

Abstract
Apoptosis of osteoblasts caused by glucocorticoids has been identified as an important contributor to the development of osteoporosis. Tanshinone IIA (Tan), an active ingredient extracted from the rhizome of the Salvia miltiorrhiza Bunge (Danshen), has been reported to cast positive effects on osteoporosis. However, the precise mechanisms accounting this action remain elusive. In this study, by using osteoblastic MC3T3-E1 cells as a model, we confirmed the protective effects of Tan against dexamethasone (Dex)-induced cell apoptosis and further clarified its molecular mechanism of action. Our results showed that treatment with Dex caused cell injury, increased cytosol cytochrome c level and Nox expression, induced apoptosis in caspase-9-dependent manner, and enhanced reactive oxygen species (ROS) production. Tan attenuated these deleterious consequence triggered by Dex. Moreover, Dex-induced ROS production and cell injury were inhibited by antioxidant, NADPH oxidases inhibitors, Nox4 inhibitor, and Nox4 small interfering RNA (siRNA). Overexpression of Nox4 almost abolished the inhibitory effect of Tan on Dex-induced cell injury and apoptosis. The results also demonstrated significant involvement of Nox4 in the Dex-induced apoptosis. Nox4-derived ROS led to apoptosis through activation of intrinsic mitochondrial pathway. Additionally, we evidenced that Tan reversed Dex-induced apoptosis via inactivation of Nox4. The present findings suggest that inhibition of Nox4 may be a novel therapeutic approach of Tan to prevent against glucocorticoids-induced osteoblasts apoptosis and osteoporosis.
Keywords: Tanshinone IIA, glucocorticoids, osteoporosis, apoptosis, Nox4
Introduction
Under normal condition, bone can be reformed often to maintain bone volume and calcium homeostasis after bone injury [1]. Osteoporosis is a calcium and metabolic disorder associated with decreased bone mineral density, disordered bone architecture and enhanced bone fragility, leading to an increased risk of fracture [2-4]. Osteoblasts and osteoclasts are two kinds of cells, which play important roles in the regulation of bone formation and resorption. Osteoblasts, originated in the bone marrow, can build up the matrix of bone and contribute to bone formation [1]. Indeed, osteoporosis is caused by a failure of bone homeostasis, which is due to the loss of osteoblastic activity and/or increase in osteoclastic activity [5]. Although increasing numbers of investigators have unraveled the many cytokines, signaling pathways and transcription factors may be responsible for the differentiation of osteoblasts [6,7], the underlying mechanisms contributing to osteoporosis are not well understood.
Glucocorticoids (e.g. prednisone, dexamethasone (Dex)) are widely used clinically for their unsurpassed anti-inflammatory and immunomodulatory effects in a variety of disorders including inflammatory, pulmonary, gastrointestinal and autoimmune diseases [1,8]. However, the therapeutic applications of glucocorticoids are greatly limited by substantial adverse side effects, especially osteoporosis, which due to high-dose and long-term exposure of glucocorticoids [9,10]. It is now clear that high-dose and long-term intake of glucocorticoids acts as a major contributor for osteoblasts and osteocyte apoptosis [11,12]. Although the positive correlation between glucocorticoids and osteoporotic status has been well characterized in many studies, the molecular mechanisms remain elusive.
Oxidase stress is caused by an imbalance between the scavenging activities of intracellular antioxidant and the production of highly reactive oxygen species (ROS) [13]. Physiological levels of ROS are helpful to maintain normal cellular function [14], while excessive accumulation of ROS results in cell injury and death [15]. Oxidative stress has been known to exhibit serious deleterious effects on osteoblasts and subsequently contributes to osteoporosis, in which the ROS production is obviously increased [16]. Moreover, clinical studies showed that the decreased bone mineral density was associated with the increased oxidase stress [17]. Maggio et al. also observed a significant reduction of antioxidant level in plasma isolated from the elderly patients with osteoporosis [16]. Mechanically, increasing evidence suggests that oxidative stress may lead to increase osteoclasts activity and promote apoptosis of osteoblasts [18,19]. Therefore, it is widely accepted that oxidase stress is a crucial participant in the pathogenesis of osteoporosis. Over the past decades, some researches have been focused on a variety of natural components isolated from oriental medicinal herbs, such as Salvia miltiorrhiza Bunge (Danshen), for their antioxidant and functional properties. Tanshinone IIA (Tan) is a major effective compound of Danshen, and has been widely used clinically for the prevention and treatment of cardiovascular disorder. Tan has diverse biological effects, including improvement of microcirculation and vasodilation, anti-inflammatory and free radical scavenging [20]. Previously, it was reported that Tan exerted the inhibitory influence on oxidative stress and attenuated the deleterious effects via Wnt/FoxO3a signaling in osteoblasts [21]. Although it is known that the beneficial actions of Tan are in part due to its antioxidant activities, the functional targets and molecular mechanisms of its biological effects in osteoblasts remain elusive.
Therefore, the purpose of this study was to test the hypothesis that Tan antagonizes glucocorticoids-induced apoptosis through the inhibition of ROS production in MC3T3-E1 cells and that the underlying mechanism accounting for this effect. Our study may provide a novel strategy for prevention against glucocorticoids-induced osteoporosis.
Materials and methods
Reagents
Alpha Minimum Essential Medium (α-MEM), dexamethasone (Dex), 2,5-diphenylterazolium bromide (MTT), 4’,6’-diamidino-2-phenylindole (DAPI), N-acetyl-L-cysteine (NAC), mitochondrial electron transport complex inhibitor rotenone, xanthine oxidase inhibitor allopurinol, specific Nox4 inhibitor plumbagin, and 2’,7’-dichlorofluorenscin diacetate (DCFH-DA) were obtained from Sigma-Aldrich (St Louis, MO, USA). Fetal Bovine Serum (FBS), penicillin-streptomycin.
