

SUMMARY
The role of internal dynamics in enzyme function is highly debated. Specifically, how small changes in structure far away from the reaction site alter protein dynamics and overall enzyme mechanisms is of wide interest in protein engineering. Using RNase A as a model, we demonstrate that elimination of a single methyl group located >10 Å away from the reaction site significantly alters conformational integrity and binding properties of the enzyme. This A109G mutation does not perturb structure or thermodynamic stability, both in the apo and ligand-bound states. However, significant enhancement in conformational dynamics was observed for the bound variant, as probed over nano- to milli-second time scales, resulting in major ligand repositioning. These results illustrate the large effects caused by small changes in structure on long-range conformational dynamics and ligand specificities within proteins, further supporting the importance of preserving wild-type dynamics in enzyme systems that rely on flexibility for function.
Graphical Abstract
INTRODUCTION
Knowledge of how proteins and enzymes function has broad implications for both fundamental and applied research. Increasing evidence suggests that proteins exist as inter-converting conformers and sample distinct conformations enabled by structural fluctuations that occur over a wide range of time scales (Frauenfelder, 1998; Frauenfelder et al., 1991). Conformational sub-states sampled by proteins have been correlated with promoting function such as binding and catalysis in numerous enzyme systems (Agarwal, 2005; Baldwin et al., 2012; Beach et al., 2005; Cole and Loria, 2002; Eisenmesser et al., 2005; Hanoian et al., 2015; Klinman, 2015; Kohen, 2015; Wang et al., 2006). Advances in biophysical and computational techniques now provide significant means to probe and gain atomistic insights into the dynamics of proteins over a wide range of time scales.
Ribonuclease A (RNase A, EC 3.1.27.5), which catalyzes the hydrolysis of single stranded RNA, has been studied extensively for over five decades and has served as a model for countless biochemical and biophysical analyses. RNase A is a small monomeric protein composed of 124 amino acids with a basic pI of 8.6. It is characterized by a V-shaped kidney structure formed by a core of β-sheets surrounded by loops and α-helices, with the active site lying in the cleft at the bottom of the inverted β-sheet (Wlodawer et al., 1988). This cleft contains subsites for interactions with nucleotide bases (B1, B2, B3) and phosphoryl groups (P0, P1, P2) of single-stranded RNA substrates (Gagne and Doucet, 2013). B1 and B2 subsites preferentially bind to pyrimidine and purine bases, respectively, with a marked preference for adenine in the B2 subsite (Cuchillo et al., 2002; Raines, 1998). Residues His12, Lys41 and His119 in the P1 subsite form the catalytic triad and are crucial for catalytic activity (Pares et al., 1991). In addition to these catalytic residues, a number of residues in the surrounding environment are essential to the optimal ribonucleolytic activity of the enzyme, where changes in the chemistry are known to alter substrate specificity, binding and optimal catalysis.
In recent years, NMR studies have investigated the conformational dynamics of RNase A, suggesting a link between flexibility and enzyme catalysis (Cole and Loria, 2002; Doucet et al., 2011; Doucet et al., 2009; Khirich and Loria, 2015; Watt et al., 2007). The rate constant for conformational exchange (kex) in RNase A was shown to correlate with the rate of enzyme turnover (kcat), implicating long-range secondary structure motions with substrate turnover (Gagne and Doucet, 2013; Loria et al., 2008). Long-range residue mutations have also been shown to drastically alter the catalytic activity, demonstrating the importance of correlated residue sector dynamics in this enzyme. Alanine at position 109 is located on the β5-strand within the V1 domain and is part of the hydrophobic core of the enzyme. This residue is 88% conserved within members of the RNase A superfamily (Smith and Raines, 2006) and 100% conserved among the eight canonical members of human RNases (Sorrentino, 2010). While it has been associated with the chain-folding initiation site (Torrent et al., 2001), other studies suggest a more direct involvement of this residue in the catalytic reaction, with Ala109 potentially making van der Waals interactions with the catalytic residue, His119. Using a relaxation-compensated Carr-Purcell-Meiboom-Gill (15N-CPMG) NMR experiment, Cole and Loria showed that Ala109 experiences millisecond (ms) motions in the apo form of RNase A (Cole and Loria, 2002). This is further supported by NMR titration studies, which demonstrated that the milli-second dynamics of Ala109 is affected by the presence of ligands (Gagne et al., 2012; Toiron et al., 1996). Mutation of A109 to a glycine has been shown to slightly lower the conformational stability of RNase A (Smith and Raines, 2006), suggesting that flexibility at this position may be important for enzyme function.
In this study, we mutated alanine for a glycine at position 109 to characterize the effect of the mutation on the global structure, dynamics, and catalytic activity in RNase A. We determined the crystal structure of the mutant in the apo form and in complex with two single nucleotide ligands, 5′-AMP and 3′-UMP, which bind specifically to the purine and pyrimidine binding sites in WT. These ligands are end-products of the catalytic reaction and are known to bind to the catalytic site of ribonucleases (Raines, 1998). Conformational dynamics of the WT and A109G variant over the nano- to milli-second time scales were characterized using MD simulations and NMR 15N-CPMG experiments. We show that there is no significant difference in the three-dimensional structure of the mutant relative to the WT enzyme in the apo and ligand-bound states. Nonetheless, the mutation induces a repositioning of the 5′-AMP purine, resulting in altered binding affinity caused by changes in the dynamic behavior of the enzyme. Despite minimal effects on the structure, the mutation considerably affects the rate of conformational exchange for residues in the V1 domain, altering loop 4 dynamics on the nano- to milli-second time scales. These results highlight the importance of preserving WT conformational dynamics in enzyme systems that rely on flexibility for function. While single mutational replacements often have neutral effects on protein structure, they can significantly alter function by perturbing long-scale conformational dynamics and by disrupting important motional sectors on multiple time scales.
RESULTS
X-ray crystallography and structural comparison between RNase A and A109G
We determined crystal structures of the A109G mutant in its apo form (PDBID 4WYN), as well as in complex with two single nucleotides adenosine-5′-monophosphate (5′-AMP) (PDBID 4WYP) and uracil-3′-monophosphate (3′-UMP) (PDBID 4WYZ) that bind to the purine and pyrimidine subsites, respectively, to characterize the effect of the mutation on the three-dimensional structure of RNase A (Figure 1). Removal of the methyl group from the side chain at position 109 did not exert major effects on the mutant enzyme structure relative to the WT enzyme in both the apo and the two ligand-bound forms (Figure 2A). The crystals of the A109G mutant of RNase A belong to the orthorhombic crystal system and contain two enzyme molecules in the asymmetric unit (Table 1). Our results show that 3′-UMP binds to the canonical B1 subsite in the catalytic groove of the mutant enzyme in both monomers (Figures 1A & 2C). While most of the hydrogen bonding interactions between 3′-UMP and the enzyme were conserved, a closer examination showed that an additional interaction with His119 was acquired in the mutant enzyme, presumably due to a slight repositioning of the phosphate group (Figure 2C).
