

Abstract
Chronic myeloid leukemia (CML) can be contextualized as a disease of unregulated self-renewal of stem cells which exist in a quiescent state and are instructed to differentiate and mobilize to circulation under pathologic circumstances leading to tumor invasion and metastasis. Here we found that matrix metalloproteinase-9 (MMP-9), induced by TGF-β1, upregulated s-KitL and s-ICAM-1, permitting the transfer of c-kit+ hematopoietic stem cells (HSCs) from the quiescent to proliferative niche in CML. Further study showed that this MMP-9 production was raised by CML specific BCR/ABL+ oncogene mediated TGF-β1. Besides, phosphatidylinositol-3 kinase (PI3K)/Akt/nuclear factor (NF)-κB signaling pathway was evidenced to govern this stem cell recruitment in CML pathogenesis. Overall, our observations defined a novel critical role for TGF-β1 induced PI3K/Akt/NF-κB signaling pathway in the recruitment of the malignant cells in CML by releasing s-KitL and s-ICAM-1 and this was through a distinct PI3K/Akt/NF-κB signaling pathway.
Keywords: Chronic myeloid leukemia (CML), mesenchymal stem cell (MSC), hematopoietic stem cell (HSC), matrix metalloproteinase-9 (MMP-9), TGF-β1, s-KitL, s-ICAM-1
Introduction
Compelling research suggests that a population of cancer stem cells (CSCs) is able to regenerate or self-renew resulting in therapeutic resistance and disease progression; this is particularly true in chronic myeloid leukemia (CML) [1-3]. Local secretion of proteases has been implicated in this tumor-stroma crosstalk. Matrix metalloproteinase-9 (MMP-9) is one of them which has the preferential ability to degrade denatured collagens (gelatin) and collagen type IV, the 2 main components of basement membranes and therefore plays a critical role in tumor progression and metastasis [4]. Previous studies have demonstrated localization of MMP-9 on the plasma membrane of various tumor cells [5-7], recently, the role of MMP-9 in CML pathogenesis has became a focus of attention [8-11]. But the research is mainly focusing on the MMP-9 inducing molecules [12-14] or the effect of the treatment of MMP-9 inhibitors [15]. However, it has become clear that the role of MMP-9 in CML is much more complex than one of the simple extracellular matrix (ECM) degradation [16]. In clinical trials, many direct MMP-9 inhibitors have not been successfully in treating cancers, therapeutic inhibition of MMP-9 may be achieved by other indirect means such as targeting the signal transduction pathways that regulate MMP-9 expression [17]. Moreover, the regulation of MMP-9 is found to be involved in multiple pathways induced by different kinds of cytokines according to different cell types and illness [18,19]. Therefore, it is necessary to verify a specific MMP-9 induced pathway in a given cell type.
Therefore, in the present investigation, we aimed to determine the mechanism underlying malignant HSCs recruitment and mobilization. We found that under steady state conditions quiescent c-kit+ HSCs reside in a niche in close contact with stromal cells. Membrane-bound cytokines, such as m-KitL and m-ICAM-1 not only convey survival signals, but support the adhesion of stem cells to the stroma. CML specific oncogene induced TGF-β1 activated PI3K/Akt/NF-κB/MMP9 signaling pathway resulting in the release of s-KitL and s-ICAM-1, enhanced recruitment and mobilization of tumor stem cells to the peripheral circulation.
Material and methods
Regents
The following antibodies and reagents were used: antiphospho-Akt (Ser473; Calbiochem, San Diego, CA); anti-MMP-9 (Chemicon, Temecula, CA), anti-Akt1/2, mouse immunoglobulin G (IgG) agarose beads, anti-IKKα agarose beads, anti-IKKα, anti-IKKβ, and protein A/G agarose beads (Santa Cruz Biotechnology, CA); wortmannin and MG-132; SH-5 (Alexis Biochemicals, San Diego, CA); phosphoglycogen synthase kinase (GSK)-3α/β (Ser21/9) antibody, GSK-3 fusion protein, kinase buffer, and adenosine 5’-triphosphate (ATP; Cell Signaling Technology, Beverly, MA); elution buffer (Pierce Biotechnology, Rockford, IL); and nuclear extract kit, TransAMTM NF-κB transcription factor assay kit (Active Motif, Carlsbad, CA).
Patients samples
20 patients with newly diagnosed CML (12 male and 8 female, aged 17-63 years) were recruited in this study. All were Ph+ patients with CML in chronic phase as revealed by bone marrow histology and cytogenetic analysis and the immunophenotype of thawed cells was quite variable. None were treated with hydroxyurea or interferon before. The control samples were healthy donors with the equal 20 samples (12 male and 8 female, aged 21-60 years). Their immunophenotype was also quite variable and they were all Ph+ negative in BM at donation. All the bone marrow samples were collected after obtaining informed consent according to procedures approved by the Ethics Committee at the 309 Hospital of Peoples Liberation Army.
Cell preparations and culture conditions
Isolation and culture of bone marrow-derived BCR/ABL+ MSCs from CML patients were performed as described previously with some modifications [19-21]. Briefly, mononuclear cells were separated by a Ficoll-Paque gradient centrifugation (specific gravity 1.077 g/mL; Nycomed Pharma AS, Oslo, Norway) and the sorted cells were plated at concentrations of 1 cell/well by limiting dilution in a total of 96×10 wells coated with fibronectin (Sigma, St Louis, MO) and collagen (Sigma) for each patient. Culture medium was Dulbecco modified Eagle medium and Ham F12 medium (DF12) containing 40% MCDB-201 medium complete with trace elements (MCDB) (Sigma), 2% fetal calf serum (FCS; Gibco Life Technologies, Paisley, United Kingdom), 1× insulin transferrin selenium (Gibco Life Technologies), 10-9 M dexamethasone (Sigma), 10-4 M ascorbic acid 2-phosphate (Sigma), 20 ng/mL interleukin-6 (Sigma), 10 ng/mL epidermal growth factor (Sigma), 10 ng/mL platelet-derived growth factor BB (Sigma), 50 ng/mL fetal liver tyrosine kinase 3 (Flt-3) ligand (Sigma), 30 ng/mL bone morphogenetic protein-4 (Sigma), and 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco Life Technologies) at 37°C and a 5% CO2 humidified atmosphere. Culture media were changed every 4 to 6 days.
