

Abstract
Background: An evolutionarily conserved gene, the CDGSH iron sulfur domain 2 (CISD2), functions to control mammalian life span and regulates human cells proliferation. However, the role of CISD2 in HCC remains unclear. This study was aimed at investigating the expression pattern and clinicopathological significance of CISD2 in patients with HCC. Methods: The mRNA and protein expression levels of CISD2 were analyzed in six HCC lines and eight paired hepatic cancer tumors by real-time PCR, Western blotting and immunohistochemical staining. Statistical analysis was used to evaluate the clinicopathological significance of CISD2 expression. Short hairpin RNA interfering approach was employed to suppress endogenous CISD2 expression in hepatic cancer cells to determine its role in proliferation. Results: CISD2 expression in liver cancer cell lines and tissues was significantly up-regulated at both the RNA and protein levels compared with that in normal cells and adjacent non-tumorous liver tissues (ANT). CISD2 was an independent prognostic factor for poor prognosis. It was correlated with tumor size (P=0.001), number of tumors (P=0.003), surgical margin (P=0.006), hepatitis B surface antigen (HBsAg) infection (P=0.002) and recurrence (P<0.001) of liver cancer. Multivariate analysis suggested that CISD2 expression was an independent prognostic indicator for the survival of patients with HCC. HCC patients with high CISD2 expression displayed a shorter overall survival and a higher recurrence rate than those with low CISD2 expression (P<0.05, respectively). Additionally, stable down-expression of CISD2 in hepatoma cells suppressed cell proliferation in vitro. Similarly, an in vivo assay showed that CISD2 down-regulation in hepatoma cells inhibited remarkably tumorigenic potential in tumor size and weight. Conclusions: CISD2 protein may serve as a candidate prognostic marker and a novel therapeutic target for HCC and play an important role in promoting proliferation and enhanced progression of HCC.
Keywords: CDGSH iron sulfur domain 2, proliferation, hepatocellular carcinoma, prognostic marker, therapeutic target
Introduction
Hepatocellular carcinoma (HCC) is one of the most frequent malignant diseases worldwide. With an estimated 748,000 newly diagnosed cases per year and a low five-year survival rate, HCC is the third leading cause of cancer-related death worldwide [1]. Presently, hepatic resection and liver transplantation are still the most curative therapeutic choices for HCC patients. However, the 5-year survival rate of HCC patients after curative resection is only 35%-43% due to recurrence and metastasis after operation [2]. Hence, it is of great significance to identify effective early markers and therapeutic targets for HCC.
The newly discovered protein, CDGSH iron sulfur domain 2 (CISD2) as a member of a newly discovered family of iron sulfur (FeS) proteins , encoded by CISD2 genes, was demonstrated to play a key role in maintaining the mitochondrial integrity localized to the outer mitochondrial membrane, whose deficiency can cause mitochondrial dysfunction accompanied by cell death with autophagic features [3,4]. Additionally, CISD2 was defined by a unique CDGSH amino acid sequence in their FeS cluster-binding domain, which also appears in endoplasmic reticulum (ER) where it interacts with BCL-2 to regulate autophagy and Ca2+ homeostasis [5-7]. The ratio of the amount of CISD2 protein in the mitochondria versus ER is estimated to be approximately 6:1 [8]. Interestingly, CISD2 gene locates on human chromosome 4q, where a genetic component for longevity maps is essential for mammalian life span control [9,10]. In addition, CISD2 deficiency shortens life span, and persistent expression of CISD2 is able to extend healthy lifespan [8,11], while the essential value of detection of genomic amplification of 4q in the progression of HCC has been affirmed in several studies [12]. Furthermore, CISD2 as the second causative gene was suggested to be responsible for Wolfram syndrome 2 (WFS2), a mitochondrial-mediated disorder, which accounted for severe blindness, hearing deficiencies, diabetes and even a lower life expectancy [13-15]. Moreover, CISD2 expression was significantly upregulated in human epithelial breast cancer cell lines, suggesting that CISD2 was central to breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor progress [16,17]. Intriguingly, impaired mitochondrial function and activation of autophagy in human breast cancer cells by suppression expression of CISD2 resulted in suppressed proliferation of cancer cells [16]. However, there has been no study to investigate the protein expression level of CISD2 in HCC. Our aim was to find out a potential biomarker for HCC, thus providing a better understanding of the molecular pathophysiology and better methods for detection, diagnosis, and classification of HCC.
Materials and methods
Samples and patients
The current retrospective study enrolled 196 patients diagnosed with primary HCC who underwent surgical resection at Department of Hepatobiliary Surgery, the First Affiliated Hospital, Sun Yat-sen University from March 2008 to September 2013. The mean patient age was 42.00±7.84 (range 17-70). Eight paired samples of HCC tissues and their corresponding adjacent non-tumorous liver tissues (ANT) were collected for quantitative polymerase chain reaction (qPCR) and western blotting analysis. All the paraffin-embedded tissues and fresh tissues were obtained with the consent of each patient for research purposes. The Institutional Ethical Board (IRB) in the First Affiliated Hospital of Sun Yat-sen University approved the protocol. The clinical information was summarized in Table 1.
Table 1.
Clinicopathological characteristics of patient samples and expression of CISD2 in liver cancer and correlation between CISD2 expression and clinicopathological characteristics of hepatic cancer patients (n=196)
| Characteristics | Total (n= 196) | CISD2 | Chi-square test | Fisher’s exact test | ||
|---|---|---|---|---|---|---|
|
| ||||||
| No or Low expression | High expression | |||||
|
| ||||||
| 87 (44.4%) | 109 (55.6%) | P-value | P-value | |||
| Gender | Male | 156 | 68 (43.6) | 88 (56.4) | 0.087 | 0.077 |
| Female | 40 | 19 (47.5) | 21 (52.5) | |||
| Age (years) | <60 | 155 | 71 (45.8) | 84 (54.2) | 0.121 | 0.098 |
| ≥60 | 41 | 16 (39.0) | 25 (61.0) | |||
| HBsAg | Negative | 18 | 16 (88.9) | 2 (11.1) | 0.002 | 0.001 |
| Positive | 178 | 71 (39.9) | 107 (60.1) | |||
| AFP | <200 ng/ml | 94 | 45 (47.9) | 49 (52.1) | 0.084 | 0.079 |
| ≥200 ng/ml | 102 | 42 (41.2) | 60 (58.8) | |||
| Edmonson grade | I–II | 135 | 58 (43.0) | 77 (57.0) | 0.965 | 0.929 |
| III–IV | 61 | 29 (47.5) | 32 (52.5) | |||
| Tumor size | <5 cm | 50 | 41 (82.0) | 9 (18.0) | 0.001 | 0.000 |
| ≥5 cm | 146 | 46 (31.5) | 100 (68.5) | |||
| Number of tumors | Single | 111 | 78 (70.3) | 33 (29.7) | 0.003 | 0.003 |
| Multiple | 85 | 9 (10.6) | 76 (89.4) | |||
| Liver cirrhosis | No | 72 | 33 (45.8) | 39 (54.2) | 0.173 | 0.135 |
| Yes | 124 | 54 (43.5) | 70 (56.5) | |||
| Tumor encapsulation | No | 91 | 43 (47.3) | 48 (52.7) | 0.401 | 0.364 |
| Yes | 105 | 44 (41.9) | 61 (58.1) | |||
| Surgical margin | <1 cm | 82 | 65 (79.3) | 17 (20.7) | 0.006 | 0.003 |
| ≥1 cm | 114 | 22 (19.3) | 92 (80.7) | |||
| Vascular invasion | No | 138 | 63 (45.7) | 75 (54.3) | 0.288 | 0.226 |
| Yes | 58 | 24 (41.4) | 34 (58.6) | |||
| One year Recurrence | No | 148 | 79 (53.4) | 69 (46.6) | 0.009 | 0.007 |
| Yes | 48 | 8 (16.7) | 40 (83.3) | |||
| Five year Recurrence | No | 59 | 36 (61.0) | 23 (39.0) | 0.000 | 0.000 |
| Yes | 137 | 51 (37.2) | 86 (62.8) | |||
Cell lines
Human hepatocyte cells (L02) and HCC cell lines, including Huh7, SK-Hep1, Hep3B, HepG2, MHCC-97L and SNU423, were cultured at 37°C in 5% CO2 in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics in a humidified incubator. These seven cell lines were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) [18].