Lipofectamine2000 reagent and all siRNA duplexes were obtained from Invitrogen (Carlsbad, CA, USA). NADPH oxidases inhibitor diphenyleneiodonium (DPI), apocynin, specific caspase-9 inhibitor Z-LEHD-FMK, specific caspase-8 inhibitor Z-IETD-FMK and specific caspase-3 inhibitor Z-VAD-FMK were purchased from Calbiochem (Darmstadt, Germany). Tanshinone IIA (Tan, 99.0% purity assayed by high performance liquid chromatography) was obtained from Yunnan Plant Pharmaceutical Factory (Yunnan, China) and was dissolved in dimethyl sulfoxide (DMSO).
Cell culture and treatment
Murine pre-osteoblasts MC3T3-E1 (CCL-240™) was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA), and maintained in α-MEM supplemented with 10% heatinactivated FBS, 100 U/ml penicillin and 100 U/ml streptomycin at 37°C in 5% CO2 atmosphere. In selected experiments, MC3T3-E1 cells were incubated with various concentrations of Tan with or without Dex treatment for 24 h. In the experiments with caspase inhibitors, cells were pretreated with Z-LEHD-FMK (10 μM), Z-IETD-FMK (20 μM) or Z-VAD-FMK (25 μM) for 30 min before Dex stimulation. To investigate the involvement of ROS source, MC3T3-E1 cells were pretreated with NAC (10 mM), DPI (10 μM), apocynin (100 μM), rotenone (10 μM), allopurinol (100 μM) or plumbagin (10 μM) for 30 min before exposure to Dex.
Nox4 specific small interfering RNA (siRNA) transfection
MC3T3-E1 cells were transiently transfected using Lipofectamine2000 with either Nox4 specific siRNA (sense 5’-GTAGGAGACTGGACAGAAC-3’, antisense 5’-GTTCTGTCCAGTCTCCTAC-3’) or control siRNA (Negative, sense 5’-TTCTCCGAACGTGTCACGT-3’, antisense 5’-ACGTGACAGTTCGGAGAA-3’) according to the manufacturer’s instructions. After transfection for 24 h, the transfected cells were then treated with Dex for another 24 h.
Adenovirus infection
Nox4 plasmid was constructed by inserting the full-length cDNA of Nox4 into the pCMV-Tag2 expression vector between BamHI and XhoI restriction sites. The plasmid was confirmed by DNA sequencing. Nox4 adenovirus (Ad-Nox4) was generated using AdMax adenovirus packaging system (Microbix Biosystems Inc., Ontario, Canada) according to the manufacturer’s instructions. Titers of the purified virus were assayed using by p24 ELISA kit (Cell Biolabs, San Diego, CA, USA). A negative control (Lacz) was obtained from Clotech (Mountain View, CA, USA). To study the role of Nox4 in Tan action, cells were infected with Ad-Nox4 or Laz at 50 multiplicity of infection (MOI) for 24 h before Tan and Dex treatment.
MTT assay
Cell viability was measured by MTT assay as previously described [21]. MC3T3-E1 at a density of 5×103 cells/well were seeded in 96-well plates for overnight and then processed with different treatment as mentioned above. The fresh medium containing 0.5 mg/mL MTT reagent was added, and then incubated at 37°C. After 4 h, the medium and MTT were removed, dimethyl sulfoxide (DMSO) was added to dissolve the formazan crystals, and the absorbance of each well was measured at 570 nm by a plate reader.
TUNEL assay
Cells were fixed with 3:1 methanol: acetic acid solution and then permeabilized using 70% ethanol for 30 min. Cells was washed with phosphate buffer saline (PBS), followed by incubation with (TdT-UDP nick end labeling) TUNEL reaction reagent (DeadEnd fluorometrictunel system, Promega, Madison, WI, USA) according to the manufacturer’s instructions. Cells were then treated with DAPI for nucleus staining and viewed using the Zeiss Axiplan2 fluorescence microscope (München, Germany). The number of TUNEL positive cells (red) and number of DAPI positive cells were counted using ImageJ software (NIH, Bethesda, Maryland, USA). The percentage of positively cells was expressed as (TUNEL positive cells/Total number of cells) ×100.
Western blot analysis
MC3T3-E1 cells were collected by scraping and lysed in lysis buffer (Cell Signaling Technology, Inc., Beverly, MA, USA). The protein content was determined by Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA) according to the manufacturer’s instructions. Equal proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electro-transferred to a PVDF membrane (Millipore, Billerica, MA, USA). The membranes were then probed with primary antibodies to Bcl-2, Bax, PARP (Santa Cruz Biotechnology, Santa Cruz, CA, USA), cytochrome, caspase-9, caspase-8, caspase-3, β-actin (Cell Signaling Technology) and Nox4 (Abcam, Cambridge, UK) overnight, followed by incubation of corresponding secondary antibodies (Beyotime, Jiangsu, China). Bounded antibodies were detected by ECL Western Blotting Substrate (Thermo Fisher Scientific, Waltham, MA, USA) and analyzed by ImageJ software. Data of the each group was expressed as fold change vs. that of control group (labeled as “1.00”).
ROS detection
To measure the amounts of intracellular ROS in MC3T3-E1 cells, DCFH-DA probe was used as recommended by the manufacturer. Cells were incubated with DCFH-DA (5 μM) at 37°C for 30 min in dark. All samples were analyzed with at least three biological replicates, and images from each replicate were taken with a Zeiss Axiplan2 fluorescence microscope. The fluorescence intensity was calculated by ImageJ software.
