriptase (Promega) with large RNA (5 μg). The cDNA was used to amplify the DEK gene and the β-actin gene, which served as an endogenous control. The PCR was performed as follows: 94°C for 3 min, followed by 40 cycles of 94°C for 30 s, 58°C for 30 s and 72°C for 30 s. Real-time PCR was performed as described above.
Western blot
The cells were harvested 48 h later after transfection, and protein was separated from the cytoplasmic protein using Nonidet P40 (NP-40) and RIPA. All proteins were resolved on a 6% SDS-denaturing polyacrylamide gel and then transferred onto a nitrocellulose membrane. The membranes were incubated with anti-DEK or anti-GAPDH with Blotto overnight at 4°C. The membranes were then washed and incubated with a horseradish peroxidase-conjugated secondary antibody. Protein expression was assessed by enhanced chemiluminescence after exposure to chemiluminescent film. Band intensity was quantified using the Lab Works Image Acquisition and Analysis Software (UVP, Upland, CA, USA).
Statistical analysis
Data are expressed as the means ± standard deviation (SD), and P ≤ 0.05 is considered to be statistically significant using the Students t-test.
Results
MiR-592 suppresses the long-term proliferation of the HCC cell line HepG2
To determine the role of miR-592 in tumor cell proliferation, the MiR-592 mimics and a miR-592 antisense oligomer were used to overexpress and block miR-592 expression respectively in HepG2 cells (Figure 1A, 1B). By using a MTT assay, overexpression of miR-592 was demonstrated to reduce HepG2 cell growth compared to control (Figure 1C). The colony formation rate of HepG2 cells transfected with miR-592 was significantly lower by approximately 70% than that of the control group (Figure 1D). In contrast, HepG2 cells transfected with anti-miR-592 showed enhanced proliferation activity in the colony formation assay (Figure 1E). These results indicated that miR-592 suppressed the cell proliferation of HepG2 cells.
Figure 1.
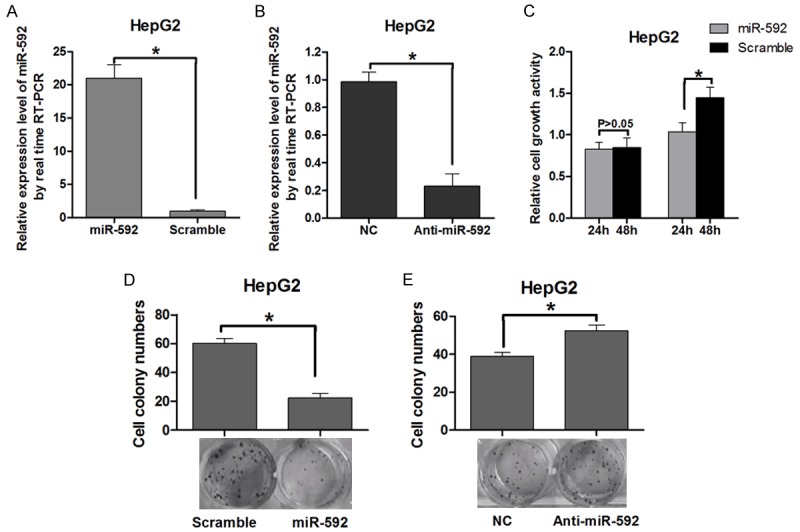
Overexpression of miR-592 suppresses cell growth in vitro. A and B: Measurement of miR-592 expression levels by real-time RT-PCR. Small RNA was extracted from HepG2 cells transfected with miR-592 mimics or control vector, miR-592 ASO (ASO-592) or control oligonucleotides, and U6 snRNA served as an endogenous normalizer. The relative miR-592 expression level (mean ± SD) is shown (P < 0.05). C: Measurement of cell growth by the MTT assay. After HepG2 cells were transfected with the miR-592 mimics or control vector, the MTT assay was used to determine the relative cell growth activity at 24 and 48 h post-transfection. The histogram shows the mean ± SD of three independent experiments (P < 0.05). The relative cell growth activity was normalized to the growth activity of HepG2 cells in the control groups. D: Effect of miR-592 on cell proliferation as evaluated by a colony formation assay. HepG2 cells transfected with miR-592 mimics or control vector were seeded in 12-well plates. On the 10th day after seeding, the number of colonies was counted, and the colony formation rate was calculated (P < 0.05). E: HepG2 cells transfected with Anti-miR-592 were seeded in 12-well plates. On the 10th day after seeding, the number of colonies was counted (P < 0.05).