Real-time PCR (qPCR)
Total RNA from cultured cells and fresh tissues were extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The extracted RNA was pretreated with RNase-free DNase, and 2 μg RNA from each sample was used for cDNA synthesis, primed with random hexamers. For the PCR amplification of CISD2 cDNA, an initial amplification step using CISD2-specific primers was performed with denaturation at 95°C for 10 min, followed by 28 denaturation cycles at 95°C for 60 s, primer annealing at 58°C for 30 s and a primer extension phase at 72°C for 30 s. Upon the completion of the cycling steps, a final extension step at 72°C for 5 min was performed before the reaction mixture was stored at 4°C [19]. qPCR was then employed to quantify the increasing amounts of CISD2 mRNA in HCC cell lines relative to the hepatocyte cells (L02) and in each of the primary HCC tissues relative to the paired ANT. Each sample was tested in triplicate. Expression data were normalized to the geometric mean of the expression level of the housekeeping gene GAPDH to control the variability in expression levels. Primer Express v 2.0 software (Applied Biosystems) designed the primers. The CISD2 primer sequences were 5’-GCAAGGTAGCCAAGAAGTGC-3’ and 5’-CCCAGTCCCTGAAAGCATTA-3’. Primers for GAPDH were 5’-TTGAGGTCAATGAAGGGGTC-3’ and 5’-GAAGGTGAAGGTCGGAGTCA-3’.
Western blotting
Cells at 70 to 80% confluence were washed twice with icecold phosphate-buffered saline (PBS) and lysed on ice in radio immunoprecipitation assay buffer (RIPA; Cell Signaling Technology, Danvers, MA, USA) containing a complete protease inhibitor cocktail (Roche Applied Sciences, Mannheim, Germany). Fresh tissue samples were ground to a powder in liquid nitrogen and lysed using SDS-PAGE sample buffer. Equal protein samples (20 μg) were separated on 10.5% SDS polyacrylamide gels and transferred to PVDF membranes (Immobilon P, Millipore, Bedford, MA, USA). Membranes were blocked with 5% fat-free milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 h at room temperature. Membranes were incubated with anti-CISD2 antibody (1:1000, Proteintech, 13318-1-AP) overnight at 4°C and then with horseradish peroxidase-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology, SC-2004). The ECL prime Western blotting detection reagent (Amersham) was used to detect CISD2 expression, according to the manufacturer’s instructions. Alpha-Tubulin (Sigma, Saint Louis, MO) was used as a loading control.
IHC analysis
IHC analysis was used to study altered protein expression in 196 human HCC tissues. Briefly, 4 um-thick paraffin sections of the HCC tissue from the patient were baked at 65°C for 30 min and then deparaffinized with xylene and rehydrated. Submerging the sections into EDTA antigenic retrieval buffer and then microwaving were used for antigen retrieval. The samples were then treated with 3% hydrogen peroxide in methanol to quench endogenous peroxidase activity, followed by incubation with 1% bovine serum albumin to block nonspecific binding. Sections were then incubated with anti-CISD2 rabbit polyclonal antibody (1:100, Proteintech, 13318-1-AP) overnight at 4°C. Normal goat serum was used as a negative control. After washing, the tissue sections were then incubated with a biotinylated anti-rabbit secondary antibody (Abcam), followed by further incubation with streptavidin-horseradish peroxidase complex (Abcam). The tissue sections were immersed in 3-amino-9-ethyl carbazole and counterstained with 10% Mayer’s hematoxylin, dehydrated and mounted in Crystal Mount. Two observers who were blinded to the histopathological features and patient data of the samples evaluated the degree of immunostaining of formalin-fixed, paraffin-embedded sections independently. The scores given by the two independent investigators were averaged and were based on both the proportion of positively stained tumor cells and the intensity of staining. The proportion of tumor cells was scored as follows: 1 (<10% positive tumor cells), 2 (10-50% positive tumor cells), 3 (50-75% positive tumor cells) and 4 (>75% positive tumor cells). The intensity of staining was graded according to the following criteria: 0 (no staining), 1 (weak staining = light yellow), 2 (moderate staining = yellow brown) and 3 (strong staining = brown). The staining index was calculated as the product of the proportion of positive cells times the staining intensity score (range from 0 to 12). Cutoff values for CISD2 were chosen on the basis of a measure of heterogeneity using the log-rank test with respect to overall survival (OS). An optimal cutoff value was identified as follows: A staining index score of ≥6 was used to define tumors with high CISD2 expression and ≤4 indicated low CISD2 expression [20].
Vectors and retroviral infection
To silence endogenous CISD2, two ShRNA oligonucleotides were cloned into the pSuper-retro-puro vector to generate pSuper-retro-CISD2-ShRNA(s), respectively. Retroviral production and infection were performed as previously described [21]. The cloning primers and ShRNA oligonucleotides used were performed as previously described [16,22]. The stable HCC cell lines, including SK-Hep1 and HepG2, expressing CISD2 shRNA, were selected for 10 days with 0.5 μg/ml puromycin 48 h after infection. After 10-day selections, the cell lysates prepared from the pooled population of cells in sample buffer were fractionated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the detection of CISD2 protein level.
MTT viability assay
The viability of the cells was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich, St. Louis, MO) assay. A total of 1×103 cells per well were plated in 96-well with triplicate wells for each transfection, and incubated for 24 h in 100 ml culture media. MTT (500 mg/ml) was added to the cells and cultivated for another 4 h. After the medium was aspirated, the cells were dissolved by dimethyl sulfoxide (Sigma-Aldrich, St. Louis, MO). Absorbance of the formazan product was measured by an enzyme-linked immunosorbent assay reader. Each assay was repeated three times.
Anchorage-independent growth ability assay
Five hundred cells were trypsinized and suspended in 2 ml complete medium plus 0.3% agar (Sigma). The agar-cell mixture was plated on top of a bottom layer with 1% complete medium agar mixture. After 10 days, viable colonies that contained 50 cells or were larger than 0.5 mm were counted. The experiment was performed for three independently times for each cell line.
Colony formation assay
Cells were plated in 6-well plated (5×102 cells) and cultured for 10 days. The colonies were stained with 1% crystal violet for 30 seconds after fixation with 4% formaldehyde for 5 minutes. Colonies were counted and the results were shown as the fold change compared to vector control cells.