Figure 1.

Schematic representation of the mutant A109G in complex with 3′-UMP and 5′-AMP. Positioning of the ligands in the density map for (a) 3′-UMP (PDB 4WYZ) and (b,c) 5′-AMP (PDB 4WYP) on the 3D structure of the mutant A109G. The positioning of the 5′-AMP in c represents a low occupancy. Key residues (Gln11, His12, Lys41, Thr45, Asn71, Gln83, Gln111, His119 and Asp121) are show in gray stick representation. Nitrogen, oxygen and phosphate atoms are colored blue, red and orange, respectively.
Figure 2.

Structural comparison between WT RNase A and mutant A109G. Schematic overlays of (a) apo forms of WT RNase A (grey, PDB 7RSA) and A109G (orange, PDB 4WYN), (b) 5′-AMP-bound WT RNase A (grey, PDB 1Z6S), soaked A109G (orange, PDB 4WYP), A109G monomer B corresponding to the low occupancy position of 5′-AMP (cyan, PDB 4WYP) (c) 3′-UMP-bound WT RNase A (grey, PDB 1O0N) and A109G (orange, PDB 4WYZ). The A109G site is indicated using green sticks representation. 5′-AMP and 3′-UMP are also displayed using stick representation. (d) Overlay of 1H-15N HSQC spectra for the WT RNase A (red) and A109G mutant (green). (e) Mapping of chemical shift differences (Δδ) resulting from the A109G mutation on the primary structure of RNase A. 1H and 15N composite chemical shift differences (Δδ) were calculated according to the following equation (Grzesiek et al., 1996): Δδ (ppm) = [(Δδ2HN + ΔδN/25)/2]½. The inset shows residues with Δδ > 0.05 ppm (blue spheres) highlighted on the three-dimensional structure of A109G (PDB 4WYN). (f) Temperature unfolding for WT (green) and A109G mutant (blue) RNase A determined by circular dichroism.
Table 1.
Crystallographic and structure refinement statistics for the apo, 3′-UMP-, and 5′-AMP-bound forms of RNase A-A109G.
| Crystal | Apo RNase A-A109G | Complex with 3′-UMP | Complex with 5′-AMP |
|---|---|---|---|
| Data Collection
| |||
| Space group | P21212 | P21212 | P21212 |
| Unit cell parameters (Å) | a = 76.2 | a = 76.5 | a = 76.8 |
| b = 51.4 | b = 51.8 | b = 51.7 | |
| c = 58.2 | c = 57.9 | c = 58.2 | |
| Resolution (Å) | 1.80 | 1.45 | 1.50 |
| Total reflections | 321,513 | 374,299 | 603,029 |
| Unique reflections | 21,146 | 39,580 | 35,341 |
| Rsym (%) | 17.7 (22.7) | 13.3 (74.8) | 14.2 (43.4) |
| Rpim (%) | 13.4 (21.5) | 6.8 (4.5) | 6.8 (22.0) |
| CC1/2 (%) | (88.1) | (50.3) | (87.3) |
| I/σI | 6.3 (2.4) | 11.7 (1.4) | 10.6 (4.1) |
| Completeness (%) | 97.7 (91.0) | 93.1 (56.9) | 94.6 (87.0) |
| Redundancy | 2.2 (1.5) | 4.6 (3.0) | 4.8 (4.5) |
|
| |||
| Structure Refinement
| |||
| Rwork/Rfree (%) | 0.145/0.203 | 0.127/0.170 | 0.151/0.186 |
| Protomers per ASU | 2 | 2 | 2 |
| Amino acids per ASU | 249 | 249 | 249 |
| Ligands per ASU | 0 | 2 | 2 |
| Solvent molecules | 382 | 589 | 425 |
| RMSDs | |||
| Bond lengths (Å) | 0.011 | 0.010 | 0.010 |
| Bond angles (°) | 1.288 | 1.445 | 1.297 |
| Average B factor (Å2) | 18.6 | 19.1 | 17.3 |
| Ramachandran | |||
| Favored/allowed (%) | 98.0/2.0 | 97.7/2.3 | 97.0/3.0 |
In striking contrast, we found a variation in the position of 5′-AMP in the catalytic pocket of the A109G mutant, with the ligand adopting two distinct orientations (Figures 1B–C). While it is abundantly documented that the adenine base of single-stranded RNA substrates bind to the B2 subsite in RNase A (Gagne and Doucet, 2013), our crystal structure shows that the base of 5′-AMP faces the solvent and not the B2 subsite when bound to monomer A (Figures 1B & 2B). In this particular complex, the adenine and ribose rings of 5′-AMP were rotated ~180° relative to the canonical conformation observed in the complex with the wild-type enzyme. Instead of being in the B2 subsite, the adenine ring sits atop the side chain of Lys41. Consequently, hydrogen bonds between the purine base and residues Asn67, Gln69 and Asn71, and the stacking interaction between the purine base and the imidazole ring of His119 were absent in the crystal of the A109G mutant. Intriguingly, 5′-AMP was bound in a distinct orientation in the second monomer of the asymmetric unit, which we shall refer to as monomer B (Figures 1C & cyan in 2B). During later stages of the structure refinement, relatively strong positive peaks in the mFo-DFc difference electron density map were observed near subsites B2 and P1 in monomer B. We interpreted the density as adenine ring and phosphate, and thus modeled and refined 5′-AMP. Because the occupancy of this particular ligand refined to 0.76, it is reasonable to suggest that this alternate orientation is not a preferred conformation of 5′-AMP by the A109G mutant of RNase A.
Chemical shift variations between RNase A and A109G
An overlay of the NMR 1H-15N HSQC spectra of both the WT and A109G RNases showed variations in the chemical shifts for a number of residues (Figure 2D). Chemical shift variations were calculated as differences between shifts of the mutant and WT RNases, with variations >0.05 ppm being considered significant. Most residues displaying significant variations were found in close proximity to the mutation site (Figure 2E). These include Asn71 and Asp121, residues important for interaction with the ligands in the B2 and P1 subsites, respectively, and residues in loop 4 (residues 65–72), which are highly dynamic (Gagne et al., 2012) and have been shown to be important for the binding of the substrate (Loria et al., 2008), showed significant variations relative to WT. Residues Ser18 and Cys95, located in the flexible loops 1 and 2, respectively, in addition to Asn27 (at the end of loop 1) exhibited large variations in chemical shifts while being positioned away from the mutation site. It remains unclear why these distant residues experience such high chemical shift variations, but since our X-ray studies show that they do not reflect major structural changes, they could result from long-range dynamic perturbations previously observed between the active site and distant loops in RNase A, primarily in loop 1 (Doucet et al., 2011; Doucet et al., 2009; Watt et al., 2007).