Apoptosis analysis by flow cytometry
MC3T3-E1 cell apoptosis was detected by the FITC-Annexin V Apoptosis Detection Kit (Beyotime) by flow cytometry according to the manufacturer’s protocol. Briefly, the cells were washed and trypsinized. After centrifugation, cells were harvested and suspended in a binging buffer for Annexin V-FITC and propidium iodide (PI) staining at room temperature in dark for 15 min. The apoptotic cells were counted by flow cytometry (BD Bioscience, Franklin Lakes, NJ, USA) and apoptosis percentage was reflected by Annexin V/PI ratio.
Statistical analysis
In this study, each experiment was conducted at least three times. Data were expressed as mean ± SEM from n independent experiments. Statistical analyses were performed using one-way ANOVA analysis or an unpaired two-tailed Student t test by SPSS16.0 software. (SPSS, Inc., Chicago, IL, USA). Value of P<0.05 were considered significant statistically.
Results
Tan reversed Dex-induced cytotoxicity and apoptosis in osteoblasts
In this study, MC3T3-E1 osteoblastic cell line was used as a cell model to simulate glucocorticoids-induced osteoporosis and examine the protective effects of Tan. Firstly, the effect of Dex on cell viability was evaluated by MMT assay. As shown in Figure 1A, the growth of MC3T3-E1 was significantly inhibited by Dex (0.125-4 μM) in a dose-dependent manner. The maximal inhibition was observed in cells treated with 1 μM Dex. To examine the safety for clinical use of Tan on MC3T3-E1 cells, the cells were exposed to Tan from 0.001 to 1000 μM for 24 h. The results showed that Tan alone had no cytotoxicity toward MC3T3-E1 cells at concentration less than 10 μM, while higher doses (≥100 μM) exhibited slight inhibition on cell growth (Figure 1B). Thus, the concentrations less than 100 μM were used to investigate the protective effects of Tan against Dex-inhibited cell viability. Treatment with Tan dose-dependently blocked the cytotoxic effect of Dex with the IC50 around 1 μM (Figure 1C and 1D). Therefore, in subsequent experiments, Dex at 1 μM concentration and Tan at 1 mM concentration were used, respectively. In agreement with the cell viability assay, the TUNEL assay showed that Tan attenuated Dex-induced apoptotic cell death (Figure 1E). The proportion of apoptotic cells was increased from 9.2±0.4% to 44.6±8.1 after treatment with Dex, while this elevation was significantly inhibited to 14.5±2.0% after exposure to 1 μM Tan (Figure 1F). Collectively, these data demonstrate the protective role of Tan against Dex-induced cytotoxicity and apoptosis in MC3T3-E1 cells.
Figure 1.
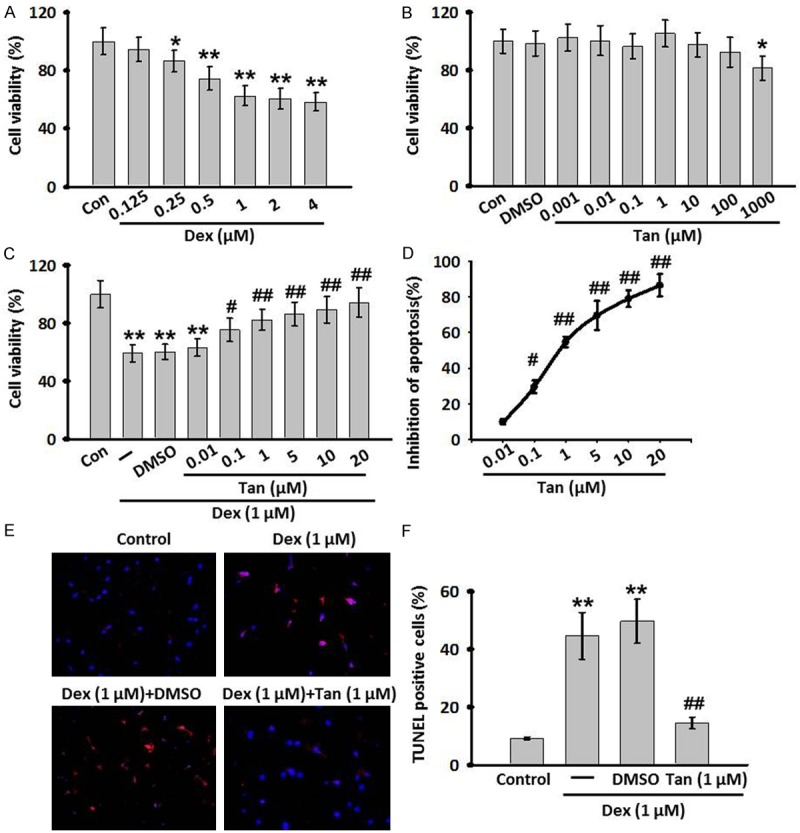
Cell viability response to various concentrations of dexamethasone (Dex) treatment and the effects of Tan omDex-induced cell injury. (A, B) MC3T3-E1 cells were treated with various contractions of Dex (0.125-4 μM) (A) or Tanshinone IIA (Tan, 0.001-1000 μM) (B) for 24 h. Cell viability was determined by MTT assay. (C) The Dex induced decrease in MC3T3-E1 cells viability was improved by Tan at various concentrations. (D) The concentraction-response cruve of anti-apoptotic effect of Tan in MC3T3-E1 cells (IC50=9.646 μM). (E) TdT-mediated dUTP nick-end labeling (TUNEL) (red) and DAPI staining (blue) of MC3T3-E1 cells following co-incubation of Dex and Tan for 24 h. (F) The percentage of TUNEL positive cells was calculated. All data are presented as mean ± SEM. *P<0.05, **P<0.01 vs. control; #P<0.05, ##P<0.01 vs. Dex treatment alone, n=6.