RT-PCR
RNA isolation and reverse transcription were performed as previously described [32]. Oligonucleotide primer sequences were as follows: β-actin (264 bp), forward: 5’-GAGACCTTCAACACCCCAGCC-3’; reverse: 5’-AATGTCACGCACGATTTCCC-3’; s-KitL (263 bp), forward: 5’-AGAGGTCTCAGAAGGGACCG-3’, reverse: 5’-GGGCCATACAGGACACGAAG-3’; s-ICAM-1 (167 bp), forward: 5’-TGCTTTTTCCAGGGGTGTGTT-3’, reverse: 5’-TACTTCCTGCACTAATTTGGCA-3’; TGFβ1 (198 bp), forward: 5’-GGAAACCCACAACGAAATCTATGAC-3’, reverse: 5’-TTGCTGAGGTATCGCCAGGAAT-3’; TGFβ2 (213 bp), forward: 5’-CGCCAAGGAGGTTTACAAAATAGAC-3’, reverse: 5’-TCAATCCGTTGTTCAGGCACTCT-3’; TGFβ3 (233 bp), forward: 5’-CTGGCGGAGCACAACGAACT-3’, reverse: 5’-AGGATATCTCCATTGGGCTGAAAG-3’; MMP-9 (201 bp), forward: 5’-TCCCCATCGCCATCCCC-3’ reverse: 5’-CACCATGGCCTCGGCTGG-3’. To all the above genes, amplification was performed under the same cycling conditions (1 minute at 94°C, 50 seconds at 57°C, 1 minute at 72°C), except the number of cycles that were specified for each gene (31 for s-KitL, s-ICAM-1, TGFβ1, TGFβ2 and TGFβ3, 32 for MMP-9).
Co-culture assay
Flk1+CD31-CD34- MSCs were isolated from healthy donors and CML patients were cultured in T25 bottle respectively and regulated the number to 1×106/bottle. They were radiated 10 Gy/unit 1.5 hours later and then mononuclear cells from cord blood were collected and cultured with the MSCs in T25 bottle to form the co-culture system. Collected 2.5 ml medium and filled 2.5 ml H5100 into it a week later. Centrifuged the HSCs and collected supernatant, using the 1640 to regulate the cell number into 1×106/well. Put CFU-mix 300 μl/well into 24 well plate and collected the supernatant 100 μl/well for further assay. After a 14-day incubation at 37°C in 5% CO2, CFU-GMs were scored as aggregated of more than 50 cells.
Western blot and immunopreciptation
MSCs were harvested at specific times after treatment with regents as indicated in each experiments and then were mixed with loading buffer and separated, after electrophoresis, proteins were transferred to polyvinyl difluoride membranes (Pall Filtron) using a semidry blotting apparatus (Pharmacia) and probed with mouse monoclonal antibodies to indicated amount, followed by incubation with peroxidase-labeled secondary antibodies. Detection was performed by the use of a chemiluminescence system (Amersham) according to the manufacturer’s instructions. Then membrane was striped with elution buffer and reprobed with antibodies against the nonphosphorylated protein as a measure of equal loading. Controls for the immnoprecipitation used the same procedure, except agarose beads contained only mouse IgG.
T lymphocyte activity assay
Cells isolation and culture were performed as above mentioned and we divided them into 3 groups: TS: PHA induced T lymphocyte; TS+ healthy MSCs: the healthy MSCs and PHA induced T lymphocyte; TS+Flk1+ MSC: Flk1+ MSC and PHA induced T lymphocyte. The proportion of MSCs and T lymphocytes was 1:10; CD69, CD25 and CD44 were examined 12, 24 and 72 hrs later respectively.
Fluorescence activated cell sorting (FACS)
For immunophenotype analysis, expanded clonal cells were stained with antibodies against Flk1, CD29, CD31, CD34, CD44, CD45, CD105, HLA-ABC, vWF (all from Becton Dickinson Immunocytometry Systems, Mountain View, CA). For intracellular antigen detection, cells were first fixed in 2% paraformaldehyde (Sigma) for 15 minutes at 4°C and permeabilized with 0.1% saponin (Sigma) for 1 hour at room temperature. Cells were washed and labeled with fluorescein isothiocyanate (FITC) conjugated secondary goat antimouse, goat antirabbit, or sheep antigoat antibodies (Sigma), then washed and analyzed using a FACS Calibur flow cytometer (Becton Dickinson, San Jose, CA).
FISH analysis
We cultured BCR/ABL+ MSCs from male CML patients (n = 12) and Y chromosome was detected using a probe (CEP Y Spectrum Red; Vysis, Downers Grove, IL) according to the manufacturer’s instructions. Normal cells showed 2 red able signals and 2 green bar signals. BCR/ABL+ MSCs showed a single red and a single green signal representing normal able and bar genes and the yellow signal representing fusion of able and bar genes.
RNA-I experiments
The is-RNA sequence targeting human MMP-9 chosen in this study (from mRNA sequence; Nitrogen online) corresponds to the coding region 377-403 relative to the first nucleotide of the start codon (target = 5’-AAC ATC ACC TAT TGG ATC CAA ACT AC-3’). Computer analysis using the software developed by Ambion Inc. confirmed this sequence to be a good target. si-RNAs were 21 nucleotides long with symmetric 2-nucleotide 3’overhangs composed of 2’-deoxythymidine to enhance nuclease resistance. The si-RNAs were synthesized chemically and high pressure liquid chromatography purified (Genset, Paris, France). Sense si-RNA sequence was 5’-CAUCACCUAUUGGAUCCAAdTdT-3’. Antisense si-RNA was 5’-UUGGAUCCAAUAGGUGAUGdTdT-3’. For annealing of si-RNAs, mixture of complementary single stranded RNAs (at equimolar concentration) was incubated in annealing buffer (20 mM Tris-HCl pH 7.5, 50 mM NaCl, and 10 mM MgCl2) for 2 minutes at 95°C followed by a slow cooling to room temperature (at least 25°C) and then proceeded to storage temperature of 4°C. Before transfection, cells cultured at 50% confluence in 6-well plates (10 cm2) were washed two times with OPTIMEM 1 (Invitrogen) without FCS and incubated in 1.5 ml of this medium without FCS for 1 hour. Then, cells were transfected with MMP-9-RNA duplex formulated into Mirus TransIT-TKO transfection reagent (Mirus Corp, Interchim, France) according to the manufacturer’s instructions. Unless otherwise described, transfection used 20 nM RNA duplex in 0.5 ml of transfection medium OPTIMEM 1 without FCS per 5×105 cells for 6 hours and then the medium volume was adjusted to 1.5 ml per well with RPMI 2% FCS. SilencerTM negative control 1 si-RNA (Ambion Inc.) was used as negative control under similar conditions (20 nM). The efficiency of silencing is 80% in our assay.
Enzyme-linked immunoadsorbent assays
This was carried out according to the manufacturer’s (Oncogene Research Products) recommendations. Results were compared with those obtained with serially diluted solutions of commercially purified controls. Briefly, anti-human cytokine antibodies (R & D Systems, Minneapolis, MN) was added at 0.4 μg/ml in 0.05 M bicarbonate buffer (pH 9.3) to 96-well, U-bottom, polyvinyl microplates (Becton Dickinson and Co., Oxnard, CA) and the cell number was 1×105/100 μl. After incubation overnight at 4°C, the plates were washed and blocked with 1% gelatin for 1 hour. Samples (50 μl) or standard protein diluted in 0.5% gelatin were added to the wells. After incubation for 1 hour at 37°C, the plates were washed again, and 50 ng/ml biotinylated antimouse antibody (R & D Systems) was added for 1 hour at 37°C. The plates were then washed and incubated with streptavidin-HRP for 1 hour at 37°C. After washing, 0.2 mM ABTS (Sigma Chemical Co.) was added to the wells, and after 10 minutes, the colorimetric reaction was measured at 405 nm with an ELISA reader VERSAmax (Molecular Devices, Sunnyvale, CA).