miR-592 directly target DEK in HepG2 cells
Based on the miR-592-induced suppression of the proliferation of a HCC cell line, we have assumption that miR-592 inhibited the malignancy of HCC cells by regulating oncogenes and/or genes involved in cell proliferation or apoptosis. Thus, bioinformatic analyses (TargetScan, miRanda and miRDB) were used to identify potential target genes of miR-592. DEK was predicated to have two putative miR-592 binding site within its 3’-UTR (Figure 2A) and thus was chosen for further study. To illuminate whether miR-592 directly regulates DEK, a luciferase reporter system was used to identify the target site in the DEK 3’-UTR. The 3’-UTR region containing either the predicted miR-592 binding site or a mutant version of the binding site was fused downstream of the luciferase gene following a stop codon (pGL3/luciferase-DEK-3’-UTR or pGL3/luciferase-DEK-3’-UTR-mut). The HepG2 cells were transfected with the reporter vector along with miR-592 mimics or a control vector. The fluorescence intensity in miR-592 transfected cells was significantly lower than in the control group (Figure 2B). In contrast, the luciferase intensity increased when treated with the miR-592 ASO (Figure 2D). To further confirm the miR-592 binding site, the pGL3-DEK-3’-UTR-mut-1 plasmid and the pGL3-DEK-3’-UTR-mut-2 plasmid were constructed, which contained a mutated version of the predicted miR-592 binding site (Figure 2A). The results suggested luciferase intensity from the mutated DEK 3’-UTR group was not affected by the amount of miR-592 (Figure 2C, 2E). Taken together, these results indicate that miR-592 binds directly to the 3’-UTR of DEK, thereby repressing gene expression.
Figure 2.
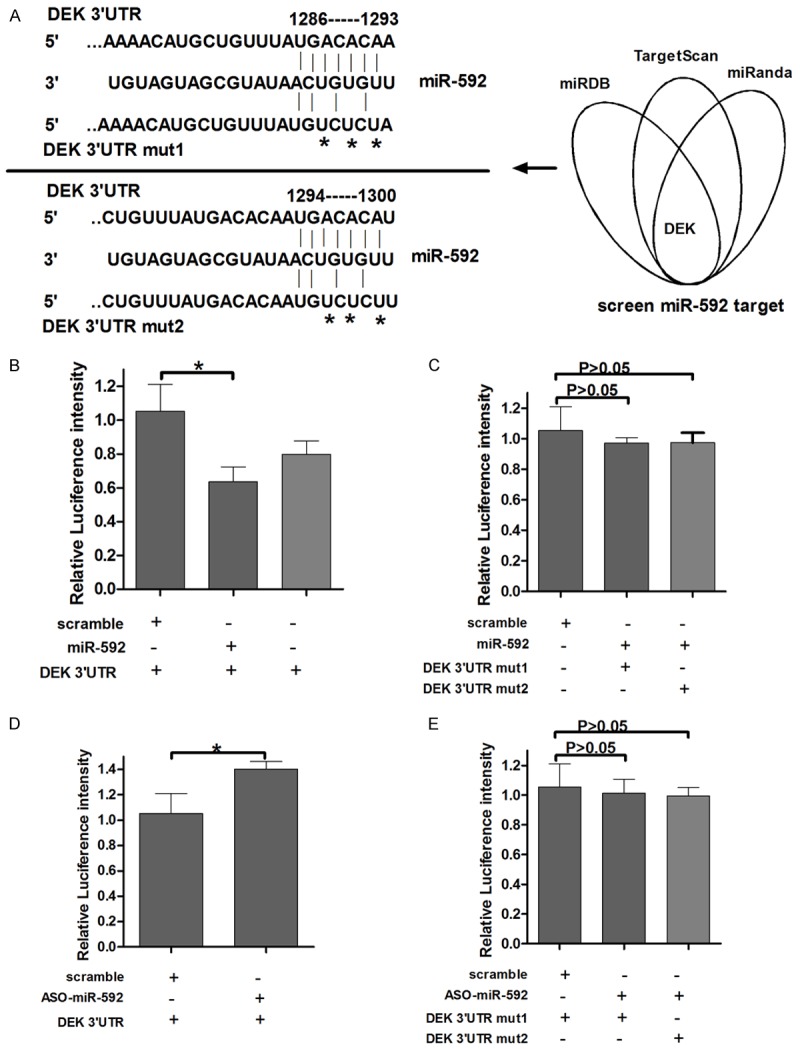