Xenografted tumor model
Male BALB/c nude mice (5~6 weeks of age, 18~20 g) were purchased from the Vital River Laboratories (Beijing, China), and were housed in barrier facilities on a 12-hour light/dark cycle. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Sun Yat-sen University. The BALB/c nude mice were randomly divided into 6 groups (n=6/group). The stable cells (5×106) including ShRNA-vector, CISD2 shRNA1 and 2-transduced, Sk-Hep1 and HepG2 cells were injected subcutaneously into the dorsal right flank of ten groups of 4-week old female Balb/c nude mice, respectively [23]. Tumors were examined twice weekly; length and width measurements were obtained with calipers, and tumor volumes were calculated using the equation (L×W2)/2. On day 30, animals were killed; tumors were excised, weighed.
Results
Elevated expression of CISD2 in hepatic cancer cells and tissues at both mRNA and protein levels
The expressions of both the CISD2 protein and mRNA were markedly up regulated in multiple hepatic cancer cell lines, including Huh7, SK-Hep1, Hep3B, HepG2, MHCC-97L, and SNU423 in comparison with those in the normal liver cell line L02 (Figure 1A, 1B). Furthermore, comparative analysis showed CISD2 protein levels were differentially up regulated more than 2-fold in all eight hepatic cancer samples compared with the matched normal ANT (Figure 2A). Moreover, qPCR analysis revealed that the mRNA expression level of CISD2 was also over expressed in HCC tissues compared with that in the paired normal ANT (Figure 2B). CISD2 up regulation in these clinical samples was further verified by IHC analysis (Figure 2C). As shown in Figure 2C, CISD2 was highly expressed in HCC tissues. In contrast, no signals or only weak signals were detected in the matched adjacent noncancerous tissues. Taken together, our results indicated CISD2 was upregulated in hepatic cancer.
Figure 1.
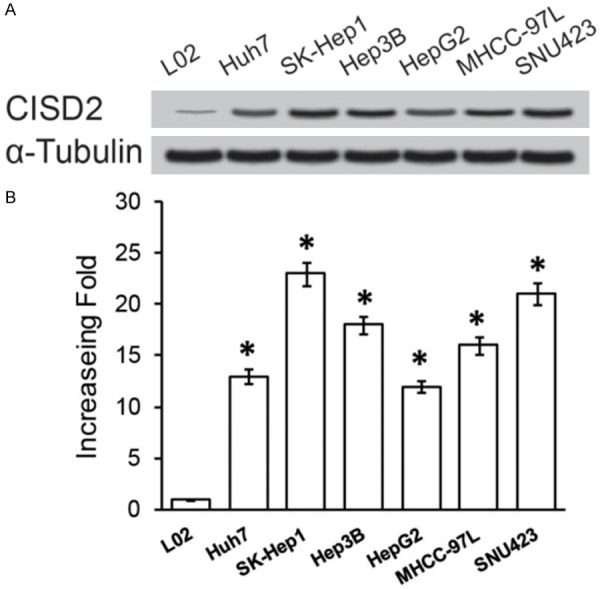
Overexpression of CISD2 mRNA and protein in HCC cell lines. Western blotting (A) and qPCR (B) examined the expression of CISD2 mRNA and protein in HCC cell lines (Huh7, SK-Hep1, Hep3B, HepG2, MHCC-97L and SNU423) and human hepatocyte cells (L02). Expression levels were normalized against α-Tubulin, respectively. Error bars represent the standard deviation of the mean (SD) calculated from three parallel experiments. *P<0.05.
Figure 2.
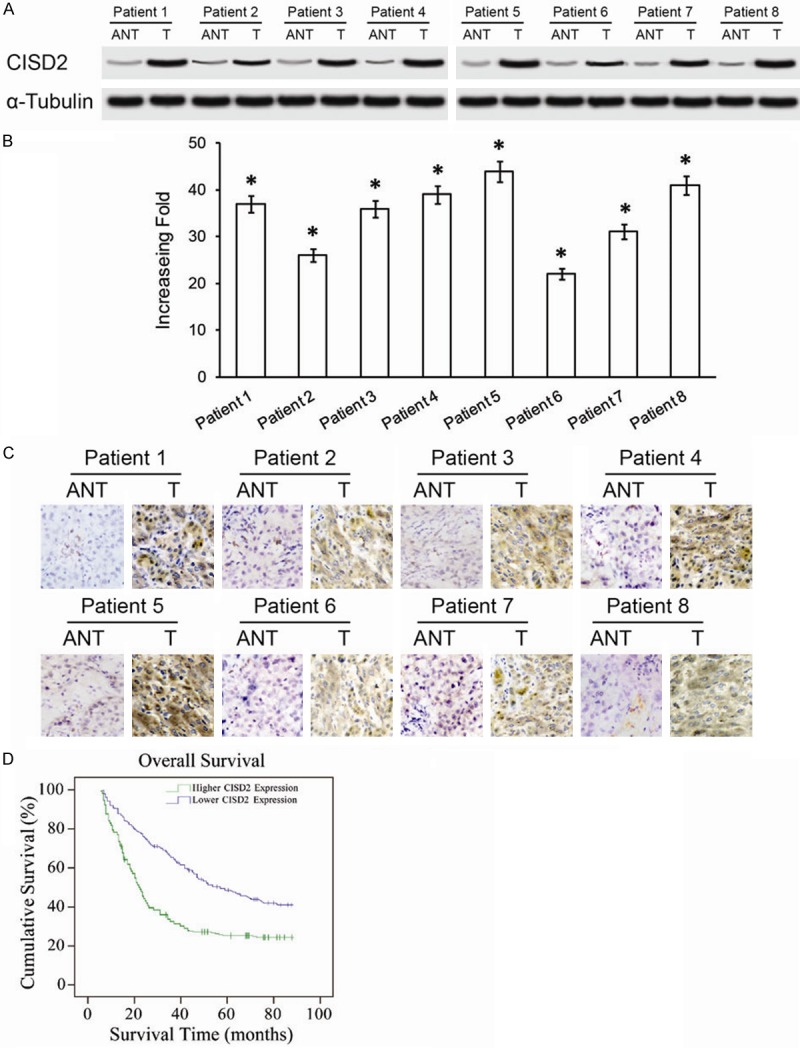
Overexpression of CISD2 mRNA and protein in primary HCC tissues. A. Western blotting of CISD2 protein expression in eight pairs of matched HCC cancer (T) and adjacent nontumor liver tissues (ANT). B. Average T/N ratios of CISD2 mRNA expression in paired liver cancer (T) and adjacent noncancerous tissues (N) were quantified using qPCR and normalized against α-Tubulin. Error bars represent the standard deviation of the mean (SD) calculated from three parallel experiments. C. Immunohistochemical assay of CISD2 protein expression in eight pairs of HCC tissues. D. Kaplan-Meier curves with univariate analyses (log-rank) for HCC patients with low CISD2 expression versus high CISD2 expression. The cumulative 5-year survival rate was 46.2% in the low CISD2 protein expression group (n=87), but only 24.5% in the high-expression group (n=109) *P<0.05.