Preparation of nuclear extracts and the electrophoretic mobility shift assay (EMSA)
BCR/ABL+ MSCs or cells pre-treated as indicted were harvested, and then nuclear extracts were prepared as described previously. Oligonucleotides corresponding to the downstream NF-kB binding sequences 5’-AGT TGA GGG GAC TTT CCC AGG C-3’ were synthesized, annealed and end-labeled with [g-32P] ATP using T4 poly-nucleotide kinase, and EMSA was performed as described previously [22].
Akt kinase assay
Akt kinase assay was performed as described previously with some modifications [22]. MSC were harvested at specific times after treatment with reagents and lysed in lysis buffer. After equalizing the protein to 1.0 mg/200 μl in a 1.5-ml Eppendorf tube, 10 μl Akt antibody was added, and the samples were rotated overnight at 4°C. Then 20 μl protein A/G agarose bead slurry was added, and the samples were rotated at room temperature for 2 hours. The beads were washed twice with lysis buffer and kinase buffer, and the samples were suspended in 50 μl kinase buffer supplemented with 1 μl 10 mM ATP and 1 μg GSK-3 fusion protein and incubated at 30°C for 30 minutes. The phosphorylation of GSK-3 fusion protein was analyzed by Western blot with phosphor-GSK-3α/β (Ser2119) antibodies.
NF-κB p65 activation assay
MSC were harvested at specific times after treatment with reagents, and the nuclear extracts and the activation assay of p65 were performed according to the instructions of the manufacturer of the TransAMTM/NF-κB kit. Briefly, 2 μg/well nuclear extract protein was added to a microtiter plate coated with a specific oligonucleotide of p65. The coated plate was then incubated for 1 hour at room temperature with mild agitation, after which a primary antibody recognizing the p65 was added, and the plate was incubated for an additional 1 hour at room temperature. Anti-IgG horseradish peroxidase-conjugated secondary antibody was then added, and the plate was incubated for 1 hour at room temperature. At the end of the hour, the developing and stop solution were added, and an optical density of 450 nm (OD450) was read on a Wallac Victor 1420 multilabel counter (Perkin Elmer Life Sciences, Shelton, CT). Three duplicates were done for each sample.
Statistical analysis
Results are expressed as mean ± standard deviation. Data were analyzed using the unpaired two-tailed student’s t test and the log rank test. P values of <0.05 were considered significant.
Results
BCR/ABL+ MSCs promoted recruitment of HSCs through s-KitL and s-ICAM-1 upregulated by MMP-9
Co-culture of BCR/ABL+ MSCs and normal MSCs with HSCs showed that the number of GM-CFU (colony forming unit-common precursor of granulocyte and monocyte) in BCR/ABL+ MSCs with HSCs group was lesser than those in healthy donors with HSCs group and lost the support at the 3rd week comparing with the control group, and in the longer period of co-culture, BCR/ABL+ MSCs group showed more clusters and less aggregates (Figure 1A). Further studies showed that MMP-9, s-KitL and s-ICAM-1 in BCR/ABL+ MSCs were higher than those in healthy donor MSCs evidenced by Western and PCR (Figure 1B). The results indicated that BCR/ABL+ MSCs secreted abnormally higher MMP-9, s-KitL and s-ICAM-1 comparing with the control group. To investigate whether the s-KitL and s-ICAM-1 were MMP-9 dependent, we used transfection of a double stranded RNA that targeted the MMP-9 mRNA into bMSC to deplete the corresponding mRNA and protein and this extinction did result in downregulation of s-KitL and s-ICAM-1 illustrated by Western blot, PCR and ELISA (Figure 1C, 1D).
Figure 1.

MMP-9 mediated release of KitL enhanced HSCs proliferation (A) Co-culture of BCR/ABL+ MSCs and healthy MSCs with HSCs repectively showed the number of GM-CFC (colony forming unit-common precursor of granulocyte and monocyte) in BCR/ABL+ MSCs with HSCs group formed more clusters and aggregates than the control one. (B) Supernatants from the co-culture were also collected and MMP-9 was analyzed by ELISA. Columns represent cytokine concentration mean ± SD of 3 different experiments, *P<0.05. (C) MSCs from BCR/ABL+ CML and healthy donors expressed different levels of MMP-9 and KitL assessed by Western blot and RT-PCR. The results were representative of at least 3 independent experiments. (D) Western blot and RT-PCR assayed the level of KitL of CML MSCs, CML-MMP-9 (i) MSCs and healthy donor MSCs respectively. Columns represent cytokine concentration mean ± SD of 3 different experiments, *P<0.05.
Abl kinase upregulated TGF-β1 in CML
To investigate the mechanism involved in MMP-9 synthesis, we first assayed the expression of TGF-β in CML and healthy donors from mRNA and protein level respectively. As TGF-β has 3 isotype, we examined the expression of them to ensure if there existed any differences and the results indicated that TGF-β1 was apparently higher than TGF-β2 and TGF-β3 as shown by ELISA, PCR and Western blot (Figure 2A, 2B) which inspired us to do further research on TGF-β1. We then examined the relationship between CML specific BCR/ABL oncogene and TGF-β1. Intimab, inhibitor of BCR/ABL oncogene, blocked the induction of TGF-β1 in a dose dependent manner (Figure 2C). Furthermore, the effect of various doses of TGFβ1 on MMP-9 production by CML MSCs was assessed to correlate the MMP-9 levels with the induction of signaling components leading to the synthesis of the enzyme (Figure 2D). Results from ELISA (Figure 2E) showed TGF-β1 upregulated MMP-9 production both in CML and healthy donors. Those results demonstrated that BCR/ABL oncogene and TGF-β1 were involved in the regulation of MMP-9 production.
Figure 2.