DEK is a direct target of miR-592. A: The predicted miR-592 binding site on the DEK mRNA 3’-UTR and the deletion mutation at the miR-592 “seed region” binding site, involving three nucleotides, on the DEK mRNA 3’-UTR are shown. B and C: HepG2 cells were transfected with the wild type (Wt. UTR) or mutated version (Mut. UTR) of the Luciferase-DEK 3’-UTR reporter vector as well as the miR-592 mimics or control vector (Ctrl). The miR-592 mimics reduced the intensity of the fluorescence from the luciferase-DEK 3’-UTR vector, but the mutation of the miR-592 binding site restored the intensity of the luciferase fluorescence. D and E: HepG2 cells were transfected with the Wt. UTR or Mut. UTR reporter vector as well as anti-miR-592 or the control oligonucleotides. The relative luciferase intensity is shown.
DEK is negatively regulated by miR-592
miRNAs may suppress the expression of target genes through translational repression or degradation of the target transcript. The results of qRT-PCR indicated that overexpression of miR-592 does not affect the DEK mRNA expression compared with the control group (Figure 3A, 3B). Furthermore, overexpression of miR-592 reduced DEK protein expression (Figure 3C). In contrast, blockage of miR-592 enhanced DEK protein expression (Figure 3D). Taken together, these results indicated that miR-592 regulates endogenous DEK expression through mRNA translational repression.
Figure 3.
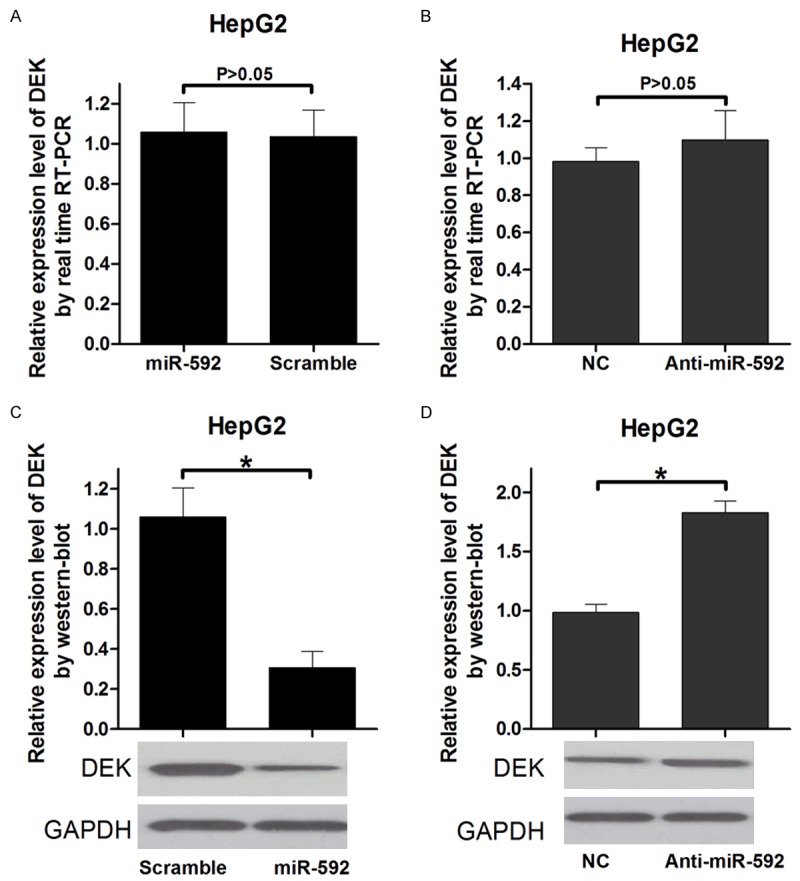
MiR-592 regulates DEK mRNA expression via transcript degradation and at the translational level. A and B: Measurement of DEK expression levels by RT-PCR. RNA was extracted from HepG2 cells transfected with the miR-592 mimics or control vector, Anti-592 or control oligonucleotides. The endogenous expression levels of the GAPDH mRNA were used for normalization, and the relative DEK expression levels are shown. (P < 0.05). C and D: Measurement of DEK expression levels by Western blot analysis. Protein was extracted from HepG2 cells transfected with the miR-592 mimics or control vector, Anti-592 or control oligonucleotides. The endogenous expression levels of the GAPDH protein were used for normalization, and the relative DEK protein expression levels are shown (P < 0.05).