CISD2 expression and the clinical characteristics of HCC
To confirm the prognostic significance of CISD2 for the early diagnosis and prediction of outcome in HCC, we analyzed the correlation between expression of CISD2 and clinicopathological and survival parameters. An immunohistochemistry analysis revealed that CISD2 was abundantly accumulated in the cytoplasm of hepatic cancer cells. Statistical analysis revealed CISD2 levels were strongly expressed in 55.6% (109/196) of patients with HCC and was strongly associated with tumor size (P=0.001), number of tumors (P=0.003), surgical margin (P=0.006), hepatitis B surface antigen (HBsAg) infection (P=0.002) (Table 1). The relationship between the clinicopathological features of HCC and CISD2 expression is summarized in Table 1. However, there was no apparent relationship between CISD2 expression and other clinicopathological parameters, including serum level of alpha fetoprotein (AFP) (≥200 ng/ml), differentiation status (Edmondson grade), liver cirrhosis, tumor encapsulation, and vascular invasion (All P>0.05).
High CISD2 expression is significantly associated with poor prognosis of patients with HCC
Kaplan-Meier analysis and the log-rank test were used to calculate the effect of CISD2 expression on survival. The log-rank test showed that the survival time was significantly different between the low and high CISD2 expression groups. Moreover, Kaplan-Meier survival curves and the log-rank test survival analysis showed the overall survival of patients with high levels of CISD2 was significantly poorer than patients with low levels of CISD2 (P<0.001 Figure 2D). The cumulative 5-year survival rate was 46.2% (95% CI 39.1-49.9%) in the low CISD2 group, whereas it was only 23.5% (95% CI 19.7-28.5%) in the high CISD2 group.
Further univariate and multivariate analyses were employed to compare the associations of CISD2 expression with other clinicopathological parameters. The Cox regression model revealed CISD2 expression (relative risk: 1.630, CI 1.050-2.876, P=0.025), tumor size (relative risk: 1.207, CI 1.031-1.419, P=0.016), surgical margin (relative risk: 1.669, CI 1.047-2.587, P=0.019) and recurrence (relative risk: 3.264, CI 1.543-6.430, P=0.008) were independent prognostic factors that influenced survival (Table 2). Thus, CISD2 may be a useful maker for predicting the overall survival of HCC patients. Collectively, these data demonstrated CISD2 was linked to the clinical progression of HCC and might represent a valuable prognostic marker for HCC.
Table 2.
Univariate and multivariate analyses of various prognostic parameters in patients with hepatic cancer Cox-regression analysis
| 5 year survival | Low expression | 46.2% (39.1%~49.9%) | ||||
|---|---|---|---|---|---|---|
| High expression | 23.5% (19.7%~28.5%) | |||||
|
| ||||||
| No. patients | Univariate analysis | Multivariate analysis | ||||
|
|
||||||
| P | Regression coefficient (SE) | P | Relative risk | 95% confidence interval | ||
| CISD2 | ||||||
| Low expression | 87 | 0.029 | 0.248 (0.243) | 0.025 | 1.630 | 1.050-2.876 |
| High expression | 109 | |||||
| Tumor size | ||||||
| <5 cm | 50 | 0.003 | 0.112 (0.075) | 0.016 | 1.207 | 1.031-1.419 |
| ≥5 cm | 146 | |||||
| Surgical margin | ||||||
| <1 cm | 82 | 0.015 | 0.256 (0.241) | 0.019 | 1.669 | 1.047-2.587 |
| ≥1 cm | 114 | |||||
| One year Recurrence | ||||||
| No | 148 | 0.007 | 0.237 (0.211) | 0.008 | 3.264 | 1.543-6.430 |
| Yes | 48 | |||||
CISD2 protein expression level in hepatic cancer significantly correlated with patient survival time (P<0.001); the correlation coefficient was -0.252 (-0.251), indicating that higher levels of CISD2 expression correlated with shorter survival time.
CISD2 promotes HCC proliferation in vitro
Since CISD2 protein was overexpressed in SK-Hep1 and HepG2 cells. We infected SK-Hep1 and HepG2 cells with empty vector alone and the two shRNAs that suppressed endogenous CISD2 expression stably, and then measured the effect of loss of CISD2 on proliferation. Since the shRNA vector contained a puromycin resistance gene, we created the stable cell lines and first measured the expression of CISD2. As expected, both short hairpins RNA1/2 (shRNAs) effectively knocked down the level of endogenous CISD2 protein in the CISD2-silence SK-Hep1 and HepG2 cells (Figure 3A). To investigate the effect of CISD2 expression on cell proliferation, we assessed the proliferation rates of shRNA-vector cells and CISD2-silenced SK-Hep1 cells and HEPG2 cells with MTT assays (Figure 3B). The silence of CISD2 drastically reduced the proliferation rate of cells infected with CISD2-siRNA compared with the rate of the control cells at days 5 (P<0.05) (Figure 3B). In consistence with these results, colony formation assays were dramatically inhibited by the ablation of CISD2 expression, where the proliferation rate of CISD2 silenced SK-Hep1 and HepG2 cells was significantly lower than the corresponding vector-control cells (Figure 3C). Moreover, In order to further confirm the aforementioned results, depletion of endogenous CISD2 in the stabled SK-Hep1 cells and HepG2 cells also caused significant inhibition of their anchorage-independent growth ability, as indicated by reduction in colony number and colony size on soft agar (P<0.05, Figure 3D).
Figure 3.
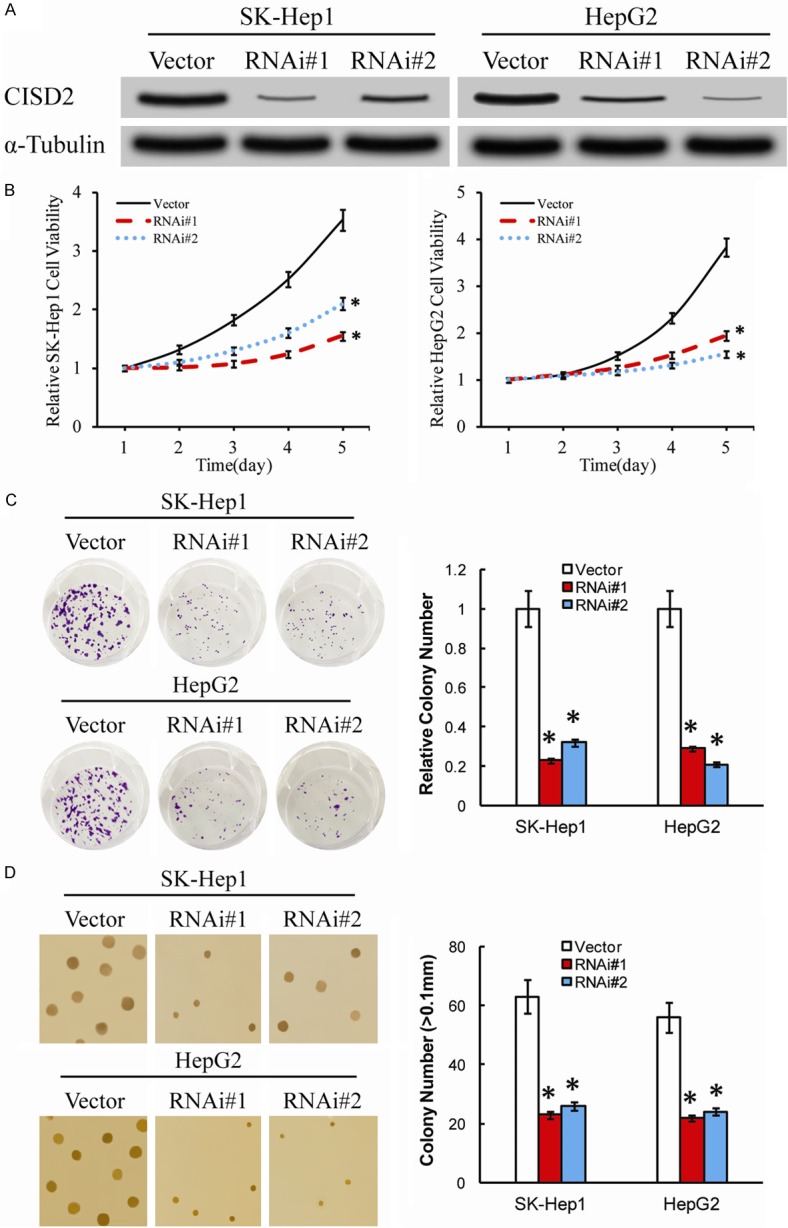
CISD2 plays a key role in HCC cell proliferation and tumorigenicity in vitro. A. Western blotting analysis of CISD2 expression in the ShRNA-vector, CISD2-silenced, SK-Hep1 and HepG2 cells. α-Tubulin was used as a loading control. B. MTT assay indicated that the growth rate decreased in CISD2-silenced cells compared with that in ShRNA-vector cells. The absorbance at day 1-5 was normalized to the absorbance at day 0 used as control (100%). C. Colony formation assays show that growth rates are decreased in CISD2-silenced cells. The number of colonies was quantified in the colony formation assay. Each bar represents the mean ± SD of three independent experiments. D. Anchorage-independent growth assays of CISD2-silenced cells and ShRNA-vector cells. The number of colonies with a diameter larger than 0.1 mm was quantified after 10 days of culture. *P<0.05.