Tan inhibited Dex-induced MC3T3-E1 cells apoptosis through mitochondria-dependent pathway
The apoptotic pathway is mainly regulated by the anti-apoptotic protein Bcl-2 and the pro-apoptotic protein Bax. The balance between anti- and pro-apoptotic proteins appears to determine survival or death of cells. In Figure 2A and 2B, incubation with Dex for 24 h significantly decreased Bcl-2 expression, whereas enhanced Bax expression. However, these alternations induced by Dex were almost reversed after Tan treatment. Moreover, Dex significantly increased cytosol cytochrome c levels, indicating that Dex induces the release of cytochrome c from mitochondria to cytosol in MC3T3-E1 cells. However, this elevation was alleviated after Tan treatment. We next examined the cleavage of caspase-9/-8/-3 and PARP following co-incubation of Dex and Tan. Western blot analysis revealed that Dex resulted in activation of caspase-9/-3 and subsequent cleavage of PARP, which was effectively blocked by incubation of Tan. Of note, Dex produced no significant effect on the cleavage of caspase-8, indicating capase-8 may not be the important initiator caspase in Dex-induced apoptosis in MC3T3-E1 cells. To further confirm these phenomena in MC3T3-E1 cells, we used specific caspase inhibitors and measured their effects on cell viability by MTT assay. Inhibitor experiments showed that caspase-9 inhibitor Z-LEHD-FMK and caspase-3 inhibitor Z-VAD-FMK remarkably inhibited the decrease of cell viability induced by Dex, but caspase-8 inhibitor Z-IETD-FMK produced no effects (Figure 2C). Together, these results suggest that intrinsic mitochondrial pathway may be the major mechanism responsible for the apoptotic effects of Dex in MC3T3-E1 cells.
Figure 2.
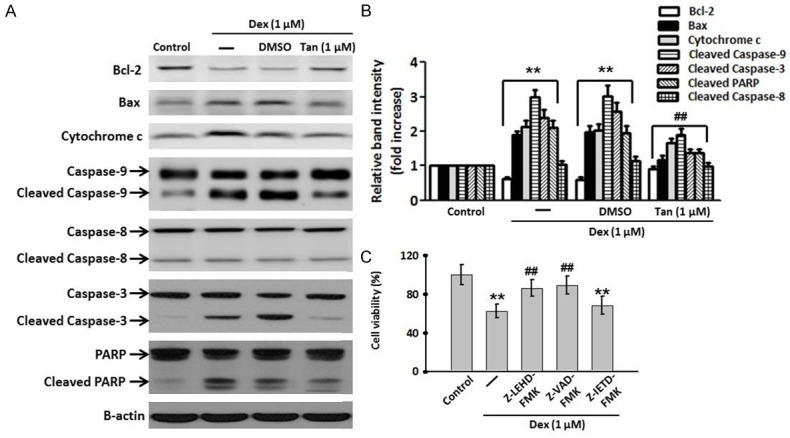
Tan attenuated Dex-induced apoptosis through the inhibition of the intrinsic pathway in MC3T3-E1 cells. A. The cells were co-incubated with Dex and Tan for 24 h. The Bcl-2 and Bax expression, cytosol cytochrome c level, the active forms of caspase-9/8/3, and the cleavage product of PARP were determined by western blot analysis. Representative results of four independent experiments are shown. B. Densitometric analysis of apoptosis-related protein expression was performed. C. MC3T3-E1 cells were pretreated with caspase-9 inhibitor Z-LEHD-FMK (10 μM), caspase-3 inhibitor Z-IETD-FMK (20 μM) or caspase-8 inhibitor Z-VAD-FMK (25 μM) for 30 min before Dex stimulation. Cell viability was assessed by MMT assay. Data were presented asmean ± SEM. **P<0.01 vs. control; ##P<0.01 vs. Dex treatment alone, n=4.
Dex-induced ROS production is Nox4 dependent
We next used the ROS-sensitive fluorescent probe, DCFH-DA, to examine the effects of Dex on ROS production in MC3T3-E1 cells. As compared with the control group, treatment of MC3T3-E1 cells with Dex resulted in an obvious increase in the production of ROS. This elevation was significantly inhibited after pretreatment with the antioxidant N-acetyl-L-cysteine (NAC, 10 μmol/L). NADPH oxidase, mitochondrial electron transport chain and xanthine oxidase are the main sources for the production of intracellular ROS [22]. Hence, various inhibitors against these sources were used to investigate their role in Dex-induced ROS production. Pretreatment with specific inhibitors against NADPH significantly suppressed Dex-induced the increases of ROS production, but the inhibitors against mitochondria and xanthine oxidase had no effects on the increased production of ROS, suggesting the source of ROS production by Dex may be ascribed to a NADPH origin. Interestingly, NADPH oxidase 4 (Nox4) inhibitor plumbagin remarkably ablated the stimulatory effects of Dex on ROS production (Figure 3A and 3B). To further validate the key role of Nox4 in Dex-induced ROS production, we used siRNA to knockdown Nox4 expression. The expression of Nox4 in MC3T3-E1 treated with Nox4 specific inhibitor or siRNA was, as expected, significantly decreased (Figure 3C). As showed in Figure 3D and 3E, down-regulation of Nox4 prevented Dex-induced increase in ROS production. These data indicate that Nox4-derived ROS production may be correlated withDex-induced apoptosis in MC3T3-E1 cells.