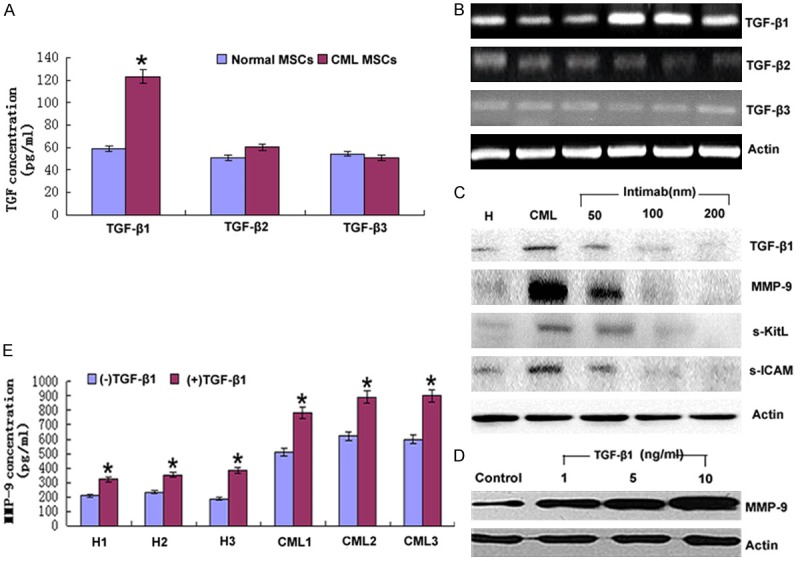
BCR/ABL tyrosine kinase induced TGF-β1 upregulated MMP-9 in CML. (A) The supernatants from BCR/ABL+ MSC and healthy donors MSCs were collected to do ELISA to examine the expression of the 3 isotype of TGF-β and the results were representative of at least 3 independent experiments. *P<0.05. (B) RT-PCR analyzed the mRNA level of 3 isotype of TGF-β in BCR/ABL+ MSCs and healthy donors. The results were representative of at least 3 independent experiments. (C) BCR/ABL+ MSCs were treated with the indicated amount of Intimab for 2 hours, an inhibitor of BCR/ABL oncogene and then cell lysates were analyzed by Western blot with GAPDH as a measure of equal sample-loading amount. (D) BCR/ABL+ MSCs were treated with the indicated amount of TGF-β1 and then cell lysates were analyzed by Western blot with GAPDH as a measure of equal sample-loading amount and (E) Supernatants were also collected and MMP-9 was analyzed by ELISA. Columns represent cytokine concentration mean ± SD of 3 different experiments, * P<0.05.
TGF-β1 upregulated MMP-9 production via PI3K/Akt/NF-κB
To determine the involvement of PI3K signaling pathway in BCR/ABL+ MSCs, we initially examined the effect of PI3K activity on the induction of MMP-9 by TGF-β1. The addition of Wortmanin, inhibitor of PI3K, SH-5, inhibitor of Akt and MG-132, inhibitor of NF-κB, suppressed the MMP-9 synthesis induced by TGF-β1 respectively (Figure 3A). We then analyzed nuclear extraction by EMSA for DNA binding activity of NF-κB. As shown by Figure 3B, lane 1, 2 and 3 represented the results of nuclear extraction of BCR/ABL+ MSCs, BCR/ABL+ MSCs with 5 ng/ml TGF-β1 pre-culturing for 2 hours and healthy donors respectively and the results indicated that NF-κB did involved in TGF-β1 induced MMP-9 production. We at the same time analyzed total cytoplasmic NF-κBp65 and phosphoralated NF-κBp65 in the presence or absence of TGF-β1 (5 ng/ml) treatment using Western blot and ELISA (Figure 3C) and the results showed that TGF-β1 upregulated phosphoralated NF-κBp65 while did little effects on total NF-κBp65 production.
Figure 3.
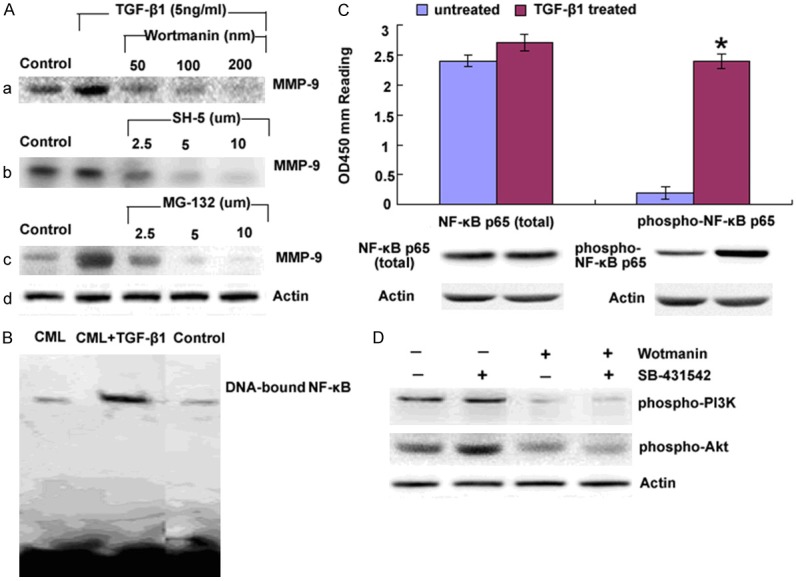
TGF-β1 upregulated MMP-9 production via PI3K/Akt/NF-κB. A. BCR/ABL+ MSCs were treated with (a) Wortmanin (b) SH-5 and (c) MG-132 for 1 hour prior to the addition of TGF-β1 as the indicated amounts. Culture supernatants were harvested after 48 hours and analyzed for MMP-9 by Western blot, Me2SO was also added to one of the cultures as a control. B. Cell nuclear extracts were prepared and the DNA bounding ability of NF-κBp65 was examined by EMSA. C. Nuclear and cytoplasmic protein were collected respectively to assay the total and phosphorylated NF-κBp65 by Western blot and ELISA (PathScan phospho-NF-κBp65 Sandwich ELISA kit) in the presence or absence of TGF-β1 treatment. OD450 reading was shown in the top figure and the corresponded Western blot using phospho-NF-κBp65 (Ser536) rabbit mAb (right panel) or NF-κBp65 antibody (left panel) was shown in the bottom figure. Columns represent cytokine concentration mean ± SD of 3 different experiments, *P<0.05. D. Protein from BCR/ABL+ MSC treated with Wortmanin, an inhibitor of PI3K and SB-431542, an inhibitor of Smad respectively and phosphorylated PI3Kand Akt were examined by Western blot.
Discussion
Chronic myeloid leukemia is a clonal hematopoietic stem cell disorder characterized by the t (9;22) chromosome translocation and resultant production of the constitutively activated BCR/ABL tyrosine kinase [23]. Although Interferon-α, Intimab(a BCR/ABL tyrosine kinase inhibitor) and stem cell transplantations are the standard therapeutic options, transplant-related morbidity from graft-versus-host disease and mortality rates of 10% to 20% have greatly reduced the allogeneic hematopoietic cell transplantation in clinics [24], while interferon-α is only effective in some patients to some degree and chemotherapeutic intervention does not result in prolonged overall survival [25,26] and the reason is possibly due to some unknown biology of the CML cancer stem cells [27].