Thermal unfolding of RNase A and A109G
Characterization of the WT and A109G apo forms was also performed using circular dichroism (CD) at 25°C. Both proteins exhibited identical spectra characteristic of α-helical and β-sheet structures (not shown) (Robertson and Baldwin, 1991; Seshadri et al., 1994). Thermal unfolding was performed by increasing the temperature up to 80°C, with changes in the secondary and tertiary structures monitored using CD at 210 nm. A melting temperature (Tm) of 63.9 ± 0.4°C was observed for the WT protein, consistent with values reported previously (Robertson and Baldwin, 1991; Seshadri et al., 1994). A comparable melting temperature of 61.4 ± 1.1°C was observed for the A109G mu tant, suggesting that the mutation does not induce significant effects on the stability of the protein (Figure 2F).
Binding kinetics of WT RNase A and A109G variant
We used isothermal titration calorimetry (ITC) to characterize the kinetics of binding of two single nucleotide product analogs 5′-AMP and 3′-UMP for WT and the A109G variant (Table 2). The thermodynamic parameters and the dissociation constant Kd for the binding of 3′-UMP showed minimal change for the mutant (WT-Kd (3′-UMP) = 67 ± 6 μM; A109G-Kd (3′-UMP) = 50 ± 8 μM), suggesting that binding of the pyrimidine ligand to the B1 subsite is not affected by the mutation. These results are consistent with the minor changes observed in the 3D structure. In striking contrast, binding of 5′-AMP led to greater than four-fold reduction in the binding affinity (WT-Kd (5′-AMP) = 105 ± 1 μM; A109G-Kd (5′-AMP) = 440 ± 26 μM). These results suggest that binding of the purine ligand to the B2 subsite is significantly affected in the mutant. The reduction in Kd, corresponding to higher energy required for binding to the ligand, is consistent with the displacement of 5′-AMP from the B2 subsite, observed from the 3D structure of the mutant (Figure 1B–C).
Table 2.
Dissociation rate constant(Kd) for WT RNase A and A109G variant determined for the binding of single nucleotide product analogs 3′-UMP and 5′-AMP at 25°C.
| Kd (μM) | Kd (μM) | |
|---|---|---|
| 3′-UMP | 5′-AMP | |
|
| ||
| WT | 67 ± 6 | 105 ± 1 |
| A109G | 50 ± 8 | 440 ± 26 |
Conformational dynamics on the microsecond-to-millisecond time scale
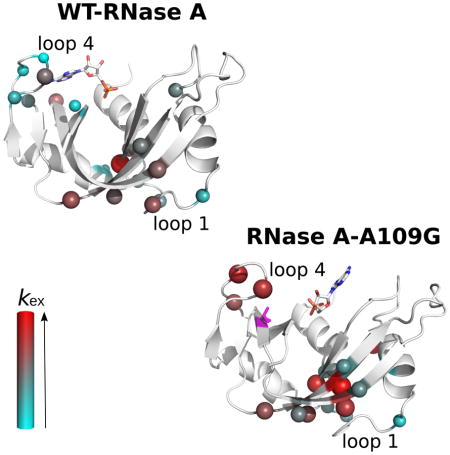
Many residues in RNase A experience conformational exchange that can be described by a two-state global motion inter-conversion process with an exchange rate constant (kex) of ~1700 s−1. In an effort to provide insight into the microsecond-to-millisecond (μs-ms) dynamic changes induced by the A109G replacement, we performed relaxation-compensated Carr-Purcell-Meiboom-Gill (15N-CPMG) experiments on the apo form and in complex with the two ligands (5′-AMP and 3′-UMP) (Figure 3). In the free forms, a total of 17 residues showed conformational exchange in both the WT and A109G variant. Residues experiencing motion on this time scale show clear relaxation dispersion profiles that are localized to clusters 1 and 3 in both proteins, as defined previously (Gagne et al., 2012). The global exchange rate (kex) of 1621 ± 152 s−1 calculated for the free WT RNase A is consistent with results previously reported (Doucet et al., 2011; Doucet et al., 2009; Watt et al., 2007). The A109G variant displayed enhanced global conformational exchange; a shift attributed to increased ms dynamics in the loop 4 environment (cluster 1) (Figure 3, Table 3). Indeed, while a few residues of loop 4 in A109G either become rigid or experience motions outside the 15N-CPMG time frame, global kex for the dynamic residues of this important active-site loop that are involved in purine recognition (Lys66, Gln69, Asn71) is significantly increased in A109G (kex = 3649 ± 473 s−1) relative to WT (kex = 1548 ± 82 s−1) (Table 3).
Figure 3.

Effects of 5′-AMP and 3′-UMP binding on the millisecond dynamics of WT RNase A and mutant A109G. Millisecond time scale (ms) conformational exchange experienced by apo, 5′-AMP- and 3′-UMP-bound forms of WT RNase A and mutant A109G. Motions were probed by 15N-CPMG relaxation dispersion experiments at 500 MHz and 800 MHz (298 K). Residues were considered for further analysis only if the difference in measured R2 (1/Tcp) values at fast (Tcp = 0.625 ms) and slow (Tcp = 10 ms) pulsing rates was greater than 2 s−1, similar to previous reports (Cole and Loria, 2002; Doucet et al., 2009; Gagne et al., 2012). Blue spheres: residues showing 15N-CPMG dispersion profiles with ΔR2 (1/Tcp) > 2 s−1; orange spheres: residues showing no conformational exchange relative to the apo form (i.e., dampened ms dynamics upon ligand binding) or outside of the 15N-CPMG time frame; green spheres: residues gaining conformational exchange [ΔR2 (1/Tcp) > 2] upon ligand binding; gray spheres: assigned residues in the apo form that cannot be assigned in the ligand-bound form due to line broadening; purple spheres: position 109. Residues are highlighted on the 3D structure of WT RNase A (apo form: PDB 7RSA; 3′-UMP-bound: PDB 1O0N; 5′-AMP-bound: PDB 1Z6S) and mutant A109G (apo form: PDB 4WYN; 3′-UMP-bound: PDB 4WYZ; 5′-AMP-bound: PDB 4WYP). Insets show representative dispersion curves acquired at 500 MHz (red) and 800 MHz (black) for a subset of loop 4 (cluster 1) in apo forms of WT and A109G. Residue clusters are identified according to (Gagne et al., 2012).