Figure 3.

Nox4 was involved in Dex-induced ROS production. (A) MC3T3-E1 cells were treated with Dex for 24 h with or without a 30-min pre-treatment with the following inhibitors: NAC (10 mM), DPI (10 μM), apocynin (100 μM), rotenone (10 μM), allopurinol (100 μM) or plumbagin (10 μM). Representative images for cells loaded with DCFH-DA (5 μmol/L) was captured with a fluorescence microscopy. (B) Quantitative analysis of DCF fluorescence intensity in (A). (C) The cells were transfected with Nox4 siRNA for different concentrations (5, 10, 20, 80 nM) or negative siRNA (Negative) for 24 h. The efficiencies of siRNA were detected by western blot. (D) MC3T3-E1 cells were transiently transfected with Nox4 siRNA (20 nM) for 24 h, and then treated with Dex for another 24 h. Intracellular ROS was detected by DCFH-DA fluorescent dye. (E) Quantitative analysis of DCF fluorescence intensity in (D). Data were presented asmean ± SEM. **P<0.01 vs. control; ##P<0.01 vs. Dex treatment alone, n=4.
Inhibition of Nox4 attenuates Dex-induced decrease of cell viability
To investigate whether the Dex-induced increase in ROS production causally contributes to MC3T3-E1 cells apoptosis, cells were pre-treated with different antioxidants following Dex treatment, and MTT assay was performed. In agreement with the above results, MTT assay showed that NAPDH oxidase inhibitors, DPI or apocynin, could reverse the inhibition of Dex on the growth of MC3T3-E1 cells, while the inhibitors against mitochondria and xanthine oxidase produced no effects (Figure 4A). To further explore the role of Nox4 in Dex-induced cell apoptosis, we treated MC3T3-E1 cells with Nox4 specific inhibitor or siRNA. As showed in Figure 4A and 4B, Dex-induced decrease in cell viability was obviously suppressed after Nox4 inhibition, suggesting that the reduction in Nox4-derived ROS production can translate into an inhibition in Dex-induced MC3T3-E1 cells apoptosis.
Figure 4.

Nox4-derived ROS was involved in Dex-induced cell injury in MC3T3-E1 cells. A. The cells were pre-treated with NAC (10 mM), DPI (10 μM), apocynin (100 μM), rotenone (10 μM), allopurinol (100 μM) or plumbagin (10 μM) for 30 min and then treated with Dex for another 24 h. Cell viability was examined by MTT assay. B. MC3T3-E1 cells were transfected with Nox4 siRNA for 24 h before exposure to Dex for another 24 h. MTT assay was performed to determine the effect of Nox4 siRNA on Dex-induced cell injury. All data are presented as mean ± SEM. **P<0.01 vs. control; ##P<0.01 vs. Dex treatment alone, n=4.
Tan suppresses Dex-induced cell apoptosis through inhibiting Nox4 expression
Based on the critical role of Nox4 in Dex-induced apoptosis in MC3T3-E1 cells, we speculated that Tan inhibited cell apoptosis through the regulation of Nox4 expression. To verify this assumption, cells were treated with Dex for 24 h in the presence or absence of Tan, and then the expression of Nox4 was measured. Western blot showed that Dex treatment caused a significant increase in Nox4 protein expression. However, this induction was almost abolished after Tan treatment (Figure 5A). To further explore whether the inhibition of Tan on Dex-induced apoptosis results from its capacity to decrease Nox4 expression, we used Nox4 adenovirus (Ad-Nox4) to overexpress Nox4 expression in MC3T3-E1 cells. Western blot analysis of overexpression efficiency showed that Ad-NOx4 effectively increased the expression of Nox4. As compared with control group, at 25, 50, and 100 MOI, the Nox4 expression was elevated by 1.56±0.21-fold, 2.32±0.33-fold, and 2.24±0.27-fold, respectively (Figure 5B). The Nox4 expression reached the maximal expression level at 50 MOI, and 50 MOI of Ad-Nox4 was then used in the following experiments. Following overexpression of Nox4, the inhibitory effect of Tan on Dex-decreased cell viability was abrogated (Figure 5C). Moreover, MC3T3-E1 cells apoptosis was analyzed by Annexin V-FITC/PI flow cytometry quantitatively (Figure 5D). Dex significantly increased the early and late apoptotic rate to 7.3 and 14.2%, respectively, which could be remarkably decreased by Tan. In line with results of MTT assay, the effect of Tan on apoptosis was dramatically reversed after Nox4 overexpression (Figure 5E), demonstrating that the suppression of Nox4 expression may underlies, at least in part, the protective role of Tan against Dex-induced cell apoptosis.
Figure 5.

Tan attenuated Dex-induced cell apoptosis through inhibiting Nox4 expression. A. MC3T3-E1 cells were co-incubated with Dex and Tan for 24 h. The expression of Nox4 was determined by western blot. B. The cells were infected with Ad-Nox4 for different MOI (25, 50, 100 MOI) or Laczfor 24 h. The expression of Nox4 was detected by western blot. C. Nox4 adenovirus (Ad-Nox4, 50 MOI) was transfected for 24 h in prior to the co-incubation of MC3T3-E1 cells with Dex and Tan. Cell viability was tested by MTT assay. D. Cell apoptosis was determined by Annexin V/PI staining. E. Quantitative analysis of the percentage of apoptotic cells. Data were presented asmean ± SEM. **P<0.01 vs. control; ##P<0.01 vs. Dex; &&P<0.01 vs. Tan + Dex, n=4.