CISD2 promotes the tumorigenicity of HCC in vivo
On the foundation of the results in vitro assays, the biologic role of CISD2 in HCC progression was further examined using an in vivo tumor model to determine whether CISD2 expression could enhance the tumorigenicity of HCC in vivo. As shown in Figure 4A, the tumors formed by SK-Hep1/CISD2-ShRNA and HepG2/CISD2-ShRNA cells grew at a dramatically slower rate than shRNA-vector SK-Hep1 and HepG2 tumors. Additionally, the CISD2–silenced SK-Hep1 tumors were smaller, in both size and weight, than the control SK-Hep1 tumors (Figure 4B, 4C). Similarly, the tumors formed by shRNA-vector HepG2 cells were larger and had higher tumor weights than the tumors formed by CISD2-silenced HepG2 cells (Figure 4B, 4C). Collectively, these results were consistent with the data in vitro and suggest that CISD2 plays a significant role in the tumorigenicity of HCC cells in vitro and in vivo.
Figure 4.
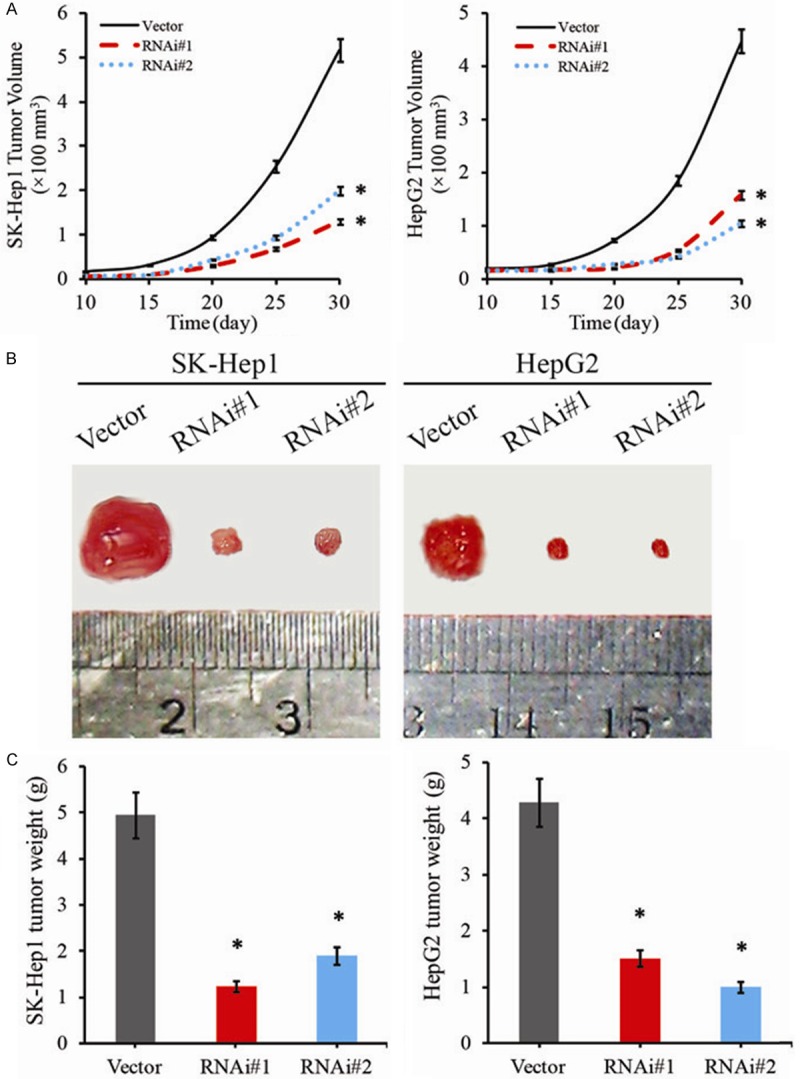
Downregulation of CISD2 repressed proliferation and tumorigenicity in vivo. Effect of CISD2 on tumor formation in nude mouse xenograft model. A. Subcutaneous tumor growth curve of CISD2 shRNA#1 and #2-transduced SK-Hep1 and HepG2 cells in nude mice was compared with ShRNA-vector SK-Hep1 and HepG2 cells in nude mice (n=6). Tumor volumes were measured on the indicated days. B. A representative picture of tumor growth in nude mice subcutaneously inoculated with ShRNA-vector, CISD2 shRNA#1 and #2-transduced, SK-Hep1 and HepG2 cells. C. Mean tumor weights. Each bar represents the mean ± SD of three independent experiments. *P<0.05.
Discussion
A recapitulation of our results, we first detected the expression of CISD2 in 196 cases of clinical paraffin-embedded HCC tissues, 6 HCC cell lines and 8 matched clinical fresh HCC tissues. IHC results showed that the positive signaling of CISD2 was observed in the cytoplasm of hepatocytes. Moreover, CISD2 was up regulated in six HCC cell lines as well as in 8 matched clinical fresh tissues at both the mRNA and protein levels. Furthermore, our study provided the first evidence that high expression of CISD2 protein was correlated with recurrence and poor prognosis in HCC. Additionally, the immunostaining analysis showed that the expression level of CISD2 protein in histological sections was also significantly correlated with tumor size, number of tumors, surgical margin and even reduced survival time of patients with HCC. Multivariate analysis revealed that CISD2 expression might be an independent prognostic indicator of survival in HCC patients. In addition to indicating the clinical significance of CISD2, we also illustrated that down-expression of CISD2 in HCC cells significantly suppressed tumor proliferation in vitro and in vivo. The effective suppression of CISD2 expression reduced HCC cells proliferation in vitro according to the results of MTT assays, anchorage-independent growth ability assays and colony formation assays, and then further inhibited tumorigenesis and tumor progression in vivo. According to these findings, we considered that CISD2 had the potential to act as a novel predictor for the prognosis of HCC.