Our laboratory has identified the Flk-1+CD34-CD31- MSCs in CML as the cancer stem cells, based on this concept, we first examined the biological characteristics of them. FISH analysis of the Flk-1+CD34-CD31- MSCs derived from CML bone marrow showed that they were BCR/ABL+ which indicated that this oncogene had already mutated at the very stem cell level. More importantly, as normal MSCs have the ability to inhibit T lymphocytes at the stage of G0/G1, while our research showed that the ability of CML-derived MSCs on T lymphocytes activity inhibition became attenuated. This indicated on one hand, the immune regulatory ability of CML MSCs became decreased, on the other hand, as the basis of immunological cells to destroy the tumor cells was to recognize the cell surface molecules and the abnormalities of these molecules would lead to false recognization and cleanse of malignant cells which was called immune evasion, so, in CML, there must exist molecules deformities. Meanwhile, co-culture assay indicated CML-derived MSCs existed adhesion deficiency, as HSCs, and other stem cells including cardiac (Orlic et al., 2001), endothelial (Peichev et al., 2000), and epithelial cells express c-Kit, the receptor for KitL, suggesting a common signaling cascade may govern their proliferation and recruitment. Besides, a lot of blood disease such as ALL, CLL and MDS are found to express high level of s-ICAM-1 and clinical researches indicated the level of it correlated directly with disease stage, prognosis and survival period [28-30]. All these inspired us to further examine the s-KitL and s-ICAM-1, and both of them were higher in CML than normal donor. So, we believed that enhanced expression of s-ICAM-1 facilitated on the one hand, tumor cells immune evasion by blocking the ICAM/LFA connection and made them fail to be recognized by T lymphocyte and NK cells; on the other hand, degraded the adhesion of c-kit+ HSCs with bone marrow matrix and destroy the protective screen between hematopoietic region (endothelial cells and matrix) and peripheral circulation, while at the same time, increased s-KitL promoted c-kit+ HSCs in CML recruitment and mobilization, resulting in extramedullary leukemia cells infiltration. Besides, it further confirmed our belief that purified true leukemia stem cells could provide a target for immune-based therapies and biologic response modifiers [31].
MMP-9 is important for maintaining normal hematopoietic process, abnormally higher MMP-9 will degrade extra-cellular matrix and change the cytokines and molecules that have bearing with HSCs adhesion and motivation. In recent years, MMP-9 is found to do effect on cells recruitment and differentiation [32-34]. In the present investigation, we evidenced that MMP-9 had already expressed at the cancer stem cell level which was the first time as other documents concerned mainly about the leukemia cells. We also found that the s-KitL and s-ICAM-1 was MMP-9 dependent proved by RNA interference in Flk1+ CML MSCs.
So, to further investigate the mechanism behind MMP-9 production in BCR/ABL+ CML, we examined the expression of the 3 isotypes of TGFβ in CML, TGF-β1 was abnormally higher in BCR/ABL+ CML comparing with that in healthy donors. Thereby, in the next research, we focused on the relationship between BCR/ABL oncogene and TGF-β1. We found that Intimab, inhibitor of BCR/ABL oncogene, blocked the induction of TGF-β1 in a dose dependent manner. Further evidence shows that TGF-β1 upregulated MMP-9 also in a dose dependent manner. All these data indicated that BCR/ABL oncogene induced TGF-β1 which upregulated MMP-9 production in CML. While, one intricate aspect about TGF-β1 is that although Smad is regarded as the classic regulating pathway, more and more evidence shows that some distinct responses are also involved in its regulation which can be classified as Smad independent pathway, such as Ras [35], Rho proteins [36], extracellular signaling kinase [37] and p38 [38]. Recently, phosphatidylinositol 3-kinase (PI3K) is found to be involved in TGF-β1 signaling positively or negatively at least in some cell types as both stimulation and inhibition effects have been described [39-41]. To investigate whether TGF-β1 was Smad dependent or independent in BCR/ABL+ MSCs, we used Smad and PI3K inhibitor respectively as indicated and the results showed that it was directed by PI3K instead of Smad in CML. Actually, TGF-β and PI3K are involved in many cell lines [42-45].
It was reported that PI3K was activated by tyrosine kinase [46-48] and our results further verified that in CML, it was activated by tyrosine kinase induced TGF-β1. Following its recruitment to these receptors in the plasma membrane, PI3K is activated and phosphorylates to generate the second messenger PIP3, whose levels are tightly regulated by the action of phosphatases. PIP3 does not activate Akt directly but instead, appears to recruit Akt to the plasma membrane and to alter its conformation to allow subsequent phosphorylation by the phosphoinositide-dependent kinase [49,50]. When the capability for PI3K to produce PIP3 was blocked in BCR/ABL+ MSCs by wortmannin, MMP-9 production was inhibited which indicated that the PI3K pathway had a central role in regulation of CML MMP-9 synthesis.
Furthermore, when SH-5 blocked Akt phosphorylation specifically, MMP-9 production was also inhibited. These data showed that PIP3 mediated MMP-9 production in BCR/ABL+ MSC, primarily through Akt and not other downstream components of PI3K containing a PH domain. MG-132 inhibition of MMP-9 production indicated that NFκB was also involved in this pathway which coordinated with the reports of other cell lines [51].
Three members of the IKK family have been isolated, and these are now referred to as IKK1/IKKα, IKK2/IKKβ and IKK3/IKKγ [52]. Expression and the ratio of IKKα to IKKβ vary among cell types. NFκB, in the cells with a high proportion of IKKα to IKKβ, is sensitive to Akt activity [53]. Our data showed that IKKα was predominant in human BCR/ABL+ MSCs, which was apparently different from that in healthy donors where the 3 subunits presented no big differences. Based on this data, we hypothesized that Akt might bound to IKKα constitutively and their phosphorylation might occur following with TGF-β1 stimulation. Immnoprecipitation confirmed our idea which showed that the phosphorylation of Akt was regulated by PI3K and Akt subsequently phosphorylated IKKα which regulated the activation of NF-κB.
Finally, the IκB proteins are degraded rapidly by the proteasome complex once ubiquitinated, thereby freeing NF-κB, which then enters the nucleus, binds to DNA, and activates transcription [54]. It had been shown that Akt regulated NF-κB activation directly through the activation of IKKα and our findings from 2 aspects, EMSA and Western blot, demonstrated that the PI3K/Akt pathway also regulated IκB degradation and activation of NF-κB in BCR/ABL+ MSC stimulated by TGF-β1.
Generally speaking, as what is schematically represented in Figure 4, we demonstrated a decisive checkpoint for MMP-9 to rapidly release the stem cell-active cytokines s-KitL and s-ICAM-1, thereby directing tumor cell immune evasion and mobilization to extramedullary infiltration. What more, the mechanism underlying this niche switch depended on the upregulaion of CML specific BCR/ABL oncogene mediated PI3K/Akt/IKKα/NF-κB signaling pathway. A intriguing aspect of this study is the illustration of stem cell-stromal cell interactions being amplified by membrane-anchored cytokines which are mediated by a specific oncogene induced signaling pathway. Modulation of local cytokines change the HSCs fates provide promising hope for clinical targeting therapy.
Figure 4.
