

Abstract:
Targeting of the high-affinity thrombin receptor protease-activated receptor-1 (PAR1) on platelets represents an exciting strategy to curb the pro-thrombotic complications of cardiac surgery without interfering with the hemostatic benefits of thrombin in the coagulation cascade. The first dedicated PAR1 antagonist to complete safety trials this year has justified expectations, showing no increased risk of bleeding when added to standard anti-platelet therapy but halving major adverse cardiovascular events after percutaneous coronary intervention. In the setting of cardiothoracic surgery with cardiopulmonary bypass, an FDA-approved drug already exists with anti-PAR1 properties: aprotinin has been shown to inhibit thrombin-induced platelet activation in vitro and clinically, through sparing of PAR1 receptor cleavage and activation. Because aprotinin also exerts anti-fibrinolytic effects through blockade of plasmin, this indicates a subtle clinical mechanism of action that is simultaneously anti-thrombotic yet hemostatic. PAR1 antagonists would also be expected to exert anti-inflammatory properties through targeting of PAR1 on endothelium, and this principle has been validated in vitro for aprotinin and newer peptidomimetric antagonists. PAR1 antagonism is likely to remain an active and exciting area of research in cardiac surgery, with newer generations of PAR1 antagonists and recombinant aprotinin variants entering clinical development.
Keywords: cardiac surgery, thrombin, receptor, antagonist
PAR RECEPTORS: SENSORS OF INJURY
Protease-activated receptors (PARs) use a weird and wonderful ligand receptor activation mechanism that allows them to sense changes in the proteolytic milieu. Whereas other receptors recognize ligands carried in solution phase, the PARs receptors carry their ligand (a hexapeptide motif) within their own receptor exodomain. The hexapeptide ligand, however, remains inaccessible to the receptor binding pocket until unveiled by cleavage with a serine protease (1). The newly created N terminus (with the hexapeptide now at the end) folds back into the body of the receptor and docks within the binding pocket (2). From then on, downstream signaling through G proteins and cell activation is similar to other G protein-coupled receptors of the same seven-transmembrane superfamily.
This unique activation mechanism allows PARs to sense the presence of serine proteases in the environment, not just thrombin. Because PARs receptors are found on all cells of the vasculature and the vessel wall, they provide a critical sensing mechanism allowing the body to respond to surgery and cardiopulmonary bypass (CPB; which is known to activate a range of critical serine proteases, including thrombin, kallikrein, plasmin, tryptase, elastase, and others) (3). Three of the four PAR receptors (PAR1, -3, and -4) are cleaved by the serine protease activity of thrombin and can therefore be considered thrombin receptors (4). PAR1 is the high affinity thrombin receptor and PAR4 is the low affinity thrombin receptor on platelets. PAR3 is poorly understood but may be an important thrombin receptor on vascular cells. PAR2 is the odd one out, because it is not a thrombin receptor, being cleaved instead by trypsin, mast cell tryptase, or the ternary coagulation complex of factor Xa-VIIa-TF.
Although PAR1 is recognized for being the high-affinity thrombin receptor, and thus of critical importance to platelet involvement in thrombosis, it should be remembered that other serine proteases, notably trypsin, kallikrein, and low concentrations of plasmin, can also cleave and activate PAR1 (3,5). This is important when considering the effect of serine protease inhibitors in cardiac surgery.
The reason there is so much excitement about the use of thrombin receptor antagonists in cardiac surgery is that they promise to abrogate the pro-thrombotic actions of thrombin on platelets while leaving the coagulation cascade largely untouched—the hope is that thrombotic events can be eliminated without causing undue risk of bleeding (6).