Hepatocellular carcinoma (HCC) is one of the most lethal malignancies worldwide [24]. Although numerous biomarkers have been studied in past decades, the serological detection of HCC recurrence mainly relies on the traditional marker AFP [25,26], functioning as a tool for risk assessment, diagnostic index, and prognostic surveillance. However, the positive predictive value (PPV) of AFP is not satisfied [27]. Furthermore, as the secreted proteins originated from tumors, AFP does not possess therapeutic value for HCC patients. In attempting to determine the molecular mechanisms of human cancers over recent decades, scientists have highlighted a few candidate molecular markers for early detection. New biomarkers in hepatocellular carcinoma, such as des-c-carboxyprothrombin (DCP), glypican 3, osteopontin, and Golgi protein 73 (GP73), have been identified and demonstrated to be of some clinical utility [28-31]. Although early studies explored the potential of vitamin K2, retinoid, chemotherapy, and recently, sorafenib, none of the studies reported successful outcomes [24]. So far, there is no promising biomarker possessing both prognostic and therapeutic value for HCC patients [32]. In our study, multivariate analysis revealed that CISD2 expression might be an independent prognostic indicator of survival in HCC patients. According to recent advancements in imaging modalities and due to the prevalence of surveillance systems for hepatocellular carcinoma (HCC), early detection of HCC has been achieved, and the proportion of patients who can receive surgical resection is increasing. Although tumors seem to be treated completely, recurrence of HCC has been frequently observed at an annual incidence rate of approximately 20%, and 5 years after surgery, 70% of cases are complicated by tumor recurrence, comprising true recurrence [33]. To achieve long survival of patients with HCC, detection and suppression of recurrence is mandatory [34]. Multivariate analysis showed that a tumor diameter >5 cm, the absence of a tumor capsule and the presence of microvascular invasion were correlated with early recurrence, whereas cirrhosis and alpha-fetal protein >400 μg/l were independent risk factors contributing to late recurrence [35]. Additionally, HBV reactivation was also reported as a major risk factor for HCC recurrence [36-38]. As treatment strategies are very limited for recurrent liver cancer, the upregulated CISD2 identified in our study is a potential marker and target for formulating treatment strategies. In this study, in patients who demonstrated high expression of CISD2, but were not detected with surgical margin invasion, the 5 recurrent rate reached 78.9%, whereas the rate of recurrence was only 58.6% (P<0.05) in the group of low expression of CISD2. Moreover, the recurrence rates at one year in patients with CISD2 over expression and under expression (36.7% and 9.1%, respectively) indicated that HCC recurrence is more likely to occur in patients with over expressed CISD2. Given that the reported potential biomarkers in HCC patients were limited to a certain extent, it was highly likely that the “real” accuracy of predicting the 5-year recurrent rate by investigating CISD2 expression level could be as high 78.9%.
Previous independent studies have demonstrated that over expression of CISD2 could act as a novel diagnostic or therapeutic target in breast cancer and cervical cancer [16,17]. Interestingly, CISD2 as a multifunctional member of the iron sulfur (FeS) proteins family [13] was also amplified in breast cancer, while inhibiting CISD2 can significantly suppressed the growth of the breast cancer cells, suggesting that CISD2 a promising mitochondrial target for cancer therapy with the diagnostic and prognostic significances in breast cancer [16]. The significant suppression of the breast cancer cell proliferation and tumor growth resulted from down-regulation of CISD2, the mechanism of which is associated with damage to the mitochondria. Additionally, another report had provided the evidence that high expression of CISD2 protein was correlated with poor prognosis and pelvic lymph node metastases in early-stage cervical cancer through the clinical analysis [17]. Consistent with these reports, we found that CISD2 was significantly up regulated in HCC cell lines and tissues compared with the L02 cells and their matched adjacent normal tissues, respectively, and was strongly correlated with number of tumors and recurrence in clinical samples of HCC (P<0.001; P=0.001, respectively). In addition, we demonstrated that silencing CISD2 decreased the cells proliferation, tumorigenesis and tumor progression of HCC in vivo and in vitro. Suppressing CISD2 resulted in diminished spare respiratory capacity of mitochondria and enhanced glycolytic activity, whereas CISD2 over expression led to an increased spare respiratory capacity of mitochondria and decreased glycolytic activity in cancer cells [16]. Moreover, suppression of CISD2 expression resulted in decreased mitochondrial membrane potential, increased mitochondrial iron levels, increased mitochondrial accumulation of ROS and activated autophagy. Thus, we also indicated that the expression of CISD2 has an important influence on the development of HCC tumor and function through the mitochondria. The oncogenic potential of CISD2 was not only correlated with recurrence and poor prognosis in HCC but also suggested by its ability to promote HCC cell proliferation and tumor growth in vitro and in vivo. All these findings indicate that CISD2 was associated with the aggressive biological behavior of HCC cells and it might be a potential biomarker for HCC diagnosis and prognosis [39].
The molecular mechanism of CISD2 expression in HCC cells also deserves further exploration. As mentioned above, CISD2 was involved in the progression of breast cancer by regulating mitochondrial function. Recently, studies showed that mitochondria play an important role in the transformation process of cells, including the biosynthetic and energetic metabolism of cancer cells, and were involved in the modulation of apoptosis and autophagy [40,41]. Consequently, we indicated that CISD2 also worked in HCC cells by mediating mitochondria function or some kind of pathway that related to mitochondria. As previous reported [16], the proliferation of breast cancer cells was significantly influenced by mitochondria-related autophagy. At the same time, in another study CISD2 was found to be related to invasion and metastasis, and autophagy was a double-edged sword in tumor metastasis [42]. Therefore, another mechanism might be involved since mitochondria-related autophagy alone was not sufficient to explain CISD2’s involvement in invasion and metastasis. Some studies reported that mitochondrial homeostasis was essential for the regulation of bioenergetics, cell proliferation, cell death and autophagy [43]. Liu et al. [17] hypothesized that CISD2, which is primarily located on the mitochondria, might modulate BCL2 to inhibit the mitochondria-mediated apoptosis pathway in cancer cells. CISD2 inhibition of apoptosis in HCC cells might facilitate cell proliferation and tumorgenesis. That might be the cause of CISD2’s association with tumor genesis and progression in HCC. In addition, some studies demonstrated that many mutations that increase the activity of nuclear factor-κB (NF-κB) transcription factors (for example, Ras) could enhance the expression of BCL2 [44,45]. Mayo et al. [44] reported that NF-κB could activate BCL2. Thus, NF-κB as the upstream factor for BCL2 controls BCL2 protein expression. In addition, constitutive NF-κB activation in HCC cancer is correlated with tumor progression and aggression, as well as poor prognoses [45]. Recent studies showed that NF-κB was involved in inducing hepatocarcinogenesis in HCC, which might enhance cancer cell proliferation and promote tumor progress [45-47]. Accordingly, we thought that CISD2 might also be associated with the upstream control of BCL2 and NF-κB. Perhaps the NF-κB pathway was involved in the hepatocarcinogenesis induced by CISD2. The detail mechanism of CISD2’s association with NF-κB requires further investigation.