Table 3.
Conformational exchange rates (kex) for WT RNase A and mutant A109G at 25°C.
| kex (s−1) | |||
|---|---|---|---|
| Cluster 1 (loop 4)1 | Cluster 3 | Global | |
| WT (apo) | 1548 ± 82 | 1438 ± 125 | 1621 ± 152 |
| A109G (apo) | 3649 ± 473 | 1115 ± 119 | 3145 ± 1651 |
|
| |||
| WT (5′-AMP) | 687 ± 41 | 1398 ± 108 | 750 ± 111 |
| A109G (5′-AMP) | 1580 ± 143 | 1669 ± 279 | 3328 ± 543 |
|
| |||
| WT (3′-UMP) | 1136 ± 46 | 1639 ± 128 | 1375 ± 50 |
| A109G (3′-UMP) | 2234 ± 523 | 1189 ± 154 | 1463 ± 216 |
RNase A dynamic residue clusters are defined as in ref. (Gagne et al., 2012).
A total of 16 and 22 residues exhibited conformational dynamics in the 5′-AMP-bound WT and mutant RNases, respectively (Figure 3). A gain in ms dynamics is predominantly observed in clusters 1, 3 and 4 in both proteins. This is particularly true for the mutant, with residues Val43, Asn44, Thr45, Cys84, Glu86 and Ser89 showing relaxation dispersion profiles upon ligand binding (Figure 3, green spheres). Repositioning of the 5′-AMP ligand modified the network of dynamic residues in cluster 4, especially on β1 (residues 43–47), which now faces the 5′-AMP. The global kex for the A109G protein variant shows a four-fold increase relative to WT. While there is a gain in dynamics for a number of residues in cluster 3 of the mutant, we note that the average kex is comparable for both proteins. The difference in kex is quite striking for cluster 1, which shows a two-fold increase in the ms dynamics for the mutant (Table 3). In accordance with the repositioning of 5′-AMP and reduced binding affinity in the A109G variant, these results support the fact that enhanced ms dynamics in cluster 1 (loop 4) hinder the proper positioning of 5′-AMP in the B2 subsite, resulting in the repositioning of the ligand to the B1 subsite.
A major reorganization of the residues displaying significant flexibility on the ms time frame is observed in the 3′-UMP-bound RNase A, a result not seen in the A109G mutant (Figure 3). In the WT, only 6 residues preserved ms motions when compared to the apo form (blue spheres), while a gain of dynamics upon ligand binding is observed for 12 additional residues (green spheres). These additional residues are located primarily on α2 (residues 30–36 on cluster 3), a region devoid of ms dynamics in the apo form. The mutant protein shows a gain in conformational exchange for 9 residues (green spheres), in addition to 14 residues that retain the motions of the apo form (blue spheres), most of them localized in cluster 3. As observed with the free and 5′-AMP-bound forms, cluster 1 in A109G shows a two-fold increase in kex, while the average dynamics in cluster 3 are comparable to WT (Table 3).
Conformational dynamics on the nanosecond-to-microsecond time scale
Molecular dynamics (MD) simulations were performed for WT and A109G in complex with 5′-AMP and 3′-UMP. Ligands with an intact phosphodiester bond between the single nucleotides were prepared to simulate the reactant state while single nucleotides without the phosphodiester bond were used to mimic the product state. We note that MD simulation results were obtained using dinucleotide substrate and corresponding cleaved products states while the NMR relaxation dispersion experiments were performed using mononucleotide ligands. Root mean square fluctuations (RMSF) were calculated for each residue, over a 1-μs simulation trajectory for WT and the A109G variant. To quantitatively assess the difference in global conformational dynamics on the nanosecond-to-microsecond (ns-μs) time scale for the WT and A109G mutant, we characterized the top ten eigenmodes using quasi-harmonic analysis (Agarwal et al., 2004; Levy et al., 1984). Primary flexibility of the WT and A109G proteins in the reactant and product states are shown in Figure 4. On the μs time scale, the mutant showed reduction of dynamics in loop 4, while significantly enhanced dynamics was observed for α2 and part of loop 1 relative to the WT for the reactant state. In contrast, the product state showed dynamics similar to that of the WT for most of the protein. There was a marginal reduction in the conformational dynamics of loop 1 (residues 16–25) in the mutant enzyme (Figure 4), consistent with long-range chemical shift effects observed in this study and previous reports which showed that the dynamics of loop 1 are correlated with efficient product release in RNase A (Doucet et al., 2009).
Figure 4.

Root mean square fluctuations (RMSFs) for WT RNase A and A109G mutant for a 1-μs molecular dynamics simulation. Enzyme backbone flexibility was determined from the top ten eigenmodes of the RMSFs. (a–d) RMSF values are shown using tube representations for the WT (a, b) and A109G mutant (c, d) RNase A for the reactant (left) and product (right) states. Thickness of the tube corresponds to the flexibility of the residues, with thicker tubes (red end of the spectrum) representing more flexible regions and thinner tubes (blue end of the spectrum) denoting less flexible regions in the protein. (e–f) Comparison of the RMSFs as a function of sequence for the reactant (e) and product (f) states for the WT (black) and mutant (red) proteins. Loop 1 and loop 4 (labeled L1 and L4, respectively) regions are represented using dotted lines. Panels A–D depict RMSF based on Cα while panels E-F are based on all atom calculations for each residue.