Knockdown of DEK inhibits the growth of HepG2 cells in vitro
An RNAi approach was used to conform if the knockdown of DEK affects HepG2 cell growth. The siRNA expression vector, pSilencer/si-DEK, effectively reduce DEK protein levels (Figure 4A, 4B). The transient transfection of pSilencer/si-DEK into HepG2 cells indicated that knockdown of DEK could suppress cell growth in 24 h and 48 h after transfection, as determined by the MTT assay (Figure 4C). The colony formation ability of the cells transfected with the si-DEK vector was markedly lower than that of the control group (Figure 4D). These results are consistent with the finding that miR-592 overexpression can suppress cell growth in vitro, which provides further evidence that DEK is involved in the miR-592-mediated suppression of HCC cell growth. Accordingly, the identification of DEK as a target gene of miR-592 might explain, at least in part, why miR-592 could suppresses HepG2 cell growth.
Figure 4.
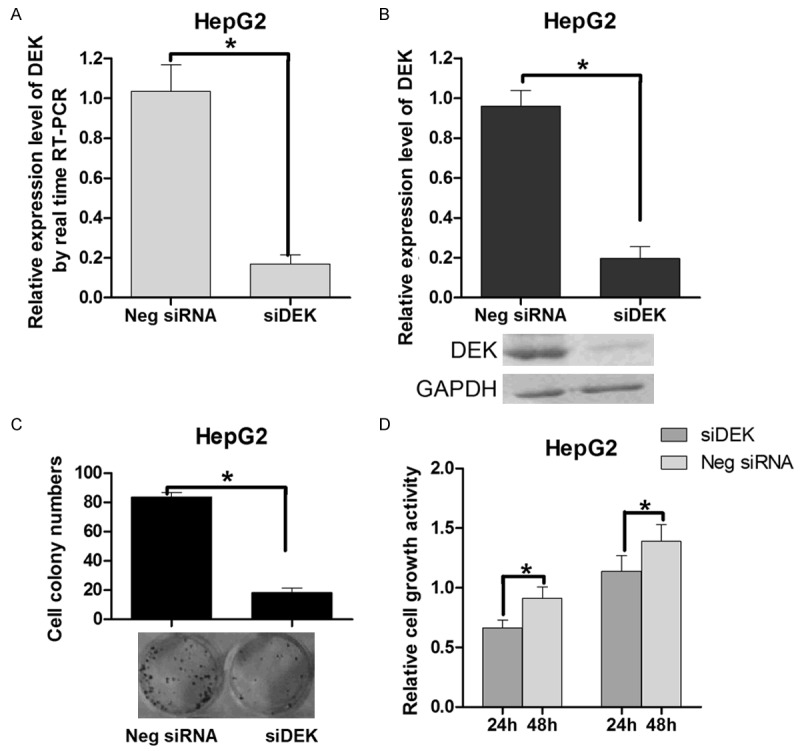
Knockdown of DEK suppresses cell growth and colony formation. A: Measurement of pSilencer/si-DEK (si-DEK) levels by real-time RT-PCR. B: DEK protein expression levels were significantly decreased in HepG2 cells transfected with si-DEK. C: Cell-independent growth activity of HepG2 cell as determined by the colony formation assay. The HepG2 cells were transfected with either the si-DEK or control vector (final concentration 5 ng/lL) and seeded in 12-well plates. On the 7th day after seeding, the number of colonies was counted. The calculated colony formation rate is shown. In the colony formation assay, the cells were stained by crystal violet, with representative pictures of the colonies being shown. D: The cell growth activity of HepG2 cells at the indicated times as determined by the MTT assay. The HepG2 cells were transfected with either the si-DEK or control vector. The relative cell growth activity is shown (P < 0.05). Values are means ± SD of triplicate experiments.