Conclusion
In conclusion, we have demonstrated an important role for CISD2 in HCC, where it was remarkably associated with poor prognosis, promoted proliferation and enhanced progression of HCC. Future studies, which were already underway in our laboratory, should address the detailed molecular mechanisms underlying the role of CISD2 in the carcinogenesis and the exact relationship between CISD2 and recurrence of HCC.
Acknowledgements
This study was supported by the grants of the Guangdong Medical Research Fund (No. A2015313), the National Natural Science Foundation of China (No. 81172039, 81302142), the Science and Technology Project of Guangdong Province (No. 2013B021800134), the Natural Science Foundation of Guangdong Province (No. 2014A030313108) and the Science and Technology Program of Guangzhou, China (2014Y2-00129). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure of conflict of interest
None.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Yang Y, Nagano H, Ota H, Morimoto O, Nakamura M, Wada H, Noda T, Damdinsuren B, Marubashi S, Miyamoto A, Takeda Y, Dono K, Umeshita K, Nakamori S, Wakasa K, Sakon M, Monden M. Patterns and clinicopathologic features of extrahepatic recurrence of hepatocellular carcinoma after curative resection. Surgery. 2007;141:196–202. doi: 10.1016/j.surg.2006.06.033. [DOI] [PubMed] [Google Scholar]
- 3.Paddock ML, Wiley SE, Axelrod HL, Cohen AE, Roy M, Abresch EC, Capraro D, Murphy AN, Nechushtai R, Dixon JE, Jennings PA. MitoNEET is a uniquely folded 2Fe 2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc Natl Acad Sci U S A. 2007;104:14342–14347. doi: 10.1073/pnas.0707189104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen YF, Kao CH, Kirby R, Tsai TF. Cisd2 mediates mitochondrial integrity and life span in mammals. Autophagy. 2009;5:1043–1045. doi: 10.4161/auto.5.7.9351. [DOI] [PubMed] [Google Scholar]
- 5.Chang NC, Nguyen M, Shore GC. BCL2-CISD2: An ER complex at the nexus of autophagy and calcium homeostasis? Autophagy. 2012;8:856–857. doi: 10.4161/auto.20054. [DOI] [PubMed] [Google Scholar]
- 6.Chang NC, Nguyen M, Germain M, Shore GC. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. Embo J. 2010;29:606–618. doi: 10.1038/emboj.2009.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang NC, Nguyen M, Bourdon J, Risse PA, Martin J, Danialou G, Rizzuto R, Petrof BJ, Shore GC. Bcl-2-associated autophagy regulator Naf-1 required for maintenance of skeletal muscle. Hum Mol Genet. 2012;21:2277–2287. doi: 10.1093/hmg/dds048. [DOI] [PubMed] [Google Scholar]
- 8.Chen YF, Kao CH, Chen YT, Wang CH, Wu CY, Tsai CY, Liu FC, Yang CW, Wei YH, Hsu MT, Tsai SF, Tsai TF. Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 2009;23:1183–1194. doi: 10.1101/gad.1779509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen YF, Wu CY, Kirby R, Kao CH, Tsai TF. A role for the CISD2 gene in lifespan control and human disease. Ann N Y Acad Sci. 2010;1201:58–64. doi: 10.1111/j.1749-6632.2010.05619.x. [DOI] [PubMed] [Google Scholar]
- 10.Puca AA, Daly MJ, Brewster SJ, Matise TC, Barrett J, Shea-Drinkwater M, Kang S, Joyce E, Nicoli J, Benson E, Kunkel LM, Perls T. A genome-wide scan for linkage to human exceptional longevity identifies a locus on chromosome 4. Proc Natl Acad Sci U S A. 2001;98:10505–10508. doi: 10.1073/pnas.181337598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu CY, Chen YF, Wang CH, Kao CH, Zhuang HW, Chen CC, Chen LK, Kirby R, Wei YH, Tsai SF, Tsai TF. A persistent level of Cisd2 extends healthy lifespan and delays aging in mice. Hum Mol Genet. 2012;21:3956–3968. doi: 10.1093/hmg/dds210. [DOI] [PubMed] [Google Scholar]
- 12.Peng SY, Ou YH, Chen WJ, Li HY, Liu SH, Pan HW, Lai PL, Jeng YM, Chen DC, Hsu HC. Aberrant expressions of annexin A10 short isoform, osteopontin and alpha-fetoprotein at chromosome 4q cooperatively contribute to progression and poor prognosis of hepatocellular carcinoma. Int J Oncol. 2005;26:1053–1061. [PubMed] [Google Scholar]
- 13.Conlan AR, Axelrod HL, Cohen AE, Abresch EC, Zuris J, Yee D, Nechushtai R, Jennings PA, Paddock ML. Crystal structure of Miner1: The redox-active 2Fe-2S protein causative in Wolfram Syndrome 2. J Mol Biol. 2009;392:143–153. doi: 10.1016/j.jmb.2009.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bu X, Rotter JI. Wolfram syndrome: A mitochondrial-mediated disorder? Lancet. 1993;342:598–600. doi: 10.1016/0140-6736(93)91416-j. [DOI] [PubMed] [Google Scholar]
- 15.Barrett TG, Bundey SE. Wolfram (DIDMOAD) syndrome. J Med Genet. 1997;34:838–841. doi: 10.1136/jmg.34.10.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sohn YS, Tamir S, Song L, Michaeli D, Matouk I, Conlan AR, Harir Y, Holt SH, Shulaev V, Paddock ML, Hochberg A, Cabanchick IZ, Onuchic JN, Jennings PA, Nechushtai R, Mittler R. NAF-1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth. Proc Natl Acad Sci U S A. 2013;110:14676–14681. doi: 10.1073/pnas.1313198110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu L, Xia M, Wang J, Zhang W, Zhang Y, He M. CISD2 expression is a novel marker correlating with pelvic lymph node metastasis and prognosis in patients with early-stage cervical cancer. Med Oncol. 2014;31:183. doi: 10.1007/s12032-014-0183-5. [DOI] [PubMed] [Google Scholar]
- 18.Wang C, Guo Y, Wang J, Min Z. The suppressive role of SOX7 in hepatocarcinogenesis. PLoS One. 2014;9:e97433. doi: 10.1371/journal.pone.0097433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye X, Guo Y, Zhang Q, Chen W, Hua X, Liu W, Yang Y, Chen G. ΒKlotho suppresses tumor growth in hepatocellular carcinoma by regulating Akt/GSK-3β/Cyclin D1 signaling pathway. PLoS One. 2013;8:e55615. doi: 10.1371/journal.pone.0055615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo Y, Chen W, Wang W, Shen J, Guo R, Gong F, Lin S, Cheng D, Chen G, Shuai X. Simultaneous diagnosis and gene therapy of immuno-rejection in rat allogeneic heart transplantation model using a T-cell-targeted theranostic nanosystem. ACS Nano. 2012;6:10646–10657. doi: 10.1021/nn3037573. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, Ou J, Guo Y, Dai T, Li X, Liu J, Xia M, Liu L, He M. TBLR1 is a novel prognostic marker and promotes epithelial-mesenchymal transition in cervical cancer. Br J Cancer. 2014;111:112–124. doi: 10.1038/bjc.2014.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tamir S, Rotem-Bamberger S, Katz C, Morcos F, Hailey KL, Zuris JA, Wang C, Conlan AR, Lipper CH, Paddock ML, Mittler R, Onuchic JN, Jennings PA, Friedler A, Nechushtai R. Integrated strategy reveals the protein interface between cancer targets Bcl-2 and NAF-1. Proc Natl Acad Sci U S A. 2014;111:5177–5182. doi: 10.1073/pnas.1403770111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo Y, Liang X, Lu M, Weng T, Liu Y, Ye X. Mammalian target of rapamycin as a novel target in the treatment of hepatocellular carcinoma. Hepatogastroenterology. 2009;57:913–918. [PubMed] [Google Scholar]