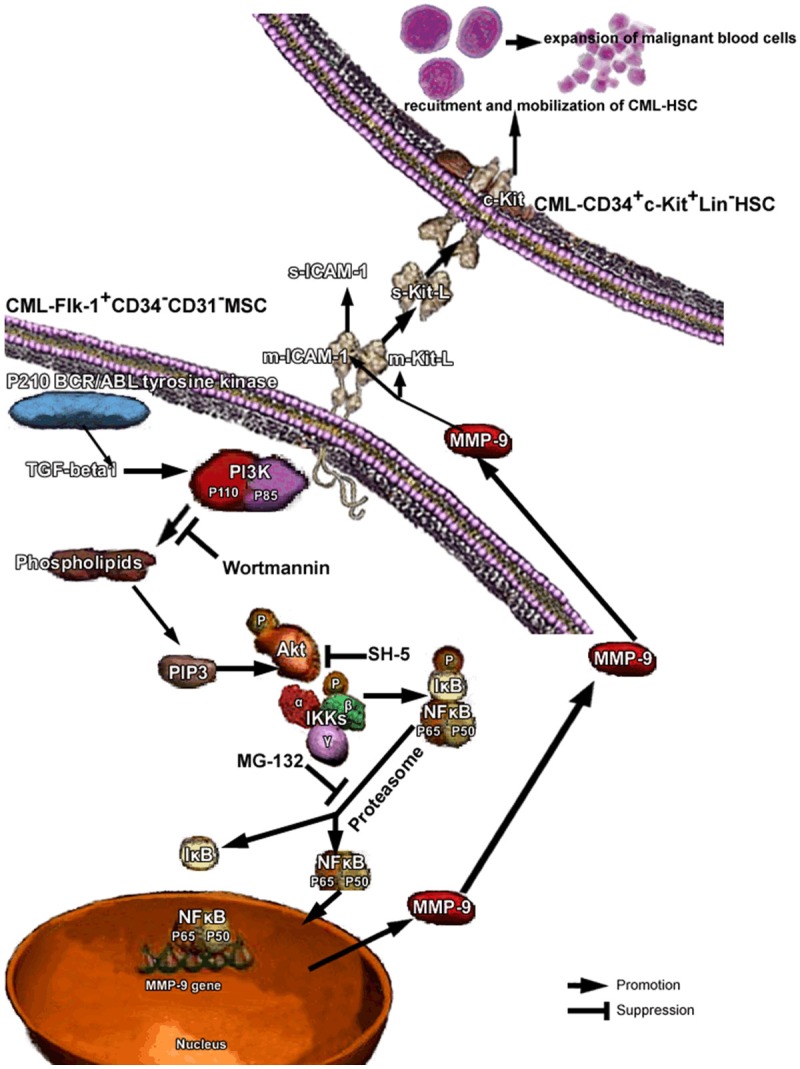
Recruitment of HSCs in CML niche requires TGF-beta1 activated PI3K/Akt/NF-κB/MMP9 signaling pathway. Under steady state conditions quiescent c-kit+ HSCs reside in a niche in close contact with stromal cells. Membrane-bound cytokines, such as mKitL and mICAM-1 not only convey survival signals, but support the adhesion of stem cells to the stroma. CML specific oncogene induced TGF-beta1 activated PI3K/Akt/NF-κB/MMP9 signaling pathway resulting in the release of s-KitL and s-ICAM-1, enhanced recruitment and mobilization of tumor stem cells to the peripheral circulation.
Disclosure of conflict of interest
None.
References
- 1.Barnes DJ, Melo JV. Primitive, quiescent and difficult to kill: the role of non-proliferating stem cells in chronic myeloid leukemia. Cell Cycle. 2006;5:2862–2866. doi: 10.4161/cc.5.24.3573. [DOI] [PubMed] [Google Scholar]
- 2.Jørgensen HG, Allan EK, Jordanides NE, Mountford JC, Holyoake TL. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+CML cells. Blood. 2007;109:4016–4019. doi: 10.1182/blood-2006-11-057521. [DOI] [PubMed] [Google Scholar]
- 3.Jørgensen HG, Copland M, Allan EK, Jiang X, Eaves A, Eaves C, Holyoake TL. Intermittent exposure of primitive quiescent chronic myeloid leukemia cells to granulocyte-colony stimulating factor in vitro promotes their elimination by imatinib mesylate. Clin Cancer Res. 2006;12:626–633. doi: 10.1158/1078-0432.CCR-05-0429. [DOI] [PubMed] [Google Scholar]
- 4.Ries C, Pitsch T, Mentele R, Zahler S, Egea V, Nagase H, Jochum M. Identification of a novel 82 kDa proMMP-9 species associated with the surface of leukaemic cells: (auto-)catalytic activation and resistance to inhibition by TIMP-1. Biochem J. 2007;405:547–58. doi: 10.1042/BJ20070191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 6.Fridman R, Toth M, Chvyrkova I, Meroueh S, Mobashery S. Cell surface association of matrix metalloproteinase-9 (gelatinase B) Cancer Metastasis Rev. 2003;22:153–166. doi: 10.1023/a:1023091214123. [DOI] [PubMed] [Google Scholar]
- 7.Stefanidakis M, Koivunen E. Cell-surface association between matrix metalloproteinases and integrins: role of the complexes in leukocyte migration and cancer progression. Blood. 2006;108:1441–1450. doi: 10.1182/blood-2006-02-005363. [DOI] [PubMed] [Google Scholar]
- 8.Paupert J, Mansat-De Mas V, Demur C. Cell-surface MMP-9 regulates the invasive capacity of leukemia blast cells with monocytic features. Cell Cycle. 2008;7:1047–53. doi: 10.4161/cc.7.8.5645. [DOI] [PubMed] [Google Scholar]
- 9.Redondo-Muñoz J, José Terol M, García-Marco JA, García-Pardo A. Matrix metalloproteinase-9 is up-regulated by CCL21/CCR7 interaction via extracellular signal-regulated kinase-1/2 signaling and is involved in CCL21-driven B-cell chronic lymphocytic leukemia cell invasion and migration. Blood. 2008;111:383–6. doi: 10.1182/blood-2007-08-107300. [DOI] [PubMed] [Google Scholar]
- 10.Redondo-Muñoz J, Escobar-Díaz E, Samaniego R. MMP-9 in B-cell chronic lymphocytic leukemia is up-regulated by alpha4beta1 integrin or CXCR4 engagement via distinct signaling pathways, localizes to podosomes, and is involved in cell invasion and migration. Blood. 2006;108:3143–51. doi: 10.1182/blood-2006-03-007294. [DOI] [PubMed] [Google Scholar]
- 11.Shishodia S, Sethi G, Konopleva M, Andreeff M, Aggarwal BB. A synthetic triterpenoid, CDDO-Me, inhibits IkappaBalpha kinase and enhances apoptosis induced by TNF and chemotherapeutic agents through down-regulation of expression of nuclear factor kappaB-regulated gene products in human leukemic cells. Clin Cancer Res. 2006;12:1828–38. doi: 10.1158/1078-0432.CCR-05-2044. [DOI] [PubMed] [Google Scholar]
- 12.Janowska-Wieczorek A, Majka M, Marquez-Curtis L, Wertheim JA, Turner AR, Ratajczak MZ. Bcr-abl-positive cells secrete angiogenic factors including matrix metalloproteinases and stimulate angiogenesis in vivo in Matrigel implants. Leukemia. 2002;16:1160–6. doi: 10.1038/sj.leu.2402486. [DOI] [PubMed] [Google Scholar]
- 13.Kaneta Y, Kagami Y, Tsunoda T, Ohno R, Nakamura Y, Katagiri T. Genome-wide analysis of gene-expression profiles in chronic myeloid leukemia cells using a cDNA microarray. Int J Oncol. 2003;23:681–91. [PubMed] [Google Scholar]
- 14.Bruchova H, Borovanova T, Klamova H, Brdicka R. Gene expression profiling in chronic myeloid leukemia patients treated with hydroxyurea. Leuk Lymphoma. 2002;43:1289–95. doi: 10.1080/10428190290026358. [DOI] [PubMed] [Google Scholar]
- 15.Ries C, Loher F, Zang C, Ismair MG, Petrides PE. Matrix metalloproteinase production by bone marrow mononuclear cells from normal individuals and patients with acute and chronic myeloid leukemia or myelodysplastic syndromes. Clin Cancer Res. 1999;5:1115–24. [PubMed] [Google Scholar]
- 16.Kim JG, Sohn SK, Kim DH, Baek JH, Lee NY, Suh JS, Chae SC, Lee KS, Lee KB. Clinical implications of angiogenic factors in patients with acute or chronic leukemia: hepatocyte growth factor levels have prognostic impact, especially in patients with acute myeloid leukemia. Leuk Lymphoma. 2005;46:885–91. doi: 10.1080/10428190500054491. [DOI] [PubMed] [Google Scholar]