In an effort to gain mechanistic insights into the structural and dynamical changes affecting enzyme function, we performed quasi-anharmonic analysis (QAA) on conformations sampled during the MD simulations to identify the conformational sub-states associated with the reactant and product landscape. This type of analysis has been previously used for other enzyme systems to provide unique insights into the role of conformational sub-states and long time-scale fluctuations (Ramanathan et al., 2012; Ramanathan et al., 2011). For WT and A109G, our results show that the wild type reactant state consists of multiple sub-states and is much more flexible, while the product state consists of a single state and appears more constrained (Figure 5). The first two QAA modes, corresponding to the global conformational fluctuations, show large amplitude in the loop regions. In the A109G mutant, the opposite behavior is observed: the reactant state appears to be much more rigid while the product state is much more flexible with multiple states (two minor sub-states are depicted for illustration purposes, however, additional states can also be characterized). Further, the global conformational fluctuations in the mutant, associated with the inter-conversion between the reactant and product states, appear to show much smaller amplitudes for the loop regions, except for loop 4 in mode 2. Note that the cyan sphere in figure 5 marks the location of the A109G mutation. We also determined changes in the correlated motions between residues using a difference of the dynamic cross-correlation maps (DCCM) between the mutant and WT for the reactant and product states. A comparison of this DCCM difference for the reactant state revealed enhanced correlated motions in the mutant, between residues 12–18 and residues 5–11, 41–59, 75–81 and 108–119, regions that include the catalytic triad residues (Figure S1). This enhanced dynamics of regions encompassing the catalytic residues may possibly contribute to the altered catalytic efficiency observed for the mutant. Further, lack of correlated motions between residues of loops 1 (residues 3–17) and 4 (residues 63–70), seen in the WT and also shown to play an important role in modulating catalysis, were observed to be absent in the A109G mutant. In contrast to the reactant state, the product state DCCM difference showed anticorrelated motions between residues. Overall, the computational studies indicate that the A109G mutant shows enhanced correlated motions in the vicinity of the active site and distal regions of the enzyme in the ground state for the bound reactant complex on the μs time-scale. Further studies are required to investigate the role of increased flexibility in the ground reactant in accessing functionally relevant conformational sub-states that have catalytically competent structural and dynamical features for the substrate cleavage (Ramanathan et al., 2014).
Figure 5.

Conformational sub-states and global fluctuations associated with reactant-product inter-conversion identified using computational simulations and QAA. Top row shows wild-type and bottom row shows the A109G variant. The sub-states are identified using a mixture of Gaussians model (McLachlan and Basford, 1988). Mode 1 (displacement along γ1 axis) corresponds to the large-scale conformational fluctuation (or primary relaxation) associated with the reactant-product sub-states inter-conversion; similarly mode 2 (displacement along γ2 axis) corresponds to the secondary conformational fluctuation.
DISCUSSION
Mutations located away from the active site have been shown to drastically alter the catalytic activity of enzymes. In this study, we characterized the effect of a single residue replacement of the alanine at position 109 to glycine on the structure, dynamics and binding affinity in RNase A. We obtained crystal structures of the A109G mutant in the apo form and in presence of two end-products of the reaction, 5′-AMP and 3′-UMP, to characterize the effect of the mutation on ligand binding. Our results show that the mutation does not significantly perturb the three-dimensional structure of the protein, with only minor differences in loop 1, which has been shown to exert long-range effects on RNase A catalysis (Doucet et al., 2011; Gagne et al., 2012).
While the structural differences between the WT and mutant enzymes were minimal, we observed a major repositioning of 5′-AMP ligand from the B2 subsite located near loop 4, to a location where the adenine base faces the solvent rather than the enzyme. As a result of this repositioning, the purine base now interacts with Lys41 instead of its usual partners (Asn67, Gln69 and Asn71). Interactions between the phosphate group of the nucleotide and residues Gln11, His12, His119 and Phe120 are preserved in the mutant (Figures 1A & 1B). It is important to note that a smaller fraction of 5′-AMP interacts with the mutant enzyme in a manner reminiscent of the canonical mode of binding. However, even in this orientation, the adenine base is not optimally positioned within the B2 subsite, but rather adopts a position partially between the B1 and B2 subsites. Apart from the His119, all other interactions are lost in this orientation of the ligand (Figures 1A & 1C). These results emphasize the large effect of a single distal residue substitution on the ligand binding affinity of the protein.
We characterized the effect of the A109G mutation on the conformational dynamics of the protein by performing a series of biophysical experiments and computational analyses. Relaxation dispersion 15N-CPMG experiments were used to extract the rate exchange (kex) for each residue experiencing conformational dynamics on the millisecond time scale. Previous studies demonstrated that 15N-CPMG provides relevant information in relation to the function of RNase A (Beach et al., 2005; Cole and Loria, 2002; Doucet et al., 2011; Gagne et al., 2012). Significant changes in the dynamic profiles are observed for the 5′-AMP-bound state of the A109G mutant, a reflection of ligand repositioning (Figure 3). We note that the 3′-UMP-bound form displays very different dynamics for the mutant protein, a change that is not reflected in the three-dimensional structure of the protein. Interestingly, chemical shift differences for the main catalytic residue His12 upon binding to both ligands are considerably affected in the mutant. His12 is known to be rigid on the fast time scale (Merlino et al., 2002), which might be a requirement for proper catalysis. These results suggest the existence of different conformational pre-equilibria between the ligand-bound forms in the WT and mutant RNase A.
We performed global fit kex calculations for residues located in pre-defined clusters corresponding to previously described dynamic sectors in RNase A (Gagne et al., 2012). kex values provide valuable information on the conformational dynamics of individual residue clusters, which can be correlated with the catalytic activity of the protein. We observed a significant increase of the kex in cluster 1 (loop 4) for the mutant in both the apo and ligand-bound forms. These results are supported by lower Rex for all residues in this cluster displaying significant conformational flexibility (Figure 3). Interestingly, similarly enhanced conformational dynamics of loop 4 (cluster 1) were also observed on the nano- to micro-second time scales probed using MD simulations (Figure 4). Further, a difference of the correlated motions between the mutant and WT revealed a lack of correlated motions between loops 1 and 4 in the mutant, suggesting a contribution of this correlated motion towards effective catalysis (Figure S1). These results suggest that the enhanced dynamics observed for loop 4 in the mutant may hinder the interaction of the ligand with its hydrogen-bonding partners (Asn67, Gln69 and Asn71) in the B2 subsite. It was previously shown that Thr45 forms hydrogen-bonding interactions with the pyrimidine base while sterically excluding the purine base from this subsite (delCardayre and Raines, 1994). As a result, the 5′-AMP adopts a position energetically unfavorable between the B1 and B2 subsites and exposed to solvent. We found no significant effect of the enhanced dynamics of loop 4 on the location of 3′-UMP within the structure of the A109G variant, demonstrating that pyrimidine binding is specific to B1 and independent from purine binding in B2.