- 24.Lu LC, Cheng AL, Poon RT. Recent advances in the prevention of hepatocellular carcinoma recurrence. Semin Liver Dis. 2014;34:427–434. doi: 10.1055/s-0034-1394141. [DOI] [PubMed] [Google Scholar]
- 25.Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, Frank PG, Casimiro MC, Wang C, Pestell RG, Martinez-Outschoorn UE, Howell A, Sotgia F, Lisanti MP. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle. 2010;9:3485–3505. doi: 10.4161/cc.9.17.12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koteish A, Thuluvath PJ. Screening for hepatocellular carcinoma. J Vasc Interv Radiol. 2002;13:S185–S190. doi: 10.1016/s1051-0443(07)61785-0. [DOI] [PubMed] [Google Scholar]
- 27.Befeler AS, Di Bisceglie AM. Hepatocellular carcinoma: Diagnosis and treatment. Gastroenterology. 2002;122:1609–1619. doi: 10.1053/gast.2002.33411. [DOI] [PubMed] [Google Scholar]
- 28.Zhang B, Yang B. Combined alpha fetoprotein testing and ultrasonography as a screening test for primary liver cancer. J Med Screen. 1999;6:108–110. doi: 10.1136/jms.6.2.108. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura S, Nouso K, Sakaguchi K, Ito YM, Ohashi Y, Kobayashi Y, Toshikuni N, Tanaka H, Miyake Y, Matsumoto E, Shiratori Y. Sensitivity and specificity of des-gamma-carboxy prothrombin for diagnosis of patients with hepatocellular carcinomas varies according to tumor size. Am J Gastroenterol. 2006;101:2038–2043. doi: 10.1111/j.1572-0241.2006.00681.x. [DOI] [PubMed] [Google Scholar]
- 30.Shang S, Plymoth A, Ge S, Feng Z, Rosen HR, Sangrajrang S, Hainaut P, Marrero JA, Beretta L. Identification of osteopontin as a novel marker for early hepatocellular carcinoma. Hepatology. 2012;55:483–490. doi: 10.1002/hep.24703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marrero JA, Romano PR, Nikolaeva O, Steel L, Mehta A, Fimmel CJ, Comunale MA, D’Amelio A, Lok AS, Block TM. GP73, a resident Golgi glycoprotein, is a novel serum marker for hepatocellular carcinoma. J Hepatol. 2005;43:1007–1012. doi: 10.1016/j.jhep.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 32.Qi X, Ng KT, Lian QZ, Liu XB, Li CX, Geng W, Ling CC, Ma YY, Yeung WH, Tu WW, Fan ST, Lo CM, Man K. Clinical significance and therapeutic value of glutathione peroxidase 3 (GPx3) in hepatocellular carcinoma. Oncotarget. 2014;5:11103–20. doi: 10.18632/oncotarget.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–1255. doi: 10.1016/S0140-6736(11)61347-0. [DOI] [PubMed] [Google Scholar]
- 34.Nouso K. How do we conquer the recurrence of HCC? J Gastroenterol. 2015;50:703–4. doi: 10.1007/s00535-014-1003-6. [DOI] [PubMed] [Google Scholar]
- 35.Cheng Z, Yang P, Qu S, Zhou J, Yang J, Yang X, Xia Y, Li J, Wang K, Yan Z, Wu D, Zhang B, Huser N, Shen F. Risk factors and management for early and late intrahepatic recurrence of solitary hepatocellular carcinoma after curative resection. HPB (Oxford) 2015;17:422–7. doi: 10.1111/hpb.12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jang JW, Kwon JH, You CR, Kim JD, Woo HY, Bae SH, Choi JY, Yoon SK, Chung KW. Risk of HBV reactivation according to viral status and treatment intensity in patients with hepatocellular carcinoma. Antivir Ther. 2011;16:969–977. doi: 10.3851/IMP1840. [DOI] [PubMed] [Google Scholar]
- 37.Ko CJ, Chien SY, Chou CT, Chen LS, Chen ML, Chen YL. Factors affecting prognosis of small hepatocellular carcinoma in Taiwanese patients following hepatic resection. Can J Gastroenterol. 2011;25:485–491. doi: 10.1155/2011/790528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ou DP, Yang LY, Huang GW, Tao YM, Ding X, Chang ZG. Clinical analysis of the risk factors for recurrence of HCC and its relationship with HBV. World J Gastroenterol. 2005;11:2061–2066. doi: 10.3748/wjg.v11.i14.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pan Z, Tang B, Hou Z, Zhang J, Liu H, Yang Y, Huang G, Yang Y, Zhou W. XAGE-1b expression is associated with the diagnosis and early recurrence of hepatocellular carcinoma. Mol Clin Oncol. 2014;2:1155–1159. doi: 10.3892/mco.2014.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tait SW, Green DR. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 41.Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Yang Z, Xie L, Xu L, Xu D, Liu X. Statins, autophagy and cancer metastasis. Int J Biochem Cell Biol. 2013;45:745–752. doi: 10.1016/j.biocel.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 43.Giorgi C, Baldassari F, Bononi A, Bonora M, De Marchi E, Marchi S, Missiroli S, Patergnani S, Rimessi A, Suski JM, Wieckowski MR, Pinton P. Mitochondrial Ca(2+) and apoptosis. Cell Calcium. 2012;52:36–43. doi: 10.1016/j.ceca.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mayo MW, Baldwin AS. The transcription factor NF-kappaB: Control of oncogenesis and cancer therapy resistance. Biochim Biophys Acta. 2000;1470:M55–M62. doi: 10.1016/s0304-419x(00)00002-0. [DOI] [PubMed] [Google Scholar]
- 45.Chou RH, Hsieh SC, Yu YL, Huang MH, Huang YC, Hsieh YH. Fisetin inhibits migration and invasion of human cervical cancer cells by down-regulating urokinase plasminogen activator expression through suppressing the p38 MAPK-dependent NF-kappaB signaling pathway. PLoS One. 2013;8:e71983. doi: 10.1371/journal.pone.0071983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chung JS, Lee S, Yoo YD. Constitutive NF-kappaB activation and tumor-growth promotion by Romo1-mediated reactive oxygen species production. Biochem Biophys Res Commun. 2014;450:1656–1661. doi: 10.1016/j.bbrc.2014.07.059. [DOI] [PubMed] [Google Scholar]
- 47.Yan S, Wang Y, Yang Q, Li X, Kong X, Zhang N, Yuan C, Yang N, Kong B. Low-dose radiation-induced epithelial-mesenchymal transition through NF-kappaB in cervical cancer cells. Int J Oncol. 2013;42:1801–1806. doi: 10.3892/ijo.2013.1852. [DOI] [PubMed] [Google Scholar]