Quantitative analysis of DEK and miR-592 expression in human HCC tissue
To determine the expression of miR-592 in HCC tumor tissues and adjacent normal tissue, real-time RT-PCR assay were performed. The results indicated that the expression levels of DEK were remarkably higher in HCC tissues than in the paired adjacent normal tissues. In contrast, MiR-592 expression levels were predominantly downregulated in HCC tissues (Figure 5).
Figure 5.
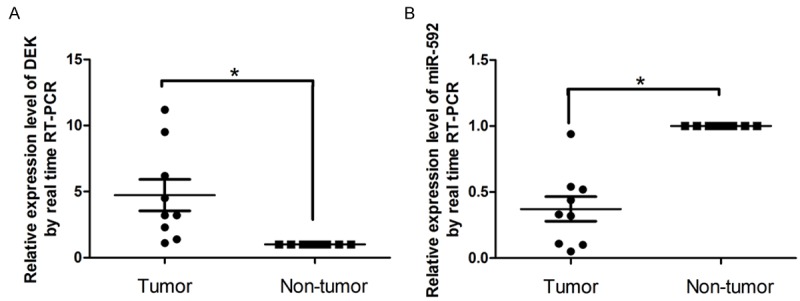
Quantitative analysis of DEK and miR-592 expression in human hepatic tissues. To test the expression of miR-592 in HCC tissues and adjacent normal hepatic tissues, a real-time RT-PCR assay was performed on the nine pairs of hepatic tissue samples, and the dysregulation fold of miR-592 and DEK was shown. In general, the DEK expression levels were significantly higher in the HCC tissues than in the matched normal hepatic tissues. The expression level of MiR-592 was predominantly downregulated in human hepatic tissues.
Discussion
Abnormal regulations of miRNAs have been linked to many human diseases, and some specific miRNAs were connected with the clinicopathological features of HCC, especially metastasis, recurrence, and prognosis [17-19]. The present study suggests that miR-592 suppresses HepG2 cell growth by directly regulating DEK. Firstly, in vitro validation experiments indicated that the expression of luciferase under the control of the DEK 3’-UTR sequence can be regulated by miR-592. This down-regulation was shown to be adjusted by the direct binding of the miRNA to an identified target site in the DEK 3’-UTR as mutation of this region wipes out this effect. Secondly, we indicated cell proliferation of HepG2 cells were suppressed by knockdown of DEK and by the overexpression of miR-592 (Figures 1, 4). Thirdly, DEK expression was inversely correlated with miR-592 levels in HCC tissues. These results suggested downregulated miR-592 could lead to the upregulated DEK in HCC then promote HCC tumorigenesis.