In conclusion, our results show that mutation of alanine at position 109 to a glycine residue does not perturb the 3D backbone structure of RNase A in the apo form and in complex with two single nucleotides, 5′-AMP and 3′-UMP. Nevertheless, a major repositioning of 5′-AMP to the solvent between the B1 pyrimidine-specific and B2 purine-specific subsites is observed, supporting the role of multiple time scale changes in the dynamics of the protein induced by this single alanine to glycine replacement. While there is no significant change in structure, the mutation induced large changes in the dynamics of the mutant relative to the WT RNase A. Enhanced dynamics of distal protein regions are observed on the nano- and milli-second time scales for the mutant system determined from MD simulations and NMR relaxation experiments. In particular, conformational dynamics of loop 4 is also significantly enhanced in the apo and ligand-bound forms of the A109G mutant, offering an explanation for the modest yet significant changes observed in binding and catalysis. These results showcase the large effects caused by seemingly modest residue substitutions on the long-range conformational dynamics and residue binding specificities within proteins. This study further underlines the importance of preserving wild type dynamics in enzyme systems that rely on flexibility for function, which is of critical significance to protein engineering and enzyme design. These results highlight the effect of single-point mutation in RNase A on the shift in the conformational dynamics, and its correlation with the catalytic efficiency. As preliminary evidence already indicates, these results also provide new insights suggesting that modulating conformational dynamics may be a mechanism used by nature for regulating enzyme activity, thus providing new opportunities for developing new enzyme engineering strategies (Agarwal et al., 2012).
EXPERIMENTAL PROCEDURES
DNA Constructs
Oligonucleotide synthesis was performed by Bio Basic (Markham, ON). The WT, 372-bp bovine RNase A gene inserted into pET22b(+) and codon-optimized for Escherichia coli expression was a generous gift from J. Patrick Loria (Yale University, New Haven, CT). The A109G mutation was generated by the QuikChange site-directed mutagenesis protocol (Agilent Technologies, Mississauga, ON) using the two following complementary primers: A109G-Forward, 5′-GCGAATAAACATATTATTGTTGGCTGCGAAGGCAATCCGTATGTG-3′; A109G-Reverse, 5′-CACCTACGGATTGCCTTCGCAGCCAACAATAATATGTTTATTCGC-3′. The amplified PCR product was DpnI-treated and transformed into E. coli BL21(DE3) for all protein expression procedures. Colonies were selected and the presence of the mutation was confirmed by DNA sequencing (Génome Québec, Montréal, QC).
Protein Expression and Purification
15N-labeled samples were prepared by growing E. coli BL21(DE3) in M9 minimal medium and enzymes were purified as previously described (Doucet et al., 2009). Protein concentration was determined using an extinction coefficient of 9,800 M−1cm−1 (Sela and Anfinsen, 1957).
Crystallization and structure determination
Crystals of apo RNase A-A109G were obtained by the sitting drop vapor-diffusion method. Equal volumes of the protein solution (~30 mg/mL) and the well buffer were mixed, and the drops were incubated at room temperature (21°C). Crystals were obtained using the NeXtal PACT Suite (Qiagen). The high-resolution crystals of RNase A-A109G were obtained from either 0.1M MIB Buffer, pH 8.0 and 25% (w/v) PEG 1500; 0.1M MIB Buffer, pH 6.0 and 25% (w/v) PEG 1500; or 0.1M HEPES, pH 7.0, 0.2M NaCl and 20% (w/v) PEG 6000. The binary complex crystals were obtained by soaking apo enzyme crystals with 50 mM of either 3′-UMP (Chem-Impex International) or 5′-AMP (Sigma-Aldrich). All crystals were cryoprotected either with 15% (w/v) PEG 400 or 20% ethylene glycol prior to X-ray exposure. Data were collected at liquid nitrogen temperature at the Southeast Regional Collaborative Access Team 22-ID (SER-CAT) beamline at the Advanced Photon Source, Argonne National Laboratory. The diffraction data were processed in HKL2000 (Otwinowski and Minor, 1997). The crystal structure of apo RNase A-A109G was determined by molecular replacement in Phaser (Mccoy et al., 2007) using the wild-type bovine RNase A structure (PDB ID code 1KF5) as a search model. The refined apo RNase A-A109G was used to phase other RNase A-A109G crystals. The structure refinement was performed in Phenix (Adams et al., 2010), and the model building was done in Coot (Emsley and Cowtan, 2004; Krissinel and Henrick, 2004). All figures were produced in PyMOL (The PyMOL Molecular Graphics System, Version 1.2, Schrödinger, LLC).
NMR Experiments
All NMR experiments were carried out on Agilent 500 MHz and 800 MHz NMR spectrometers equipped with triple-resonance cold probes and pulsed-field gradients. NMR spectra were recorded at 298 K on samples containing 0.75 mM 15N-labeled RNase A (or the A109G variant) in 5 mM MES-NaOH buffer (pH 6.4) with 7 mM NaCl, 0.01% NaN3, and 10% 2H2O. Backbone resonances were assigned by standard multidimensional experiments (Cavanagh et al., 2007). Relaxation-compensated 15N-CPMG experiments were acquired in an interleaved fashion with Tcp 15N-CPMG repetition delays of 0.625, 0.714 (×2), 1.0, 1.25, 1.67, 2.0, 2.50 (×2), 3.33, 5.0, and 10 ms, using a total relaxation period of 40 ms. All NMR spectra were processed using NMRPipe (Delaglio et al., 1995), in-house scripts and analyzed with Sparky (Goddard and Kneller). Global residue fits and model analyses were performed by fitting 500 MHz and 800 MHz relaxation dispersion data to the full single-quantum 15N-CPMG equation (Manley and Loria, 2011) using GraphPad Prism 5. NMR titration experiments were performed as previously described (Gagne et al., 2012), using the single-nucleotide ligands 3′-UMP and 5′-AMP reconstituted in the NMR buffer.
Molecular Dynamics Simulations
RNase A-ligand complexes were modeled from the PDB structure 1U1B, using only the coordinates of chain A. The ligands were modeled based on a related PDB structure 1RCN. Residue 109 was computationally mutated from alanine to glycine by removing the methyl group to generate the mutant structure. Preprocessing steps and simulations were performed using the AMBER simulation package (Case et al., 2005). Simulations were performed with AMBER ff98 force-field and SPC/E water model. For system preparation, the protein-ligand complex was solvated and the system was neutralized through the addition of four Cl− counterions. After the preprocessing steps, the system was equilibrated using the protocol described previously (Agarwal, 2004). All production runs were performed at 300K under NVE (constant volume and energy) conditions. The WT and mutant structures in complex with ligands were simulated for 1 μs for each system (after 50 ns of additional equilibration). AMBER’s GPU-enabled pmemd simulation engine was used for equilibration and production runs. Backbone (Cα) and all-atom flexibility of simulation trajectories was determined from the RMSF, computed by aggregating the magnitude of displacement eigenmodes computed using the quasi-harmonic analysis (QHA) in the ptraj analysis module in AMBER. As described previously (Ramanathan and Agarwal, 2011), only the top 10 QHA modes were used in the analysis to focus on the principal dynamics or long time-scale fluctuations in the proteins.