PROMISE OF TARGETING PAR1 IN CARDIAC SURGERY
Several PAR1 antagonists are in clinical development. The most advanced, which just completed glowing safety trials for use in percutaneous coronary intervention (PCI) and is now in a 10,000 patient phase III trial, is a peptide antagonist based on the hexapeptide ligand sequence (7). This blocking peptide sits in the ligand binding pocket and prevents access to the natural ligand, even when that is generated after proteolytic cleavage of PAR1 with thrombin (8). The phase II TRA-PCI safety trial met its primary safety endpoint, showing no increase in thrombolysis in myocardial infarction (TIMI) bleeding when added to standard anti-platelet care, but showing a 46% reduction in major adverse clinical events (7). Figure 1 shows how PAR1 antagonists can block thrombotic complications by preventing platelet activation caused by thrombin, whereas they do not interfere with the hemostatic properties of thrombin in the coagulation cascade.
Figure 1.
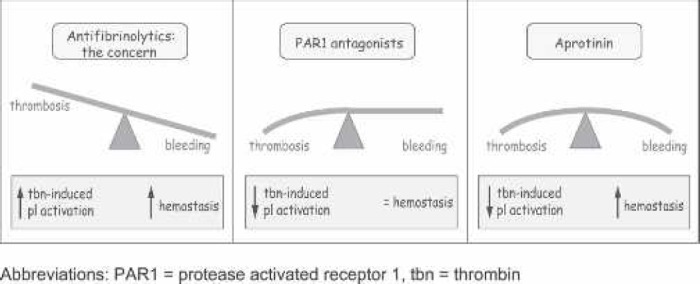
Anti-fibrinolytics and PAR1 antagonists in cardiac surgery. (Left) A concern in cardiothoracic surgery is that, although antifibrinolytics are effective at reducing bleeding, might they not also present a concomitant risk of thrombosis? (Middle) The promise of PAR1 antagonists is that they can inhibit the action of thrombin on platelets while maintaining the hemostatic properties of thrombin in the coagulation cascade. (Right) The TRA-PCI study (phase II safety trial) seems to have borne out this early promise by noting no increase in TIMI bleeding but a 46% reduction in major adverse cardiovascular events after percutaneous coronary intervention. Aprotinin exhibits anti-thrombotic properties in on- and off-pump surgery by inhibiting thrombin-induced platelet activation through PAR1, yet it exhibits simultaneous hemostatic properties by blocking plasmin in the fibrinolytic pathway. tbn, thrombin.
Although specific PAR1 antagonists have stolen the limelight in 2007, the first clinical demonstration of PAR1 antagonism came in 2004 through the use of aprotinin in cardiothoracic surgery with CPB.
PAR1 TARGETING BY APROTININ: TEACHING AN OLD DOG NEW TRICKS
Aprotinin is a broad-spectrum serine protease inhibitor first isolated from cow lung in 1936. It was shown to be a plasmin inhibitor in 1979, and its clinical anti-fibrinolytic properties were co-discovered in 1987 by groups in the United Kingdom and Holland (9,10). From the first studies in cardiothoracic surgery, aprotinin was recognized to preserve platelet function (10). Elegant electron microscopy studies showed that platelet morphology was completely preserved throughout CPB (11). The critical study into the mechanism of platelet protection came in 1998 from a study by Victor Ferraris, which showed excessive bleeding was linked to activation and degranulation of platelets through the high-affinity thrombin receptor PAR1 (12).
Given that thrombin activates PAR1 through a serine protease mechanism and that aprotinin is a serine protease inhibitor, we hypothesized that aprotinin should possess anti-thrombotic properties by preventing thrombin-induced platelet activation. This hypothesis was controversial at the time, following the ambiguous results of the IMAGE trial into graft patency (13). We first studied the effect of aprotinin on washed human platelets and were able to show a dose-dependent inhibition of thrombin-induced platelet aggregation (14). This was achieved at clinically relevant concentrations of aprotinin: 42.6 ± 21.6% inhibition at 50 KIU/mL (p = .0047), 61.0 ± 25.2% inhibition at 100 KIU/mL (p = .0001), and 86.6 ± 8.9% inhibition at 160 KIU/mL (p < .0001).