Previous study has been reported that the level of DEK was reduced by butyrate in a dose-dependent manner [20]. Other researchers have shown that DEK is a phospho-protein with several phosphorylation sites of located on chromosome 6p22.3, which most are clustered in the carboxy terminal region [21]. Despite its broad use as a marker for cellular proliferation there have been few studies that revealed the correlation between the expression of DEK and miRNAs in HCC. Our study provides evidence that the upregulation of DEK in HCC, which was confirmed by quantitative RT-PCR, may promote the oncogenic activity of DEK and thereby contribute to tumorigenesis. Because DEK is a target gene for miR-592, the suppression of miR-592 may account for the overexpression of DEK in HCC, however, other mechanisms cannot be eliminated. DEK as a putative diagnostic marker and candidate for drug development will also be discussed [22].
In conclusion, DEK was definite at higher levels in HCC tissue compared with normal hepatic tissue, and miR-592 was verified to be a direct modulator of DEK. The overexpression of miR-592 suppressed the expression of DEK, leading to the inhibition of HCC cell growth. These finding obviously indicate that DEK expression is partially regulated by miR-592 in HCC cells. The repression of miR-592 in HCC could contribute to the malignant phenotype by allowing high levels of DEK to be detected. Thus, the identification of miR-592 and its target gene, DEK, in HCC provides a basis for future investigations, to explore the role of this regulatory relationship in the physiology and pathophysiology of this intriguing protein. In addition, the miR-592/DEK axis dynamic may have diagnostic and/or therapeutic value in the future.
Acknowledgements
We thank the support by funding of Jilin Provincial Education Department Science and technology research project (2015347).
Disclosure of conflict of interest
None.
References
- 1.Wang W, Zhao LJ, Tan YX, Ren H, Qi ZT. Identification of deregulated miRNAs and their targets in hepatitis B virus-associated hepatocellular carcinoma. World J Gastroenterol. 2012;18:5442–5453. doi: 10.3748/wjg.v18.i38.5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu T, Tang H, Lang Y, Liu M, Li X. MicroRNA-27a functions as an oncogene in gastric adenocarcinoma by targeting prohibitin. Cancer Lett. 2009;273:233–242. doi: 10.1016/j.canlet.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 3.Moriyama T, Ohuchida K, Mizumoto K, Yu J, Sato N, Nabae T, Takahata S, Toma H, Nagai E, Tanaka M. MicroRNA-21 modulates biological functions of pancreatic cancer cells including their proliferation, invasion, and chemoresistance. Mol Cancer Ther. 2009;8:1067–1074. doi: 10.1158/1535-7163.MCT-08-0592. [DOI] [PubMed] [Google Scholar]
- 4.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 5.Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, Stephens RM, Okamoto A, Yokota J, Tanaka T, Calin GA, Liu CG, Croce CM, Harris CC. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–198. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 6.Hou YY, Cao WW, Li L, Li SP, Liu T, Wan HY, Liu M, Li X, Tang H. MicroRNA-519d targets MKi67 and suppresses cell growth in the hepatocellular carcinoma cell line QGY-7703. Cancer Lett. 2011;307:182–190. doi: 10.1016/j.canlet.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Ménard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 8.Iorio MV, Visone R, Di Leva G, Donati V, Petrocca F, Casalini P, Magri E, Pedriali M, Fabbri M, Campiglio M, Ménard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007;67:8699–8707. doi: 10.1158/0008-5472.CAN-07-1936. [DOI] [PubMed] [Google Scholar]