- 17.Yoon SO, Shin S, Lee HJ, Chun HK, Chung AS. Isoginkgetin inhibits tumor cell invasion by regulating phosphatidylinosito 3 kinase/Akt dependent matrix metalloproteinase-9 expression. Mol Cancer Ther. 2006;5:2666–75. doi: 10.1158/1535-7163.MCT-06-0321. [DOI] [PubMed] [Google Scholar]
- 18.Anand P, Sundaram C, Jhurani S, Kunnumakkara AB, Aggarwal BB. Curcumin and cancer: an “old-age” disease with an “age-old” solution. Cancer Lett. 2008;267:133–64. doi: 10.1016/j.canlet.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 19.Zhang B, Liu R, Shi D, Liu X, Dou X, Zhu X, Lu C, Liang W, Liao L, Zenke M, Zhao RC. Mesenchymal stem cells induce mature dendritic cells into a novel Jagged-2-dependent regulatory dendritic cell production. Blood. 2009;113:46–57. doi: 10.1182/blood-2008-04-154138. [DOI] [PubMed] [Google Scholar]
- 20.Reyes M, Lund T, Lenvik T, Aguiar D, Koodie L, Verfaillie CM. Purification and ex vivo expansion of postnatal human marrow mesodermal progenitor cells. Blood. 2001;98:2615–25. doi: 10.1182/blood.v98.9.2615. [DOI] [PubMed] [Google Scholar]
- 21.Guo H, Fang B, Zhao RC. Hemangioblastic characteristics of fetal bone marrow-derived Flk1(+)CD31(-)CD34(-) cells. Exp Hematol. 2003;31:650–613. doi: 10.1016/s0301-472x(03)00087-0. [DOI] [PubMed] [Google Scholar]
- 22.Lu YB, Wahl LM. Production of matrix metalloproteinase-9 by activated human monocytes involves a phosphatidylinositol-3 kinase/Akt/IKK/NF-κB pathway. J Leuk Bio. 2005;78:259–65. doi: 10.1189/jlb.0904498. [DOI] [PubMed] [Google Scholar]
- 23.Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, Werb Z, Bissell MJ. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–7. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karanes C, Nelson GO, Chitphakdithai P, Agura E, Ballen KK, Bolan CD, Porter DL, Uberti JP, King RJ, Confer DL. Twenty years of unrelated donor hematopoietic cell transplantation for adult recipients facilitated by the National Marrow Donor Program. Biol Blood Marrow Transplant. 2008;14:8–15. doi: 10.1016/j.bbmt.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 25.Martin MG, Dipersio JF, Uy GL. Management of the advanced phases of chronic myelogenous leukemia in the era of tyrosine kinase inhibitors. Leuk Lymphoma. 2009;50:14–23. doi: 10.1080/10428190802517765. [DOI] [PubMed] [Google Scholar]
- 26.Martinelli G, Soverini S, Iacobucci I, Baccarani M. Intermittent targeting as a tool to minimize toxicity of tyrosine kinase inhibitor therapy. Nat Clin Pract Oncol. 2009;6:68–9. doi: 10.1038/ncponc1276. [DOI] [PubMed] [Google Scholar]
- 27.Catriona H, Jamieson Y. Chronic myeloid leukemia stem cell. Hematology Am Soc Hematol Educ Program. 2008;34:436–42. doi: 10.1182/asheducation-2008.1.436. [DOI] [PubMed] [Google Scholar]
- 28.Pelletier SD, Hong DS, Hu Y, Liu Y, Li S. Lack of the adhesion molecules P-selectin and intercellular adhesion molecule-1 accelerate the development of BCR/ABL-induced chronic myeloid leukemia-like myeloproliferative disease in mice. Blood. 2004;104:2163–2171. doi: 10.1182/blood-2003-09-3033. [DOI] [PubMed] [Google Scholar]
- 29.Martin-Henao GA, Quiroga R, Sureda A, González JR, Moreno V, García J. L-selectin expression is low on CD34+ cells from patients with chronic myeloid leukemia and interferon-a up-regulates this expression. Haematologica. 2000;85:139–146. [PubMed] [Google Scholar]
- 30.Wertheim JA, Forsythe K, Druker BJ, Hammer D, Boettiger D, Pear WS. BCR-ABL-induced adhesion defects are tyrosine kinase-independent. Blood. 2002;99:4122–4130. doi: 10.1182/blood.v99.11.4122. [DOI] [PubMed] [Google Scholar]
- 31.Fiore E, Fusco C, Romero P, Stamenkovic I. Matrix metalloproteinase 9 (MMP-/gelatinase B) proteolytically cleaves ICAM-1 and participates in tumor cell resistance to natural killer cell-mediated cytotoxicity. Oncogene. 2002;21:5213–5223. doi: 10.1038/sj.onc.1205684. [DOI] [PubMed] [Google Scholar]
- 32.Darai E, Stefanidakis M, Koivunen E. Cell-surface association between matrix metalloproteinases and integrins: role of the complexes in leukocyte migration and cancer progression. Blood. 2006;108:1441–1450. doi: 10.1182/blood-2006-02-005363. [DOI] [PubMed] [Google Scholar]
- 33.Molica S, Vitelli G, Levato D, Giannarelli D, Vacca A, Cuneo A, Cavazzini F, Squillace R, Mirabelli R, Digiesi G. Increased serum levels of matrix metalloproteinase-9 predict clinical utcome of patients with early B-cell chronic lymphocytic leukemia. Eur J Haematol. 2003;10:373–378. doi: 10.1034/j.1600-0609.2003.00064.x. [DOI] [PubMed] [Google Scholar]
- 34.Kamiguti AS, Lee ES, Till KJ, Harris RJ, Glenn MA, Lin K, Chen HJ, Zuzel M, Cawley JC. The role of matrix metalloproteinase 9 in the pathogenesis of chronic lymphocytic leukaemia. Br J Haematol. 2004;125:128–140. doi: 10.1111/j.1365-2141.2004.04877.x. [DOI] [PubMed] [Google Scholar]