Discussion
Osteoporotic fracture, one of the most aggressive bone diseases, is the leading cause of morbidity and mortality, particularly in elderly women and men [23]. Accumulating evidence has indicated that glucocorticoids-induced osteoblasts apoptosis is proposed as an important cause for osteoporosis. For example, clinical observation showed that over 30% patients who chronically exposed to high-dose glucocorticoids could develop osteoporosis, and this was mainly resulted from the increased osteoblasts apoptosis [11,12]. MC3T3-E1 cells, an osteoblastic precursor cell line, are the most-used cell type in the study of osteoblast apoptosis and differentiation [13]. Therefore, in the present study, we used MC3T3-E1 cells to investigate the effect of Dex on osteoblasts apoptosis and the related underlying mechanisms. Indeed, our findings showed that Dex could inhibit cell viability dose-dependently and induce apoptosis in MC3T3-E1 cells. 1 μM Dex caused the maximal inhibition on the growth of MC3T3-E1 cells. The final concentration of Dex at 1 μM is consistently reported to significantly decrease cell viability in MC3T3-E1 cells in many previous studies [1,24,25].
It has been known that apoptotic process is usual associated with the abnormal expression of Bcl-2 family, which consists of the anti-apoptotic protein Bcl-2 and the pro-apoptotic protein Bax [26]. An increase in Bax expression results in a lower concentration of Bcl-2, driving the cells towards apoptosis [27]. On other hand, this imbalance between Bax and Bcl-2 increases the mitochondrial membrane permeability, leading to the release of cytochrome c from mitochondria to cytosol and the activation of caspase pathway [28]. There are two major apoptotic pathways: the extrinsic death receptor-mediate pathway and the intrinsic pathway. Each apoptotic pathway activates its own initiator caspase (caspase-9 or caspase-8), which in turn promotes the cleavages of caspase-3 and PARP [29]. In the present study, we demonstrated that Dex resulted in a decrease in Bcl-2, with an increase in Bax, concomitantly with the release of cytochrome c, and then activated caspase-9/3 and PARP. It is worthy to note that Dex produced no effects on caspase-8 activation, indicating Dex activates the intrinsic mitochondrial pathway rather than the extrinsic pathway. Furthermore, caspase-9 and caspase-3 inhibitors but not caspase-8 inhibitor, remarkably reversed the inhibition of cell viability induced by Dex, demonstrating that Dex induces MC3T3-E1 cells apoptosis mainly through intrinsic apoptotic pathway.
Nowadays, increasing numbers of investigators have unraveled the action mechanisms of glucocorticoids-induced apoptosis in osteoblasts. For instance, activation of Bim, GSK 3β and p38 might account for apoptosis-inducing effects of glucocorticoids in osteoblasts [7,30]. It also reported that glucocorticoids induced apoptosis in osteoblasts through the regulation of 11b-hydroxysteroid dehydrogenase type 2, leading to reduce the bone formation [31]. On the other hand, Rauch A et al showed that suppression of cytokines such as interleukin 11 via interaction with monomeric GR and AP-1 contributed to the pro-apoptotic effects of Dex on osteoblasts [6]. Additionally, studies have found that, high reactive oxygen species (ROS) play a critical role in the development and progression of osteoporosis [21,25]. Consistently, in the present study, we evidenced that Dex significantly induced ROS production in osteoblasts. Furthermore, in the experiment with antioxidant NAC, Dex-induced ROS production and cell injury were obviously suppressed, further demonstrating that oxidase stress exerts an important functional role in Dex-induced apoptosis. However, the intracellular sources of ROS involved in this process are still unknown. It has been well documented that NADPH oxidase, mitochondrial electron transport chain and xanthine oxidase are the main sources of intracellular ROS [22,29]. In the experiments with various inhibitors against these sources, we found that the inhibition of NADPH oxidase with DPI or apocynin almost completely suppressed intracellular ROS production and apoptotic cell death in Dex-treated MC3T3-E1 cells. Notably, inhibition of mitochondrial electron transport chain and xanthine oxidase with specific inhibitors had no effects on ROS production and apoptosis, suggesting that NADPH oxidase is responsible for the increased ROS production and apoptosis following Dex stimulation.
NAPDH oxidases (Noxs) the major source of ROS, have been suggested to be activated in osteoblasts by various stimuli such as indoxyl sulfate, high-cholesterol, oxidation protein products, diet and ethanol [32-35]. To date, five Nox isoforms (Nox1, Nox2, Nox3, Nox4 and Nox5) have been identified. Notably, previous study reported that Nox1, Nox2, and Nox4, but not Nox3, Nox5, were found expressed abundantly in osteoblasts [36]. Here, we evidenced that Nox4 expression was also up-regulated by Dex stimulation. Furthermore, either inhibition of Nox4 with Plumbagin or knockdown of Nox4 with siRNA markedly suppressed the ROS production and cell injury induced by Dex, indicating the important involvement of Nox4 in the Dex-induced apoptosis in osteoblasts. Cautions should be noted that we examined the protein expression of Nox4 only, but did not determine the changes of other Noxs expressions. This is the limitation of the current study, and more experiments should be performed to further determine whether Nox4 was a specific target of Dex.