- 9.Wan HY, Guo LM, Liu T, Liu M, Li X, Tang H. Regulation of the transcription factor NF-kappaB1 by microRNA-9 in human gastric adenocarcinoma. Mol Cancer. 2010;9:16. doi: 10.1186/1476-4598-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.von Lindern M, Fornerod M, van Baal S, Jaegle M, de Wit T, Buijs A, Grosveld G. The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia-specific dek-can mRNA. Mol Cell Biol. 1992;12:1687–1697. doi: 10.1128/mcb.12.4.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wise-Draper TM, Allen HV, Jones EE, Habash KB, Matsuo H, Wells SI. Apoptosis inhibition by the human DEK oncoprotein involves interference with p53 functions. Mol Cell Biol. 2006;26:7506–7519. doi: 10.1128/MCB.00430-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ganesan J, Ramanujam D, Sassi Y, Ahles A, Jentzsch C, Werfel S, Leierseder S, Loyer X, Giacca M, Zentilin L, Thum T, Laggerbauer B, Engelhardt S. MiR-378 controls cardiac hypertrophy by combined repression of mitogen-activated protein kinase pathway factors. Circulation. 2013;127:2097–2106. doi: 10.1161/CIRCULATIONAHA.112.000882. [DOI] [PubMed] [Google Scholar]
- 13.Privette Vinnedge LM, Ho SM, Wikenheiser-Brokamp KA, Wells SI. The DEK oncogene is a target of steroid hormone receptor signaling in breast cancer. PLoS One. 2012;7:e46985. doi: 10.1371/journal.pone.0046985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shibata T, Kokubu A, Miyamoto M, Hosoda F, Gotoh M, Tsuta K, Asamura H, Matsuno Y, Kondo T, Imoto I, Inazawa J, Hirohashi S. DEK oncoprotein regulates transcriptional modifiers and sustains tumor initiation activity in high-grade neuroendocrine carcinoma of the lung. Oncogene. 2010;29:4671–4681. doi: 10.1038/onc.2010.217. [DOI] [PubMed] [Google Scholar]
- 15.Soares LM, Zanier K, Mackereth C, Sattler M, Valcarcel J. Intron removal requires proofreading of U2AF/3’ splice site recognition by DEK. Science. 2006;312:1961–1965. doi: 10.1126/science.1128659. [DOI] [PubMed] [Google Scholar]
- 16.Lin LJ, Chen LT. The role of DEK protein in hepatocellular carcinoma for progression and prognosis. Pak J Med Sci. 2013;29:778–782. doi: 10.12669/pjms.293.3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong QW, Ching AK, Chan AW, Choy KW, To KF, Lai PB, Wong N. MiR-222 overexpression confers cell migratory advantages in hepatocellular carcinoma through enhancing AKT signaling. Clin Cancer Res. 2010;16:867–875. doi: 10.1158/1078-0432.CCR-09-1840. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Lee AT, Ma JZ, Wang J, Ren J, Yang Y, Tantoso E, Li KB, Ooi LL, Tan P, Lee CG. Profiling microRNA expression in hepatocellular carcinoma reveals microRNA-224 up-regulation and apoptosis inhibitor-5 as a microRNA-224-specific target. J Biol Chem. 2008;283:13205–13215. doi: 10.1074/jbc.M707629200. [DOI] [PubMed] [Google Scholar]
- 19.Gramantieri L, Fornari F, Callegari E, Sabbioni S, Lanza G, Croce CM, Bolondi L, Negrini M. MicroRNA involvement in hepatocellular carcinoma. J Cell Mol Med. 2008;12:2189–2204. doi: 10.1111/j.1582-4934.2008.00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kondoh N, Wakatsuki T, Ryo A, Hada A, Aihara T, Horiuchi S, Goseki N, Matsubara O, Takenaka K, Shichita M, Tanaka K, Shuda M, Yamamoto M. Identification and characterization of genes associated with human hepatocellular carcinogenesis. Cancer Res. 1999;59:4990–4996. [PubMed] [Google Scholar]
- 21.Sanden C, Ageberg M, Petersson J, Lennartsson A, Gullberg U. Forced expression of the DEK-NUP214 fusion protein promotes proliferation dependent on upregulation of mTOR. BMC Cancer. 2013;13:440. doi: 10.1186/1471-2407-13-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riveiro-Falkenbach E, Soengas MS. Control of tumorigenesis and chemoresistance by the DEK oncogene. Clin Cancer Res. 2010;16:2932–2938. doi: 10.1158/1078-0432.CCR-09-2330. [DOI] [PMC free article] [PubMed] [Google Scholar]