- 35.Yue J, Mulder KM. Transforming growth factor-h signal transduction in epithelial cells. Pharmacol Ther. 2001;91:1–34. doi: 10.1016/s0163-7258(01)00143-7. [DOI] [PubMed] [Google Scholar]
- 36.Mucsi I, Skorecki KL, Goldberg HJ. Extracellular signal-regulated kinase and the small GTP-binding protein, Rac, contribute to the effects of transforming growth factor-h1 on gene expression. J Biol Chem. 1996;271:16567–72. doi: 10.1074/jbc.271.28.16567. [DOI] [PubMed] [Google Scholar]
- 37.Munshi HG, Wu YI, Mukhopadyay S. Differential regulation of membrane type 1-matrix metalloproteinase activity by ERK 1/2 and p38 MAPK-modulated tissue inhibitor of metalloproteinase expression controls transforming growth factor-h1-induced pericellular collagenolysis. J Biol Chem. 2004;279:39042–50. doi: 10.1074/jbc.M404958200. [DOI] [PubMed] [Google Scholar]
- 38.Kim ES, Kim MS, Moon A. TGF-h-induced upregu-lation of MMP-2 and MMP-9 depends on p38 MAPK, but not ERK signaling in MCF10A human breast epithelial cells. Int J Oncol. 2004;25:1375–82. [PubMed] [Google Scholar]
- 39.Horowitz JC, Lee DY, Waghray M. Activation of the pro-survival phosphatidylinositol 3-kinase/AKTpathway by transforming growth factor-h1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J Biol Chem. 2004;279:1359–67. doi: 10.1074/jbc.M306248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor b-medicated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803–10. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- 41.Kim WK, Hwang SY, Oh ES, Piao HZ, Kim KW, Han IO. TGF-beta1 represses activation and resultant death of microglia via inhibition of phosphatidylinositol 3-kinase activity. J Immunol. 2004;172:7015–23. doi: 10.4049/jimmunol.172.11.7015. [DOI] [PubMed] [Google Scholar]
- 42.Rahimi RA, Andrianifahanana M, Wilkes MC, Edens M, Kottom TJ, Blenis J, Leof EB. Distinct roles for mammalian target of rapamycin complexes in the fibroblast response to transforming growth factor-beta. Cancer Res. 2009;69:84–93. doi: 10.1158/0008-5472.CAN-08-2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Assinder SJ, Dong Q, Mangs H, Richardson DR. Pharmacological Targeting of the Integrated AKT, PTEN and TGF-{beta} Pathways in Prostate Cancer. Mol Pharmacol. 2009;75:429–36. doi: 10.1124/mol.108.053066. [DOI] [PubMed] [Google Scholar]
- 44.Xiao YQ, Freire-de-Lima CG, Schiemann WP. Transcriptional and translational regulation of TGF-beta production in response to apoptotic cells. J Immunol. 2008;181:3575–85. doi: 10.4049/jimmunol.181.5.3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kattla JJ, Carew RM, Heljic M. Protein kinase B/Akt activity is involved in renal TGF-beta1-driven epithelial-mesenchymal transition in vitro and in vivo. Am J Physiol Renal Physiol. 2008;295:F215–25. doi: 10.1152/ajprenal.00548.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miraoui H, Oudina K, Petite H, Tanimoto Y, Moriyama K, Marie PJ. Fibroblast growth factor receptor2 promotes osteogenic differentiation in mesenchymal cells via extracellular-related kinase and protein kinase c signalling. J Biol Chem. 2008;284:4897–904. doi: 10.1074/jbc.M805432200. [DOI] [PubMed] [Google Scholar]
- 47.Rexer BN, Engelman JA, Arteaga CL. Overcoming resistance to tyrosine kinase inhibitors: Lessons learned from cancer cells treated with EGFR antagonists. Cell Cycle. 2009;8:18–22. doi: 10.4161/cc.8.1.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Papakonstanti EA, Zwaenepoel O, Bilancio A, Burns E, Nock GE, Houseman B, Shokat K, Ridley AJ, Vanhaesebroeck B. Distinct roles of class IA PI3K isoforms in primary and immortalised macrophages. J Cell Sci. 2008;121:4124–33. doi: 10.1242/jcs.032763. [DOI] [PubMed] [Google Scholar]
- 49.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 50.Williams MR, Arthur JS, Balendran A, van der Kaay J, Poli V, Cohen P, Alessi DR. The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defi ned in embryonic stem cells. Curr Biol. 2000;10:439–448. doi: 10.1016/s0960-9822(00)00441-3. [DOI] [PubMed] [Google Scholar]
- 51.Lu Y, Wahl LM. Production of matrix metalloproteinase-9 by activated human monocytes involves a phosphatidylinositol-3 kinase/Akt/IKK/NF-κB pathway. J Leuk Bio. 2005;78:259–65. doi: 10.1189/jlb.0904498. [DOI] [PubMed] [Google Scholar]
- 52.Neri LM, Borgatti P, Capitani S, Martelli AM. The nuclear phosphoinositide 3-kinase/AKT pathway: a new second messenger system. Biochim Biophys Acta. 2002;1584:73–80. doi: 10.1016/s1388-1981(02)00300-1. [DOI] [PubMed] [Google Scholar]
- 53.Gustin JA, Ozes ON, Akca H, Pincheira R, Mayo LD, Li Q, Guzman JR, Korgaonkar CK, Donner DB. Cell type-specific expression of the IκB kinases determines the significance of phosphati-dylinositol 3-kinase/Akt signaling to NF-κB activation. J Biol Chem. 2004;279:1615–1620. doi: 10.1074/jbc.M306976200. [DOI] [PubMed] [Google Scholar]
- 54.Ben-Neriah Y. Regulatory functions of ubiquitination in the im-mune system. Nat Immunol. 2002;3:20–26. doi: 10.1038/ni0102-20. [DOI] [PubMed] [Google Scholar]