Antioxidants, such as medicinal agents, natural phytochemicals and healthy food, have been shown to play a beneficial role in oxidative stress-related diseases. Tan is one of the main active components in Danshen and widely used for the treatment of various cardiovascular and cerebrovascular diseases [21]. In addition to its functions in cardiovascular systems, recent studies have been carried out to elucidate the molecular mechanisms which account for the protective action of Tan in glucocorticoids-inducedosteoporosis, such as stimulation of osteogenesis, depression of adipogenesis [37], elevation of Runx2 and β-catenin mRNA expression [38] and regulation of Wnt/FoxO3a signaling pathway [21]. In our study, we found that Tan attenuated Dex-induced cell injury in a dose-dependent manner. The IC50 of Tan in MC3T3-E1 cells was compared with those reported in a previous study [21]. Next, we also evidenced that the intrinsic mitochondrial pathway was involved in the protective action of Tan. Importantly, overexpression of Nox4 almost completely abolished the inhibitory effects of Tan on Dex-induced apoptosis in MC3T3-E1 cells. These findings were first to our knowledge to demonstrate that Tan attenuated Dex-induced cell injury and apoptosis via inactivation of Nox4.
In summary, our data of the present study reveal for the first time that Nox4 plays an important role in the Dex-induced ROS. The elevation in Nox4-derived ROS production subsequently activates intrinsic mitochondrial pathway, leading to cell apoptosis. Moreover, Tan effectively inhibits Dex-induced cell apoptosis through inactivation of Nox4. This study suggests that Nox4 may be a potential target to ameliorate glucocorticoids-induced apoptosis, and provides a novel mechanism by which Tan prevents against osteoporosis.
Disclosure of conflict of interest
None.
References
- 1.Lin H, Wei B, Li G, Zheng J, Sun J, Chu J, Zeng R, Niu Y. Sulforaphane reverses glucocorticoid-induced apoptosis in osteoblastic cells through regulation of the Nrf2 pathway. Drug Des Devel Ther. 2014;8:973–982. doi: 10.2147/DDDT.S65410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hsu WL, Chen CY, Tsauo JY, Yang RS. Balance control in elderly people with osteoporosis. J Formos Med Assoc. 2014;113:334–339. doi: 10.1016/j.jfma.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 3.Raisz LG. Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J Clin Invest. 2005;115:3318–3325. doi: 10.1172/JCI27071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sambrook P, Cooper C. Osteoporosis. Lancet. 2006;367:2010–2018. doi: 10.1016/S0140-6736(06)68891-0. [DOI] [PubMed] [Google Scholar]
- 5.Reid IR. Anti-resorptive therapies for osteoporosis. Semin Cell Dev Biol. 2008;19:473–478. doi: 10.1016/j.semcdb.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Rauch A, Seitz S, Baschant U. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. 2010;11:517–531. doi: 10.1016/j.cmet.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Yun SI, Yoon HY, Jeong SY, Chung YS. Glucocorticoid induces apoptosis of osteoblast cells through the activation of glycogen synthase kinase 3beta. J Bone Miner Metab. 2009;27:140–148. doi: 10.1007/s00774-008-0019-5. [DOI] [PubMed] [Google Scholar]
- 8.Gudbjornsson B, Juliusson UI, Gudjonsson FV. Prevalence of long term steroid treatment and the frequency of decision making to prevent steroid induced osteoporosis in daily clinical practice. Ann Rheum Dis. 2002;61:32–36. doi: 10.1136/ard.61.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Angeli A, Guglielmi G, Dovio A. High prevalence of asymptomatic vertebral fractures in post-menopausal women receiving chronic glucocorticoid therapy: a cross-sectional outpatient study. Bone. 2006;39:253–259. doi: 10.1016/j.bone.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 10.Buehring B, Viswanathan R, Binkley N, Busse W. Glucocorticoid-induced osteoporosis: an update on effects and management. J Allergy Clin Immunol. 2013;132:1019–1030. doi: 10.1016/j.jaci.2013.08.040. [DOI] [PubMed] [Google Scholar]
- 11.den Uyl D, Bultink IE, Lems WF. Advances in glucocorticoid-induced osteoporosis. Curr Rheumatol Rep. 2011;13:233–240. doi: 10.1007/s11926-011-0173-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinstein RS. Clinical practice. Glucocorticoid-induced bone disease. N Engl J Med. 2011;365:62–70. doi: 10.1056/NEJMcp1012926. [DOI] [PubMed] [Google Scholar]
- 13.Jung WW. Protective effect of apigenin against oxidative stress-induced damage in osteoblastic cells. Int J Mol Med. 2014;33:1327–1334. doi: 10.3892/ijmm.2014.1666. [DOI] [PubMed] [Google Scholar]
- 14.Rached MT, Kode A, Xu L. FoxO1 is a positive regulator of bone formation by favoring protein synthesis and resistance to oxidative stress in osteoblasts. Cell Metab. 2010;11:147–160. doi: 10.1016/j.cmet.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H. Trends in oxidative aging theories. Free Radical Biol Med. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 16.Maggio D, Barabani M, Pierandrei M. Marked decrease in plasma antioxidants in aged osteoporotic women: results of a cross-sectional study. J Clin Endocrinol Metab. 2003;88:1523–1527. doi: 10.1210/jc.2002-021496. [DOI] [PubMed] [Google Scholar]