Isothermal Titration Calorimetry (ITC)
WT RNase A and mutant A109G enzymes were prepared in 20 mM MES, pH 6.4 at a concentration of 0.2 mM. Freshly prepared 3′-UMP and 5′-AMP were reconstituted in the same buffer at a concentration of 3 mM (15-fold higher than the protein concentration). All experiments were performed on a MicroCal MCS microcalorimeter (GE Healthcare, Piscataway, NJ) at 25°C. A minimum of three repeats was performed for each ligand/enzyme combination. A total of 16 injections (2.5 μL each) were used for the experiment, with a delay of 130 seconds between each injection. Values for n, ΔH, ΔS andKd were calculated by fitting ΔQ for each injection to a sigmoidal curve in Origin 7.0 (OriginLab, Northampton, MA) using a one-site model, as previously described (Freyer and Lewis, 2008).
Circular dichroism spectroscopy (CD)
CD spectra were recorded using a Jasco J-815 spectropolarimeter equipped with a thermostated cell holder. Proteins were reconstituted at a concentration of 50 μM in phosphate-buffered saline solution (PBS, pH 7.4) in the mid-UV (170–290 nm) in a quartz cuvette with 0.1-cm optical path length. CD spectra were recorded at a scan speed of 100 nm/min, a 2-nm bandwidth, and a response time of 2 sec at a temperature of 25°C. The sample compartment was pur ged with nitrogen, and spectra were averaged over 5 scans. The contribution of the solvent to the spectra was subtracted using the Spectral Management software (Jasco). Ellipticity at 210 nm was measured at 2.5°C interva ls, with 5 seconds equilibration time, up to 80°C. The denaturation temperature (Tm), corresponding to the temperature by which half of the tertiary structure at a specific wavelength (210 nm) is denatured, was determined using Spectral Analysis (Jasco Analytical Instruments).
Supplementary Material
Highlights.
A long-range mutation affects conformational integrity and binding in RNase A
The mutation induces major repositioning of the purine ligand but not pyrimidine
Enhanced dynamics in distal regions are observed on the catalytic time scale
Significant enhancement in loop dynamics induces major ligand repositioning
Acknowledgments
The authors thank Tara Sprules and Sameer Al-Abdul-Wahid of the Quebec/Eastern Canada High Field NMR Facility (McGill University) for their excellent technical assistance, in addition to Laurie-Anne Charest for her help with NMR titration experiments. This work was supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) under award number R01GM105978 (to N.D. and P.K.A.) and a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant under award number RGPIN 402623-2011 (to N. D.). D.G. holds a NSERC Alexander Graham Bell Canada Graduate Scholarship. N.D. holds a Fonds de Recherche Québec – Santé (FRQS) Research Scholar Junior 1 Career Award.
Footnotes
AUTHOR CONTRIBUTIONS
D.G. and N.D. conceived and designed the research. D.G. produced and purified all enzymes, in addition to performing and analyzing all the biophysical and NMR experiments. R.L.F. and M.S. performed and analyzed all crystallographic experiments. C.N. and P.K.A. performed and analyzed all molecular dynamics simulations. D.G., C.N., P.K.A and N.D. wrote the manuscript. All authors reviewed and contributed to the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal PK. Computational Studies of the mechanism of cis/trans isomerization in HIV-1 catalyzed by cyclophilin A. Proteins: Structure, Function and Bioinformatics. 2004;56:449–463. doi: 10.1002/prot.20135. [DOI] [PubMed] [Google Scholar]
- Agarwal PK. Role of protein dynamics in reaction rate enhancement by enzymes. J Am Chem Soc. 2005;127:15248–15256. doi: 10.1021/ja055251s. [DOI] [PubMed] [Google Scholar]
- Agarwal PK, Geist A, Gorin A. Protein dynamics and enzymatic catalysis: investigating the peptidyl-prolyl cis-trans isomerization activity of cyclophilin A. Biochemistry. 2004;43:10605–10618. doi: 10.1021/bi0495228. [DOI] [PubMed] [Google Scholar]
- Agarwal PK, Schultz C, Kalivretenos A, Ghosh B, Broedel SE. Engineering a hyper-catalytic enzyme by photoactivated conformation modulation. J Phys Chem Lett. 2012;3:1142–1146. [Google Scholar]
- Baldwin AJ, Walsh P, Hansen DF, Hilton GR, Benesch JL, Sharpe S, Kay LE. Probing dynamic conformations of the high-molecular-weight alphaB-crystallin heat shock protein ensemble by NMR spectroscopy. J Am Chem Soc. 2012;134:15343–15350. doi: 10.1021/ja307874r. [DOI] [PubMed] [Google Scholar]
- Beach H, Cole R, Gill ML, Loria JP. Conservation of mus-ms enzyme motions in the apo- and substrate-mimicked state. J Am Chem Soc. 2005;127:9167–9176. doi: 10.1021/ja0514949. [DOI] [PubMed] [Google Scholar]
- Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. Journal of computational chemistry. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh J, Fairbrother WJ, Palmer AG, Rance M, Skelton NJ. Protein NMR Spectroscopy: Principles and Practice. Elsevier Academic; San Diego: 2007. [Google Scholar]
- Cole R, Loria JP. Evidence for flexibility in the function of ribonuclease A. Biochemistry. 2002;41:6072–6081. doi: 10.1021/bi025655m. [DOI] [PubMed] [Google Scholar]
- Cuchillo CM, Moussaoui M, Barman T, Travers F, Nogues MV. The exo- or endonucleolytic preference of bovine pancreatic ribonuclease A depends on its subsites structure and on the substrate size. Protein science : a publication of the Protein Society. 2002;11:117–128. doi: 10.1110/ps.13702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. Journal of biomolecular NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- delCardayre SB, Raines RT. Structural determinants of enzymatic processivity. Biochemistry. 1994;33:6031–6037. doi: 10.1021/bi00186a001. [DOI] [PubMed] [Google Scholar]
- Doucet N, Khirich G, Kovrigin EL, Loria JP. Alteration of hydrogen bonding in the vicinity of histidine 48 disrupts millisecond motions in RNase A. Biochemistry. 2011;50:1723–1730. doi: 10.1021/bi1018539. [DOI] [PubMed] [Google Scholar]
- Doucet N, Watt ED, Loria JP. The flexibility of a distant loop modulates active site motion and product release in ribonuclease A. Biochemistry. 2009;48:7160–7168. doi: 10.1021/bi900830g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Frauenfelder H. The energy landscape in non-biological and biological molecules. Nature Structural Molecular Biology. 1998;5:757–759. doi: 10.1038/1784. [DOI] [PubMed] [Google Scholar]