We next examined whether aprotinin could inhibit PAR1 activation clinically (15). This study confirmed that (i) thrombin was generated during passage of blood through the bypass circuit; (ii) platelets were activated by thrombin because of cleavage of PAR1; (iii) high-dose (Hammersmith dose) aprotinin prevented platelet activation through PAR1 without affecting net thrombin generation; and (iv) the mechanism of PAR1 protection was by preventing proteolytic cleavage of PAR1. In vitro, the mechanism is definitively through targeting of thrombin-induced PAR1 activation. Clinically, we cannot rule out the possibility that aprotinin may also target plasmin and kallikrein, both of which can cleave and activate PAR1, in addition to thrombin.
This clinical study therefore revealed a subtle “anti-thrombotic yet hemostatic mechanism” of action for aprotinin when used in cardiothoracic surgery (Figure 1): anti-thrombotic by virtue of preventing thrombin-induced platelet activation and hemostatic by virtue of antifibrinolytic targeting of plasmin. Thus, like the more modern peptidomimetric PAR1 antagonists, this opportunistic PAR1 antagonist is able to exert anti-thrombotic properties without increasing the risk of bleeding. Better still, because of its additional targeting of plasmin in the fibrinolytic pathway, aprotinin simultaneously delivers anti-thrombotic and hemostatic properties. This is an exceptionally useful pharmacologic profile for a compound used primarily as a hemostatic agent in cardiothoracic surgery. Similar anti-thrombotic yet hemostatic properties of aprotinin have been observed in animal models of thrombosis and clinically in off-pump surgery (16,17). Meta-analyses of the randomized trials have borne out that aprotinin does not add risk to graft patency but significantly lowers the risk of stroke (18). A possible mechanism contributing to stroke protection is through reduced perioperative platelet activation by thrombin (19). Another contributory mechanism would be through reduced thrombin activation of endothelium, which is expected to yield anti-inflammatory and anti-thrombotic drug effects (20).
CONCLUSIONS
Clinical phase II trials in 2007 seem to have borne out anticipated anti-thrombotic benefits of PAR1 antagonism not linked to an increased risk of bleeding. The first clinical demonstration of PAR1 antagonism, however, came from earlier work using the anti-fibrinolytic agent aprotinin. This possesses PAR1 antagonistic properties by virtue of blocking proteolytic activation of PAR1 by thrombin. It is anticipated that PAR1 antagonism will remain an active field for further development in cardiothoracic surgery with CPB, because it holds the prospect of reducing thrombotic complications without incurring a concomitant bleeding risk or even while realizing a simultaneous antifibrinolytic hemostatic benefit.
REFERENCES
- 1.Vu T-KH, Hung DT, Wheaton VI, Coughlin SR.. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–68. [DOI] [PubMed] [Google Scholar]
- 2.Vu T-KH, Wheaton VI, Hung DT, Charo I, Coughlin SR.. Domains specifying thrombin-receptor interaction. Nature. 1991;353:674–7. [DOI] [PubMed] [Google Scholar]
- 3.Parry MA, Myles T, Tschopp J, Stone SR.. Cleavage of the thrombin receptor: identification of potential activators and inactivators. Biochem J. 1996;320:335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landis RC.. Protease activated receptors: clinical relevance to hemostasis and inflammation. Hematol Oncol Clin North Am. 2007;21:103–13. [DOI] [PubMed] [Google Scholar]