- 17.Basu S, Michaelsson K, Olofsson H, Johansson S, Melhus H. Association between oxidative stress and bone mineral density. Biochem Biophys Res Commun. 2001;288:275–279. doi: 10.1006/bbrc.2001.5747. [DOI] [PubMed] [Google Scholar]
- 18.Lee DH, Lim BS, Lee YK, Yang HC. Effects of hydrogen peroxide (H2O2) on alkaline phosphatase activity and matrix mineralization of odontoblast and osteoblast cell lines. Cell Biol Toxicol. 2006;22:39–46. doi: 10.1007/s10565-006-0018-z. [DOI] [PubMed] [Google Scholar]
- 19.Suda N, Morita I, Kuroda T, Murota S. Participation of oxidative stress in the process of osteoclast differentiation. Biochim Biophys Acta. 1993;1157:318–323. doi: 10.1016/0304-4165(93)90116-p. [DOI] [PubMed] [Google Scholar]
- 20.Li YH, Xu Q, Xu WH, Guo XH, Zhang S, Chen YD. Mechanisms of protection against diabetes-induced impairment of endothelium-dependent vasorelaxation by Tanshinone IIA. Biochim Biophys Acta. 2015;1850:813–823. doi: 10.1016/j.bbagen.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 21.Yang Y, Su Y, Wang D. Tanshinol attenuates the deleterious effects of oxidative stress on osteoblastic differentiation via Wnt/FoxO3a signaling. Oxid Med Cell Longev. 2013;2013:351895. doi: 10.1155/2013/351895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forstermann U. Oxidative stress in vascular disease: causes, defense mechanisms and potential therapies. Nature clinical practice. Nat Clin Pract Cardiovasc Med. 2008;5:338–349. doi: 10.1038/ncpcardio1211. [DOI] [PubMed] [Google Scholar]
- 23.Johnell O, Kanis JA, Oden A. Predictive value of BMD for hip and other fractures. J Bone Miner Res. 2005;20:1185–1194. doi: 10.1359/JBMR.050304. [DOI] [PubMed] [Google Scholar]
- 24.Kim BY, Yoon HY, Yun SI. In vitro and in vivo inhibition of glucocorticoid-induced osteoporosis by the hexane extract of Poncirus trifoliata. Phytother Res. 2011;25:1000–1010. doi: 10.1002/ptr.3373. [DOI] [PubMed] [Google Scholar]
- 25.Lin H, Gao X, Chen G. Indole-3-carbinol as inhibitors of glucocorticoid-induced apoptosis in osteoblastic cells through blocking ROS-mediated Nrf2 pathway. Biochem Biophys Res Commun. 2015;460:422–427. doi: 10.1016/j.bbrc.2015.03.049. [DOI] [PubMed] [Google Scholar]
- 26.Besbes S, Mirshahi M, Pocard M, Billard C. New dimension in therapeutic targeting of BCL-2 family proteins. Oncotarget. 2015;6:12862–12871. doi: 10.18632/oncotarget.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sochalska M, Tuzlak S, Egle A, Villunger A. Lessons from gain- and loss-of-function models of pro-survival Bcl2 family proteins: implications for targeted therapy. FEBS J. 2015;282:834–849. doi: 10.1111/febs.13188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gimenez-Cassina A, Danial NN. Regulation of mitochondrial nutrient and energy metabolism by BCL-2 family proteins. Trends Endocrinol Metab. 2015;26:165–175. doi: 10.1016/j.tem.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yeh CC, Chang JZ, Yang WH, Chang HH, Lai EH, Kuo MY. NADPH oxidase 4 is involved in the triethylene glycol dimethacrylate-induced reactive oxygen species and apoptosis in human embryonic palatal mesenchymal and dental pulp cells. Clin Oral Investig. 2015;19:1463–1471. doi: 10.1007/s00784-014-1370-7. [DOI] [PubMed] [Google Scholar]
- 30.Espina B, Liang M, Russell RG, Hulley PA. Regulation of bim in glucocorticoid-mediated osteoblast apoptosis. J Cell Physiol. 2008;215:488–496. doi: 10.1002/jcp.21335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Brien CA, Jia D, Plotkin LI. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145:1835–1841. doi: 10.1210/en.2003-0990. [DOI] [PubMed] [Google Scholar]
- 32.Wassmann S, Laufs U, Muller K. Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2002;22:300–305. doi: 10.1161/hq0202.104081. [DOI] [PubMed] [Google Scholar]
- 33.Zhong ZM, Bai L, Chen JT. Advanced oxidation protein products inhibit proliferation and differentiation of rat osteoblast-like cells via NF-kappaB pathway. Cell Physiol Biochem. 2009;24:105–114. doi: 10.1159/000227818. [DOI] [PubMed] [Google Scholar]
- 34.Muteliefu G, Enomoto A, Jiang P, Takahashi M, Niwa T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol Dial Transplant. 2009;24:2051–2058. doi: 10.1093/ndt/gfn757. [DOI] [PubMed] [Google Scholar]
- 35.Liberman M, Bassi E, Martinatti MK. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler Thromb Vasc Biol. 2008;28:463–470. doi: 10.1161/ATVBAHA.107.156745. [DOI] [PubMed] [Google Scholar]
- 36.Kanazawa I, Yamaguchi T, Yamauchi M, Sugimoto T. Rosuvastatin increased serum osteocalcin levels independent of its serum cholesterol-lowering effect in patients with type 2 diabetes and hypercholesterolemia. Intern Med. 2009;48:1869–1873. doi: 10.2169/internalmedicine.48.2645. [DOI] [PubMed] [Google Scholar]
- 37.Cui L, Liu YY, Wu T, Ai CM, Chen HQ. Osteogenic effects of D+beta-3,4-dihydroxy-phenyl lactic acid (salvianic acid A, SAA) on osteoblasts and bone marrow stromal cells of intact and prednisone-treated rats. Acta Pharmacol Sin. 2009;30:321–332. doi: 10.1038/aps.2009.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cui L, Li T, Liu Y. Salvianolic acid B prevents bone loss in prednisone-treated rats through stimulation of osteogenesis and bone marrow angiogenesis. PLoS One. 2012;7:e34647. doi: 10.1371/journal.pone.0034647. [DOI] [PMC free article] [PubMed] [Google Scholar]