- Frauenfelder H, Sligar S, Wolynes P. The Energy Landscapes and Motions of Proteins. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- Freyer MW, Lewis EA. Isothermal titration calorimetry: experimental design, data analysis, and probing macromolecule/ligand binding and kinetic interactions. Methods in cell biology. 2008;84:79–113. doi: 10.1016/S0091-679X(07)84004-0. [DOI] [PubMed] [Google Scholar]
- Gagne D, Charest LA, Morin S, Kovrigin EL, Doucet N. Conservation of flexible residue clusters among structural and functional enzyme homologues. J Biol Chem. 2012;287:44289–44300. doi: 10.1074/jbc.M112.394866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagne D, Doucet N. Structural and functional importance of local and global conformational fluctuations in the RNase A superfamily. FEBS J. 2013 doi: 10.1111/febs.12371. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Kneller DG. Sparky 3. University of California; San Francisco: [Google Scholar]
- Grzesiek S, Stahl SJ, Wingfield PT, Bax A. The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR. Biochemistry. 1996;35:10256–10261. doi: 10.1021/bi9611164. [DOI] [PubMed] [Google Scholar]
- Hanoian P, Liu CT, Hammes-Schiffer S, Benkovic S. Perspectives on electrostatics and conformational motions in enzyme catalysis. Acc Chem Res. 2015;48:482–489. doi: 10.1021/ar500390e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khirich G, Loria JP. Complexity of protein energy landscapes studied by solution NMR relaxation dispersion experiments. J Phys Chem B. 2015;119:3743–3754. doi: 10.1021/acs.jpcb.5b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman JP. Dynamically achieved active site precision in enzyme catalysis. Acc Chem Res. 2015;48:449–456. doi: 10.1021/ar5003347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohen A. Role of dynamics in enzyme catalysis: substantial versus semantic controversies. Acc Chem Res. 2015;48:466–473. doi: 10.1021/ar500322s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Levy RM, Karplus M, Kushick J, Perahia D. Evaluation of the configurational entropy for proteins - Application to molecular-dynamics simulations of an alpha-helix. Macromolecules. 1984;17:1370–1374. [Google Scholar]
- Loria JP, Berlow RB, Watt ED. Characterization of enzyme motions by solution NMR relaxation dispersion. Accounts of chemical research. 2008;41:214–221. doi: 10.1021/ar700132n. [DOI] [PubMed] [Google Scholar]
- Manley G, Loria JP. NMR insights into protein allostery. Archives of biochemistry and biophysics. 2011 doi: 10.1016/j.abb.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan GJ, Basford KE. Mixture models: Inference and applications to clustering. Applied Statistics 1988 [Google Scholar]
- Merlino A, Vitagliano L, Ceruso MA, Di Nola A, Mazzarella L. Global and local motions in ribonuclease A: a molecular dynamics study. Biopolymers. 2002;65:274–283. doi: 10.1002/bip.10225. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pares X, Nogues MV, de Llorens R, Cuchillo CM. Structure and function of ribonuclease A binding subsites. Essays Biochem. 1991;26:89–103. [PubMed] [Google Scholar]
- Raines RT. Ribonuclease A. Chem Rev. 1998;98:1045–1066. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- Ramanathan A, Agarwal PK. Evolutionarily Conserved Linkage between Enzyme Fold, Flexibility, and Catalysis. PLoS Biology. 2011;9:e1001193. doi: 10.1371/journal.pbio.1001193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan A, Savol A, Burger V, Chennubhotla CS, Agarwal PK. Protein conformational populations and functionally relevant substates. Accounts of chemical research. 2014;47:149–156. doi: 10.1021/ar400084s. [DOI] [PubMed] [Google Scholar]
- Ramanathan A, Savol AJ, Agarwal PK, Chennubhotla CS. Event detection and sub-state discovery from biomolecular simulations using higher-order statistics: application to enzyme adenylate kinase. Proteins. 2012;80:2536–2551. doi: 10.1002/prot.24135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan A, Savol AJ, Langmead CJ, Agarwal PK, Chennubhotla CS. Discovering conformational sub-states relevant to protein function. PLoS One. 2011;6:e15827. doi: 10.1371/journal.pone.0015827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson AD, Baldwin RL. Hydrogen exchange in thermally denatured ribonuclease A. Biochemistry. 1991;30:9907–9914. doi: 10.1021/bi00105a014. [DOI] [PubMed] [Google Scholar]
- Sela M, Anfinsen CB. Some spectrophotometric and polarimetric experiments with ribonuclease. Biochim Biophys Acta. 1957;24:229–235. doi: 10.1016/0006-3002(57)90186-5. [DOI] [PubMed] [Google Scholar]
- Seshadri S, Oberg KA, Fink AL. Thermally denatured ribonuclease A retains secondary structure as shown by FTIR. Biochemistry. 1994;33:1351–1355. doi: 10.1021/bi00172a010. [DOI] [PubMed] [Google Scholar]
- Smith BD, Raines RT. Genetic selection for critical residues in ribonucleases. J Mol Biol. 2006;362:459–478. doi: 10.1016/j.jmb.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Sorrentino S. The eight human “canonical” ribonucleases: Molecular diversity, catalytic properties, and special biological actions of the enzyme proteins. Febs Lett. 2010;584:2194–2200. doi: 10.1016/j.febslet.2010.04.018. [DOI] [PubMed] [Google Scholar]
- Toiron C, Gonzalez C, Bruix M, Rico M. Three-dimensional structure of the complexes of ribonuclease A with 2′,5′-CpA and 3′,5′-d(CpA) in aqueous solution, as obtained by NMR and restrained molecular dynamics. Protein science : a publication of the Protein Society. 1996;5:1633–1647. doi: 10.1002/pro.5560050817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrent J, Rubens P, Ribo M, Heremans K, Vilanova M. Pressure versus temperature unfolding of ribonuclease A: an FTIR spectroscopic characterization of 10 variants at the carboxy-terminal site. Protein science : a publication of the Protein Society. 2001;10:725–734. doi: 10.1110/ps.43001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Tharp S, Selzer T, Benkovic SJ, Kohen A. Effects of a distal mutation on active site chemistry. Biochemistry. 2006;45:1383–1392. doi: 10.1021/bi0518242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt ED, Shimada H, Kovrigin EL, Loria JP. The mechanism of rate-limiting motions in enzyme function. Proc Natl Acad Sci U S A. 2007;104:11981–11986. doi: 10.1073/pnas.0702551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodawer A, Svensson LA, Sjolin L, Gilliland GL. Structure of phosphate-free ribonuclease A refined at 1.26 A. Biochemistry. 1988;27:2705–2717. doi: 10.1021/bi00408a010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.