- 5.Oikonomopoulou K, Hansen KK, Saifeddine M, et al. Proteinaseactivated receptors, targets for kallikrein signaling. J Biol Chem. 2006;281:32095–112. [DOI] [PubMed] [Google Scholar]
- 6.Derian CK, Maryanoff BE, Zhang HC, Andrade-Gordon P.. Therapeutic potential of protease-activated receptor-1 antagonists. Expert Opin Investig Drugs. 2003;12:209–21. [DOI] [PubMed] [Google Scholar]
- 7.Moliterno DJ.. Results of a Multinational Randomized, Double-Blind, Placebo-Controlled Study of a Novel Thrombin Receptor Antagonist SCH 530348 in Percutaneous Coronary Intervention. American College of Cardiology Meeting, New Orleans, LA, March 24, 2007. [Google Scholar]
- 8.Andrade-Gordon P, Maryanoff BE, Derian CK, et al. Design, synthesis, and biological characterization of a peptide-mimetic antagonist for a tethered-ligand receptor. Proc Natl Acad Sci USA. 1999;96:12257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Royston D, Bidstrup BP, Taylor KM, Sapsford RN.. Effect of aprotinin on need for blood transfusion after repeat open-heart surgery. Lancet. 1987;2:1289–91. [DOI] [PubMed] [Google Scholar]
- 10.van Oeveren W, Jansen NJ, Bidstrup BP, et al. Effects of aprotinin on hemostatic mechanisms during cardiopulmonary bypass. Ann Thorac Surg. 1987;44:640–5. [DOI] [PubMed] [Google Scholar]
- 11.Mohr R, Goor DA, Lusky A, Lavee J.. Aprotinin prevents cardiopulmonary bypass-induced platelet dysfunction. A scanning electron microscope study. Circulation. 1992;86:II405–9. [PubMed] [Google Scholar]
- 12.Ferraris VA, Ferraris SP, Singh A, et al. The platelet thrombin receptor and postoperative bleeding. Ann Thorac Surg. 1998;65:352–8. [DOI] [PubMed] [Google Scholar]
- 13.Alderman EL, Levy JH, Rich JB, et al. Analyses of coronary graft patency after aprotinin use: Results from the International Multicenter Aprotinin Graft Patency Experience (IMAGE) trial. J Thorac Cardiovasc Surg. 1998;116:716–30. [DOI] [PubMed] [Google Scholar]
- 14.Poullis M, Manning R, Laffan M, Haskard DO, Taylor KM, Landis RC.. The antithrombotic effect of aprotinin: Actions mediated via the proteaseactivated receptor 1. J Thorac Cardiovasc Surg. 2000;120:370–8. [DOI] [PubMed] [Google Scholar]
- 15.Day JR, Punjabi PP, Randi AM, Haskard DO, Landis RC, Taylor KM.. Clinical inhibition of the seven-transmembrane thrombin receptor (PAR1) by intravenous aprotinin during cardiothoracic surgery. Circulation. 2004;110:2597–600. [DOI] [PubMed] [Google Scholar]
- 16.Khan TA, Bianchi C, Voisine P, Sandmeyer J, Feng J, Sellke FW.. Aprotinin inhibits protease-dependent platelet aggregation and thrombosis. Ann Thorac Surg. 2005;79:1545–50. [DOI] [PubMed] [Google Scholar]
- 17.Poston RS, White C, Gu J, et al. Aprotinin shows both hemostatic and antithrombotic effects during off-pump coronary artery bypass grafting. Ann Thorac Surg. 2006;81:104–10. [DOI] [PubMed] [Google Scholar]
- 18.Sedrakyan A, Treasure T, Elefteriades JA.. Effect of aprotinin on clinical outcomes in coronary artery bypass graft surgery: A systematic review and meta-analysis of randomized clinical trials. J Thorac Cardiovasc Surg. 2004;128:442–8. [DOI] [PubMed] [Google Scholar]
- 19.Jurk K, Jahn UR, Van AH, et al. Platelets in patients with acute ischemic stroke are exhausted and refractory to thrombin, due to cleavage of the seven-transmembrane thrombin receptor (PAR-1). Thromb Haemost. 2004;91:334–44. [DOI] [PubMed] [Google Scholar]
- 20.Day JRS, Taylor KM, Lidington EA, et al. Aprotinin inhibits proinflammatory activation of endothelial cells by thrombin through the protease-activated receptor 1. J Thorac Cardiovasc Surg. 2006;131:21–7. [DOI] [PubMed] [Google Scholar]